
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Utilization of cyclocarbonated lignin as a bio-based cross-linker for the preparation of poly(hydroxy urethane)s†
Anika Salanti a,
Luca Zoia*a,
Michele Maurib and
Marco Orlandia
a,
Luca Zoia*a,
Michele Maurib and
Marco Orlandia
aDepartment of Earth and Environmental Sciences, University of Milano-Bicocca, Piazza della Scienza, 1 Milan, I-20126 Italy. E-mail: luca.zoia@unimib.it; Tel: +39 02-6448-2709
bDepartment of Materials Science, University of Milano-Bicocca, Via R. Cozzi, 55 Milan, I-20125 Italy
First published on 10th May 2017
Abstract
In this work, cyclocarbonated modified lignin, the most abundant reservoir of aromatic moieties in nature, was used as a green bio-based cross-linker on polyhydroxy urethane formulations. As a preliminary study, a complete 13C NMR characterization of the aminolysis products of cyclocarbonated lignin with 1,12-diaminododecane was provided. The best reaction conditions found for the aminolysis reaction were applied for the preparation of thermoset resins. Cyclocarbonated lignin was then reacted with 1,12-diaminododecane, acting as a chain extender, in the presence of poly(ethylene glycol) bis cyclic carbonate, a soft segment used to tune the resin properties. Different (total cyclocarbonates)/(total amines) equivalent ratios and diverse poly(ethylene glycol)/lignin stoichiometry were tested. The evolution in time of the molecular weight distribution of a selected composition was followed by gel permeation chromatography until the solubility in THF was appreciable. Moreover, some significant resin samples were subjected to DSC analysis to evaluate favourable changes in the glass transition temperature and compared to blank experiments that did not include lignin in the composition.
Introduction
The increasing demand for more environmentally friendly polymers to reduce the dependence on exhaustible fossil resources has prompted research towards partially or, more desirably, totally bio-based polymers. The development of bioplastics is growing fast and it is urged by a deeper awareness of the concerns arisen by the prolonged pollution associated to the continuous consumption of petrochemical feedstocks and the need to meet the requirements of green chemistry. The study of alternative sources for the engineering of bio-based polymers is highly desirable and diverse solutions exploiting raw material of natural origin are currently under investigation.The origin of polyurethanes (PU) dates back to 1937, when Otto Bayer discovered the polyisocyanate-polyaddition process1 for the production of a substitute of rubber during World War II. Nowadays, commercial bio-based polyurethanes can be obtained by reacting bio-based polyols (obtained from castor oil, soybean oil, sunflower oil and alike) with synthetic diisocyanates. The global bio-based PU market was 1534 tons in 2012![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2 against a purely petroleum-based PU production estimated in 14950 ktons in the same year.3 Despite promising growth prospects, polyurethanes incorporating polyols of vegetal origin still suffer the disadvantage of being made from highly toxic diisocyanate such as methylene diphenyl diisocyanate (MDI) and tolylene-2,4-diisocyanate (TDI),4 which account for about 95% of the whole production of rigid and flexible foams. It is clear that a major breakthrough towards an authentic green synthesis of PUs should embrace the opportunity to exploit natural resources even for the preparation of synthons of the carbamate group avoiding the resort to diisocyanates. In this respect, the most popular alternative pathway is the polyaddition of bis-5-membered cyclic carbonates and diamines, a process leading to the formation of poly(hydroxy urethane)s (PHUs) bearing primary and secondary hydroxyls on the carbon chain.5–7 Among the main advantages of this approach, beyond the non-utilization of diisocyanates, there is the possibility for CO2 fixation (if the 5-membered cyclic carbonate is obtained by reaction between epoxides and CO2), and the improvement of the thermal and chemical resistance provided by the additional hydroxyl functionalities. Bio-based building blocks can be attained from a number of natural sources, such as vegetable oils, polyols, and phenols. Bio-based bis-cyclic carbonates were obtained by epoxidation and following carbonation of terminal and internal double bonds of a variety of plant seed oils, such as sunflower, linseed and soybean oil.8,9 However, the flexible nature of vegetable oils often results in poor mechanical and thermal properties of the resulting PHU, requiring the use of more rigid scaffolds to match the performance of traditional polyurethane resins. In this respect, the utilization of stiffer monomers like terpenes were investigated and a bis-cyclic carbonate was successfully prepared from epoxidized limonene and employed for the preparation of PHU resins10 with improved properties. Different polyols obtained from the biodiesel industry and from the refinery of starch and sugars, such as glycerol, pentaerythritol and trimethylolpropane were epoxidized, carbonated and cured with diamines to provide PHUs with satisfying mechanical properties, especially when a higher functionality was involved.11,12 Phenol-based bis-cyclic carbonates were synthesized by an epichlorohydrin mediated glycidylation reaction and subsequent CO2 fixation of bisphenols obtained through the para–para condensation between cresol and formaldehyde13 or by a chemo-enzymatic synthesis of ferulic acid derivatives,14 whereas vanillin, a product of the catalytic conversion of lignin, is especially regarded as a promising building block for monomers synthesis over a wide range of different kind of polymers.15
2 against a purely petroleum-based PU production estimated in 14950 ktons in the same year.3 Despite promising growth prospects, polyurethanes incorporating polyols of vegetal origin still suffer the disadvantage of being made from highly toxic diisocyanate such as methylene diphenyl diisocyanate (MDI) and tolylene-2,4-diisocyanate (TDI),4 which account for about 95% of the whole production of rigid and flexible foams. It is clear that a major breakthrough towards an authentic green synthesis of PUs should embrace the opportunity to exploit natural resources even for the preparation of synthons of the carbamate group avoiding the resort to diisocyanates. In this respect, the most popular alternative pathway is the polyaddition of bis-5-membered cyclic carbonates and diamines, a process leading to the formation of poly(hydroxy urethane)s (PHUs) bearing primary and secondary hydroxyls on the carbon chain.5–7 Among the main advantages of this approach, beyond the non-utilization of diisocyanates, there is the possibility for CO2 fixation (if the 5-membered cyclic carbonate is obtained by reaction between epoxides and CO2), and the improvement of the thermal and chemical resistance provided by the additional hydroxyl functionalities. Bio-based building blocks can be attained from a number of natural sources, such as vegetable oils, polyols, and phenols. Bio-based bis-cyclic carbonates were obtained by epoxidation and following carbonation of terminal and internal double bonds of a variety of plant seed oils, such as sunflower, linseed and soybean oil.8,9 However, the flexible nature of vegetable oils often results in poor mechanical and thermal properties of the resulting PHU, requiring the use of more rigid scaffolds to match the performance of traditional polyurethane resins. In this respect, the utilization of stiffer monomers like terpenes were investigated and a bis-cyclic carbonate was successfully prepared from epoxidized limonene and employed for the preparation of PHU resins10 with improved properties. Different polyols obtained from the biodiesel industry and from the refinery of starch and sugars, such as glycerol, pentaerythritol and trimethylolpropane were epoxidized, carbonated and cured with diamines to provide PHUs with satisfying mechanical properties, especially when a higher functionality was involved.11,12 Phenol-based bis-cyclic carbonates were synthesized by an epichlorohydrin mediated glycidylation reaction and subsequent CO2 fixation of bisphenols obtained through the para–para condensation between cresol and formaldehyde13 or by a chemo-enzymatic synthesis of ferulic acid derivatives,14 whereas vanillin, a product of the catalytic conversion of lignin, is especially regarded as a promising building block for monomers synthesis over a wide range of different kind of polymers.15
As a general observation, the thermal and mechanical properties of PHUs resins obtained after the curing process with diamines are strongly dependent upon the chemical characteristic of each reactant. More rigid systems of either the cyclic carbonate or the curing agent, i.e., aromatic or cyclic structures, generally provide thermoset polymers with better properties; the same applies to the degree of functionality of the involved reactants.
In concordance with the illustrated approaches, the purpose of the presented work is the valorisation of lignin as a macromonomer to be employed as structural component in epoxy and PHU resins. Lignin represents the most abundant aromatic polymer in the world and is currently recovered as a side-product of many lignocellulose processing industries and related facilities. An extensive exploitation of lignin in specialty products is hampered by the widely varying chemical and morphological structure of the macromolecule, which in turn is highly dependent upon the specific process used to isolate it. Nevertheless, by virtue of its cross-linked structure and great amount of reactive phenolic and aliphatic hydroxyl functionalities, possible applications of lignin are particularly searched by the scientific community.
The conversion of lignin into high-value polymeric materials has been recently reviewed by Upton and Kasko.16 Several publications deal with the partial or total substitution of the polyol acting as hard segment in traditional polyurethane formulations by lignin. But to the best of the authors' knowledge, the utilization of lignin as a macromonomer in the synthesis of non-isocyanate poly(hydroxy urethane)s is so far unexplored. In fact, as a molecule rich in phenolic and aliphatic hydroxyl functionality, lignin can easily replace the macropolyol component of polyurethanes and be reacted with a diisocyanate,17–19 even in its native form or after a chemical functionalization aimed at the generation of new reactive sites. Moreover, lignin can be combined in different ratios with other polyols acting as soft segments to afford polyurethanes with tailored properties.20–22 Obviously, a limit of this approach is still the utilization of toxic diisocyanates.
In the presented work, bio-based poly(hydroxy urethane) resins were prepared exploiting cyclocarbonated lignin as both a hard segment and a cross-linker. The preparation and chemical characterization of cyclocarbonated lignin derivatives23 and a perspective utilization of lignin as a cross-linker24 has already been reported by the authors. The aminolysis reaction of cyclocarbonated lignin in the presence of excess 1,12-diaminododecane (DAD) was at first investigated to find the best experimental conditions for the formation of the β-hydroxycarbamate derivative. The lignin derivative obtained according to the best reaction conditions found was thoroughly characterized by FT-IR and 13C NMR spectroscopy. The formed carbamate moieties were also quantified by means of quantitative 13C NMR analysis. Cyclocarbonated lignin was then inserted as a cross-linking agents into PHU resins compositions and reacted with 1,12-diaminododecane (DAD) as chain extender in the presence of poly(ethylene glycol) bis cyclic carbonate (PEGCC) as soft segment to modulate the resin characteristics. Given the characteristic stiff aromatic structure, lignin also acted as a substitute of hard segments in the ensuing polymers. Different equivalent ratios of total cyclic carbonate groups vs. total amine functionality, as well as different soft segment vs. hard segment equivalent ratios were investigated. The evolution in time of the molecular weight distributions of a selected formulation was followed by gel permeation chromatography (GPC) until the solubility in THF was appreciable. Some significant resin samples were also subjected to DSC analysis to compare their glass transition temperature to blank experiment not involving lignin in the formulation.
Experimental section
Reagents and materials
All the reagents and solvents used in this work were purchased from Sigma-Aldrich and used as received. The lignin used was supplied during the project ProLignin, EraNets WoodWisdom/Bioenergy, 2012-14. It is a herbaceous lignin (purity 98.2%, total phenolic content 3.55 mmol g−1) obtained from a soda pulping process and sold under the trademark Protobind 1000®.1-Allyl-3-methylimidazolium bromide ([amim]Br) synthesis
N-Methylimidazole (50 mL, 0.627 mol) was poured in a two-neck 250 mL round bottom flask equipped with a magnetic stirrer, a thermometer and a dropping funnel placed in an ice bath. Allyl bromide (57 mL, 0.659 mol) was added dropwise to N-methylimidazole. The internal temperature of the system was carefully maintained between 40–60 °C during the addition, halting the flow of allyl bromide if needed. At the end of the addition, when the temperature ceased to rise, the ice bath was removed and the mixture kept under gentle stirring overnight. [amim]Br was washed twice with fresh aliquots of n-hexane to remove unreacted allyl bromide and then exsiccated under vacuum at 80° for 6 hours. The resulting product was an orangish, honey-like viscous ionic liquid at room temperature.Lignin cyclocarbonation
The cyclocarbonation of lignin was accomplished according to the best reaction conditions found in a previous report.23 500 mg of epoxidized lignin and 5.5 g of [amim]Br were placed into a screw top glass vial equipped with a magnetic stirrer and the mixture heated with a heat gun (metabo H 16-500, 2 heating cycles of approximately 45 s) and stirred overnight at room temperature to achieve complete solubility. The sample was gently loaded into the pressure vessel and the reactor purged with CO2 for 10 min. The pressure was then loaded up to 20 bars and the reactor heated to 80 °C. After 20 hours carbonated lignin was precipitated out in excess cold water, filtered on a sintered glass crucible (size 4), thoroughly washed with deionized water, and lyophilized. 510 mg of cyclocarbonated lignin were recovered, accounting for 93% yield calculated on cyclocarbonate moieties.In dry aminolysis reaction of cyclocarbonated derivatives with 1,12-diaminododecane
400 mg of 1,12-diaminododecane (DAD, mp 67–69 °C) were melted into a desiccated 10 mL round bottom flask under gentle stirring at 80 °C. After liquefaction, 100 mg of acetylated cyclocarbonated lignin were added and rapidly solubilized by the amine. During the 30 minutes required for complete reaction, 100 μL of distilled water were added in two aliquots (50 μL each) to the experiments, to both catalyze the aminolysis reaction and keep the mixture flowing. At the end of the reaction period, aliquots of hot acidic water (HCl 0.1 M, 5 mL at a time) were added to the flask until complete solubilization of the aminolysis product and of excess DAD was achieved. Each aliquot was transferred into a 100 mL Erlenmeyer flask when saturated (at eye inspection, the firsts water aliquots become milky when DAD cannot be further dissolved), the final pH should be around 3. The aqueous solution containing the aminolysis derivative and the unreacted diamine was kept under stirring until room temperature was reached; precipitation of components was not observed. 1 g of NaCl was then added to increase the ionic strength of the solution; as a result, the aminolysis products precipitated out while excess DAD remained in solution. The precipitates were filtered on a sintered glass crucible (size 4) and washed with acidic brine. The solid was then recollected by dissolution in ethanol (ca. 15 mL); this step is also useful to remove residual traces of NaCl. Finally, the solution was filtered, ethanol rotary evaporated and the product exsiccated in vacuum. Aminolysis products were obtained in 95% apparent yield. About 100 mg of the crude were acetylated to improve the solubility in DMSO-d6 and analyzed by 13C NMR.General procedure for the preparation of poly(hydroxy)urethanes
In a typical experiment, 100 mg of poly(ethylene glycol) bis cyclic carbonate (PEGCC, Mn ≈ 500; carbonate equivalent weight ≈ 250 g eq.−1; functionality: 2) were put into a 8 mL screw top glass vial equipped with a magnetic stirrer. Poly(ethylene glycol) bis cyclic carbonate was prepared by cycloaddition reaction of CO2 on poly(ethylene glycol) diglycidyl ether according to the same conditions described for the preparation of cyclocarbonated lignin. A certain amount of cyclocarbonated lignin (LignCC, Mn ≈ 2400 estimated by GPC-UV; cyclocarbonate content = 1.35 mmol g−1 calculated by quantitative 13C NMR) was then added, as according to the PEGCC/LignCC stoichiometry reported in Table 1, along with 250 μL of tetrahydrofuran (THF). After dissolution, a specific amount of 1,12-diaminododecane (DAD, MW = 200.4 g mol−1; amine equivalent weight = 100.2 g eq.−1; functionality: 2) was added to the solution, as in accordance to the total cyclocarbonate groups/DAD equivalent ratios described in Table 1. The vial was then placed in an oil bath and heated up to 80 °C (also to favour the melting of DAD) for 20 h. The resulting polyaddition product was finally cured in oven at 80 °C for 20 h. Selected resin samples were subjected to DSC analysis and compared to a reference blank. Furthermore, the evolution in time of the molecular weight distribution of a selected polymer was monitored by GPC analysis. A sensible blank reference experiment where cyclocarbonated lignin was absent was also prepared and benzoylated in pyridine/benzoyl chloride to gain UV response.:1
| Entry | PEGCC/LignCC equivalent ratio | LignCC (%) | Appearance | Tg (°C) |
|---|---|---|---|---|
| 1 | Only PEGCC, blank | 0.0 | Viscous liquid | −35.5 |
| 2 | 40:1 |
4.4 | Ductile mass | — |
| 3 | 20:1 |
8.3 | Ductile mass | — |
| 4 | 15:1 |
10.6 | Rubbery mass | — |
| 5 | 10:1 |
15.3 | Rubbery mass | −27.0 |
| 6 | 4:1 |
29.9 | Hard mass | −15.7 |
| 7 | 2:1 |
44.4 | Hard mass | — |
| 8 | 1:1 |
58.8 | Hard mass | — |
Lignin acetylation
If required, lignin acetylation was performed overnight in a pyridine–acetic anhydride solution (1:1 v/v, 4 mL). After stripping with ethanol, toluene, and chloroform (25 mL × 3, each solvent), each sample was dried under vacuum. Acetylated lignin samples were dissolved in DMSO-d6 for 13C NMR experiments.
FT-IR analysis
Fourier transform infrared spectroscopy was used for a rapid screening of the reaction outcome. The analyses were performed on a Nicolet iS10 spectrometer (Thermo Scientific) equipped with a Smart iTR™ Total Attenuated Reflectance (ATR) sampling accessory (total scan 16, range 4000–700 cm−1, resolution 1 cm−1).13C NMR analysis
Broadband decoupled 13C-NMR spectra were recorded on a Bruker Avance 500 MHz spectrometer at room temperature on 800 μL samples dissolved in deuterated dimethylsulfoxide (DMSO-d6). The chemical shifts were referred to the methoxyl group signal (56 ppm). For qualitative evaluations relaxation delay of 4 s was used between the scans (90° pulse angle). Line broadening of 0.8 Hz was applied to FIDs before Fourier transform. For each spectrum, typically 6000 scans were accumulated. Quantitative 13C NMR spectra were acquired for the β-hydroxycarbamate lignin derivative in the presence of dimethylformamide (5 μL, 99.8%) as internal standard. Relaxation delay of 10 s was applied and approximately 10000 scans were accumulated. The assignment of predominant signals was based on literature data.23,25
GPC analysis
GPC analyses were performed on a Waters 600 E liquid chromatography system connected to a HP1040 ultraviolet UV detector set at 240 nm. The injection port was a Rheodyne loop valve equipped with a 20 μL loop. The GP-column system was composed of a sequence of an Agilent PL gel 5 μm, 500 Å, and an Agilent PL gel 5 μm, 104 Å. Samples were dissolved in tetrahydrofuran (THF, 1 mg mL−1) and analyzed at a flow rate of 1 mL min−1 using THF as eluent (Fluka 99.8%). PL polymer standards of polystyrene from Polymer Laboratories were used for calibration.DSC analysis
Differential scanning calorimetry (DSC) was performed with a Mettler Toledo Stare thermal analysis system equipped with a liquid N2 low-temperature apparatus. The experiments were run under nitrogen atmosphere. Samples of selected polymers were sealed into 40 μL aluminium pans under dry conditions, quenched to −80 °C for 3 min and then heated to 150 °C at 20 °C min−1 rate. The samples were kept at 150 °C for 3 min and then cooled down to −80 °C at 20 °C min−1 rate after which they were re-heated to 150 °C at the same rate. Indium was used as standard for temperature and heat flow calibration. The glass transition temperatures (Tg) were obtained as the mid-point of the step transition in the calorimetric curve.Results and discussion
Aminolysis reaction of cyclocarbonate groups on lignin
The outcome of the aminolysis reaction exerted by 1,12-diaminododecane (DAD) on the cyclocarbonated derivative of lignin was followed by FT-IR as changes in the intensity of the characteristic cyclocarbonates band at 1790 cm−1 are easily appreciated. At first, different wet reaction environment, including solvents of diverse characteristics such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), dimethylacetamide (DMAc), methanol, water, PEG400, acetone, acetonitrile and tetrahydrofuran (THF), either or not in the presence of different catalysts including tetrabutylammonium bromide (TBAB), potassium iodide (KI), 1,5-diazabicyclo(5.4.0)undec-5-ene (DBU), diphenylthiourea (DPTU), trimethylamine (TEA), [amim]Br and water, and thereof combinations were tested. The choice of solvents and catalysts, as well as the reaction parameters, was made according to the most popular conditions described in literature.5 Typically, 50 mg of cyclocarbonated lignin (1.56 mmol g−1, ca. 80 μmol of cyclic carbonate moieties) were dissolved in 5 mL of a specific solvent and reacted with 200 mg of 1,12-diaminododecane (DAD, 1 mmol) in presence of 10–20% mol mol−1 of a specific catalyst for 20 h at 80 °C. Depending upon the experiment, the lignin derivative was recovered after precipitation in either diisopropyl ether or water and then soaked in a methanol/water mixture (1:1 v/v) to remove unreacted amine. The ATR-FTIR screening of the recovered products showed that the cyclocarbonated lignin samples were mostly unaffected by the treatment with DAD. An appreciable variation of the cyclic carbonate carbonyl band at 1790 cm−1 was accomplished only in DMF, using TEA as catalyst, and in water, without the assistance of any catalyst. The infrared spectra of reference cyclocarbonated lignin and β-hydroxycarbamate lignin derivatives obtained by solvent treatment are reported in Fig. 1. As evident on account of a residual band at 1790 cm−1, the aminolysis reaction performed in DMF/TEA did not run to completion whereas a complete conversion was achieved in water medium. In this case, the cyclic carbonates band disappears while a new absorption at 1700 cm−1 (±5 cm−1), associated to hydrogen bonded carbonyls in PUs and PHUs,26,27 become dominant. The prevailing formation of associated species is not surprising: the carbonyl oxygen can easily be involved in hydrogen bonds with the N–H hydrogen of both the carbamate group and the terminal amino group of DAD, as well as with the O–H hydrogen of both the β-hydroxycarbamate group and the aliphatic hydroxyls of lignin. On the other hand, the ability of water to act as solvent and catalyst in the aminolysis reaction of cyclic carbonates has been investigated by Ochiai et al.,28 who demonstrated that a greater hydrophobicity of the cyclic carbonate structure slowed down the rate of the competitive hydrolysis reaction. In the present work, water obviously did not act as solvent but it is undeniable that it played a crucial catalytic role in directing the reaction towards the formation of the β-hydroxycarbamate lignin derivative.
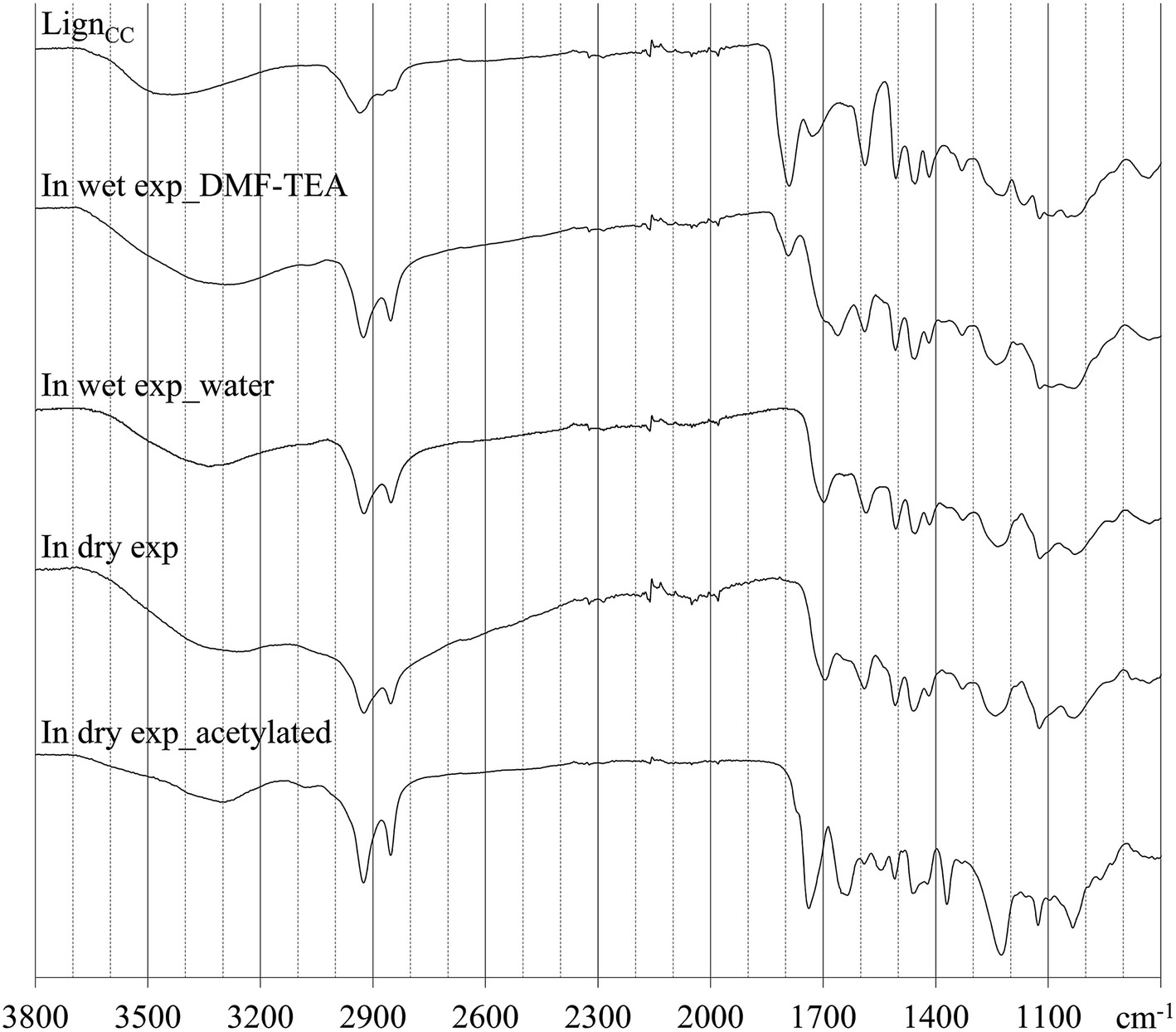 | ||
| Fig. 1 Infrared spectra of (top to bottom): cyclocarbonated lignin (reference), in wet aminolysis experiment performed in DMF using TEA as catalyst, in wet aminolysis experiment performed in water, in dry aminolysis experiment, and in dry aminolysis experiment after acetylation. | ||
Despite the satisfying result, the β-hydroxycarbamate lignin derivatives recovered showed a substantial insolubility in all the solvents suitable for an analytical characterization, perhaps due to strong inter- and intramolecular association. It was therefore decided to perform the aminolysis reaction in dry conditions, exploiting DAD as solvent for cyclocarbonated lignin and using a small amount of water to catalyze the reaction. To further favour disaggregation of the ensuing product, acetylated cyclocarbonated lignin was employed as starting material. The aminolysis reaction was found to be complete also in dry reaction conditions: residual peaks ascribable to cyclic carbonates were not detected, the major absorption in the carbonyl region was placed around 1700 cm−1 whereas a strong signal related to O–H and N–H bond stretching was observed. It was noticed that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of carbonyl esters (ca. 1730 cm−1) was no more detected, likely due to the occurrence of hydrolysis reactions promoted by the alkaline environment created by DAD and the high temperature involved. The β-hydroxycarbamate lignin derivative was once again acetylated to maximize its solubility in DMSO-d6 for 13C NMR analysis. The IR spectra before and after acetylation are reported in Fig. 1. After acetylation, the principal bands in the carbonyl IR region were placed around 1735 and 1625 cm−1, in agreement with the formation of O-acetyl and N-acetyl ester groups, respectively, while a residual band centred around 3300 cm−1, generated by the N–H stretching of carbamates and mono-acetylated terminal amino groups of DAD, was observed.
O stretching of carbonyl esters (ca. 1730 cm−1) was no more detected, likely due to the occurrence of hydrolysis reactions promoted by the alkaline environment created by DAD and the high temperature involved. The β-hydroxycarbamate lignin derivative was once again acetylated to maximize its solubility in DMSO-d6 for 13C NMR analysis. The IR spectra before and after acetylation are reported in Fig. 1. After acetylation, the principal bands in the carbonyl IR region were placed around 1735 and 1625 cm−1, in agreement with the formation of O-acetyl and N-acetyl ester groups, respectively, while a residual band centred around 3300 cm−1, generated by the N–H stretching of carbamates and mono-acetylated terminal amino groups of DAD, was observed.
Fig. 2 reports the 13C NMR spectra of the acetylated aminolysis derivative and of acetylated cyclocarbonated lignin, taken as reference, while Scheme 1 depicts reactants and products of the aminolysis reaction, providing particular emphasis to the formation of primary and secondary derivatives ensuing from a different regioselectivity of the addition of DAD to the cyclocarbonate group.
 | ||
| Fig. 2 13C NMR spectra recorded in DMSO-d6 of acetylated cyclocarbonated lignin and acetylated cyclocarbonated lignin after aminolysis reaction with DAD (above). Both the spectra are referred to the methoxyl signal at 56.0 ppm. The intensities of the two intervals reported (175–45 ppm with the exclusion of the aromatic region, and the span between 42 and 20 ppm) have been modified to better appreciate characteristic absorbances of each region. Selected expansions of the overlapped spectra (below) relating to different carbonyl regions and to the acetyl carbonyl region are normalized on the methoxyls signal. Assignment based on carbon atoms numbering reported in Scheme 1. | ||
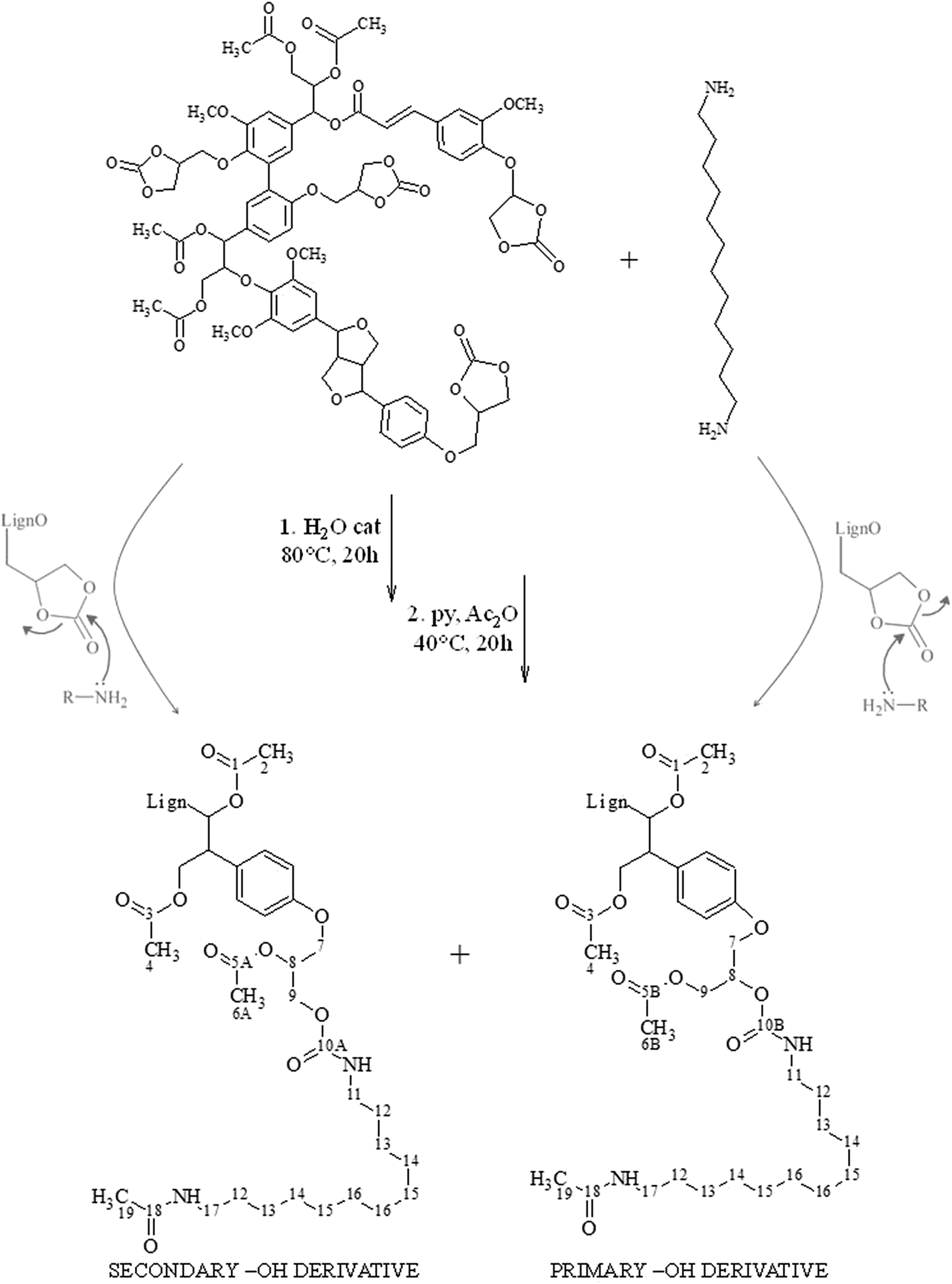 | ||
| Scheme 1 Reaction scheme for the polyaddition reaction between a representative fragment of the cyclocarbonated lignin derivative used in this work and DAD. The orientation of the nucleophilic attack exerted by the amine group of DAD and related orientation of the cyclic carbonate ring opening determines the formation of a β-hydroxycarbamate derivative bearing either a secondary or a primary alcoholic group. | ||
A thorough interpretation of the main 13C NMR signals of cyclocarbonated lignin has already been reported by the authors.23 It is highlighted the presence of dimethyl carbonate (peaks at 54.8 and 155.9 ppm) and dimethylformamide (peaks at 31.0, 36.1, and 162.6 ppm), employed as internal standard for the quantification of cyclic carbonate groups22 and β-hydroxycarbamate groups, respectively.
According to Fig. 2, after the aminolysis reaction, the characteristic peaks of the cyclic carbonate groups (66.1 ppm, 68–72 ppm, 73–75 ppm, and 155.1 ppm) disappeared, revealing intense signals in the aliphatic carbons region: at 26.7 ppm and between 29.0–29.2 ppm are placed the peaks of the C3 carbon atoms (labelled as number 13 in Scheme 1) and of the C2, C4, C5, and C6 carbon atoms (labelled as number 12, 14, 15 and 16 in Scheme 1) of the symmetric diamine, respectively. Given the reaction conditions employed, the addition of acetyl groups was considered only for one of the terminal amino group of DAD. Accordingly, the partially overlapped signal at 38.7 ppm was assigned to the α-carbon of DAD adjacent to the acetylated terminal amino group (labelled as number 17 in Scheme 1) whereas, according to literature data,25 the signal pertaining to the α-carbon of DAD adjacent to the amino group involved in the cyclocarbonate ring opening was tentatively located under the solvent signal (carbon atom number 11 in Scheme 1). Given the complex chemical structure of lignin, which reverberates in moderate variations of the chemical shifts of differently surrounded carbon atoms, a rigorous assignment of the C-3 chain ensuing from the ring opening of cyclic carbonates is not feasible and just a certain number of peaks ascribable to this unit were recognized between 75 and 65 ppm (carbon atoms number 7, 8 and 9 in Scheme 1).
Other interesting sections of the 13C NMR spectrum of the acetylated aminolysis derivative are highlighted and reported as expansions in the lower part of Fig. 2, where they were also overlapped to the reference spectrum of acetylated cyclocarbonated lignin (grey line) for comparison. The overlapped spectra are normalized on the methoxyls signal.
The acetyl methyl region is characterized by three distinct signals (Fig. 2, below, right side). At 20.7–20.9 ppm was recognized the characteristic broad peak of –CH3 groups pertaining to acetylated primary and secondary aliphatic alcohols in lignin (carbon atoms number 2 and 4 in Scheme 1). In fact, this same peak is also detected in the reference spectrum of cyclocarbonated lignin. Characteristic signals of the acetylated β-hydroxycarbamate derivative are found at 21.4 and 22.4 ppm, instead. The signal placed at 21.4 ppm was assigned to the methyl groups of the acetylated β-hydroxycarbamate alcoholic moiety, both of primary and secondary structure (atoms number 6A and 6B in Scheme 1); at 22.8 ppm were instead recognized the methyl groups of acetamides (atoms number 19 in Scheme 1).
Concerning the acetyl carbonyl region (Fig. 2, below, left side), centred at 170.3 and 170.0 ppm was recognized the absorption of primary and secondary O-acetyl carbonyl carbons, respectively, originating from the acetylation of both the aliphatic hydroxyls of lignin and the primary and secondary –OH ensuing from the ring opening reaction of cyclic carbonates (labelled as atoms number 1, 3 and 5 in Scheme 1). It is worth noticing that the intensity of both these peaks is higher if compared to the reference profile of acetylated cyclocarbonated lignin (grey line) and, particularly, that the population of the signal pertaining to the carbonyls of acetylated secondary hydroxyls are larger. Therefore, new alcoholic functionalities are formed after the addition of DAD to cyclic carbonates and, in agreement with several publications,28–30 the formation of secondary hydroxyl groups was predominantly observed. Moreover, at 169.2 ppm is found a sharp peak related to carbonyl carbon atoms arising from N-acetylation (atoms number 18 in Scheme 1).
The 13C NMR spectrum of the aminolysis derivative is also characterized by two, partially overlapped signals placed at 155.7 and 156.0 ppm (Fig. 2, below, centre), accounting for carbamate carbonyls provided by β-hydroxycarbamate derivatives bearing either primary or secondary hydroxyl groups, respectively. The stronger intensity of the peak placed at 156.0 ppm is in solid agreement with the favoured formation of the secondary derivative, as already reported above. It is also highlighted the complete disappearance of the cyclic carbonate CO signal after aminolysis (the peak at 155.9 ppm in the grey reference profile belongs to the internal standard used for cyclocarbonates quantification).
Finally, the amount of hydroxycarbamate groups was assessed through quantitative 13C NMR analysis using DMF as internal standard. The integrations of the carbonyl carbons peaks at 162.6 ppm (DMF) and 155.7–156.0 ppm (β-hydroxycarbamate derivatives) were considered for the calculation. The theoretical value of carbamate units, expressed as mmol g−1, was calculated applying a weight correction,23 accounting for the molecular weight increment of the resulting aminolysis derivative, to the experimental value of cyclic carbonate groups content as obtained by quantitative 13C NMR performed on acetylated cyclocarbonated lignin. The theoretical value accordingly retrieved was 0.74 mmol g−1. The experimental value calculated was 0.52 mmol g−1, which was lower than expected. A possible explanation of this discrepancy can be found in a characteristic consequence of the Nuclear Overhauser Effect (NOE), i.e., H-decoupled 13C NMR experiments return signals whose intensity is not proportional to the actual population. In particular, peaks originated by carbon atoms bound to one or more hydrogen atoms are stronger than the ones arising from quaternary carbon atoms. Therefore, the formamido group of DMF probably was not the internal standard of election for the quantitative evaluation of carbamates, despite the substantial similarity in the two structures. Other, more suitable internal standards based on the acetamido group were not taken into consideration because of a partial to complete overlap of the CO signal of interest with the acetyl carbonyl groups of the aminolysis derivative.
Poly(hydroxy urethane) resins preparation and characterization
Various PHU resins, containing cyclocarbonated lignin as co-macromonomer, were prepared in accordance with the compositions reported in Table 1. Cyclocarbonated lignin (LignCC) was utilized as both hard segment and cross-linker, DAD served as a chain extender, while poly(ethylene glycol) bis cyclic carbonate (PEGCC) was employed as a soft segment to modulate the physical property of the resin (Scheme 2). Since cyclic carbonates and amine groups reacts according to a 1:1 stoichiometry, this ratio was kept fixed while a number of resins involving different PEGCC/LignCC ratios were synthesized to evaluate the performance of cyclocarbonated lignin as a cross-linking agent. Polymeric compositions comprising up to 58.8% cyclocarbonated lignin were prepared according to the described method.
 | ||
| Scheme 2 Reactants, products, and experimental conditions used for the preparation of lignin cross-linked PHU resins (cyclocarbonated lignin, Mn ≈ 2400, cyclic carbonates content 1.35 mmol g−1; poly(ethylene glycol) bis cyclic carbonate, Mn ≈ 500, functionality 2; 1,12-diaminododecane, MW 200.4 g mol−1, functionality 2). Reticulation points highlighted in grey. | ||
The physical appearance of the recovered polymers (Fig. 3) was undoubtedly related to a different degree of reticulation of the resin constituents, which is in turn a function of the amount of lignin inserted in the formulation. Where higher PEGCC/LignCC ratios were employed (Table 1, entries 2 and 3), a viscous liquid or a ductile gel was recovered; for medium PEGCC/LignCC ratios (Table 1, entries 4 and 5) the result was a rubbery gel which was not impaired by treatment with solvents; for lower PEGCC/LignCC ratios (Table 1, entries 6 to 8) a hard gel, completely insoluble in analytical solvents, was obtained. In agreement with the absence of a cross-linking component, the polymer recovered from the reference experiment excluding lignin from the composition (Table 1, entry 1) was a viscous liquid.
 | ||
| Fig. 3 Physical appearance of representative resins formulation containing different percentage of cyclocarbonated lignin as cross-linker. | ||
DSC analyses were performed on three selected samples (entries 4, 5, and 8) to assess significant changes in the thermal behaviour of the obtained PHUs connected to the addition of cyclocarbonated lignin. All thermograms (provided as ESI†) displayed a broad evaporation phenomenon during the first heating cycle around 80 °C, which was related to the presence of residual solvent (THF). Furthermore, a sharp endothermic peak characteristic of a melting process was detected around 40 °C in all cases. Interestingly, the same endothermic process was found again during the second heating cycle of the reference, linear PHU (entry 8), but it was absent when cyclocarbonated lignin was contemplated in the polymeric composition (entries 4 and 5), demonstrating a different self-organization of the polymeric chains. The reticulating action exerted by lignin is furtherly proven by the trend of the glass transition temperatures of the examined resins (Table 1) during the second heating ramp. It was assumed that volatile components with possible plasticizing effect had been eliminated during the first cycle, thus the glass transition temperature is only influenced by the effectiveness of lignin as cross-linker. The detected Tg values increased as a function of the lignin content, going from −35.5 °C of the reference sample not including lignin (entry 8) to −15.7 °C of the lower PEGCC/LignCC equivalent ratio (entry 5), passing through the intermediate temperature −27.0 °C of the higher PEGCC/LignCC equivalent ratio (entry 4). The comparison between the thermal profile of the first and the second heating cycle pointed out a discrepancy between the measured Tg concerning entry 5. In particular, the Tg detected during the first heating was rather lower than the Tg measured during the second heating (−21.2 °C vs. −15.7 °C, respectively), suggesting the need for more drastic reaction condition (higher temperatures) to push the reaction to completion. It is also worth noticing that the exothermic peak connected to the recrystallization of the system (around −20 °C) became more and more less defined with increasing lignin content during the cooling cycle, as a confirmation of the stiffer structure of the cross-linked resin.
Additionally, the evolution in time of the molecular weight distribution was followed for one of the resin composition resulting in a rubbery gel (entry 5). GPC monitoring was performed over 8 hours, after this reaction period the resulting resin was for the most part insoluble in THF, indicating a further increase in the average molecular weight. The GPC chromatograms of the most indicative samplings, along with reference chromatograms of cyclocarbonated lignin and of the corresponding blank experiment (entry 8), are reported in Fig. 4. A progressive shift of the chromatogram towards higher molecular weights is observed over the whole examined reaction period, indicating a gradual consumption of the cyclic carbonate groups. The peak molecular weight (Mp) of the ensuing PHU shifted from ca. 500 g mol−1 at zero reaction time (reference cyclocarbonated lignin) to ca. 15000 g mol−1 after 8 hours reaction. As a final observation, it was detected that the molecular weight distributions of the last sampling and the benzoylated reference blank were quite similar. However, it should be reminded that the last sampling did not coincide with the end of the reaction period, at which the polymer was found as a THF insoluble gel.
 | ||
| Fig. 4 Molecular weight profile evolution of experiment 5 (cyclocarbonates tot/DAD tot 1:1 eq. ratio; PEGCC/LignCC 10:1 eq. ratio) after 30 min, 2, 5, and 8 h reaction. At the end of the established reaction period (20 h) the mixture was found as a THF insoluble gel. The molecular weight distributions of cyclocarbonated lignin and of a corresponding benzoylated blank experiment are reported as reference. | ||
In light of the discussed results, it was therefore concluded that cyclocarbonated lignin is able to act as a reticulating agent in PHUs formulation. Polymers with tailored properties, ranging from a soft elastomer to a thermoset resin passing through a thermoset elastomer, can be obtained by tuning the PEGCC/LignCC ratio.
References
- I. G. FaberIndustrie, Verfahren zur Herstellung von Polyurethanen, German patent 728,981, 1942.
- Bio-Based Polyurethane (PU) Market Analysis By Product (Rigid Foams, Flexible Foams, CASE), By End-Use (Furniture & Interiors, Construction, Automotive, Footwear) And Segment Forecasts To 2020, Published: January 2015, San Francisco, CA, available over internet at http://www.grandviewresearch.com/industry-analysis/bio-based-polyurethane-industry.
- Polyurethane (PU) Market Analysis By Product (Rigid Foam, Flexible Foam, Coatings, Adhesives & Sealants, Elastomers), By End-use (Furniture and Interiors, Construction, Electronics & Appliances, Automotive, Footwear, Packaging) And Segment Forecasts To 2020, Published: October 2014, San Francisco, CA, available over internet at http://www.grandviewresearch.com/industry-analysis/polyurethane-pu-market.
- J. M. Peters, R. L. Murphy, L. D. Pagnotto and J. L. Whittenberger, Respiratory impairment in workers exposed to “safe” levels of toluene diisocyanate (TDI), Arch. Environ. Health, 1970, 20(3), 364–367 CrossRef CAS PubMed.
- L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Isocyanate-free routes to polyurethanes and poly (hydroxy urethane)s, Chem. Rev., 2015, 115(22), 12407–12439 CrossRef CAS PubMed.
- O. Figovsky, L. Shapovalov, A. Leykin, O. Birukova and R. Potashnikova, Recent advances in the development of non-isocyanate polyurethanes based on cyclic carbonates, PU Magazine, 2013, 10(4), 1–9 Search PubMed.
- G. Rokicki, P. G. Parzuchowski and M. Mazurek, Non-isocyanate polyurethanes: synthesis, properties, and applications, Polym. Adv. Technol., 2015, 26(7), 707–761 CrossRef CAS.
- S. Samanta, S. Selvakumar, J. Bahr, D. S. Wickramaratne, M. Sibi and B. J. Chisholm, Synthesis and Characterization of Polyurethane Networks Derived from Soybean-Oil-Based Cyclic Carbonates and Bioderivable Diamines, ACS Sustainable Chem. Eng., 2016, 4(12), 6551–6561 CrossRef CAS.
- A. Boyer, E. Cloutet, T. Tassaing, B. Gadenne, C. Alfos and H. Cramail, Solubility in CO2 and carbonation studies of epoxidized fatty acid diesters: towards novel precursors for polyurethane synthesis, Green Chem., 2010, 12(12), 2205–2213 RSC.
- M. Bähr, A. Bitto and R. Mülhaupt, Cyclic limonene dicarbonate as a new monomer for non-isocyanate oligo-and polyurethanes (NIPU) based upon terpenes, Green Chem., 2012, 14(5), 1447–1454 RSC.
- M. Fleischer, H. Blattmann and R. Mülhaupt, Glycerol-, pentaerythritol- and trimethylolpropane-based polyurethanes and their cellulose carbonate composites prepared via the non-isocyanate route with catalytic carbon dioxide fixation, Green Chem., 2013, 15(4), 934–942 RSC.
- J.-M. Bernard, Method for preparing polyhydroxy-urethanes, US Pat. 8,017,719, 2011.
- Q. Chen, K. Gao, C. Peng, H. Xie, Z. K. Zhao and M. Bao, Preparation of lignin/glycerol-based bis (cyclic carbonate) for the synthesis of polyurethanes, Green Chem., 2015, 17(9), 4546–4551 RSC.
- R. Ménard, S. Caillol and F. Allais, Chemo-enzymatic synthesis and characterization of renewable thermoplastic and thermoset isocyanate-free poly (hydroxy) urethanes from ferulic acid derivatives, ACS Sustainable Chem. Eng., 2017, 5, 1446–1456 CrossRef.
- M. Fache, E. Darroman, V. Besse, R. Auvergne, S. Caillol and B. Boutevin, Vanillin, a promising biobased building-block for monomer synthesis, Green Chem., 2014, 16(4), 1987–1998 RSC.
- B. M. Upton and A. M. Kasko, Strategies for the conversion of lignin to high-value polymeric materials: review and perspective, Chem. Rev., 2015, 116(4), 2275–2306 CrossRef PubMed.
- A. Gandini, M. N. Belgacem, Z. X. Guo and S. Montanari, in Chemical Modification, Properties, and Usage of Lignin, ed. T. Q. Hu, Kluwer Academic/Plenum Publishers, Springer, US, New York, 2002, ch. 4, p. 57 Search PubMed.
- H. Chung and N. R. Washburn, Improved lignin polyurethane properties with Lewis acid treatment, ACS Appl. Mater. Interfaces, 2012, 4(6), 2840–2846 CAS.
- S. Laurichesse, C. Huillet and L. Avérous, Original polyols based on organosolv lignin and fatty acids: new bio-based building blocks for segmented polyurethane synthesis, Green Chem., 2014, 16(8), 3958–3970 RSC.
- D. Feldman, in Chemical modification, properties, and usage of lignin, ed. T. Q. Hu, Kluwer Academic/Plenum Publishers, Springer, US, New York, 2002, ch. 5, p. 81 Search PubMed.
- Y. Li and A. J. Ragauskas, Kraft lignin-based rigid polyurethane foam, J. Wood Chem. Technol., 2012, 32(3), 210–224 CrossRef CAS.
- X. Pan and J. N. Saddler, Effect of replacing polyol by organosolv and kraft lignin on the property and structure of rigid polyurethane foam, Biotechnol. Biofuels, 2013, 6(1), 12 CrossRef CAS PubMed.
- A. Salanti, L. Zoia and M. Orlandi, Chemical modifications of lignin for the preparation of macromers containing cyclic carbonates, Green Chem., 2016, 18(14), 4063–4072 RSC.
- A. Salanti, L. Zoia, S. Zanini and M. Orlandi, Synthesis and characterization of lignin–silicone hybrid polymers as possible consolidants for decayed wood, Wood Sci. Technol., 2016, 50(1), 117–134 CrossRef CAS.
- Spectral Database of Organic Compounds, National Institute of Advanced Industrial Science and Technology, Japan, available over Internet at http://sdbs.db.aist.go.jp.
- E. Yilgör, E. Burgaz, E. Yurtsever and I. Yilgör, Comparison of hydrogen bonding in polydimethylsiloxane and polyether based urethane and urea copolymers, Polymer, 2000, 41(3), 849–857 CrossRef.
- E. K. Leitsch, G. Beniah, K. Liu, T. Lan, W. H. Heath, K. A. Scheidt and J. M. Torkelson, Nonisocyanate thermoplastic polyhydroxyurethane elastomers via cyclic carbonate aminolysis: critical role of hydroxyl groups in controlling nanophase separation, ACS Macro Lett., 2016, 5(4), 424–429 CrossRef CAS.
- B. Ochiai, Y. Satoh and T. Endo, Nucleophilic polyaddition in water based on chemo-selective reaction of cyclic carbonate with amine, Green Chem., 2005, 7(11), 765–767 RSC.
- H. Tomita, F. Sanda and T. Endo, Model reaction for the synthesis of polyhydroxyurethanes from cyclic carbonates with amines: substituent effect on the reactivity and selectivity of ring-opening direction in the reaction of five-membered cyclic carbonates with amine, J. Polym. Sci., Part A: Polym. Chem., 2001, 39(21), 3678–3685 CrossRef CAS.
- B. Nohra, L. Candy, J. F. Blanco, Y. Raoul and Z. Mouloungui, Aminolysis reaction of glycerol carbonate in organic and hydroorganic medium, J. Am. Oil Chem. Soc., 2012, 89(6), 1125–1133 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03416d |
| This journal is © The Royal Society of Chemistry 2017 |