DOI:
10.1039/C7RA03588H
(Paper)
RSC Adv., 2017,
7, 28009-28015
Semisynthesis of esters and oxime esters/sulfonates from furyl-ring-based acetylation derivatives of fraxinellone as insecticidal agents†
Received
30th March 2017
, Accepted 23rd May 2017
First published on 26th May 2017
Abstract
In continuation of our program to discover new natural-product-based crop protection agents, we prepared a series of esters and oxime esters/sulfonates from furyl-ring-based acetylation derivatives as pesticidal agents by structural modification of fraxinellone, a biorenewable degraded limonoid isolated from Meliaceae and Rutaceae plants. The structural assignment was based on the spectroscopic and X-ray analysis data. Their insecticidal activity was evaluated against a crop-threatening agricultural insect pest, Mythimna separata Walker. Among all derivatives, compounds 9a and 10i exhibited the most promising pesticidal activity. Their structure–activity relationships were also discussed.
Introduction
Oriental armyworm (Mythimna separata Walker) is a terrible lepidopteran insect pest, and its infestations are hard to control.1,2 In 2012, approximately 4 million hectares of crops were completely lost in China due to the intermittent outbreaks of M. separata.3 Obviously, lots of chemical pesticides had to be extensively applied to deal with insect pest outbreaks, however, it ultimately led to insect pests resistance, and human health and environmental problems.4–7 Consequently, research and development of new potential alternatives to effectively and selectively control insect pests is extremely desirable.
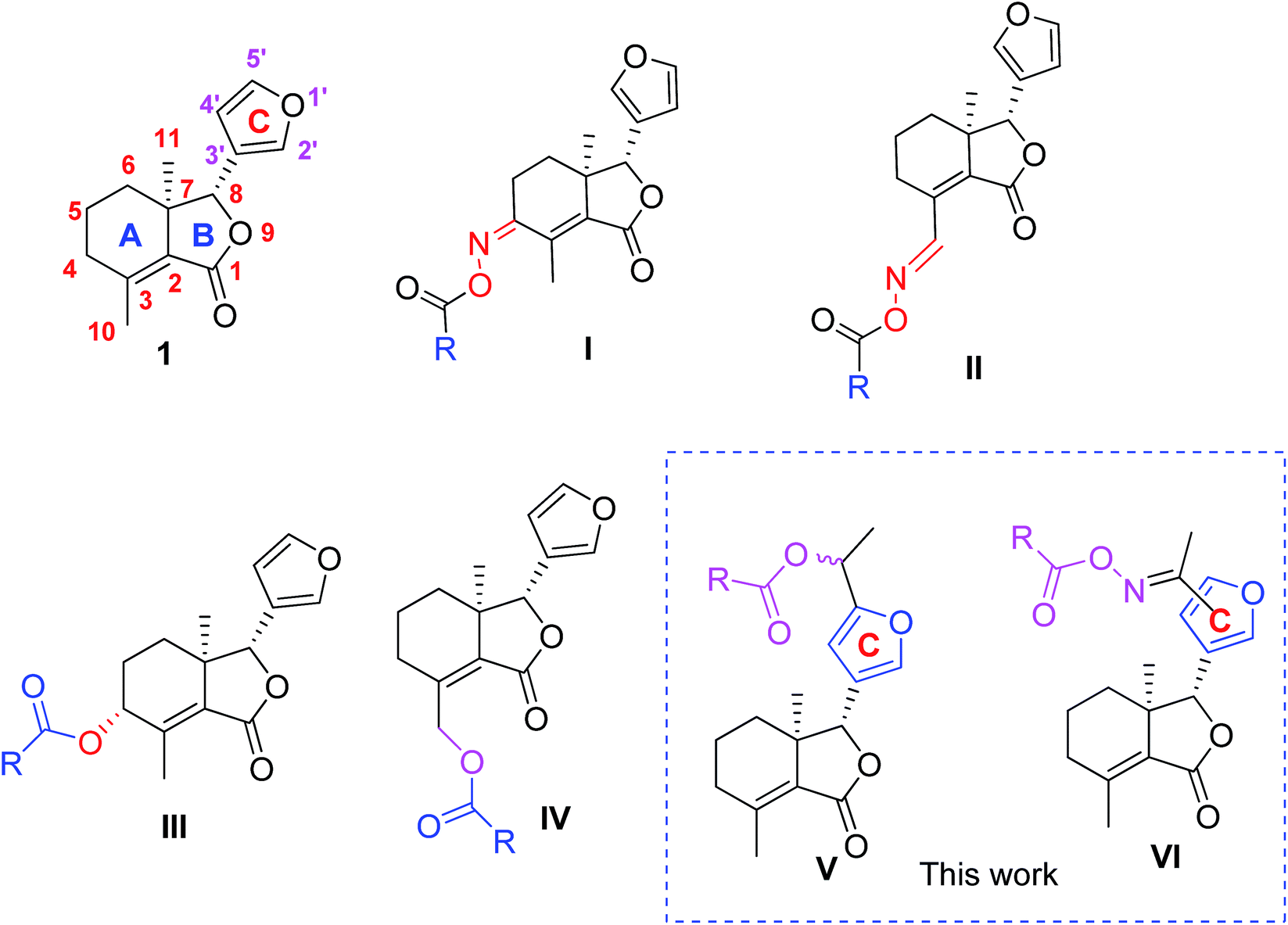
Fraxinellone (1, Fig. 1), a biorenewable degraded limonoid, is isolated from many Meliaceae and Rutaceae plants, and exhibits a variety of interesting properties, including the antiinflammatory bowel disease,8 neuroprotective,9 and insecticidal activities.10–13 In our previous reports, compound 1 was modified at its C-4/C-10 positions, and some fraxinellone-based oxime esters14 (I and II, Fig. 1) and esters15 (III and IV, Fig. 1) displayed higher insecticidal activity than toosendanin against pre-third-instar larvae of Mythimna separata. In addition, to the best of our knowledge, little attention has been paid to the structural modifications on the C-ring (furyl-ring) of compound 1 as pesticidal agents. Based upon the above results, and in continuation of our program to discover biorenewable fraxinellone-based pesticides, herein we wanted to prepare a series of new esters/oxime esters from furyl-ring-based acetylation derivatives of fraxinellone (V and VI, Fig. 1) as insecticidal agents against M. separata in vivo.
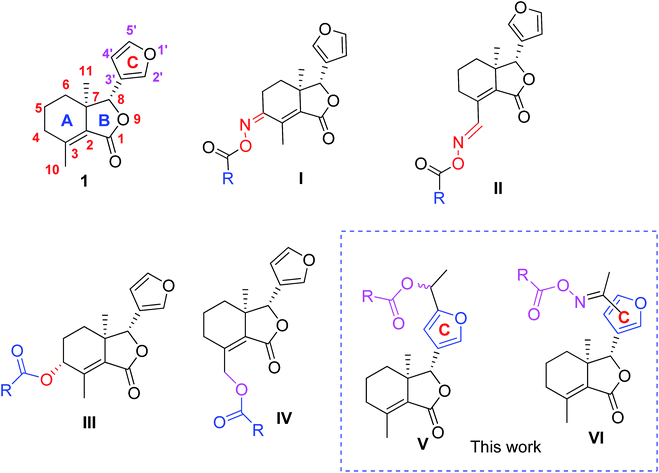 |
| | Fig. 1 Chemical structures of fraxinellone (1), and its derivatives (I–VI). | |
Materials and methods
General
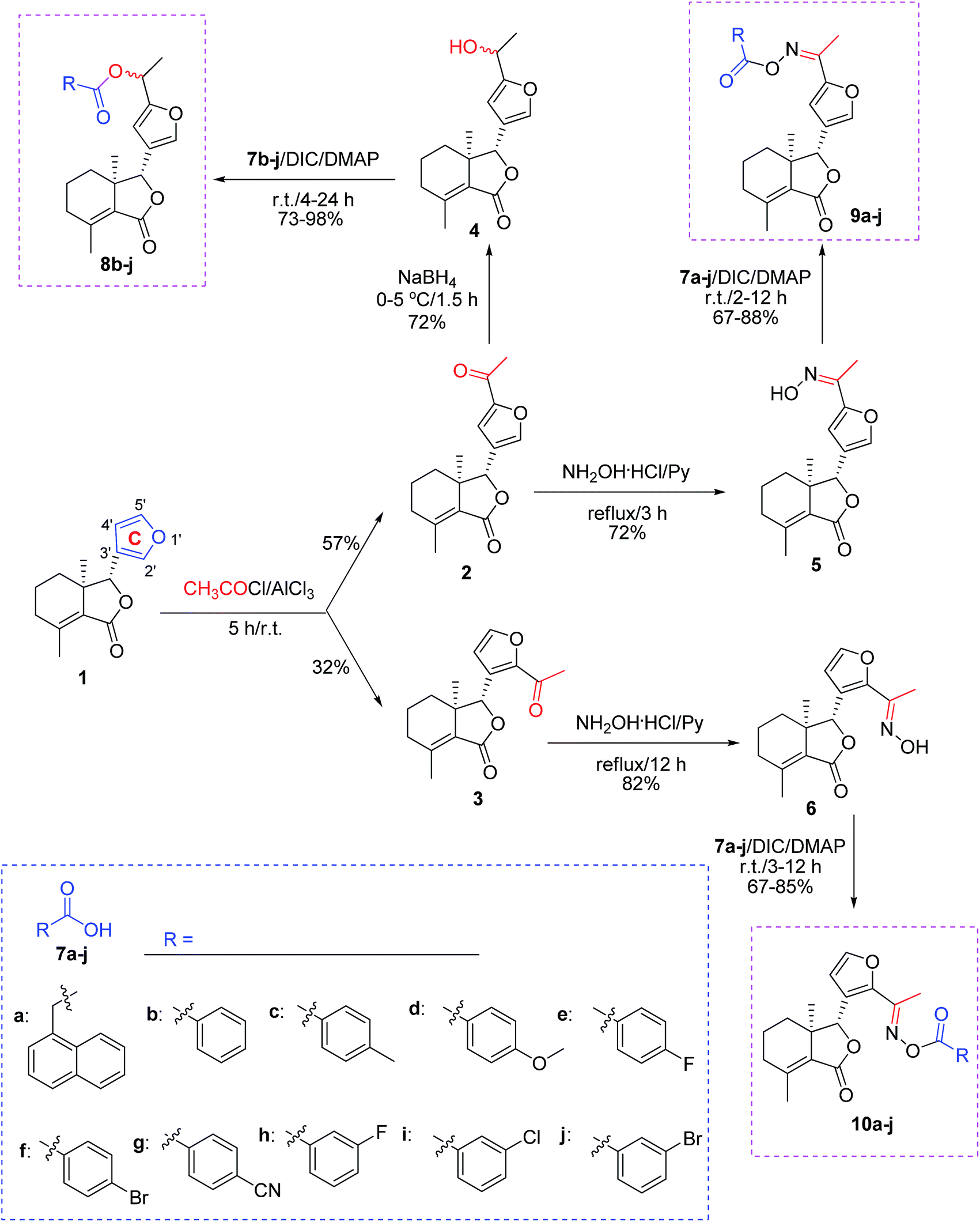
All chemical reagents were purchased and utilized without further purification. Melting point (mp) was determined using the XT-4 digital mp apparatus. Optical rotation was measured using an Autopol III automatic polarimeter. Infrared (IR) spectra were measured by a TENSOR 27 spectrometer. Proton nuclear magnetic resonance spectra (1H NMR) were measured with the Avance 400 or 500 MHz equipment. 5′-Acetylfraxinellone (2) and 2′-acetylfraxinellone (3) were prepared according to our previous paper.16
Synthesis of compound 4
To a stirred solution of 2 (0.5 mmol) in methanol at 0–5 °C, NaBH4 (1 mmol) was slowly added. After addition, the reaction mixture was stirred at 0–5 °C for 1.5 h. Then the solvent was removed, and the residue was dissolved in CH2Cl2 (50 mL). The mixture was washed by brine (20 mL). The organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Finally, the crude product was purified by preparative thin-layer chromatography (PTLC) to give 4 as a white solid in 72% yield.
Data for 4 (isomer α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :β = 1:1). Mp 96–98 °C; [α]20D = −27 (c 3.0 mg mL−1, acetone); IR cm−1: 3402, 2933, 2896, 1748, 1673, 1671, 1205, 978; 1H NMR (400 MHz, CDCl3) δ: 7.40 (s, 1H, H-2′), 6.20 (s, 1H, H-4′), 4.86–4.89 (m, 1H, –OH(CH)CH3), 4.83 (s, 1H, H-8), 2.12–2.31 (m, 2H, H-4), 2.11 (s, 3H, H-10), 2.03 (s, 1H, –OH), 1.71–1.87 (m, 3H, H-5, 6), 1.55 (s, 1.5H, –OH(CH)CH3), 1.53 (s, 1.5H, –OH(CH)CH3), 1.40–1.46 (m, 1H, H-6), 0.87 (s, 3H, H-11). HRMS (ESI): calcd for C16H21O4 ([M + H]+), 277.1434; found, 277.1433.
:β = 1:1). Mp 96–98 °C; [α]20D = −27 (c 3.0 mg mL−1, acetone); IR cm−1: 3402, 2933, 2896, 1748, 1673, 1671, 1205, 978; 1H NMR (400 MHz, CDCl3) δ: 7.40 (s, 1H, H-2′), 6.20 (s, 1H, H-4′), 4.86–4.89 (m, 1H, –OH(CH)CH3), 4.83 (s, 1H, H-8), 2.12–2.31 (m, 2H, H-4), 2.11 (s, 3H, H-10), 2.03 (s, 1H, –OH), 1.71–1.87 (m, 3H, H-5, 6), 1.55 (s, 1.5H, –OH(CH)CH3), 1.53 (s, 1.5H, –OH(CH)CH3), 1.40–1.46 (m, 1H, H-6), 0.87 (s, 3H, H-11). HRMS (ESI): calcd for C16H21O4 ([M + H]+), 277.1434; found, 277.1433.
General procedure for synthesis of compounds 5 and 6
A mixture of 2 or 3 (0.5 mmol), hydroxylamine hydrochloride (0.75 mmol), and pyridine (2 mmol) in absolute ethanol (10 mL) was refluxed. When the reaction was complete according to TLC analysis, the solvent was removed under reduced pressure. Finally, the crude product was purified by PTLC to give 5 (72% yield) or 6 (82% yield) as a white solid.
Data for 5. Mp 176–178 °C; [α]20D = −25 (c 3.8 mg mL−1, acetone); IR cm−1: 3265, 2961, 2922, 2855, 1746, 1264, 1049, 901; 1H NMR (400 MHz, CDCl3) δ: 7.49 (s, 1H, H-2′), 6.59 (s, 1H, H-4′), 4.87 (s, 1H, H-8), 2.17–2.32 (m, 6H, –CH3, –OH and H-4), 2.13 (s, 3H, H-10), 1.72–1.84 (m, 3H, H-5, 6), 1.42–1.48 (m, 1H, H-6), 0.87 (s, 3H, H-11). HRMS (ESI): calcd for C16H20O4N ([M + H]+), 290.1387; found, 290.1387.
Data for 6. Mp 186–188 °C; [α]20D = 88 (c 4.1 mg mL−1, acetone); IR cm−1: 3300, 2957, 2925, 2852, 1727, 1670, 1272, 1218, 984; 1H NMR (500 MHz, CDCl3) δ: 7.43 (s, 1H, H-5′), 7.31 (s, 1H, –OH), 6.61 (d, J = 1.5 Hz, 1H, H-4′), 5.59 (s, 1H, H-8), 2.21–2.26 (m, 4H, –CH3 and H-4), 2.08–2.16 (m, 4H, H-4, 10), 1.75–1.77 (m, 1H, H-5), 1.58–1.68 (m, 3H, H-5, 6), 0.92 (s, 3H, H-11). HRMS (ESI): calcd for C16H20O4N ([M + H]+), 290.1387; found, 290.1385.
General procedure for synthesis of compounds 8b–j, 9a–j and 10a–j
A mixture of the corresponding acid (7, 0.28 mmol), N,N′-dicyclohexylcarbodiimide (DCC, 0.28 mmol), 4-dimethylaminopyridine (DMAP, 0.04 mmol), and 4, 5 or 6 (0.2 mmol) in dry CH2Cl2 (10 mL) was stirred at room temperature. When the reaction was complete according to TLC analysis, the mixture was diluted by CH2Cl2 (30 mL), washed by HCl (0.1 mol L−1, 15 mL), 5% NaHCO3 (15 mL) and brine (15 mL), dried over anhydrous Na2SO4, concentrated in vacuo, and purified by PTLC to give products 8b–j, 9a–j and 10a–j. The example data of 8b–e, 9a–e and 10a–e were described as follows, whereas the data of 8f–j, 9f–j and 10f–j were shown in the ESI.†
Data for 8b. Yield: 70%, colorless liquid; [α]20D = −17 (c 3.1 mg mL−1, acetone); IR cm−1: 2951, 2924, 2853, 1754, 1717, 1451, 1265, 1205, 1048; 1H NMR (400 MHz, CDCl3) δ: 8.04 (d, J = 8.0 Hz, 2H, –Ph), 7.54 (t, d, J = 7.6 Hz, 1H, –Ph), 7.41–7.45 (m, 3H, –Ph, H-2′), 6.35 (s, 1H, H-4′), 6.16–6.21 (m, 1H, –O(CH)CH3), 4.84 (s, 1H, H-8), 2.24–2.31 (m, 2H, H-4), 2.12 (s, 3H, H-10), 1.79–1.83 (m, 3H, H-5, 6), 1.71 (d, J = 2.4 Hz, 1.5H, –OH(CH)CH3), 1.69 (d, J = 2.4 Hz, 1.5H, –OH(CH)CH3), 1.40–1.47 (m, 1H, H-6), 0.86–0.87 (m, 3H, H-11). HRMS (ESI): calcd for C23H28O5N ([M + NH4]+), 398.1962; found, 398.1959.
Data for 8c. Yield: 81%, colorless liquid; [α]20D = −18 (c 3.2 mg mL−1, acetone); IR cm−1: 2952, 2923, 2853, 1756, 1716, 1267, 1047; 1H NMR (400 MHz, CDCl3) δ: 7.92 (d, J = 8.0 Hz, 2H, –Ph), 7.43 (s, 1H, H-2′), 7.22 (d, J = 8.0 Hz, 2H, –Ph), 6.33 (d, J = 2.4 Hz, 1H, H-4′), 6.14–6.19 (m, 1H, –O(CH)CH3), 4.84 (s, 1H, H-8), 2.40 (s, 1H, –PhCH3), 2.17–2.30 (m, 2H, H-4), 2.12 (s, 3H, H-10), 1.73–1.83 (m, 3H, H-5, 6), 1.70 (d, J = 2.4 Hz, 1.5H, –OH(CH)CH3), 1.68 (d, J = 2.4 Hz, 1.5H, –OH(CH)CH3), 1.39–1.47 (m, 1H, H-6), 0.86–0.87 (m, 3H, H-11). HRMS (ESI): calcd for C24H30O5N ([M + NH4]+), 412.2118; found, 412.2115.
Data for 8d. Yield: 98%, colorless liquid; [α]20D = −18 (c 3.2 mg mL−1, acetone); IR cm−1: 2952, 2923, 2853, 1743, 1713, 1606, 1258, 1165; 1H NMR (400 MHz, CDCl3) δ: 7.99 (d, J = 9.2 Hz, 2H, –Ph), 7.43 (s, 1H, H-2′), 6.89 (d, J = 8.8 Hz, 2H, –Ph), 6.33 (s, 1H, H-4′), 6.12–6.17 (m, 1H, –O(CH)CH3), 4.84 (s, 1H, H-8), 3.85 (s, 3H, –OCH3), 2.15–2.31 (m, 2H, H-4), 2.12 (s, 3H, H-10), 1.79–1.83 (m, 3H, H-5, 6), 1.69 (d, J = 2.4 Hz, 1.5H, –OH(CH)CH3), 1.67 (d, J = 2.4 Hz, 1.5H, –OH(CH)CH3), 1.39–1.47 (m, 1H, H-6), 0.84–0.86 (m, 3H, H-11). HRMS (ESI): calcd for C24H27O6 ([M + H]+), 411.1802; found, 411.1802.
Data for 8e. Yield: 73%, white solid, mp 158–160 °C; [α]20D = −23 (c 4.1 mg mL−1, acetone); IR cm−1: 2953, 2923, 2852, 1755, 1719, 1266, 1048, 855; 1H NMR (400 MHz, CDCl3) δ: 8.04–8.08 (m, 2H, –Ph), 7.44 (s, 1H, H-2′), 7.08–7.12 (m, 2H, –Ph), 6.35 (s, 1H, H-4′), 7.08–7.12 (m, 2H, –Ph), 6.13–6.18 (m, 1H, –O(CH)CH3), 4.84 (s, 1H, H-8), 2.16–2.31 (m, 2H, H-4), 2.13 (s, 3H, H-10), 1.79–1.83 (m, 3H, H-5, 6), 1.70 (d, J = 2.4 Hz, 1.5H, –OH(CH)CH3), 1.69 (d, J = 2.4 Hz, 1.5H, –OH(CH)CH3), 1.40–1.47 (m, 1H, H-6), 0.86–0.87 (m, 3H, H-11). HRMS (ESI): calcd for C23H27O5NF ([M + NH4]+), 416.1868; found, 416.1862.
Data for 9a. Yield: 78%, white solid, mp 89–90 °C; [α]20D = −23 (c 4.0 mg mL−1, acetone); IR cm−1: 2940, 2873, 1755, 1675, 1205, 1046, 909; 1H NMR (500 MHz, CDCl3) δ: 8.04 (d, J = 8.5 Hz, 1H, Ar–H), 8.86 (d, J = 7.5 Hz, 1H, Ar–H), 7.80 (d, J = 8.0 Hz, 1H, Ar–H), 7.43–7.56 (m, 5H, Ar–H and H-2′), 6.81 (s, 1H, H-4′), 4.82 (s, 1H, H-8), 4.27 (s, 2H, –CH2C10H7), 2.14–2.29 (m, 2H, H-4), 2.12 (s, 3H, –CH3), 2.02 (s, 3H, H-10), 1.77–1.85 (m, 2H, H-5, 6), 1.69–1.73 (m, 1H, H-5), 1.39–1.44 (m, 1H, H-6), 0.80 (s, 3H, H-11). HRMS (ESI): calcd for C28H28O5N ([M + H]+), 458.1962; found, 458.1962.
Data for 9b. Yield: 72%, white solid, mp 200–202 °C; [α]20D = −10 (c 3.4 mg mL−1, acetone); IR cm−1: 2926, 2853, 1743, 1676, 1250, 1048, 907; 1H NMR (500 MHz, CDCl3) δ: 8.10 (dd, J = 8.0, 1.5 Hz, 2H, Ar–H), 7.60–7.63 (s, 2H, Ar–H and H-2′), 7.48 (t, J = 8.0 Hz, 2H, Ar–H), 6.97 (s, 1H, H-4′), 4.89 (s, 1H, H-8), 2.46 (s, 3H, –CH3), 2.27–2.32 (m, 1H, H-4), 2.18–2.21 (m, 1H, H-4), 2.14 (s, 3H, H-10), 1.84–1.87 (m, 2H, H-5, 6), 1.73–1.77 (m, 1H, H-5), 1.44–1.49 (m, 1H, H-6), 0.87 (s, 3H, H-11). HRMS (ESI): calcd for C23H24O5N ([M + H]+), 394.1649; found, 394.1648.
Data for 9c. Yield: 64%, white solid, mp 160–162 °C; [α]20D = −28 (c 3.4 mg mL−1, acetone); IR cm−1: 2940, 2872, 1745, 1675, 1260, 1049, 911; 1H NMR (500 MHz, CDCl3) δ: 7.99 (d, J = 8.5 Hz, 2H, Ar–H), 7.61 (s, 1H, H-2′), 7.28 (d, J = 8.5 Hz, 2H, Ar–H), 6.96 (s, 1H, H-4′), 4.88 (s, 1H, H-8), 2.45 (s, 3H, –PhCH3), 2.44 (s, 3H, –CH3), 2.26–2.32 (m, 1H, H-4), 2.18–2.21 (m, 1H, H-4), 2.14 (s, 3H, H-10), 1.85–1.87 (m, 2H, H-5, 6), 1.73–1.79 (m, 1H, H-5), 1.45–1.49 (m, 1H, H-6), 0.87 (s, 3H, H-11). HRMS (ESI): calcd for C24H26O5N ([M + H]+), 408.1805; found, 408.1804.
Data for 9d. Yield: 73%, white solid, mp 150–152 °C; [α]20D = −21 (c 3.2 mg mL−1, acetone); IR cm−1: 2954, 2923, 2853, 1739, 1607, 1265, 1047, 957; 1H NMR (500 MHz, CDCl3) δ: 8.06 (d, J = 9.0 Hz, 2H, Ar–H), 7.62 (s, 1H, H-2′), 6.96 (d, J = 9.0 Hz, 2H, Ar–H), 6.95 (s, 1H, H-4′), 4.88 (s, 1H, H-8), 3.88 (s, 3H, –OCH3), 2.44 (s, 3H, –CH3), 2.17–2.32 (m, 2H, H-4), 2.14 (s, 3H, H-10), 1.84–1.88 (m, 2H, H-5, 6), 1.74–1.77 (m, 1H, H-5), 1.44–1.49 (m, 1H, H-6), 0.87 (s, 3H, H-11). HRMS (ESI): calcd for C24H26O6N ([M + H]+), 424.1755; found, 424.1754.
Data for 9e. Yield: 61%, white solid, mp 184–186 °C; [α]20D = −28 (c 3.3 mg mL−1, acetone); IR cm−1: 2934, 2871, 1748, 1673, 1601, 1257, 1047, 910; 1H NMR (500 MHz, CDCl3) δ: 8.12–8.15 (m, 2H, Ar–H), 7.61 (s, 1H, H-2′), 7.15 (t, J = 8.5 Hz, 2H, Ar–H), 6.97 (s, 1H, H-4′), 4.89 (s, 1H, H-8), 2.45 (s, 3H, –CH3), 2.19–2.32 (m, 2H, H-4), 2.14 (s, 3H, H-10), 1.85–1.87 (m, 2H, H-5, 6), 1.73–1.77 (m, 1H, H-5), 1.45–1.49 (m, 1H, H-6), 0.87 (s, 3H, H-11). HRMS (ESI): calcd for C23H23O5NF ([M + H]+), 412.1555; found, 412.1556.
Data for 10a. Yield: 75%, white solid, mp 70–71 °C; [α]20D = 82 (c 3.3 mg mL−1, acetone); IR cm−1: 2952, 2924, 2853, 1752, 1207, 908; 1H NMR (500 MHz, CDCl3) δ: 8.01 (d, J = 8.5 Hz, 1H, Ar–H), 7.87 (d, J = 8.0 Hz, 1H, Ar–H), 7.81–7.82 (m, 1H, Ar–H), 7.43–7.56 (m, 5H, Ar–H and H-5′), 6.66 (d, J = 1.5 Hz, 1H, H-4′), 5.69 (s, 1H, H-8), 4.22 (s, 2H, –CH2C10H7), 2.14–2.19 (m, 2H, H-4), 2.11 (s, 3H, –CH3), 2.09 (s, 3H, H-10), 1.87–1.93 (m, 1H, H-5), 1.68–1.72 (m, 1H, H-6), 1.50–1.60 (m, 2H, H-5, 6), 0.88 (s, 3H, H-11). HRMS (ESI): calcd for C28H28O5N ([M + H]+), 458.1962; found, 458.1956.
Data for 10b. Yield: 80%, white solid, mp 146–148 °C; [α]20D = 110 (c 3.6 mg mL−1, acetone); IR cm−1: 2961, 2922, 2910, 1754, 1449, 1247, 978; 1H NMR (500 MHz, CDCl3) δ: 8.08 (d, J = 7.0 Hz, 2H, Ar–H), 7.60 (t, J = 7.5 Hz, 1H, Ar–H), 7.55 (d, J = 1.5 Hz, 1H, H-5′), 7.48 (t, J = 8.0 Hz, 2H, Ar–H), 6.73 (d, J = 2.0 Hz, 1H, H-4′), 5.85 (s, 1H, H-8), 2.49 (s, 3H, –CH3), 2.21–2.23 (m, 2H, H-4), 2.14 (s, 3H, H-10), 2.05–2.12 (m, 1H, H-5), 1.80–1.84 (m, 1H, H-6), 1.63–1.66 (m, 2H, H-5, 6), 0.96 (s, 3H, H-11). HRMS (ESI): calcd for C23H24O5N ([M + H]+), 394.1649; found, 394.1645.
Data for 10c. Yield: 75%, white solid, mp 174–176 °C; [α]20D = 111 (c 4.3 mg mL−1, acetone); IR cm−1: 2954, 2923, 2851, 1745, 1459, 1376, 1049, 980; 1H NMR (500 MHz, CDCl3) δ: 7.96 (d, J = 8.0 Hz, 2H, Ar–H), 7.54 (d, J = 1.5 Hz, 1H, H-5′), 7.28 (d, J = 8.0 Hz, 2H, Ar–H), 6.73 (d, J = 1.5 Hz, 1H, H-4′), 5.84 (s, 1H, H-8), 2.48 (s, 3H, –CH3), 2.44 (s, 3H, –PhCH3), 2.21–2.24 (m, 2H, H-4), 2.14 (s, 3H, H-10), 2.05–2.09 (m, 1H, H-5), 1.80–1.83 (m, 1H, H-6), 1.62–1.66 (m, 2H, H-5, 6), 0.95 (s, 3H, H-11). HRMS (ESI): calcd for C24H26O5N ([M + H]+), 408.1805; found, 408.1802.
Data for 10d. Yield: 70%, white solid, mp 120–122 °C; [α]20D = 110 (c 3.4 mg mL−1, acetone); IR cm−1: 2954, 2924, 2853, 1747, 1252, 1049; 1H NMR (500 MHz, CDCl3) δ: 8.03 (d, J = 9.0 Hz, 2H, Ar–H), 7.53 (d, J = 2.0 Hz, 1H, H-5′), 6.96 (d, J = 8.5 Hz, 2H, Ar–H), 6.72 (d, J = 1.5 Hz, 1H, H-4′), 5.84 (s, 1H, H-8), 3.88 (s, 3H, –OCH3), 2.48 (s, 3H, –CH3), 2.21–2.24 (m, 2H, H-4), 2.14 (s, 3H, H-10), 2.05–2.12 (m, 1H, H-5), 1.81–1.84 (m, 1H, H-6), 1.62–1.66 (m, 2H, H-5, 6), 0.95 (s, 3H, H-11). HRMS (ESI): calcd for C24H26O6N ([M + H]+), 424.1755; found, 424.1750.
Data for 10e. Yield: 74%, white solid, mp 142–144 °C; [α]20D = 126 (c 3.0 mg mL−1, acetone); IR cm−1: 2947, 2929, 2871, 1747, 1602, 1250, 1051, 913; 1H NMR (500 MHz, CDCl3) δ: 8.09–8.11 (m, 2H, Ar–H), 7.55 (d, J = 2.0 Hz, 1H, H-5′), 7.15–7.19 (m, 2H, Ar–H), 6.73 (d, J = 1.5 Hz, 1H, H-4′), 5.83 (s, 1H, H-8), 2.49 (s, 3H, –CH3), 2.21–2.24 (m, 2H, H-4), 2.14 (s, 3H, H-10), 2.04–2.10 (m, 1H, H-5), 1.80–1.83 (m, 1H, H-6), 1.62–1.68 (m, 2H, H-5, 6), 0.96 (s, 3H, H-11). HRMS (ESI): calcd for C23H23O5NF ([M + H]+), 412.1555; found, 412.1554.
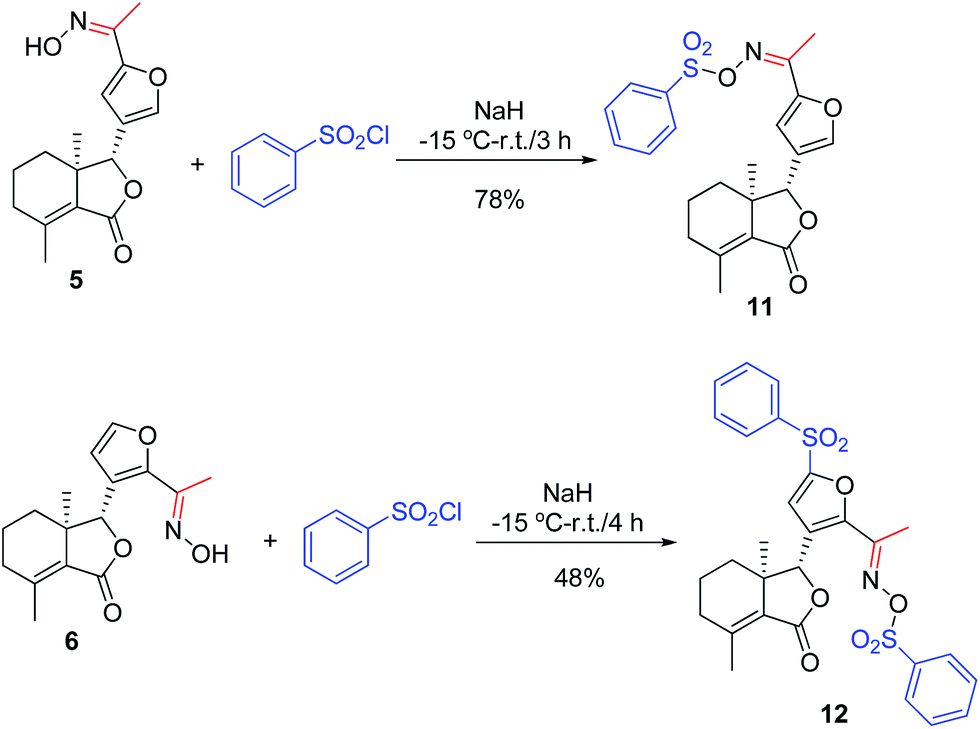
General procedure for synthesis of compounds 11 and 12
To a stirred solution of NaH (1.26 mmol) in dry tetrahydrofuran (THF, 10 mL) at −15 °C was slowly added compound 5 or 6 (0.18 mmol). After addition, the reaction mixture was stirred at −15 °C for 10 min. Then, benezenesulfochloride (0.72 mmol) were added. When the reaction was complete according to TLC analysis, saturated aqueous NaHCO3 (15 mL) was added to the mixture, which was extracted with CH2Cl2 (3 × 40 mL). Finally, the combined organic phase was dried over anhydrous Na2SO4, filtered, concentrated under reduced pressure, and purified by PTLC to give 11 (78% yield) or 12 (48% yield) as a white solid.
Data for 11. Mp 144–146 °C; [α]20D = −14 (c 3.3 mg mL−1, acetone); IR cm−1: 2952, 2924, 2851, 1752, 1450, 1185, 986; 1H NMR (400 MHz, CDCl3) δ: 8.01 (d, J = 8.8 Hz, 2H, Ar–H), 7.65 (d, J = 7.6, Hz, 1H, Ar–H), 7.54–7.60 (m, 3H, Ar–H and H-2′), 7.34 (s, 1H, H-4′), 4.89 (s, 1H, H-8), 2.19–2.34 (m, 5H, –CH3 and H-4), 2.15 (s, 3H, H-10), 1.73–1.88 (m, 3H, H-5, 6), 1.49–1.56 (m, 1H, H-6), 0.84 (s, 3H, H-11). HRMS (ESI): calcd for C22H24O6NS ([M + H]+), 430.1319; found, 430.1316.
Data for 12. Mp 100–101 °C; [α]20D = −5 (c 4.2 mg mL−1, acetone); IR cm−1: 2955, 2921, 2869, 1750, 1378, 1191, 846; 1H NMR (500 MHz, CDCl3) δ: 8.01 (d, J = 7.5 Hz, 2H, Ar–H), 7.90 (d, J = 7.5 Hz, 2H, Ar–H), 7.74 (t, J = 7.5 Hz, 1H, Ar–H), 7.66 (t, J = 7.5 Hz, 1H, Ar–H), 7.59 (t, J = 8.0 Hz, 2H, Ar–H), 7.54 (t, J = 8.0 Hz, 2H, Ar–H), 7.12 (s, 1H, H-4′), 5.27 (s, 1H, H-8), 2.21–2.26 (m, 2H, H-4), 2.12 (s, 3H, –CH3), 2.07 (s, 3H, H-10), 1.78–1.80 (m, 2H, H-5, 6), 1.58–1.61 (m, 1H, H-5), 1.47–1.52 (m, 1H, H-6), 0.57 (s, 3H, H-11). HRMS (ESI): calcd for C28H28O8NS2 ([M + H]+), 570.1251; found, 570.1240.
X-ray crystallography
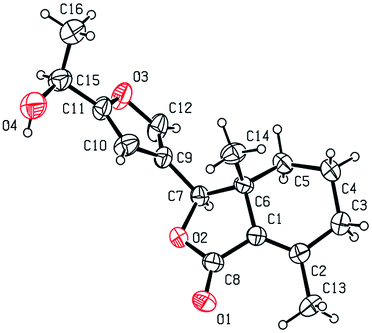
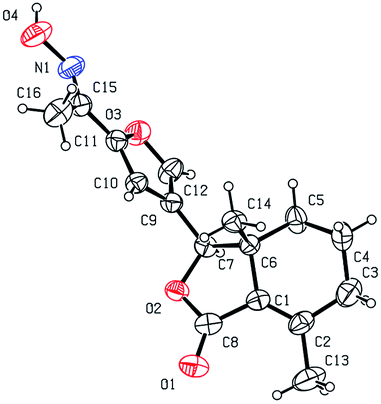
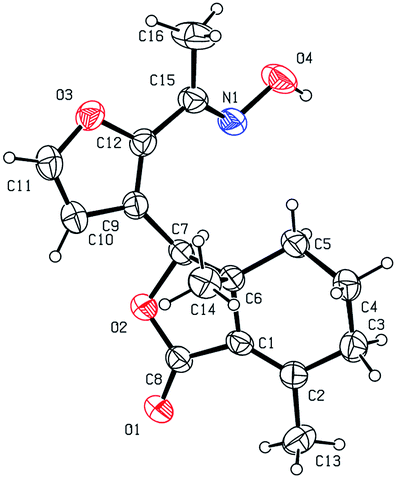
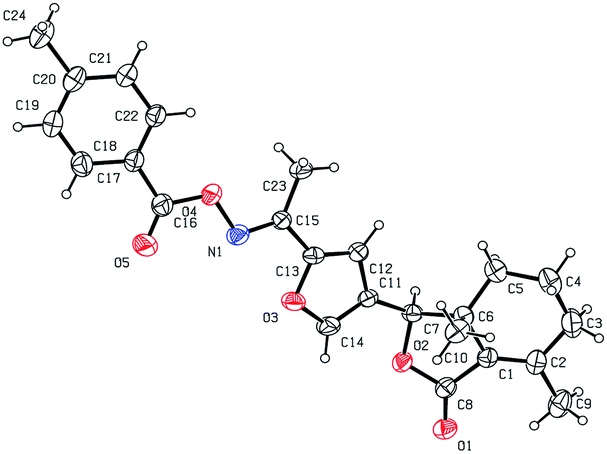
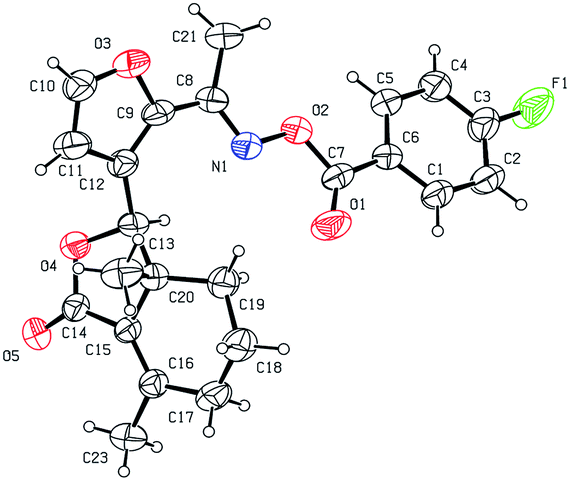
Five three-dimensional structures of compounds 4 (α-OH isomer), 5, 6, 9c and 10e were confirmed by X-ray crystallography. Crystallographic data (excluding structure factors) of 4 (α-OH isomer), 5, 6, 9c and 10e were deposited at the Cambridge Crystallographic Data Centre (CCDC†) with deposition numbers of 1524889, 1524890, 1524891, 1524894, and 1524893, respectively.
Biological assay
Growth inhibitory activity of 1, 4–6, 8b–j, 9a–j, 10a–j, 11 and 12 against Mythimna separata. Thirty early 3rd-instar larvae of M. separata were chosen as the tested insects for each compound. Solutions of 1, 4–6, 8b–j, 9a–j, 10a–j, 11, 12 and toosendanin (a positive control) were prepared in acetone at 1 mg mL−1. After dipped into the corresponding solution for 3 s, wheat leaf discs (1 × 1 cm) were taken out and dried. Wheat leaf discs were treated by acetone alone as the blank control group (CK). Several above discs were added to each culture dish (ten insects per dish). Once the discs were consumed, additional ones were added. After 48 h, the rest of compound-soaked discs was cleaned out, and the untreated ones were added till the end of pupae (temperature: 25 ± 2 °C; RH: 65–80%; photoperiod: L/D = 12/12 h). Their corrected mortality rate values were calculated as follows: corrected mortality rate (%) = (T − C) × 100/(100% − C); C is the mortality rate of CK, and T is the mortality rate of the treated M. separata.15,17
Results and discussion
Synthesis
As shown in Scheme 1, first, 5′-acetylfraxinellone (2) and 2′-acetylfraxinellone (3) were prepared by reaction of fraxinellone (1) with acetyl chloride in the presence of AlCl3.16 Then, reduction of 5′-acetylfraxinellone (2) with NaBH4 gave compound 4 (α/β = 1/1, which was determined by 1H NMR). Hydroxylamine hydrochloride reacted with 2 or 3 to easily afford the corresponding oximes 5 and 6, respectively. Finally, in the presence of N,N′-dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine (DMAP), the corresponding acid 7 reacted with 4, 5 or 6 to produce target products 8b–j, 9a–j and 10a–j, respectively. On the other hand, as described in Scheme 2, compounds 5 and 6 reacting with benezenesulfochloride was also investigated. When 5 reacted with benezenesulfochloride (4 equiv.), oxime sulfonate 11 was afforded; when 6 reacted with benezenesulfochloride (4 equiv.), interestingly, oxime sulfonate 12 containing 5′-phenylsulfonyl was produced. Their structures were well determined by melting points, optical rotation, IR, and 1H NMR. More importantly, five compounds 4 (α-OH isomer), 5, 6, 9c and 10e were further testified by X-ray crystallography (Fig. 2–6). As shown in Fig. 2, the hydroxyethyl was at the 5′-position on the furyl ring, and the OH group was in α position; as shown in Fig. 3 and 4, the corresponding oxime groups of 5 and 6 were at the 5′ and 2′-position on the furyl ring, respectively; as described in Fig. 5 and 6, the 4-methylphenylcarbonyloxy of 9c and the 4-fluorophenylcarbonyloxy of 10e were at the 5′ and 2′-position on the furyl ring, respectively.
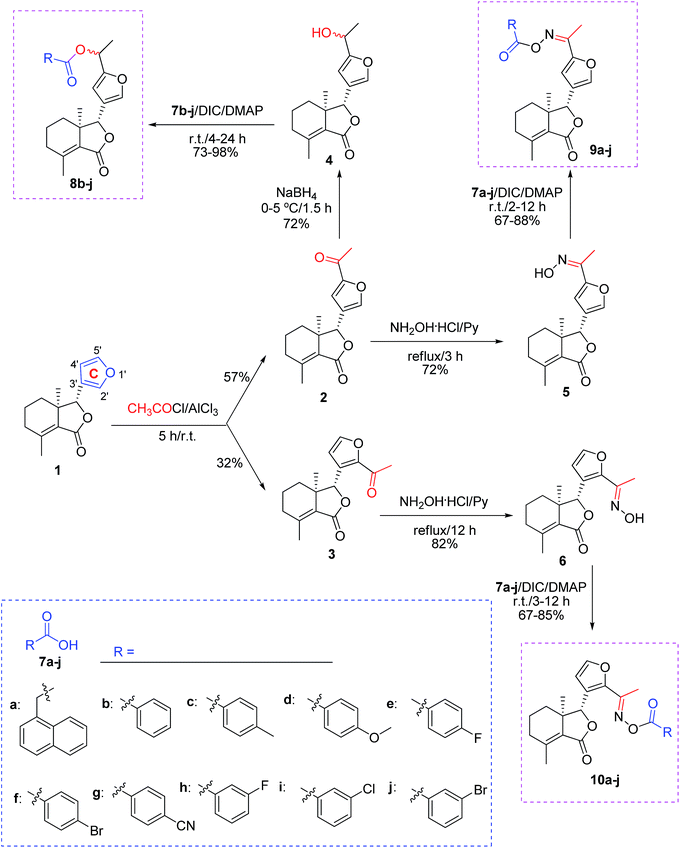 |
| | Scheme 1 Synthesis of esters (8b–j) and oxime esters (9a–j and 10a–j) from furyl-ring-based acetylation derivatives of fraxinellone. | |
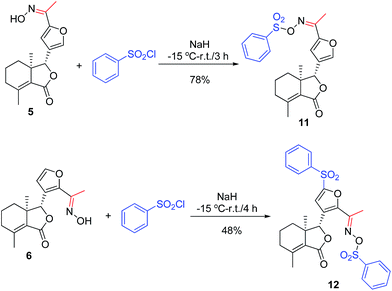 |
| | Scheme 2 Synthesis of oxime sulfonates (11 and 12). | |
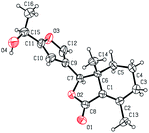 |
| | Fig. 2 X-ray crystal structure of compound 4 (α-OH isomer). | |
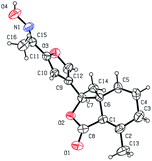 |
| | Fig. 3 X-ray crystal structure of compound 5. | |
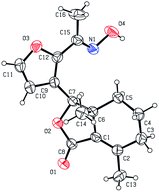 |
| | Fig. 4 X-ray crystal structure of compound 6. | |
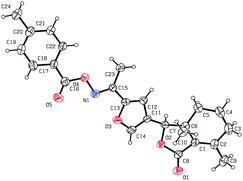 |
| | Fig. 5 X-ray crystal structure of compound 9c. | |
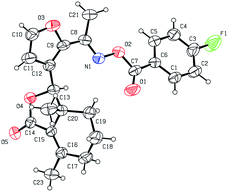 |
| | Fig. 6 X-ray crystal structure of compound 10e. | |
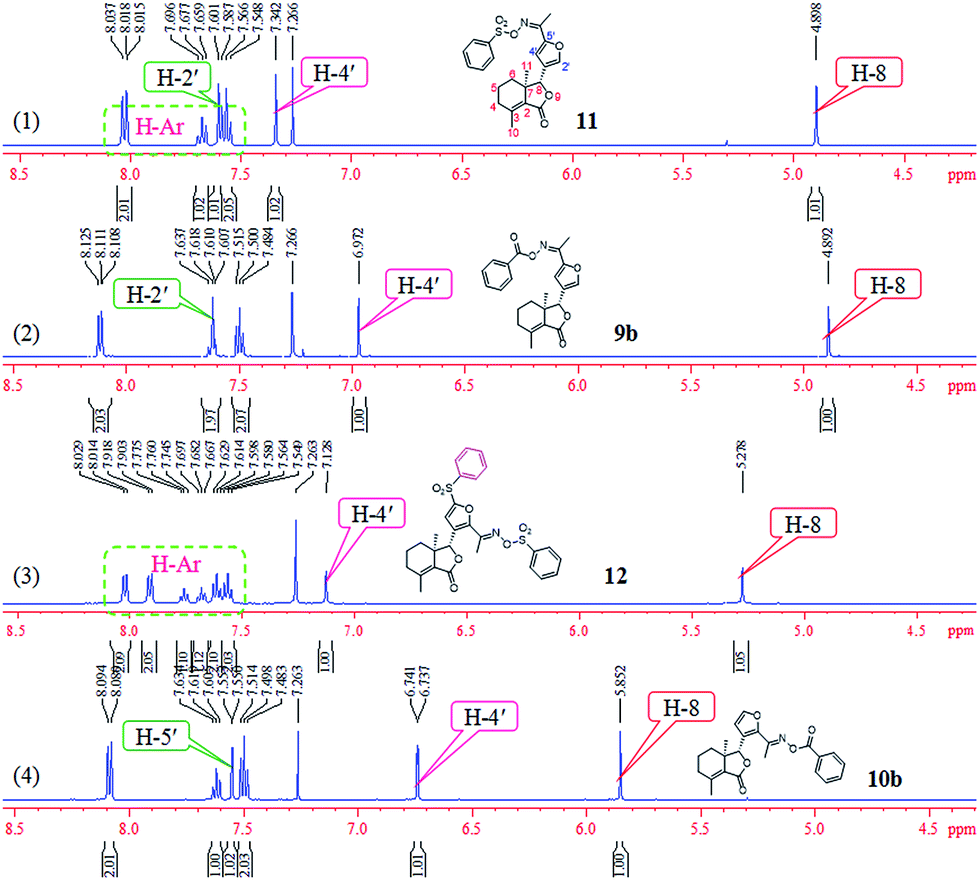
Comparison of partial 1H NMR spectra of compounds 9b, 10b, 11, and 12 was described in Fig. 7. Assignment of the position of a phenylsulfonyl group on the furyl ring of 12 was based on the chemical shift. If the phenylsulfonyl group was at the 4′-position on the furyl ring, the corresponding chemical shift of H-5′ will greater than 7.342 ppm; here the chemical shift was 7.128 ppm, which belonged to H-4′, so the phenylsulfonyl group was at the 5′-position on the furyl ring of 12.
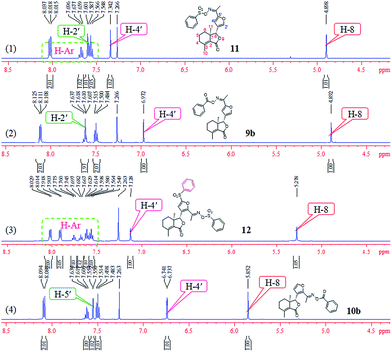 |
| | Fig. 7 Comparison of partial 1H NMR spectra of compounds 9b, 10b, 11, and 12. | |
Insecticidal activity
As described in Table 1, the growth inhibitory activity of compounds 1, 4–6, 8b–j, 9a–j, 10a–j, 11 and 12 against M. separata was tested at 1 mg mL−1. Toosendanin, a commercial insecticide derived from Melia azedarach, was used as the positive control. Leaves treated with acetone alone were used as a blank control group. Compounds 9a, 9c, 9j, 10h and 10i displayed more potent insecticidal activity than toosendanin. For example, the final mortality rates (FMRs) of 9a, 9c, 9j, 10h and 10i were 57.1%, 50.0%, 50.0%, 50.0%, and 57.1%, respectively; whereas the FMRs of 1 and toosendanin were 42.9% and 46.4%, respectively. The symptoms for the treated M. separata during the larval, pupation and adult periods were observed by the same way as our previous reports.14,15,17 For example, the dead larvae with thin and wrinkled bodies, the malformed and dead pupae, and the malformed moths also appeared during the above three stages.
Table 1 Growth inhibitory activity of compounds 1, 4–6, 8b–j, 9a–j, 10a–j, 11 and 12 against M. separata on leaves treated with a concentration of 1 mg mL−1
| Compound |
Corrected mortality rate (mean ± SD, %) |
| 10 days |
20 days |
35 days |
| 1 |
10.0 ± 0 |
13.3 ± 3.3 |
42.9 ± 3.3 |
| 4 |
13.3 ± 3.3 |
26.7 ± 3.3 |
39.3 ± 3.3 |
| 5 |
13.3 ± 3.3 |
13.3 ± 3.3 |
42.9 ± 3.3 |
| 6 |
23.3 ± 3.3 |
33.3 ± 3.3 |
46.4 ± 5.8 |
| 8b |
6.7 ± 3.3 |
10.0 ± 0 |
35.7 ± 5.8 |
| 8c |
20.0 ± 0 |
26.7 ± 3.3 |
42.9 ± 3.3 |
| 8d |
16.7 ± 3.3 |
16.7 ± 3.3 |
21.4 ± 3.3 |
| 8e |
10.0 ± 0 |
13.3 ± 3.3 |
21.4 ± 3.3 |
| 8f |
10.0 ± 5.8 |
6.7 ± 6.7 |
35.7 ± 0 |
| 8g |
26.7 ± 3.3 |
33.3 ± 6.7 |
39.3 ± 3.3 |
| 8h |
16.7 ± 3.3 |
16.7 ± 3.3 |
42.9 ± 3.3 |
| 8i |
26.7 ± 3.3 |
33.3 ± 3.3 |
42.9 ± 3.3 |
| 8j |
13.3 ± 3.3 |
13.3 ± 3.3 |
32.1 ± 3.3 |
| 9a |
20.0 ± 5.8 |
23.3 ± 6.7 |
57.1 ± 0 |
| 9b |
36.7 ± 3.3 |
36.7 ± 3.3 |
46.4 ± 5.8 |
| 9c |
16.7 ± 3.3 |
20.0 ± 5.8 |
50.0 ± 3.3 |
| 9d |
13.3 ± 3.3 |
13.3 ± 3.3 |
46.4 ± 5.8 |
| 9e |
30.0 ± 0 |
33.3 ± 3.3 |
42.9 ± 3.3 |
| 9f |
6.7 ± 3.3 |
13.3 ± 3.3 |
32.1 ± 6.7 |
| 9g |
26.7 ± 3.3 |
26.7 ± 3.3 |
39.3 ± 3.3 |
| 9h |
13.3 ± 3.3 |
16.7 ± 3.3 |
35.7 ± 0 |
| 9i |
3.3 ± 3.3 |
10.0 ± 0 |
46.4 ± 5.8 |
| 9j |
10.0 ± 0 |
10.0 ± 0 |
50.0 ± 3.3 |
| 10a |
13.3 ± 3.3 |
13.3 ± 3.3 |
46.4 ± 5.8 |
| 10b |
10.0 ± 0 |
13.3 ± 3.3 |
35.7 ± 5.8 |
| 10c |
20.0 ± 0 |
23.3 ± 3.3 |
42.9 ± 3.3 |
| 10d |
6.7 ± 3.3 |
6.7 ± 3.3 |
39.3 ± 3.3 |
| 10e |
20.0 ± 0 |
30.0 ± 0 |
42.9 ± 3.3 |
| 10f |
26.7 ± 3.3 |
26.7 ± 3.3 |
42.9 ± 3.3 |
| 10g |
23.3 ± 3.3 |
30.0 ± 0 |
42.9 ± 3.3 |
| 10h |
13.3 ± 3.3 |
26.7 ± 3.3 |
50.0 ± 3.3 |
| 10i |
13.3 ± 3.3 |
20.0 ± 0 |
57.1 ± 5.8 |
| 10j |
26.7 ± 3.3 |
30.0 ± 0 |
46.4 ± 0 |
| 11 |
6.7 ± 3.3 |
13.3 ± 3.3 |
25.0 ± 5.8 |
| 12 |
16.7 ± 6.7 |
23.3 ± 3.3 |
42.9 ± 3.3 |
| Toosendanin |
6.7 ± 3.3 |
16.7 ± 3.3 |
46.4 ± 5.8 |
| Blank control |
0 ± 0 |
0 ± 0 |
6.7 ± 3.3 |
Additionally, the structure–activity relationships of tested compounds were also observed. When compared with toosendanin, all esters (8b–j) and oxime sulfonates (11 and 12) exhibited less potent insecticidal activity. To 5′-oxime esters (9a–j), introduction of the electron-withdrawing groups on the phenyl of 9b usually resulted in the less active compounds (except 9i,j); for instance, the FMRs of 9e–h were 42.9%, 32.1%, 39.3% and 35.7%, respectively. On the contrary, to 2′-oxime esters (10a–j), compounds containing the electron-withdrawing groups generally showed more potent insecticidal activity than those containing the electron-donating ones. For example, the FMRs of 10e–j were all greater than 42.9%, especially the FMR of 10i was 57.1%. Previously, we noticed that introduction of a (1-naphthylacetyl)oxy group at the C-4 position of the podophyllotoxin derivatives could lead to promising compounds;18–20 in this paper, introduction of a (1-naphthylacetyl)oxy group to 5 or 6 also produced the potent compounds 9a and 10a, respectively. So this suggested that the (1-naphthylacetyl)oxy group could be introduced into other natural products in the future.
Conclusions
In summary, we have semisynthesized a series of esters and oxime esters/sulfonates from furyl-ring-based acetylation derivatives of fraxinellone as pesticidal agents. Especially the steric structures of five products were assigned by X-ray analysis data. Their insecticidal activity was evaluated against a crop-threatening agricultural insect pest, M. separata. Among all derivatives, especially compounds 9a and 10i exhibited the most promising pesticidal activity. Their structure–activity relationships were also observed. It will pave the way for further structural modifications of fraxinellones as biorenewable pesticidal agents for agriculture.
Acknowledgements
The present research was partly supported by National Natural Science Foundation of China (No. 31672071), and Special Funds of Central Colleges Basic Scientific Research Operating Expenses (No. 2452015096) to H. X.
Notes and references
- K. Muralidharan and I. C. Pasalu, Crop Prot., 2006, 25, 409–417 CrossRef.
- J. Y. Sun, P. Liang and X. W. Gao, Pest Manage. Sci., 2012, 68, 285–289 CrossRef CAS PubMed.
- J. Zeng, Y. Jiang and J. Liu, Plant Prot., 2013, 39, 117–121 Search PubMed.
- X. L. Wang, S. W. Wu, W. Y. Gao and Y. D. Wu, J. Econ. Entomol., 2016, 109, 68–74 Search PubMed.
- D. G. Heckel, Science, 2012, 337, 334–338 CrossRef PubMed.
- M. Q. Yu, G. Liu, Y. Y. Zhang, F. Tao, M. Xu and H. Xu, Sci. Rep., 2016, 6, 33062 CrossRef CAS PubMed.
- K. M. Mousa, M. M. Elsharkawy, I. A. Khodeir, T. N. El-Dakhakhni and A. E. Youssef, Egypt. J. Biol. Pest Control, 2014, 24, 347–351 Search PubMed.
- X. F. Wu, Z. J. Ouyang, L. L. Feng, G. Chen, W. J. Guo, Y. Shen, X. D. Wu, Y. Sun and Q. Xu, Toxicol. Appl. Pharmacol., 2014, 281, 146–156 CrossRef CAS PubMed.
- J. S. Yoon, H. Yang, S. H. Kim, S. H. Sung and Y. C. Kim, J. Mol. Neurosci., 2010, 42, 9–16 CrossRef CAS PubMed.
- Z. L. Liu, S. H. Ho and S. H. Goh, Insect Sci., 2009, 16, 147–155 CrossRef CAS.
- M. Lv, W. J. Wu and H. X. Liu, Toxins, 2014, 6, 2708–2718 CrossRef PubMed.
- M. Lü, W. J. Wu and H. X. Liu, Pestic. Biochem. Physiol., 2010, 98, 263–268 CrossRef.
- M. Lv, W. J. Wu and H. X. Liu, Pestic. Biochem. Physiol., 2008, 90, 114–118 CrossRef.
- Q. Li, X. B. Huang, S. C. Li, J. C. Ma, M. Lv and H. Xu, J. Agric. Food Chem., 2016, 64, 5472–5478 CrossRef CAS PubMed.
- Y. Guo, Y. Yan, X. Yu, Y. Wang and H. Xu, J. Agric. Food Chem., 2012, 60, 7016–7021 CrossRef CAS PubMed.
- Y. Guo, R. Yang and H. Xu, Sci. Rep., 2016, 6, 35321 CrossRef PubMed.
- X. Yu, D. Shi, X. Zhi, Q. Li, X. Yao and H. Xu, RSC Adv., 2015, 5, 31700–31707 RSC.
- H. Xu, X. Xiao, X. F. Zhao, Y. Guo and X. J. Yao, Bioorg. Med. Chem. Lett., 2011, 21, 4008–4012 CrossRef CAS PubMed.
- Z. Che, X. Yu, X. Y. Zhi, L. L. Fan and H. Xu, J. Agric. Food Chem., 2013, 61, 8148–8155 CrossRef CAS PubMed.
- L. L. Fan, Y. Guo, X. Y. Zhi, X. Yu and H. Xu, J. Agric. Food Chem., 2014, 62, 3726–3733 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization for the new compounds. CCDC 1524889–1524891, 1524894 and 1524893. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra03588h |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Ruige Yang‡a and
Hui Xu
a,
Ruige Yang‡a and
Hui Xu