
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
LTA zeolite membranes: current progress and challenges in pervaporation
I. G. Wenten
 *ab,
P. T. Dharmawijaya
b,
P. T. P. Aryanti
c,
R. R. Mukti
bd and
Khoiruddin
a
*ab,
P. T. Dharmawijaya
b,
P. T. P. Aryanti
c,
R. R. Mukti
bd and
Khoiruddin
a
aDepartment of Chemical Engineering, Faculty of Industrial Technology, Institut Teknologi Bandung, Jl. Ganesha 10, Bandung – 40132, Indonesia. E-mail: igw@che.itb.ac.id
bResearch Center for Nanosciences and Nanotechnology, Institut Teknologi Bandung, Jl. Ganesha 10, Bandung – 40132, Indonesia
cDepartment of Chemical Engineering, Jenderal Achmad Yani University, Jl. Terusan Jendral Sudirman, PO Box 148 Cimahi, Jawa Barat, Indonesia
dDivision of Inorganic and Physical Chemistry, Faculty of Mathematics and Natural Sciences, Institut Teknologi Bandung, Jl. Ganesha 10, Bandung – 40132, Indonesia
First published on 7th June 2017
Abstract
Linde Type A (LTA) zeolite-based membranes have demonstrated excellent selectivity in pervaporation due to their unique structural framework and interaction with water. The development of LTA zeolite membranes for commercial application is limited by some parameters, particularly the complexity of the membrane preparation required to produce reproducible defect-free membranes and the high costs required for the membrane materials. In addition, the high content of Al in the zeolite framework makes the LTA zeolite membrane unsuitable for acidic conditions. A number of modification techniques have been proposed to produce a thin, defect-free, and high permselectivity LTA zeolite membrane with high reproducibility. Two major approaches are generally used to produce defect-free zeolite membranes, i.e. modifying either the seeding step or the synthesis process. Since the self-supported zeolite membrane has low mechanical stability, the LTA zeolite membrane is usually synthesized on an inorganic support to give better properties. Zeolite membrane costs can be reduced by several methods such as replacing the support, manufacturing a higher flux zeolite membrane, and fabricating a polymer–zeolite membrane. One should consider, however, that changing the support can dramatically influence and even reverse the obtained separation behavior. Despite various techniques used to prepare dense LTA zeolite membranes, a facile mass production technique with a highly reproducible result remains a significant challenge. To present a clear background for LTA zeolite and its performances in pervaporation, this paper includes a brief discussion on the recent trends related to LTA zeolite membranes. Some topics are discussed, including the features inherent to LTA zeolite, the transport phenomena in zeolite structures, preparation methods of LTA zeolite membranes, and the challenges associated with preparation. Furthermore, critical issues related to LTA zeolite membranes in pervaporation will be discussed to develop the topic further.
 I. G. Wenten | I Gede Wenten received his bachelor's degree in chemical engineering from Institut Teknologi Bandung (ITB) Indonesia and his MSc and PhD degrees from DTU Denmark. He is a professor of chemical engineering and a member of the Research Center for Nanosciences and Nanotechnology, ITB. He has extensive experience in membrane technology, both at industrial and academic levels, with a career spanning more than 20 years. His research interests include membrane preparation, membrane applications, and membrane fouling. |
 P. T. Dharmawijaya | Putu Teguh Dharmawijaya received his bachelor's degree in chemical engineering from Institut Teknologi Bandung (ITB) in 2015. Afterwards, he worked as a research assistant at the Research Center for Nanosciences and Nanotechnology, ITB, under the supervision of Professor Wenten from 2015–2016. He is currently studying for an MSc in Advanced Chemical Engineering as Indonesia Endowment Fund for Education Awardee at Imperial College London. |
 P. T. P. Aryanti | Putu Teta P. Aryanti received her PhD in Chemical Engineering from Institut Teknologi Bandung (Indonesia) in 2016 under the supervision of Prof. Wenten. Her master's degree was received in 2002 from Institut Teknologi Sepuluh Nopember (ITS), Indonesia, and her bachelor's degree from Institut Teknologi Nasional Malang (ITN Malang), Indonesia, in 1997. She joined the chemical engineering department at Universitas Jenderal Achmad Yani (UNJANI), Indonesia, in 2016 and her current research interests include membrane preparation and modification. |
 R. R. Mukti | Rino R. Mukti received his PhD degree from Technische Universität München (Germany) in 2007 under the supervision of Prof. J. A. Lercher. He carried out postdoctoral research at The University of Tokyo, (Japan) in the group of Prof. T. Okubo, working on the development of hierarchical zeolite and understanding the crystallization mechanisms of novel crystal morphologies. Since 2011, he has been an assistant Professor at Institut Teknologi Bandung, Indonesia. His research interests involve the synthesis of microporous, mesoporous, and hierarchically porous crystalline materials and the development of characterization based on spectroscopy techniques. |
 Khoiruddin | Khoiruddin received his bachelor's degree from Universitas Diponegoro, Indonesia, and his master's degree from Institut Teknologi Bandung (ITB), Indonesia, in chemical engineering. He is currently a PhD student at chemical engineering, ITB, advised by Professor Wenten. His research focuses on the preparation, characterization, modification, and application of heterogeneous ion-exchange membranes. |
Introduction
Membrane technology has shown an excellent performance in separation processes.1–9 Membranes have lower energy and operational costs and are more scalable than conventional separation processes.10–12 They have developed rapidly during the last 40 years. During those years, many modifications have been developed in respect to process configuration and/or membrane materials.13–23Pervaporation is a membrane-based separation process wherein the membrane acts as a selective barrier between a feed (liquid phase) and a permeate (vapor phase). The desired components in the feed pass through the membrane by vaporization. A vacuum is maintained on the permeate side to allow the permeating components to vaporize. Separation by pervaporation is almost independent of the vapor–liquid equilibrium, because the transport depends on the sorption and mobility of the components in the membrane.24 Pervaporation is mainly used for the separation of water from organic liquids due to the azeotropic limitations of some mixtures during conventional distillation processes. Different types of membrane materials have been used for pervaporation, such as polymeric, ceramic, and composite membranes.25
Zeolite has been considered as a promising pervaporation membrane material due to its uniform and well-defined pore size, high porosity, and ability to operate under extreme conditions.26 As the most frequently produced zeolite type, LTA zeolite is a prominent candidate.27 Due to their high hydrophilicity, LTA zeolite membranes have shown excellent performances in separations related to polar molecules. In addition, their tunable pores make them sufficiently flexible for use in molecular sieving separation.28
LTA membranes were the first zeolite membranes to be used in commercial scale applications.29 In 1999, the first industrial facility for ethanol dehydration using NaA zeolite membranes was established by Mitsui Engineering and Shipbuilding Co., Japan for solvent dehydrations such as ethanol, IPA, and methanol.30 In 2011, another five plants for ethanol dehydration (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000; 60000; 80000; 100000; and 200000 L per day) were constructed by the German consortium GFT Membrane Systems GmbH Homburg/Saar, which were equipped with four-channel-alumina tubes coated with LTA zeolite membrane (on the inner wall of the alumina tube).27 Currently, there are over 100 industrial facilities based on LTA zeolite membranes.31
000; 60000; 80000; 100000; and 200000 L per day) were constructed by the German consortium GFT Membrane Systems GmbH Homburg/Saar, which were equipped with four-channel-alumina tubes coated with LTA zeolite membrane (on the inner wall of the alumina tube).27 Currently, there are over 100 industrial facilities based on LTA zeolite membranes.31
There are many reviews discussing zeolite membranes and their features.25,29,32–35 Some reviews even focused on specific applications of zeolite membranes, such as fuel cells,36 gas separation,12 membrane reactors,37 and some emerging applications.38,39 However, to the best of our knowledge, there is no review that specifically discusses LTA zeolite membranes, although they have many unique features and were the first commercially available zeolite membranes. This paper will discuss general research trends related to LTA zeolite membranes, including their application in pervaporation, industrial use, preparation, and future potential. Special attention will be given to the fabrication of LTA zeolite membranes in order to broaden their use on an industrial scale.
Principles and practice of zeolite membranes in pervaporation
Pervaporation is a membrane-based process in which a liquid-phase stream is placed in contact with a dense membrane on the feed side and where permeate is removed as vapor by applying a vacuum on the permeate side.40 The overall results are a vapour permeate and a liquid retentate. Pervaporation can be classified as vacuum pervaporation and sweep gas pervaporation, according to the process used to establish a partial pressure difference41 (Fig. 1). Mostly, vacuum pervaporation is used.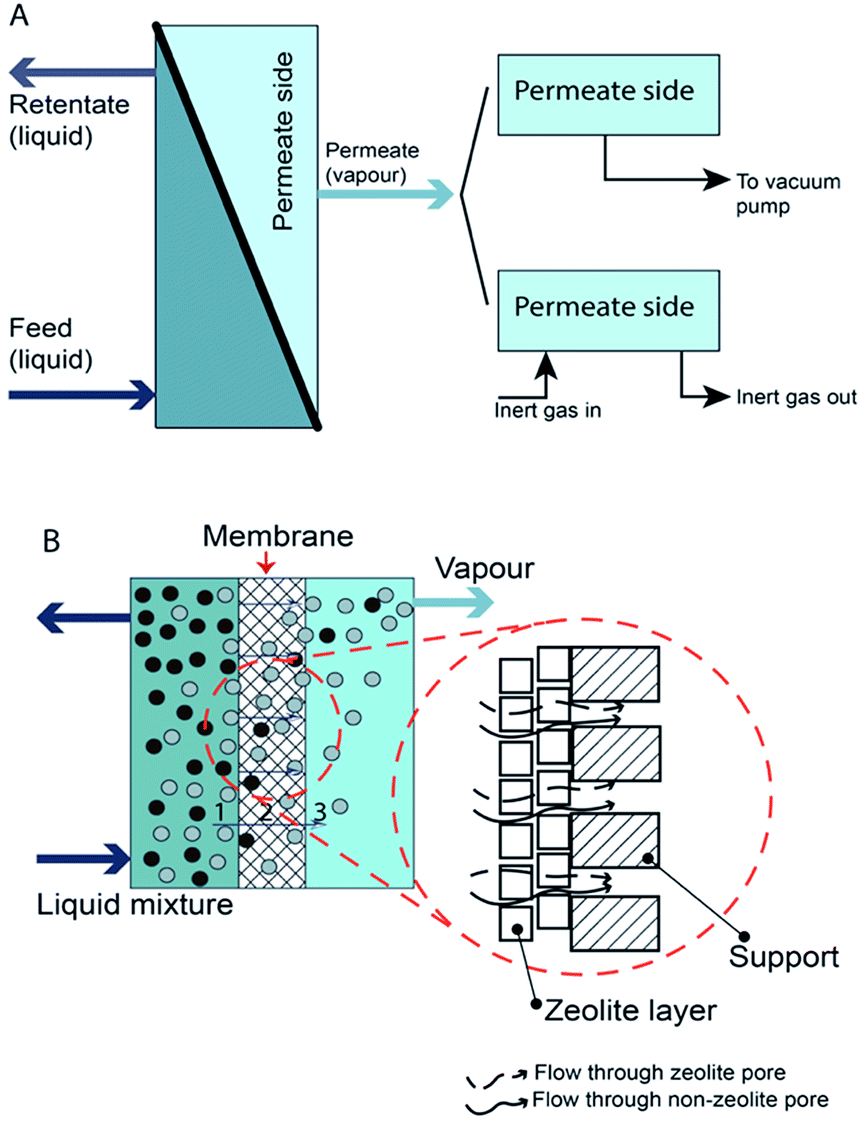 | ||
| Fig. 1 Schematic of pervaporation process. (A) Type of pervaporation and (B) mechanism of pervaporation process. | ||
Pervaporation is used to separate a liquid mixture containing two or more miscible components that have azeotropic or close-boiling point characteristics. The features of pervaporation are low temperature and pressure, ability to separate an azeotropic mixture with low energy consumption, without entrainer or contamination, and independence from vapor–liquid equilibrium.42 With these advantages, pervaporation can be used as a potential alternative to distillation. The potential applications of pervaporation include: (1) dehydration of organic-aqueous mixtures, (2) removal of the organic fraction from an organic-aqueous mixture, and (3) separation of organic–organic mixtures.43,44 Today, pervaporation is widely used in the dehydration of organic-aqueous mixtures. Furthermore, pervaporation can be a breakthrough technology in organophilic separation, pervaporation membrane reactors, and bioethanol upgrading.45
Pervaporation is considered to be one of the most useful and promising methods in the dehydration of organic solvents (e.g., alcohols, ethers, acids, and ketones) since it has the capability to separate a solution beyond the azeotropic limit. The superiority of pervaporation is mainly due to its separation principle, which is not based on differences in volatilities, as encountered in the distillation process, but on solution–diffusion (or adsorption–diffusion in zeolite-based membranes). Traditional distillation can only be used to recover pure solvents with the use of entrainers. Subsequently, an additional separation step is needed.46 Moreover, products obtained using entrainers cannot be used for several applications. Pervaporation is also more energy efficient compared with distillation. Azeotropic distillation requires 3305 kJ kg−1 ethanol, while pervaporation only requires 423 kJ kg−1 for ethanol dehydration from 95% to 99.5%.47
Separation by pervaporation comprises several steps including sorption of components into a membrane phase, permeation or diffusion of components in the membrane phase, and evaporation of components on the permeate side. In particular, this can be achieved due to the different affinities and different diffusion rates of components in the membrane.48 The mechanism is generally explained by using the solution–diffusion model.
Permeate flux and selectivity are the two most important parameters that are usually used for evaluating pervaporation performance. The flux and selectivity of the pervaporation process have been explained by Shao and Huang49 using solution–diffusion theory as follows. When the liquid feed mixture makes contact with the membrane, a thermodynamic equilibrium is reached at the membrane–solution interface. The concentrations of a component in the feed solution (Ci,feed) and membrane (Ci,m) are expressed by:
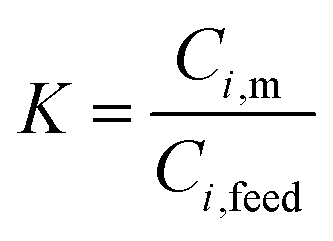 | (1) |
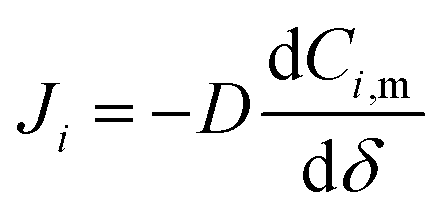 | (2) |
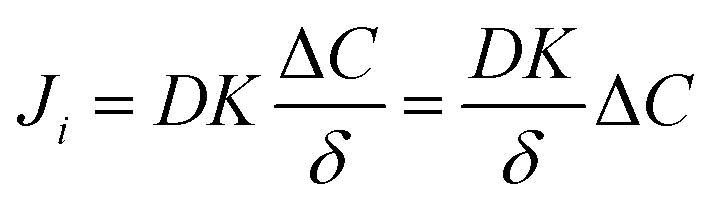 | (3) |
In eqn (3), if the concentration difference is taken as the driving force, then the permeability of the component (Pi) in the membrane phase is defined as:
| P = DK | (4) |
Meanwhile, the selectivity of the membrane is defined as:
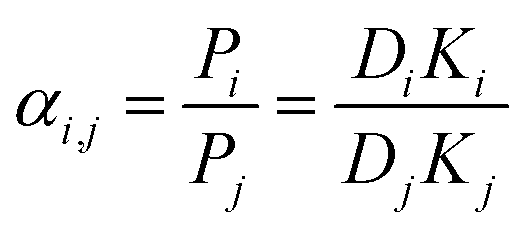 | (5) |
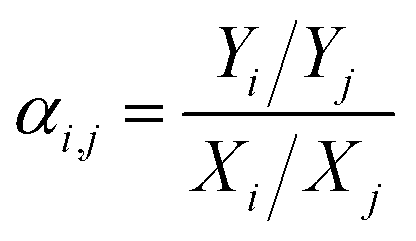 | (6) |
In pervaporation, the separation factor depends on the membrane material, the components in the feed, and also operating parameters such as feed temperature and composition as well as permeate pressure.48 To obtain both high flux and selectivity, a suitable membrane should be used. Two strategies were proposed by Jiang et al.50 to develop a suitable synthetic membrane for pervaporation. The first strategy is designing a membrane with desired physicochemical properties, while the second is macromolecular engineering of a membrane structure with the desired morphology, which is ultrathin and defect-free. Besides the performance, e.g. flux and selectivity, the membrane characteristics also determine the applicability of the pervaporation process. A hydrophilic membrane can be used for permeating water from a water–organic mixture, while a hydrophobic membrane is used for permeating organic compounds from the mixture.
The development of zeolite membranes has gained increasing interest due to their unique properties, such as well-defined pore sizes and superior mechanical, thermal, and chemical properties compared to polymeric membranes.51–53 With these properties, zeolite membranes are suitable for use in various potential applications, e.g. selective reaction membranes, catalytic membrane reactors, gas separation membranes, and pervaporation.53–55 The hydrophilic nature of zeolite makes it suitable for pervaporation processes, especially for solvent dehydration or water removal from organic/water mixtures. Zeolite membranes contain two main types of pores that are involved in pervaporation: zeolite pores and non-zeolite pores. The former are known as intracrystalline pores while the latter are intercrystalline pores. These names describe the pathways that molecules will follow during pervaporation. An intracrystalline pathway occurs within one single crystal of zeolite, while an intercrystalline pathway exists within a small channel formed by two adjacent zeolite crystals.56
During pervaporation, both types of pores influence the transport process across the membrane with their own mechanisms.26 Nevertheless, adsorption–diffusion is the primary transport mechanism and prevails in both zeolite and non-zeolite pores. However, for non-zeolite pores, there are also several mechanisms that may prevail, such as viscous flow and Knudsen diffusion. This is true especially when the size of the permeate is much larger than that of the zeolite pores.29
The adsorption process during pervaporation with a zeolite membrane is a physical phenomenon. The extent of interaction between the adsorbate and adsorbent is proportional to the energy released during adsorption. Hence, a molecule that releases more energy during adsorption will have a stronger adsorption tendency than a molecule with lower adsorption energy. This also indicates that adsorption energy is different from one molecule to another and is dependent on several factors, such as molecular weight, dipole moment, and molecular branching.29 Hence, there will be molecules that have low and high surface coverages on a zeolite membrane. Compared to other molecules, the heat of adsorption of water is unusually high (25–30 kcal mol−1) on zeolite.57 This heat of adsorption is greatly influenced by electrostatic forces because zeolites have an ionic structure and water is a polar molecule.47 Moreover, for a specific case where molecules have low surface coverage, surface coverage is inversely proportional to temperature since a higher temperature will promote desorption. Under these conditions, surface coverage during adsorption can be described by the equation below.29
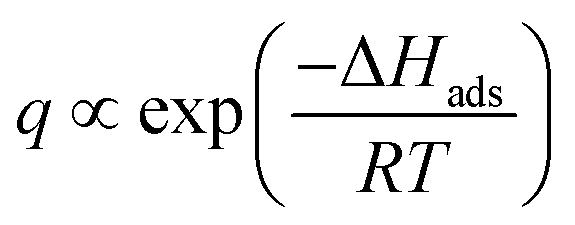 | (7) |
Aside from temperature, the surface coverage can also be affected by the hydrophilicity of the zeolite. Water will have a higher tendency to be adsorbed on a hydrophilic zeolite while organic molecules exhibit higher adsorption on hydrophobic zeolite. In this respect, water is preferentially adsorbed onto a hydrophilic zeolite surface compared with organic molecules. Moreover, this property is observed with both zeolite and non-zeolite pores in zeolite membranes.58 Hence, in the presence of a sufficient amount of water vapor, permeation of other components in the feed mixture can be significantly reduced due to the coverage of the pore surface by water molecules. However, this condition will not occur in the absence of water molecules, resulting in permeation transport that is governed by non-zeolite pores.
Accompanying adsorption, diffusion occurs because of the gradient in the chemical potential. For zeolite pores, the prevailing diffusion is configurational diffusion.29 In this respect, there are several parameters that influence the diffusion: pore diameter, molecular kinetic diameter, temperature, degree of coverage, and the presence of other components in the case of multicomponent diffusion.29
Using the adsorption–diffusion model as a basis, transport through a zeolite membrane can be modeled using the Maxwell–Stefan approach as developed by Pera-Titus and co-workers.56,59 The basic equation for the Maxwell–Stefan model for zeolite was given previously by Krishna and Wesselingh:25,60
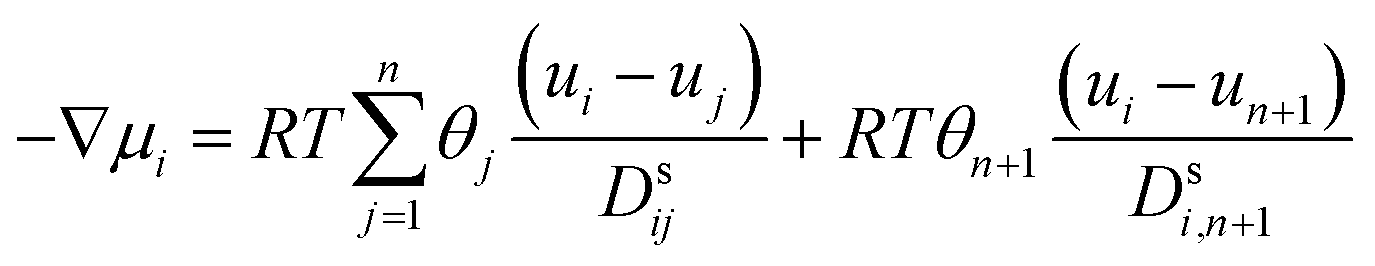 | (8) |
This is the basic equation used in many transport models proposed for permeation across zeolite membranes.56,59–62 This equation describes a process where an adsorbed molecule diffuses within a micropore where surface forces are dominant. The molecule will jump from one adsorption site to another driven by the surface chemical potential gradient (left-hand side of eqn (8)). Both terms on the right-hand side are related to friction occurring within the micropore. The first term on the right-hand side reflects friction between components i and j, while the second term denotes the friction experienced by an adsorbed molecule (molecule i) from the vacancy site. The above equation can then be simplified to the equation below.
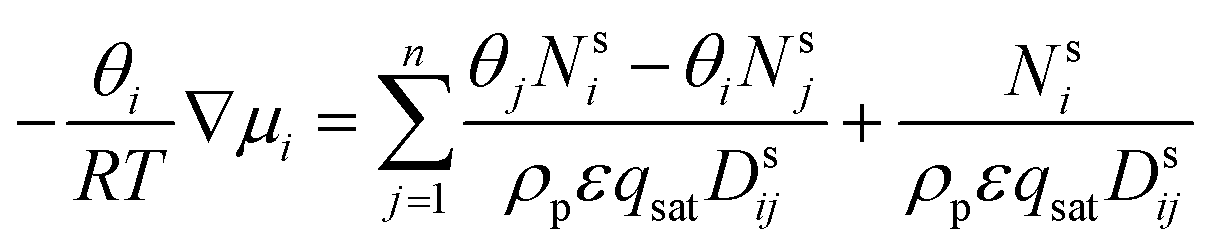 | (9) |
Further simplification of the equation can be made by ignoring the first term on the right-hand side (friction occurring between molecules). However, this condition only prevails if there is only one molecule that passes through the membrane; thus, there will be no friction between the molecule and its counterparts. In solvent dehydration using an LTA zeolite membrane, this condition exists when water vapor is the only molecule that is transported across the membrane. However, when there is competition to reach a vacant site of the LTA zeolite membrane, this term can no longer be neglected since more than one molecule is transported. And, as can be predicted, this is the case where a feed mixture contains water and a polar organic solvent, since both of these will compete for adsorption on the LTA zeolite surface.62
The Maxwell–Stefan eqn (9) above can be simplified to eqn (10). Eqn (10) denotes the linear relationship between the water flux and partial pressure and has been conventionally used by researchers to model pervaporation transport.62
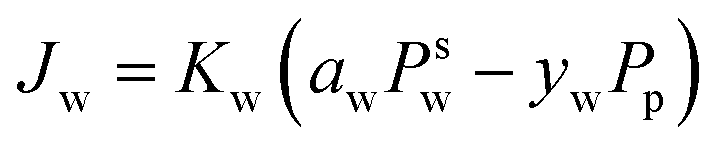 | (10) |
Recently, a specific transport mechanism in commercially available LTA zeolite membranes has been developed using a zeolite–non-zeolite pore approach.63 This model assumes that the LTA zeolite membrane has zeolite pores and very narrow and fine non-zeolite pores and that the membrane surface is dominated by zeolite pores. Furthermore, this model also assumes that zeolite crystals within the membrane structure are randomly arranged. This means that a parallel flow approach, where both zeolite and non-zeolite pores stand side by side and contribute evenly to permeate transport, can no longer be used.
During dehydration, permeation is begun by adsorption of water molecules into zeolite pores. At the beginning, the non-zeolite pores are not involved in water uptake due to the negligible available area of non-zeolite pores compared with zeolite pores and also because these pores contain condensate that hinders water molecules from passing through. Afterwards, water molecules will move from zeolite pores to non-zeolite pores, where capillary condensation occurs due to the small radius of the non-zeolite pores. This condensation phenomenon is responsible for filling up non-zeolite pores with water, resulting in inhibition of water uptake by non-zeolite pores from the feed side. Finally, upon the application of a vacuum at the permeate side, the condensate will evaporate and be transported through the support layer in which Knudsen diffusion governs the process.
Mass transfer within intercrystalline defects will be different from that within intracrystalline defects. Mass transfer across the membrane through intercrystalline defects is expected to occur by three main mechanisms: (1) Knudsen diffusion, (2) pressure-driven viscous flux, and (3) surface diffusion of adsorbed molecules by skating along the pore walls.61 However, viscous flux and Knudsen diffusion usually dominate the transfer mechanism because of large intercrystalline defect pores. Eqn (11) describes an intercrystalline flux that accounts for both viscous flux (first term on the right-hand of the equation) and Knudsen flux (second term on the right-hand of the equation).59 For practical purposes, empirical eqn (12) has been derived from eqn (11) to infer the intercrystalline porosity given the water/ethanol selectivity of a target zeolite LTA membrane, and this was proven to fit the experimental data for separation factors up to 1000.59
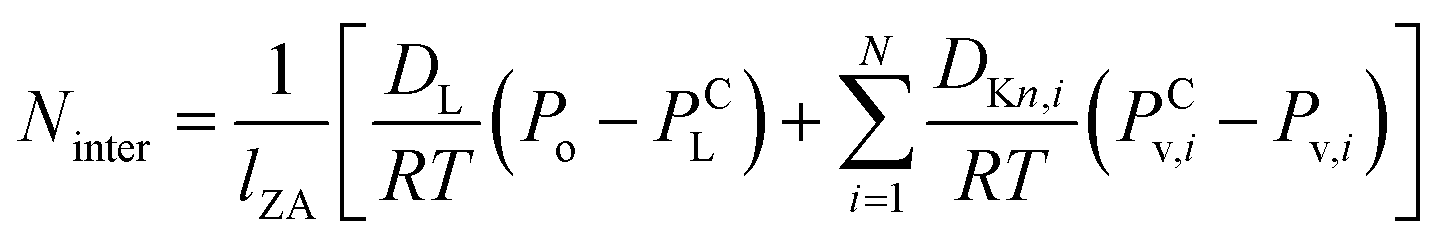 | (11) |
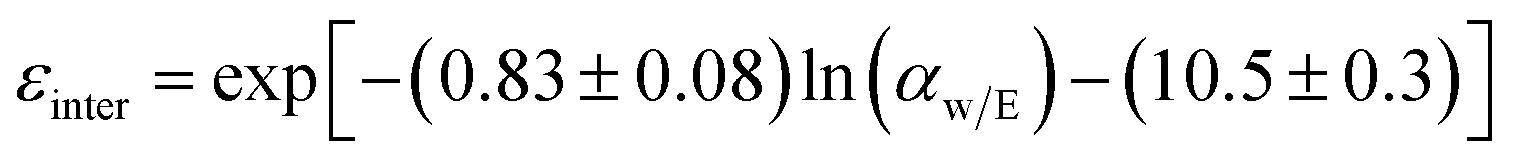 | (12) |
Fabrication of LTA zeolite membranes
Various types of zeolite membranes have been prepared and reported in the literature, such as LTA, FAU, MOR, FER, MEL, and AFI.33 Differences in the aluminium to silica ratios give the zeolite membranes unique structures and pore sizes. Zeolite can be defined as hydrated; the crystalline structure of tectoaluminosilicate is constructed from TO4 (T = tetrahedral atoms) with an oxygen atom that is shared between two adjacent tetrahedral atoms.64 In addition, zeolite also contains a cation in its structure to balance its negatively charged aluminosilicate framework. The cations can come from either alkali metals or alkaline-earth metals.64 Thus, the chemical composition of a zeolite can be represented as:| Ay/mm + [(SiO2)x(AlO2–)y]zH2O |
LTA zeolite exhibits typical single-crystal cubic shapes with smooth surfaces and angular edges.66 It usually has sizes of a few μm depending on the synthesis conditions. Usually, LTA zeolite is prepared in the Na+ form and the structure shows a three-dimensional pore network that resembles a sodalite cage, a building block similar to that of FAU and EMT zeolite.5–65 The tetrahedra are linked to form cages connected by pore openings of defined sizes. This connection results in three different cages: d4r, sodalite cage (β-cage), and LTA cage (α-cage). The negative charge on the lattice is neutralized by the positive charges of cations located within the material's pores. In the basic zeolites, these are usually univalent and bivalent metals or a combination of these. The metal cations may be replaced by acidic protons via ion-exchange to ammonium and subsequent calcination.67 Metal cation replacement will modify the pore opening from 0.4 nm in the normal Na form to 5 nm or 3 nm by ion exchange with aqueous solutions of calcium or potassium salts, respectively.29,68,69 The typical morphology of zeolite LTA membrane is shown in Fig. 2.
 | ||
| Fig. 2 Typical morphology of LTA zeolite crystal monolayer (magnification ×1200). Reprinted with permission from A. Kulak, Y. J. Lee, Y. S. Park and K. B. Yoon, Orientation-Controlled Monolayer Assembly of Zeolite Crystals on Glass and Mica by Covalent Linkage of Surface-Bound Epoxide and Amine Groups, Angew. Chem., 2000, 112, 980–983. Copyright (2000) John Wiley and Sons.72 | ||
The pore diameter of LTA zeolite is defined by an eight-membered oxygen ring with diameters between 0.23 and 0.42 nm.70 This size is close to that of a water molecule, which has a molecular diameter of about 0.275 nm.71 The LTA zeolite form has eight sodium ions located inside α-cages and four ions located in β-cages. LTA zeolite has a void volume fraction of 0.47, with an Si/Al ratio of 1.0. This high aluminum content makes it hydrophilic. Aluminum is trivalent so it requires cations to balance its charge when it is in the zeolite framework in place of Si. The localized electrostatic poles between the negatively charged framework and positively charged cations strongly attract highly polar molecules, resulting in a hydrophilic structure.29
Although the physical and chemical properties of zeolites largely depend on their structures, the morphology and size distributions of zeolite crystals also have a significant effect on their properties and applications.73 In general, LTA zeolites are extremely hydrophilic with a crystal pore size close to the size of a water molecule. Thus, LTA zeolite membranes are widely studied as pervaporative membranes for the dehydration of alcohol or other solvents.74
Primary growth method
Generally, LTA zeolite membranes are prepared by hydrothermal synthesis onto a porous support. Based on the growth of LTA crystals, the techniques can be classified as primary or secondary growth methods (Fig. 3). In primary growth, a support is coated with synthesis solution and undergoes direct hydrothermal synthesis. Meanwhile, in secondary growth, a support is seeded using zeolite crystals prior to hydrothermal synthesis. Several zeolite membranes, their preparation methods, and pervaporation performances are listed in Tables 1 and 2.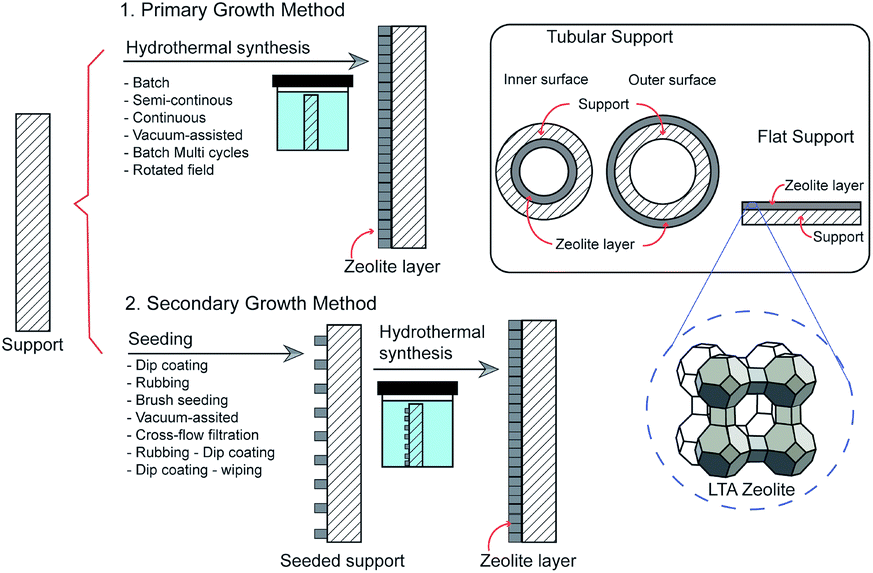 | ||
| Fig. 3 Schematic of LTA zeolite membrane preparation methods. | ||
| No. | Support material | Hydrothermal synthesis | Zeolite thickness (μm) | Pervaporation | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Reactor | T (°C); time (h) | Al2O3:SiO2:Na2O:H2O |
Feed mixture | T (°C) | Separation factor | Flux (kg m−2 h−1) | ||||
| a AC – autoclave; A + MH – aging and microwave heating; CH – conventional heating; EtOH – ethanol; IPA – isopropyl alcohol; NA – not available. | ||||||||||
| 1. | α-Alumina tube | Batch | 60; 24 | 1:5:50:1000 |
2 | EtOH/water (90%) | 75 | 5086 | 3.94 | 77 |
| 2. | α-Alumina tube | Batch; AC; A + MH | A: 50; 7; MH: 90; 25 min | 1:5:50:1000 |
5 | EtOH/water (90%) | 70 | >10000 |
0.64 | 80 |
| 3. | α-Alumina tube | Batch; MH + CH | MH: 25 min, CH: 90; 4 h | 1:5:50:1000 |
10 | IPA/water (95%) | 70 | >10000 |
1.44 | 81 |
| 4. | α-Alumina tube | Continuous flow | 80; 8 | 12:53:503:10000 |
5 | IPA/water (65%) | 90 | 41 | 17 | 79 |
| 5. | α-Alumina tube | Vacuum-assisted; AC | 90 | 1:5:50:1000 |
14 | IPA/water (95%) | 70 | 5289 | 1.12 | 74 |
| 6. | α-Alumina tube | Batch; AC; two-cycles | 65; 6 + 6 | 1:5:6:1000 |
20 | EtOH/water (90%) | 75 | 42 | ∼1 | 75 |
| 7. | UV-irradiated rutile/stainless steel (flat) | Batch | 80; 4 | 1:4.4:41.9:833.3 |
3.4 | EtOH/water (95%) | 45 | 54000 |
0.86 | 78 |
| 8. | Ceramesh (zirconia sheet on Ni/Cr alloy mesh) | Batch | 50–90; 2–48 | 1:5:55.12:104.66 |
NA | EtOH/water (90%) | 70 | 8500 | 0.82 | 105 |
| No. | Support material | Seeding | Hydrothermal synthesis | Th (μm) | Pervaporation | Ref. | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Technique | SS (μm) | SC | Reactor | T (°C); t (h) | Al2O3:SiO2:Na2O:H2O |
Feed | T (°C) | Separation factor | Flux (kg m−2 h−1) | ||||
| a AC – static autoclave; EG – ethylene glycol; EtAC – ethyl acetate; EtOH – ethanol; IPA – isopropyl alcohol; IPDC – interfacial polymerization and dip-coating; MH – microwave heating; RAC – rotating autoclave; SC – seed concentration; SS – seed size; th – thickness of zeolite layer; VTHD – varying temperature hot dip-coating.b Intermediate layer coated on the support.c First and second coating.d Selectivity. | |||||||||||||
| 1. | α-Alumina tube | Dip-coating | <1 | 0.5%-wt | Batch | 80–100; 4 | 1:2:2:150 |
4 | EtOH/water (90%) | 75 | 13000 |
4.3 | 87 |
| 2. | α-Alumina hollow fiber | Dip-coating | 0.09 | 5.5%-wt | Batch; RAC (30 rpm) | 100; 5 | 1:2:7.5:600 |
4.2–4.5 | EtOH/water (90%) | 75 | >10000 |
5.8 | 86 |
| 3. | α-Alumina tube | Rubbing | 1 | — | Semi-continuous | 90; 5 | 1:1.8:3.9:273 |
10 | EtOH/water (90%) | 125 | 3603 | 3.8 | 104 |
| 4. | α-Alumina tube | Rubbing | — | Gel | Batch; AC | 100; 3–4 | 1:2:2.2:150 |
— | EtOH/water (90%) | 75 | >10000 |
3.17 | 93 |
| 5. | α-Alumina tube | Rubbing | 0.8 | Paste | Batch | 100; 4 | 1:2:2:120 |
4–5 | EtOH/water (90%) | 75 | >10000 |
3.6 | 89 |
| 6. | α-Alumina tube | Brush seeding | 2 | 20 mg L−1 | Semi-continuous; AC | 95; 5 + 5 | 1:1.8:3.9:270 |
— | EtOH/water (90%) | 50–60 | 16222d |
0.43 | 26 |
| 7. | Rutile tube | Brush seeding | 2 | — | Continuous flow | 80–90; 7 | 1:1.8:3.9:270 |
10–20 | EtOH/water (90%) | 50 | 8538d | 0.89 | 106 |
| 8. | α-Alumina tube | Vacuum-assisted | 0.5–1.2 | 4–8 g L−1 | Batch; AC | 60; 24 | 1:5:50:1000 |
6 | IPA/water (95%) | 70 | >10000 |
1.67 | 88 |
| 9. | α-Alumina tube | Vacuum-assisted | — | — | Batch; AC | 100; 24 | 1:2:4.5:600 |
6.7 | EtOH/water (95%) | 70 | >10000 |
∼0.03 | 94 |
| 10. | α-Alumina tube | Vacuum-assisted | 0.5 | 3 g L−1 | Batch; MH | 90; 15–20 min | 1:5:50:1000 |
7.4 | EtOH/water (95%) | 70 | 6532.7 | 1.7 | 108 |
| 11. | α-Alumina tube | Vacuum-assisted | 2.8 | 0.5%-wt | Batch; AC | 100; 4 | 1:2:2:120 |
6 | EtOH/water (90%) | 75 | 17020 |
7.02 | 95 |
| 12. | α-Alumina tube | Vacuum-assisted | 1.5 | 5 g L−1 | Batch; AC | 105; 6 | 1:2:3:200 |
45 | EtAC/water (98%) | 50 | 163000 |
0.315 | 85 |
| 13. | Stainless steel tube | Colloidal seeding/sonication technique | 0.35 | 10% | Batch; AC | 90; 4 days | 1:1.8:1.84:300 |
5 | EtOH/water (90%) | 40 | 180d | 0.079 | 102 |
| 14. | α-Alumina tube | Cross-flow filtration | 2 | 20 mg L−1 | Batch; RAC (100); multi-steps | 100; 3 (4 steps or 4 × 3) | 1:2:2.1:120 |
30 | EtOH/water (92.8%) | 50 | 600d | 0.5 | 103 |
| 15. | α-Alumina tube | Cross-flow filtration | 2 | 20 mg L−1 | Semi-continuous; AC | 95; 5 + 5 | 1:2:2.2:150 |
— | EtOH/water (90%) | 50–60 | 386d | 0.73 | 26 |
| 16. | α-Alumina tube/γ-aluminab | Rubbing–dip coating | — | — | Batch | 100; 3 | 1:2:3.4:155 |
8 | EG/water (90 wt%) | 80 | 10996 |
7.16 | 98 |
| 17. | Mullite | Rubbing–dip coating | 2.1 | 1%-wt | Batch; AC | 100; 3.5 | 1:2:2:120 |
10–15 | EtOH/water (90 wt%) | 70 | >15000 |
3.26 | 96 |
| 18. | α-Alumina hollow fiber | Dip coating–wiping | 0.8 | 2%-wt | Batch; AC | 100; 3 | 1:2:3.4:155 |
3–4 | EtOH/water (90 wt%) | 75 | 10000 |
9 | 99 |
| 19. | α-Alumina/PES hollow fiber | Dip coating–wiping | 0.19 | 1%-wt | Batch; AC | 100; 4 | 1:2:3.4:155 |
3.1 | EtOH/water (90 wt%) | 75 | >10000 |
11.1 | 100 |
| 20. | α-Alumina tube | VTHD | 2 + 0.4c | 2 + 0.5c%-wt | Batch; AC | 80; 5 | 1:5:50:1000 |
4 | EtOH/water (90%) | 70 | >10000 |
2.85 | 90 |
| 21. | α-Alumina tube | IPDC | 0.15 | 0.5%-wt | Batch; RAC (10 rpm) | 100; 4 | 1:2:3.4:155 |
3.8 | EtOH/water (90%) | 75 | >10000 |
9 | 101 |
The preparation of LTA zeolite membranes using primary growth methods has been reported in the literature. Several techniques have been developed for hydrothermal synthesis, such as batch, continuous, or semi-continuous mode. Basak et al. reported the synthesis of an LTA zeolite membrane on the inner wall of a porous alumina support by a low temperature (65 °C) primary crystallization method in batch mode.75 The support is dipped in synthesis solution and heated for a given time. The support is then placed in a new synthesis solution in an inverted position and heated for the same time. The results of the study showed that the crystallization time affects the thickness of the membrane, with thicknesses of 10, 20, and 30 μm for growth times of 6 h, 12 h, and 24 h, respectively. They found that the membrane with the fewest defects was obtained after 12 h (6 h + 6 h) crystallization time. With a higher crystallization time, defects were generated because the zeolite formed a thicker layer.
Aguado et al. reported the preparation of LTA zeolite membranes on macroporous α-alumina supports that had been previously modified.76 Prior to hydrothermal synthesis, the support was coated with three types of organic modifier, i.e. poly(diallyldimethylammonium chloride) or PDDA, polyethylene glycol (PEG), and glucose. The support was immersed in organic solution and calcinated (400 °C for 3 h). This calcination is required to remove the sacrificial organic phase. The modified support is then placed in an autoclave containing solution. During the hydrothermal synthesis, the autoclave is rotated (60 rpm) to prevent the incorporation of suspended crystals into the LTA zeolite layer. They found that the modifier improves the continuity of the LTA layer and the crystal intergrowth. The positive support provided by the PDDA layer is expected to improve the adhesion between zeolite particles and the support. This proposed method provides advantages such as simple pretreatment, elimination of pre-seeding steps, and better layer formation.
The formation of a continuous and defect-free zeolite layer on a coarse macroporous support is quite challenging. Li et al.77 used a linker, 3-aminopropyltriethoxysilane (APTES) functionalized Al2O3, to improve the link between the zeolite layer and the macroporous support. The use of a linker was aimed at providing heterogeneous sites for zeolite crystals on the support surface. First, a larger size of APTES-Al2O3 (1–1.5 μm) was deposited on the surface of the support. A smaller size (0.4 μm) was deposited later. The modified support (coated) then underwent hydrothermal synthesis for 24 h at 60 °C. By using this linker, a thin (2 μm) and compact LTA zeolite layer was formed. This strategy is potentially useful for the preparation of a thin and defect-free zeolite layer on a macroporous support.
To obtain a better interaction between the synthesis solution and the support, Van den Berg et al. used UV irradiation to modify a titania/stainless steel support before hydrothermal synthesis.78 They found that UV-irradiated titania/stainless steel has higher hydrophilicity. Consequently, the interaction between the synthesis gel and the support was improved and the number of nuclei on the support was increased. Thus, a high-quality membrane was produced. The membrane has a selectivity of up to 54000 and a flux of 0.86 kg (m2 h)−1 when tested for pervaporation of an ethanol/water mixture. However, the membrane was sensitive to defect formation.
Reactant depletion occurs during the hydrothermal synthesis, which could reduce nucleation and crystal growth during this phase. To solve this problem, Aguado et al. introduced a continuous synthesis system.79 In continuous flow, the synthesis solution is continuously recirculated through the inner wall of an asymmetric alumina tubular support. The support was placed in an autoclave for in situ hydrothermal synthesis (crystallization). A laminar flow is ensured with a Reynolds number of 1–55 for flow rates of 0.10–5.00 mL min−1. This flow regime is expected to produce a homogeneous deposition of synthesis solution. The advantages of this method are that depletion of nutrients in the synthesis solution can be avoided, the composition of the reactants can be effectively controlled, and scale-up of the procedure is more feasible. Huang et al. investigated a vacuum-assisted technique for the hydrothermal synthesis process.74 Their results showed that the negative effect of the gravitational force during solution coating can be reduced by a vacuum-assisted technique. This positively impacts the formation of the zeolite layer on the support. The entire surface of the support is coated by the zeolite particles; thus, a homogeneous layer is formed. Furthermore, the rate of coating can be decreased due to continuous transportation of the crystal from the suspension. In addition, the homogeneous layer facilitates the formation of a dense, defect-free, and uniform LTA zeolite membrane.
Generally, hydrothermal synthesis is conducted in an autoclave and uses conventional heating. Li et al. reported a preparation method called “in situ aging—microwave synthesis” for the preparation of an LTA zeolite membrane on an α-alumina support.80 Basically, this method is similar to the primary growth method. Before crystallization or thermal synthesis using microwave heating, the support containing the precursor is aged using conventional heating, which is known as in situ aging. The aim of this in situ aging was to adjust the synthesis mixture and form germ nuclei. Meanwhile, microwave heating is used for nucleation and crystal growth on the support. The results of this study indicated that in situ aging is necessary for primary growth using microwave heating. A high-quality LTA zeolite membrane was successfully prepared by this method. Microwave heating could increase the crystallization rate and produce fine crystals.
Huang and Yang investigated the effect of heating during hydrothermal synthesis on a zeolite structure and the performance of the zeolite membrane.81 They compared conventional, microwave, and combined microwave/conventional heating in the preparation of an LTA zeolite membrane using a porous α-Al2O3 tube as the support. They found that uniform and small zeolite particles (2–3 μm) were formed when using only microwave heating (90 °C; 25 min). However, the membrane or zeolite layer was not continuous. When the membrane was prepared using only conventional heating (90 °C; 4 h), other types of crystals were formed and the crystal size was not uniform. In addition, the growth of the zeolite crystals was less ordered, while the surface of the membrane was very rough and loose. The membrane prepared by combining microwave heating and conventional heating had a better structure with uniform and compact LTA zeolite crystals. The zeolite layer was continuous with no intercrystalline gaps, highly intergrown, and very smooth. Microwave heating produces abundant nuclei, which leads to the formation of a uniform, pure, and dense zeolite membrane in the conventional heating process. The results showed that the separation factors (αwater/isopropanol) of the membranes were as follows: combined heating (10000) > conventional heating (171) > microwave heating (28.5). Meanwhile, the fluxes of the membranes (in kg m−2 h−1) were as follows: microwave (3.2) > conventional (1.6) > combined heating (1.4).
An interesting synthesis strategy has been developed by Huang and Caro for the preparation of LTA zeolite membranes. In the strategy, linkers were used to functionalize the support before hydrothermal synthesis.82–84 For example, a positively charged polymer, poly(diallyldimethylammonium chloride), was used as a covalent linker for the preparation of an LTA zeolite membrane on an α-Al2O3 support.82 Functionalization of the support was conducted via an adsorption process (at 60 °C for 24 h). After adsorption, the support underwent a hydrothermal synthesis process. The results showed that an oriented and uniform layer of LTA zeolite could be produced. In this case, the cationic layer facilitated homogeneous migration of LTA zeolite particles during the hydrothermal synthesis due to electrostatic interaction between the cationic polymer and zeolite particles. In another study, diisocyanates were used as covalent linkers to modify porous and non-porous supports such as an asymmetric porous α-Al2O3 disk, a non-porous glass plate, and non-porous stainless steel.83 Urethane bonds were formed on the supports as the products of reaction between isocyanate groups and surface hydroxyls. Like the cationic polymer in the previous study, the covalent linker helped to anchor the zeolite particles on the support surface during hydrothermal synthesis. A uniform and defect-free LTA zeolite membrane could thus be prepared. Covalent bonding was also achieved by treatment with 3-chloropropyltrimethoxysilane (CPTMS),84 which was deposited on the supports (0.2 mM in 10 mL toluene at 100 °C for 1 h, under argon). The modified supports were then used for preparation of the LTA zeolite layer. As in previous studies, the prepared LTA zeolite membrane was thin, dense, and defect-free. This strategy is potentially suitable for the preparation of defect-free LTA zeolite membranes without seeding.
Secondary growth method
The primary growth method is simpler than the secondary growth method because seeding is unnecessary. However, random crystallization resulting in an inhomogeneous layer is the main drawback, which has made this method less popular. The secondary growth method is considered as the most effective method for the preparation of defect-free zeolite membrane. Numerous studies have been reported on the preparation of LTA zeolite membranes using this method. In the secondary growth method, zeolite crystals are seeded on the support prior to hydrothermal synthesis. The properties of the crystal seeds, such as crystal size, thickness, density, and continuity of the seed layer, are crucial parameters that affect the separation properties of as-synthesized LTA zeolite membranes.76 The support should be homogeneously covered with a seed layer to achieve a homogeneous, continuous, and defect-free zeolite membrane. Various seeding techniques have been used in the preparation of zeolite membrane, including dip-coating, rubbing, cross-flow filtration of a suspension, and combined techniques. In hydrothermal synthesis, the seeds act as nuclei that provide sites for zeolite growth. In a seeded support, crystal nucleation and growth are separated; thus, membrane formation can be effectively controlled.74,85 Seeding provides some advantages; for example, the crystallization time can be reduced, crystal growth is ensured on the support rather than in the solution, and transformation of nuclei into other types of crystal is avoided.74,85LTA zeolite membranes are usually synthesized on the inner or outer wall of a tubular support (Fig. 2). Synthesis of a zeolite layer on the inner wall of the support is attractive because the layer can be protected from mechanical damage.86
Dip-coating is a simple seeding technique where a support is dipped into a seed suspension before the hydrothermal synthesis process.58,87 The main drawback of dip-coating is that the seed cannot closely attach to the support surface and the colloidal suspension easily dribbles when the support is withdrawn.88 Therefore, it is difficult to obtain a uniform and continuous seeding layer. This reduced the reproducibility of the dip-coating technique.89
Li and co-workers used a varying temperature hot dip-coating seeding technique (VTHD) for the synthesis of an LTA zeolite membrane on a coarse macroporous support.90 VTHD comprises three steps, i.e. (1) seeds are dip-coated on the support at a higher temperature; (2) the seeded support is rubbed carefully to remove excess seeds that are loosely packed on the surface; and (3) the support is dip-coated in a seed suspension at a lower temperature. VTHD is reported as a flexible and effective method for controlling the seed concentration, seeding temperature, and seed size. This method makes it possible to prepare a dense, thin, and defect-free LTA zeolite membrane on a coarse macroporous support by using seeds with different sizes. Larger seeds were used as filler to reduce the pore size of the support while smaller seeds were used as nuclei. This method showed relatively high reproducibility (70%). This technique has also been used to prepare MFI91 and T zeolite membranes.92
In rub-coating, zeolite crystals or seeds are directly coated on a support surface by rubbing the seeds. Basak et al. used poly(ethyleneimine) or PEI to modify an α-alumina support prior to seeding. Seeds are rubbed onto the inner wall of the modified support using a glass rod.75 The seed support is then placed or dipped in synthesis solution and heated at 65 °C for different crystallization times (i.e. 6 h, 12 h, and 24 h). The total crystallization time comprises two crystallization steps. The first is used for crystallization where the support is in a top-side down position, while the second is used for an inverted position. The results showed that the PEI coating yielded a good quality membrane with a lower crystallization time (6 h). The PEI coating increased the thickness of the membrane layer. As the crystallization time was longer, the membrane was thicker. The thicker layer is more prone to defect formation due to increased strain. However, the crystal is easily washed down when the coated support is immersed in the synthesis solution.88
Wang et al. prepared an LTA zeolite membrane by using a rub-coating method for seeding a paste containing zeolite seeds.89 They also used a binding agent derived from seed particles in a hydrogel form to fill the large holes and produce a smooth seeded surface. This method can also prevent seed particles from intruding into the pores of the support. Deposition of seed paste is potentially useful for seeding a support with large pore sizes. Therefore, a cheap, symmetric, and defect-free LTA zeolite membrane can be prepared on a support with large pores. Ma et al. used a wetting–rubbing method to coat an α-alumina support with a seed hydrogel.93 The hydrogel has a similar composition to the synthesis solution (secondary growth). The hydrogel easily formed a uniform coating. LTA membranes with separation factors of up to 10000 can be synthesized.
Vacuum-assisted seeding has been reported by several researchers to improve the deposition of seeds on a support. An LTA zeolite membrane was prepared by Huang et al. using a vacuum-assisted filtration process for seeding.88 A support is immersed in a suspension containing seeds and is connected to a vacuum pump. During vacuum filtration, water flows through the support pores and the zeolite crystals are transported and coated on the support surface. This technique can be used to coat the support surface with a smooth and uniform layer of zeolite seeds. After seeding, the coated support underwent hydrothermal synthesis. It was reported that under the optimum conditions (seed size: 500–1200 nm; seed concentration: 4–8 g L−1; vacuum pressure: 0.010–0.025 MPa; coating time: 45–180 s), a high-quality LTA zeolite membrane was successfully formed. The membrane showed a separation factor of >10000 and a flux of 1.67 kg m−2 h−1 in the pervaporation of isopropanol/water (95% at 70 °C). Cho et al. prepared an LTA zeolite membrane using vacuum-assisted filtration for seeding with zeolite nanoseeds.94 Membranes with narrow non-zeolitic pores were produced. However, an insignificant water flux in the zeolitic pores was observed. In vacuum-assisted seeding, the thickness of the seed layer is tunable depending on the seed suspension concentration, coating time, and vacuum degree.95 Moheb Shahrestani et al. synthesized an LTA zeolite membrane for pervaporation of an acetate/water mixture by a vacuum-assisted technique for seeding micron-sized zeolite powder.85 They reported that by aging the synthesis gel at room temperature for 48 h prior to hydrothermal treatment, a high purity LTA zeolite can be effectively synthesized. Prolonging the aging time produced smaller particles and an impure zeolite phase. In addition, the suspension residence time (seeding time) and concentration were shown to have a significant effect on the formation of the seed layer. The most stable and uniform layer was obtained with 90 s and 5 g L−1 seeding time and seed concentration, respectively. Despite its ability to prepare a smooth zeolite layer, the vacuum-assisted seeding technique needs auxiliary equipment; thus, it is inconvenient to operate in large-scale production.96
Beside the above-mentioned techniques, several combined techniques have been introduced for seeding.97 These were used to solve the problems associated with simpler techniques such as dip-coating and rubbing. Dip-coating requires a support with a very smooth and uniform surface to avoid defect formation. However, commercial supports contain pin-holes and dents. With rubbing, it is difficult to achieve a uniform coverage of seeds on the support surface. Liu et al. studied the influence of seeding techniques on the preparation of an LTA zeolite membrane on a tubular mullite support.96 Three seeding techniques, i.e. dip-coating, rubbing, and combined rubbing and dip-coating, were used to coat mullite with LTA seeds (2.1 μm). They found that the dip-coating technique produced well-distributed seeds on a flat area of the support surface but poor coverage on dents and pinholes. On the other hand, rubbing introduced seeds into the dents and pinholes but gave inhomogeneous coverage on the flat area. Meanwhile, the combined seeding technique could provide a better seed layer compared to the two separate techniques. A uniform distribution of seed could thus be formed.
An LTA zeolite membrane was prepared by Jafari et al. on a modified α-alumina macroporous support using a rubbing–dip coating seeding technique.98 Zeolite nanoseeds were prepared using an organic structure-directing agent, tetramethylammonium hydroxide (TMAOH), in a hydrothermal treatment. Prior to seeding, an α-alumina support was modified with γ-alumina. The γ-alumina with denser pores was used as an intermediate layer. The γ-alumina layers were coated by dip-coating of α-alumina supports in boehmite sols. Due to its denser pores, this intermediate layer could avoid penetration of nanoseeds; thus, a smooth and thin zeolite layer could be obtained. A combined rubbing and dip-coating technique was used for seeding. In this technique, the seeds were rubbed on the support surface. Afterwards, the support was dipped into a seed suspension. The results of the study indicated that an ultra-thin layer of LTA zeolite could be synthesized. The thickness of the zeolite layer was less than 8 μm. The zeolite membrane was then tested for pervaporation of an ethylene glycol/water mixture. The LTA membrane showed an excellent separation factor (10996) with a relatively high permeation flux (7.16 kg m−2 h−1).
Wang et al. used a dip coating–wiping technique for the preparation of an LTA zeolite membrane.99 After a support is dipped into the LTA zeolite suspension, it is wiped. In wiping, two fingers of one hand in a Latex glove held the support next to the joint of the support and the Teflon while anther hand rotated and pulled the Teflon tube through the two fingers towards the end of the support. They reported that a high-quality LTA zeolite membrane was synthesized. Wiping resulted in a much smaller amount of seeds on the support, but a dense membrane was produced. The LTA zeolite membrane exhibited an excellent separation performance (>10000), indicating that the amount of seeds is not the critical factor for preparing a high-quality membrane. A dip coating–wiping technique was also used by Shao et al. for the preparation of an LTA zeolite membrane on asymmetric Al2O3 home-made hollow fibers with different macrostructures.100 LTA zeolite membranes with different thicknesses were made by varying the crystallization time and the number of synthesis cycles. The study indicated that the pervaporation flux was proportional to the porosity of the support. They also recommended developing and improving the support properties to obtain a high-performance zeolite membrane with good mechanical strength. A dip coating–wiping technique was also reported by Yan et al., using ball-milled microcrystal seeds.66
Cao et al. introduced a novel seeding technique combining interfacial polymerization (IP) and a dip-coating technique for seeding.101 This technique was used to coat nanocrystals (150 nm) onto a micrometer-sized α-Al2O3 hollow fiber support. In this technique, the seed suspension dissolved in piperazine with a monomer concentration of 1 × 10−2 g mL−1 was used as an aqueous phase and a solution of trimesoyl chloride in n-hexane at 1.5 × 10−3 g mL−1 was the organic phase. First, the support was dipped into an aqueous phase solution. The coated support was then dried at room temperature (for 0–10 minutes). Afterwards, it was dipped into the organic phase to produce a polyamide (PA) phase. The PA produced by IP acts as a medium to freeze and fix the seed crystals in a suitable position. The coated support was dipped in the seed suspension. A very thin seed layer with good quality and adhesion was achieved through two cycles of dip-coating without drying between cycles. The prepared membrane exhibited an average separation factor of >10000 and a flux of ∼9.0 kg (m2 h)−1 in the pervaporation of ethanol/water (90% at 75 °C).
Holmes et al.102 used a new technique known as colloidal seeding/sonication to prepare a zeolite membrane on a stainless steel tube support (pore size: 0.5 μm). Before seeding, the stainless steel was washed with toluene and acetone to remove oil and grease. It was then immersed in a suspension of nanocrystalline zeolite (in an ultrasonic bath). Then, it was sonicated for 6 hours at ∼70 °C. The seeded support was dried and used in hydrothermal synthesis. Sonication facilitated the anchoring of the nanocrystals (about 350 nm) on the support. The membrane prepared using this procedure has a relatively thin zeolite layer (5 μm). The performance of the membrane is shown in Table 2.
Dip coating, rubbing, and vacuum-assisted seeding are typically effective for seeding the outer wall of the support. However, as previously mentioned, preparing a zeolite membrane on the inner wall of a support could be more beneficial, especially for protecting the zeolite layer from mechanical damage. Pera-Titus et al. prepared an LTA zeolite membrane using a cross-flow filtration seeding technique to coat the inner wall of a surface.103 Cross-flow filtration seeding is aimed to introduce individual LTA zeolite crystals into the pores of the support. To avoid aggregation, the zeolite crystals are stirred and kept in an ultrasound bath. By using the cross-flow method, deposition of the zeolite seeds on the surface of the support and inside the pores can be effectively controlled. By using this technique, LTA zeolite membranes were produced with selectivities of up to 600 and fluxes of 0.50 kg m−2 h−1 in the pervaporation of an ethanol/water mixture (92:8 at 50 °C).
Pera-Titus et al. compared brush-seeding and continuous filtration methods in the preparation of an LTA zeolite membrane.26 In brush-seeding, a brush is rolled over zeolite powder. The brush is moved along the inner wall of the support while rotating it. In the cross-flow filtration technique, evolution of seeding is monitored with respect to the reduction in water permeability. By using the cross-flow technique, the seeding weight gain (SWG) or the deposition of seeds can be controlled. They found that the performance of the membrane was significantly influenced by the SWG, which could be easily adjusted when using cross-flow filtration seeding.
Besides the various seeding techniques, a reactor system and hydrothermal steps were developed to improve the hydrothermal synthesis process in the secondary growth method. Lai et al. used a rotating or tumbling autoclave (30 rpm) for preparing an LTA zeolite membrane on the inner surfaces of α-alumina hollow fibers after the support was coated using a dip coating seeding technique.86 An LTA zeolite membrane with a high separation factor (up to 10000 for ethanol/water mixture separation) was obtained. The tumbling autoclave could effectively homogenize the synthesis solution in the inner tube of the hollow fiber support and the bulk solution in order to maintain sufficient primary units for membrane formation.
Pera-Titus et al. used a semi-continuous system to periodically refresh the synthesis solution.26 In semi-continuous hydrothermal synthesis, a fixed volume of synthesis solution in an autoclave is refreshed at periodic intervals. They compared three different arrangements for semi-continuous synthesis; these were: (1) membrane set in an intermediate vertical position; (2) membrane attached to inlet tube of synthesis solution; and (3) membrane attached to outlet tube of synthesis solution. The best quality membranes were prepared under the second arrangement in which the fresh solution directly renews the synthesis solution in contact with the zeolite layer. The prepared zeolite membrane showed selectivities of up to 16000 and fluxes of 0.50 kg m−2 h−1 in the pervaporation of ethanol/water mixtures (90:10 wt%; at 50 °C). In semi-continuous hydrothermal synthesis, the synthesis solution is removed from the membrane environment at periodic intervals, while at the same time an equal amount of fresh solution is added.104 This is expected to provide an effective way of controlling the preparation solution especially on depletion of reactant. To solve the problem of the temperature changing during the introduction of colder solution, an appropriately higher temperature should be chosen.104
However, semi-continuous hydrothermal synthesis has several disadvantages, such as: a relatively large volume of synthesis solution surrounding the tubular support is not employed in the synthesis of the inner-wall layer, which is not feasible for large-scale implementation; periodic pulse-renewal may sweep away the zeolite seeds and remove the amorphous gel-layer covering the support surface; and it requires two semi-continuous synthesis cycles.106 Therefore, Pera-Titus et al.106 used continuous flow hydrothermal synthesis and compared it with other reported synthesis techniques such as semi-continuous, centrifugal field (rotated), and batch. For the continuous flow system, a support was seeded with zeolite crystals using a brush-seeding technique prior to hydrothermal synthesis. The membrane prepared by continuous flow has a uniform zeolite layer with a thickness of around 10–20 μm (for 7 hours of synthesis time). The membrane showed higher selectivity (αwater/ethanol: 8538) and flux (0.89 kg m−2 h−1) than those prepared by semi-continuous (αwater/ethanol: 2444; 0.48 kg m−2 h−1) and centrifugal techniques (αwater/ethanol: 502; 0.62 kg m−2 h−1). The high flux was reported to be a result of a thinner zeolite layer (almost half) being obtained by continuous flow compared to those synthesized by other techniques. Because the synthesis solution is continuously refreshed, the supply of nutrient for zeolite formation is sufficient during the hydrothermal synthesis. Therefore, a more crystalline layer could be produced with uniform and controlled growth of zeolite. Thus, the membrane prepared by continuous flow exhibited a higher selectivity. The proposed continuous flow hydrothermal synthesis provides some advantages:106 it is economic because only the synthesis solution in the inner wall of the support (lumen side) is refreshed; the technique can be easily scaled up; it can be used to prepared a zeolite membrane on the inner wall of the multi-channel and capillaries. They also claimed that the membrane prepared by using this technique exhibited the best selectivity and fluxes reported in the literature for inner-wall zeolite membranes.
Xu et al. investigated the effect of different parameters on the synthesis of an LTA zeolite membrane, including seeding and un-seeding, synthesis time, and synthesis stages.107 The results of the study showed that the seeded support has a better quality than the unseeded support. Seeding can accelerate the crystallization of zeolite and avoid the formation of other types of zeolite. A continuous zeolite layer was formed on the seeded support after 2 h of synthesis. The quality of the membrane was further improved after 3 h of synthesis. However, synthesis times longer than 4 h resulted in an impure zeolite phase and a low-quality membrane. Furthermore, the multi-stage synthesis may improve the quality of the membrane. The best membrane was produced after 2 h of synthesis time and two stages of synthesis. By applying more than one stage, more crystals were formed. This ensured the homogeneity and continuity of the layer formation. However, when the number of stages was increased further, the formation of other types of zeolite was also increased. Consequently, the quality of the LTA zeolite membrane was reduced.
As in the primary growth method, hydrothermal synthesis using microwave heating can improve the quality of the prepared zeolite membrane and shorten the synthesis time. An LTA zeolite membrane was prepared on an α-alumina support via vacuum-assisted seeding and microwave heat treatment by Kuanchertchoo et al.108 First, the α-alumina support was coated with α-alumina (pore size 0.06 μm) as an intermediate layer using a dip-coating method, where the support was dipped into a submicron α-alumina suspension. The coated support was then dried and calcinated. For zeolite membrane synthesis, the LTA seeds were deposited on the modified support using a vacuum-assisted technique. In hydrothermal synthesis, microwave heating was applied. To improve the quality of the LTA zeolite layer, a multi-stage synthesis was carried out. The optimum conditions were a synthesis temperature of 90 °C and 15–20 minutes microwave heating with a 0.5 μm LTA crystal seed concentration of 3 g L−1 via vacuum seeding. The synthesized membrane has a flux of 1.6 kg m−2 h−1 and a separation factor of 1760 when used for pervaporation of an ethanol–water mixture (95:5). A better performance was shown by a zeolite membrane with an intermediate layer. The synthesized membrane with the intermediate layer had a smoother surface and a thinner layer due to uniform seeding on the substrate. The results showed that the advantages of microwave heating include facile synthesis, a narrow crystal size distribution, and high crystallinity.108
Problems and challenges in LTA membrane preparation
The trend in using secondary growth methods has made zeolite crystal preparation increasingly important. To provide better properties and a homogeneous layer, nanoseeds are usually used. An LTA zeolite membrane with a high separation factor and reproducibility can be prepared with small seeds (<100 nm).109 Large seeds could result in an inhomogeneous layer with defects. Small seeds can homogeneously cover the surface of the support even at low concentrations. For larger seeds, a larger concentration is needed to obtain the same coverage. Low coverage leads to inhomogeneity and defects.109 The use of small seeds can also improve the crystallization rate.97However, the preparation cost of nanocrystals is high.76 Zeolite nanocrystals are usually hydrothermally synthesized using an organic structure-directing agent (SDA) in a clear solution.110–112 This type of synthesis usually faces several problems, such as:66 (1) a low-concentration solution results in a slow crystallization rate; (2) repeated high centrifugation (usually >10000 rpm) is needed; (3) a large amount of reactants remains unused; and (4) calcination is needed to remove the organic molecules embedded in the crystal structure, leading to irreversible agglomeration of the crystal. Therefore, a simple preparation of nanoseeds is needed. For example, Yang et al. prepared submicron seeds using a ball mill.97 The seeds, which have average particle sizes of 0.3, 0.5, and 1 μm, can be produced by ball-milling of LTA zeolite powders (∼2.8 μm) with a high-energy ball mill. The result indicated that ball-milled seeds provide a thoroughly intergrown LTA zeolite membrane with a higher crystallization rate (shorter synthesis time). Nanoseeds can avoid defect formation and non-zeolitic pores.109 Another strategy is the use of Pt(NH3)·4Cl2 as a crystallization directing agent.113 Pt is also used to form a Pt/LTA catalyst for various reactions. A platinum precursor promoted nanocrystal formation by either inhibiting the crystallization rate because of [Pt(NH3)4]2+ ion adsorption on the crystal surface or by promoting the nucleation rate. However, nanoseeds may penetrate and block the pores of the support if the support has irregular pore sizes or a macroporous structure. Therefore, nanoseeds require a support with consistent pore sizes.114
Intercrystalline defect transport is linked to the Al content in LTA zeolite. LTA zeolite has a negative crystal surface due to its high Si/Al ratio. Two adjacent negatively charged surfaces will prevent a negatively charged precursor from transporting into the space between them and the crystal intergrowth is stopped. This phenomenon contributes to defect formation in LTA zeolite membranes.115 The intergrowth supporting substance (ISS) concept was developed for Al-containing LTA membranes following its success with Al-rich MFI zeolite membranes. In fact, Al-rich LTA zeolite crystals are similar to Al-rich MFI in terms of the value of the zeta potential, which shows a strong negative surface charge. This negative zeta potential can be shifted near to the isoelectric point when adsorbing HMEDA-J2 (the ISS). This may improve the intergrowth of the seed crystal on the support. By using the ISS, an LTA membrane with an improved performance, particularly its permeability and selectivity, can be obtained. Nevertheless, LTA membranes prepared with an ISS still contain defects and their permselectivities are found to be in the range of the Knudsen diffusion factor.115
Covalent modification of the support, which has been discussed previously, is another interesting strategy for producing dense and defect-free LTA zeolite membranes.82–84 A covalent linker deposited on the support could assist the homogeneous migration of zeolite particles during hydrothermal synthesis. Therefore, dense and defect-free LTA zeolite membranes could be synthesized without seeding.
In addition to efforts to prepare a dense and defect-free LTA zeolite membrane, some efforts are also devoted to ensuring uniform zeolite orientation along the support's surface. As a result of the random orientation of the precursor layer, intergrown films are often randomly oriented. In one case, preferentially oriented films of silicalite were prepared with the preferred orientation induced by secondary growth.116 In these films, most of the surface crystals were aligned with their straight and “sinusoidal” channels nearly parallel to the film surface.116
Many synthesized LTA zeolite membranes have an anisotropic pore structure. The permeability of LTA zeolite membranes is influenced by the orientation of the zeolite crystals on the support surface.31,117 Zeolite crystals oriented perpendicularly to the porous support will give high permeability.
Zeolite LTA has a cubic shape and thus only a few orientations are possible. The most popular orientation of LTA membrane is [h 0 0], i.e. with the zeolite face parallel to the support surface.118 An oriented LTA zeolite membrane surface has a neat cubical one-layer formation. On the other hand, randomly oriented LTA zeolite membranes may have any possible orientation. Although oriented zeolite membranes have been reported in several studies, controlling the orientation of the zeolite crystals remains a challenge. Therefore, identification of the conditions and possible mechanisms for the growth of zeolite membranes is required.119
Oriented LTA zeolite can be synthesized using both primary and secondary growth methods. In situ or primary growth techniques use oriented deposition, which is based on the growth mode of the zeolite crystals.120 Secondary growth methods use oriented seed particles so that they will grow facing in a specific direction. However, most researchers used randomly oriented particles in the precursor layer. As a result of the random orientation of the precursor layer, the intergrown films are usually randomly oriented.116
Boudreau et al.120 synthesized an oriented LTA zeolite membrane on a silicon wafer via electrostatic deposition. Electrostatic deposition is achieved by modifying both the support and seeds so that they carry a charge. Polymers with positive and negative end groups are then placed alternately between the support and seed particles so that the particles will attach with a specific orientation. This method produces seeds with an [h 0 0] orientation (zeolite face parallel to support surface) that will grow to form a zeolite membrane with a similar orientation. This method has been used by several authors.121,122 However, no pervaporation performance data has been reported.
Solvent dehydration using LTA zeolite membranes
LTA zeolite membranes have been tested for several applications that already use polymeric membranes or other types of zeolite membranes such as solvent dehydration, desalination, esterification, and gas separation. Studies on desalination using LTA zeolite membranes have been reported in the literature. For example, Cho et al. used an LTA zeolite membrane for seawater desalination.123 They called the process pervaporative seawater desalination. About 1.9 kg m−2 h−1 of flux (at 69 °C) and more than 99.9% salt rejection could be achieved except for boron (about 80%). Malekpour et al. studied the performance of LTA zeolite membranes in the desalination of Cs+, Sr2+, and I− ionic solutions.124 The membrane could achieve 0.14–0.29 kg m−2 h−1 of flux (at 67 °C) with more than 99% ionic rejection. In other studies, Malekpour et al. synthesized LTA zeolite membranes and used them in the desalination of Cs+, Sr2+, and MoO42− ionic solutions.125 In this work, higher water fluxes were achieved (2.2–5.3 kg m−2 h−1 for Cs+ solutions; 1.4–3.0 kg m−2 h−1 for Sr2+ solutions; and 0.9–1.6 kg m−2 h−1 for MoO42− solutions; all at 25 °C).The potential applications of LTA membranes for vapor permeation in esterification processes were indicated in several studies. Ameri et al. used LTA zeolite membranes for the removal of water from the esterification of isopropanol/propionic acid.126 This process was known as vapor-permeation-aided esterification. In this process, the maximum water flux was about 0.25 kg m−2 h−1. A similar flux was obtained by Jafar et al. in vapor permeation during ethyl lactate esterification.127 A higher water flux, about 2.2 kg m−2 h−1, was achieved by Li et al. in vapor permeation during esterification of acetic acid and n-propanol.128 Iglesia et al. used LTA zeolite membranes in the esterification of ethanol and acetic acid.129 In their study, the separation factors of water/ethanol and water/acetic acid were 32 and 18, respectively. With the assistance of vapor permeation using LTA zeolite membranes, the reaction conversion of esterification could be enhanced. This was because the removal of water could shift the equilibrium.
The use of LTA zeolite membranes for gas separation was investigated in several studies. Aoki et al. synthesized LTA zeolite membranes on α-alumina and tested them for the separation of gas mixtures.130,131 LTA zeolite membranes could achieve a separation factor of 4.8 for H2/N2 (ref. 130) and more than 160 for H2O/H2 gas mixtures.131 Xu et al. reported the use of an LTA zeolite membrane for the separation of H2/n-C4H10 mixtures. The membrane exhibited a permselectivity of 106.132 Chen et al. synthesized an LTA zeolite membrane with a permselectivity of 7.0 for H2/C3H8 separation.133
Solvent dehydration is the most widely used and studied LTA zeolite membrane application. Solvent dehydration using LTA membranes is far superior to that using other materials, especially polymeric membranes. A comparison of some solvent dehydration studies using pervaporation is shown in Table 3. LTA zeolite membranes are very effective in the separation of water/ethanol mixtures due to their high hydrophilicity and pore sizes that are between the molecular sizes of water (0.26 nm) and ethanol (0.43 nm).27 LTA zeolite membrane is highly effective in solvent dehydration with a low water content compared to polymeric membranes.134 This feature is favorable because traditional separation processes cannot further purify most solvents due to azeotrope limits or high energy use.46 Ethanol (forms azeotrope at approximately 4 wt% water) and isopropanol (forms azeotrope at approximately 12 wt% water) are examples of such solvents.135
| Membrane support | Selective layer | Separation factor | Flux (kg m−2 h−1) | Temperature (°C) | Ref. |
|---|---|---|---|---|---|
| Polyamide | Polyamide | 26 | 0.38 | 25 | 136 |
| PVA/clay MMM | PVA/clay MMM | 112 | 0.039 | N/A | 137 |
| PVA | PVA | 115 | 0.12 | 60 | 138 |
| Chitosan | Chitosan | 1791 | 0.472 | 60 | 139 |
| PSF | PVA/Na-Alg | 384 | 0.384 | 45 | 140 |
| BAPP | BAPP | 22 | 0.27 | 25 | 141 |
| PSF | PSF | 600 | 0.7–0.9 | 25 | 142 |
| Nylon-4 | Nylon-4 | 4.5 | 0.35 | 25 | 143 |
| PP | PAA | 4.9 | 0.175 | 24 | 144 |
| γ-Alumina | Silica | 50 | 0.35 | 70 | 145 |
| γ-Alumina | LTA zeolite | >10000 |
6.9 | 75 | 146 |
In 1999, the first industrial facility for ethanol dehydration using LTA zeolite membranes was established by Mitsui Engineering and Shipbuilding Co., Japan for multi-purpose dehydration of solvents such as ethanol, IPA, and methanol.30 Currently, there are over 100 industrial facilities based on LTA zeolite membranes.147 On the other hand, the relatively high cost of LTA zeolite membranes makes it difficult to broaden their use towards industrial applications.89 LTA zeolite membranes, which offer high permeability, high separation performances, and low-cost support, are of great interest and are preferable in industrial applications.77
Most of the work related to LTA zeolite membranes is targeted at solvent dehydration, especially ethanol/water, which has the potential to be used as fuel. Ethanol/water mixtures have been used to test LTA zeolite membranes prepared using various methods.77,86,94,95,148–150 Much work has been done in this area, resulting in separation factors of α > 10000 with fluxes of 6.9–12.2 kg m−2 h−1 being reported for laboratory scale experiments.66,86,90,95,100,150–152 Other organic substances that are azeotropic with water are also used, such as ethylene glycol (EG)153–155 and isopropanol (IPA).26,88 LTA zeolite membrane performances reported by several authors are shown in Fig. 4. It is clearly shown that today's LTA membranes perform well in terms of separation factors. Today, more studies are addressed to increase their fluxes while maintaining their separation factors with easy fabrication methods (Fig. 4).
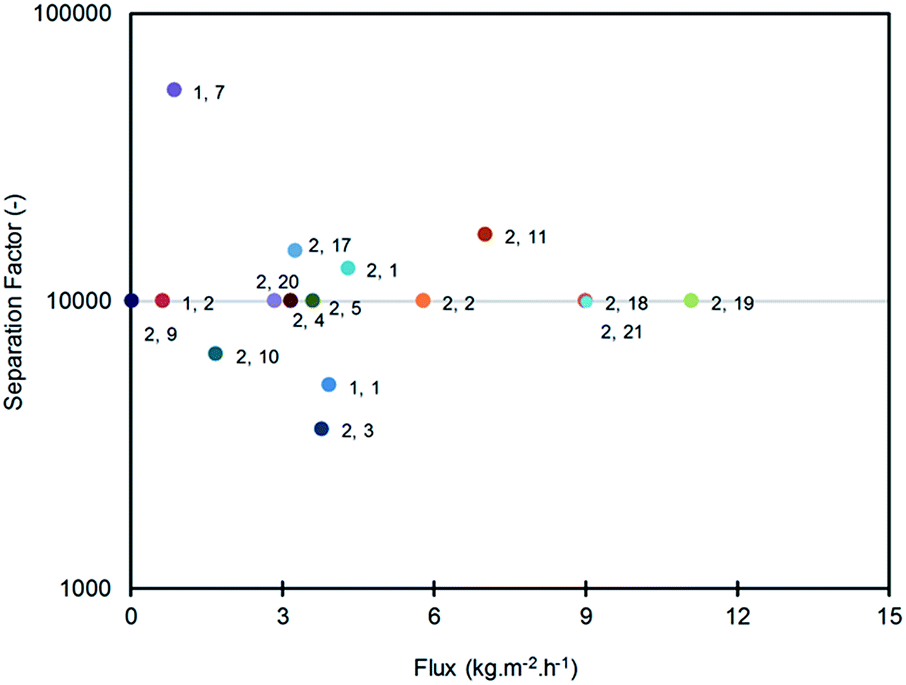 | ||
| Fig. 4 Graphical representation of membrane performance data presented in Tables 1 and 2 (data labels refer to table and number in table; (1, 2) means Table 1, no. 2). The separation factor depicted at 10000 might be higher due to instrumentation limitations. | ||
These high separation factors indicate that the permeate streams obtained contain only traces or undetectable solvents. Hydrophilic LTA zeolite is highly selective to water compared to other solvents. A very dense LTA zeolite membrane will reject solvent completely because of its 4 Å pores that can only let water through. The lower separation factors of LTA zeolite membranes are mainly caused by defects formed during preparation. Other components that are present in the zeolite layer such as polymers can provide an easier path for the solvent to pass through. Thicker membranes give additional resistance to mass transfer; therefore, additional time is allowed to reject undesired components and increase the separation factor.
Industrial scale pervaporation using LTA zeolite membranes
Although much work has been dedicated to the improvement of the separation performance of LTA zeolite membranes, the only current large-scale commercial use of zeolite membranes to the best of our knowledge is in solvent dehydration. An LTA zeolite membrane was firstly commercialized by Mitsui Engineering & Shipbuilding Co. in 1999.30 The first large scale pervaporation plant using zeolite NaA membrane was designed to dehydrate various solvents such as ethanol, IPA, and methanol. The plant is a major breakthrough in the development of LTA zeolite membranes. It proves their reliability in large scale operations. The highest reported water flux is 16 kg water per m2 per h in the pervaporation of water/i-propanol mixtures at 120 °C.31An LTA zeolite membrane used in the first commercial plant was reported by Morigami et al.30 The plant dehydrates 600 L h−1, 90 wt% waste ethanol feed to 515 L h−1, with a 99.8 wt% ethanol product. The plant uses 16 modules; each consists of 125 pieces of tubular type NaA zeolite membrane. The plant adopts two vacuum systems for more effective dehydration. Firstly, the feed is dehydrated to 1.5 wt% water at 15 mmHg permeate pressure. Then, a final dehydrated product of 0.5 wt% water is obtained at 8 mmHg permeate pressure.
Mitsui Engineering & Shipbuilding Co. has installed vapor permeation plants not only in Japan but also in Brazil and India with capacities of 3000 L per day and 30000 L per day, respectively.32 The plants dehydrate bioethanol at an operation temperature of 130 °C and an operating pressure of 600 kPa using LTA zeolite membranes. The plant successfully enhances the ethanol concentration from 93% in the feed stream to 99.7% in the retentate stream. However, the permeate stream still contains 0.5% to 1.0% ethanol.
An industrial-scale ethanol dehydration plant was installed in Hermsdorf, Germany.157 The plant was a cooperation between SMART Chemicals Development Ltd. and Inoceramic GmbH. It was tested to dehydrate an ethanol/water mixture (90/10 wt%) by pervaporation and vapor permeation. The membrane produces nearly pure water of >95% on the permeate side with a separation factor of up to 30000. The plant uses feeds from the processes of wine and grain fermentation. Bioethanol produced from these processes can be further purified up to 99.5% ethanol.
Nowadays, some companies already offer commercial LTA zeolite membranes for both industrial and laboratory use. Mitsui & Co. offer tubular NaA type zeolite membranes identical to those used in their pervaporation plant.62,134 I3 Nanotec and BNRI offer ZeoSep membrane-based compact, pre-assembled and automated pervaporation systems that can be customized to meet customers' specific process requirements.25 Inoceramic GmbH delivers NaA zeolite membranes and membrane modules for solvent dehydration by pervaporation or vapor permeation. The NaA zeolite is deposited on the internal side of tubular supports to protect it from mechanical damage and to produce optimum streaming conditions.157 Some companies offer commercial LTA zeolite membranes for solvent dehydration applications as shown in Table 4.
| Manufacturer | Pervaporation test | Membrane performance | Ref. | |
|---|---|---|---|---|
| Separation factor | Flux (kg m−2 h−1) | |||
| Busan Nanotech Research Institute (BNRI) | 10/90 wt% of water/ethanol at 75 °C | 10000–40000 |
3.5–4 | 25 |
| Inoceramic GmbH | 15/85 wt% of water/ethanol at 120 °C | N/A | 13 | 25 |
| Smart chemical company | 7/93 wt% of water/tetrahydrofuran at 55 °C | 1240 | 1 | 35 |
| Smart chemical company | 10/90 wt% of water/isopropyl alcohol | 16000 |
1.5 | 35 |
| Nanjing Jiusi Hi-Tech Co. | 80/20 wt% of water/ethylene glycol | 5000 | 4.0 | 156 |
| Mitsui Engineering & Shipbuilding Co | 10/90 wt% of water/ethanol | 10000 |
2 | 30 |
The economic issues of LTA zeolite membranes
LTA zeolite membranes have gained much attention with regard to their use in solvent—especially ethanol—dehydration. In spite of their superior performance, especially in solvent dehydration processes, the development of LTA zeolite membranes for commercial applications is limited by some parameters, particularly the high costs required for the membrane materials32,90,158 and the complexity of the preparation of reproducible defect-free membranes.87One major disadvantage of using inorganic membranes is their high cost compared to polymeric membranes. This disadvantage is also inherent in LTA zeolite membranes. The estimated cost of a zeolite membrane is about 1000–3000 € m−2,31,159,160 including a full module design. Most costs are related to the module and support and only 10–20% to the membrane itself.31 At all events, a rough estimate of the costs of a zeolite membrane without housing is 1000 € m−2 (and that with housing is in the 3000–8000 € m−2 range161). In fact, in 2000 the cost of a zeolite membrane module was already estimated to be about 2300 € m−2,31 which is comparable with metal (i.e., Pd) membranes (15000 € m−2), and it has been suggested that once in mass production and application, the cost may decline to below 1000 € m−2.161 Some economic estimations require a price of 200 € m−2 for viability, close to the cost of 40–400 € m−2 for a polymer membrane module used for processing natural gas.35 Although cost is the predominant factor regarding the application, feasibility studies for zeolite membrane-based processes are scarce. The cost that would make a zeolite membrane process profitable is strongly related to the targeted process.162 Zeolite membrane costs can be reduced by several methods, such as using a lower cost support,32 manufacturing a higher flux zeolite membrane,87 and fabricating polymer–zeolite membranes.153,155,163,164
Caro et al.32 pointed out that the price of a zeolite membrane was governed by the production cost of the supports (about 70%) rather than by that of the zeolite layer. There is a search, therefore, for economic and continuous production of membrane supports.32 Unfortunately, the use of cheaper supports leads to a lower performance zeolite membrane. This lower performance includes lower mechanical strength or a lower separation factor due to a higher defect tendency, which also lead to difficulties in the membrane's reproducibility. Some materials suggested to support zeolite membranes are coarse macroporous supports,90 porous ceramic tubes or discs,152 metal grids,165 and metal sheets.166
Li et al.90 investigated the use of a coarse macroporous support to grow an LTA zeolite membrane. The synthesis process used VTHD (see section on Secondary growth method). Low cost macroporous supports are desirable for industrial applications. Concerning this strategy, there is a trade-off between the membrane cost and its separation performance. One should consider, however, that changing the support can dramatically influence and even reverse the obtained separation behavior. This is because the properties of the support, such as thickness, porosity, tortuosity, and pore size, affect the total mass transfer in the membrane. Owing to the support resistance, this will cause a concentration polarization phenomenon.
Another way to cope with the economic aspects of LTA zeolite is the production of a higher flux membrane. By using a higher flux membrane, for a given production rate, a plant will need less membrane area to achieve the desired capacity, thus reducing the cost/product ratio, which is the more economically relevant comparison factor. Some factors used to increase the permeation flux of water through LTA zeolite membranes are: (a) thin zeolite layer; (b) high intercrystalline phase, and (c) a lower mass transfer resistance in the support.87 Increasing the membrane flux can also be achieved by using oriented zeolite membranes. Flux is a trade-off between permselectivity and mechanical strength in many cases. The processes of making higher flux LTA zeolite membranes should maintain sufficient permselectivity and mechanical strength, otherwise the cost will increase for other aspects.
Recently, Shao et al.100 showed that the main mass transfer resistance in pervaporation using LTA zeolite membranes is located in their supports. By using a high porosity support and a dense LTA zeolite layer, a high flux (11 kg m−2 h−1) and separation factor (>10000) were obtained. This shows that the achievement of high porosity with a cheap support is urgently needed to enhance LTA zeolite membrane performance in pervaporation.
In their application, LTA zeolite membranes require not only a support but also a strong module which could withstand under high temperature and pressure. The use of a polymeric component in its housing is avoided because this does not have the required stability under harsh conditions.34 Thus, the use of a polymeric component in either support or housing will negate the advantages of using an inorganic membrane. This condition is also responsible for the high LTA zeolite membrane price.
Summary and outlook
Pervaporation is a potential alternative to distillation, which is mostly used in solvent dehydration. The performance of a pervaporation process depends on the membrane material, the components in the feed, and the operating conditions. To obtain both high permeability and selectivity, a suitable membrane should be used. Besides the performance, the characteristics of the membrane also determine the applicability of the pervaporation process. For example, a hydrophilic membrane can be used for permeating water from a water–organic mixture, while a hydrophobic membrane is suitable for permeating the organic compounds. The unique properties of zeolite membranes, such as well-defined pore sizes and superior mechanical, thermal, and chemical properties compared to polymeric membranes, have attracted interest in developing zeolite membranes. The hydrophilic nature of zeolite makes zeolite membranes promising for pervaporation processes, especially for solvent dehydration.A high-quality zeolite membrane is defined by the absence of defects or pinholes at the macroscopic or mesoscopic scale. The presence of mesopores in a zeolite structure is attributed to the misdirection of zeolite growth. The flux through these defect mesopores can destroy shape selectivity and result in very low separation factors, especially in high-temperature operation.34
Pera-Titus et al.167 pointed out three main drawbacks when dealing with LTA zeolite membranes compared to other zeolite types: (1) dealumination under strongly acidic conditions, which limits their application at pH > 6; (2) membrane shrinkage due to a large amount of Al in the framework; and (3) the formation of large intercrystalline defects. The first and second problems are associated with the Al content in the LTA zeolite structure. An example of LTA zeolite membrane degradation in an acid environment has been reported by Jamieson et al.168 The degradation occurred due to the removal of Al from the zeolite framework. Preparation of an LTA zeolite membrane with a high silica content such as zeolite HOU-2,169 or LTA siliceous zeolite (Al-free) such as ITQ-29,170 may be used as one of the possible approaches to solve these problems. However, the high Si content could affect the performance of the LTA zeolite membrane in pervaporation because of the increase in membrane hydrophobicity.171,172 Therefore, if the membrane is used under acidic conditions, optimization of the Si/Al content should be made to obtain both high acid stability and high water permeability.
The main problem when preparing zeolite membranes is the presence of non-zeolite pores or intercrystalline defects. These pores can take the form of mesopores, grain boundaries, or even pinholes and cracks generated during synthesis or operation. These defects can be permeated by solvent, thus reducing the selectivity of the solvent dehydration operation.59 As stated earlier, intercrystalline defects also have a positive impact, mainly in the higher flux obtained during pervaporation. During membrane operation, flux and permselectivity are often in trade-off. The increasing number of intercrystalline defects both reduces selectivity and increases flux. With a higher flux, a lower specific membrane cost can be achieved.
Defects can usually be avoided during the synthesis process by using a controlled synthesis method so that a defect-free zeolite membrane is produced. Two major approaches used to produce defect-free zeolite membranes are modifying either the seeding step (e.g., dip-coating,158 vacuum seeding,95 and rub-coating50) or the synthesis process (e.g., microwave heating,80 combined heating,81 and continuous79).59 Some authors are also using small seed crystals that are either the direct result of synthesis151 or are ball milled.97 Another interesting strategy is covalent modification of the support for seeding-free hydrothermal synthesis.82–84 Despite various techniques being used to prepare dense LTA zeolite membranes, a facile mass production technique with a highly reproducible result remains a significant challenge.
Notes and references
- E. Drioli, A. Ali and F. Macedonio, Desalination, 2015, 356, 56–84 CrossRef CAS.
- I. G. Wenten and K. Khoiruddin, Desalination, 2016, 391, 112–125 CrossRef CAS.
- M. Purwasasmita, D. Kurnia, F. C. Mandias, K. Khoiruddin and I. G. Wenten, Food Bioprod. Process., 2015, 94, 180–186 CrossRef CAS.
- K. Khoiruddin, I. N. Widiasa and I. G. Wenten, J. Food Eng., 2014, 133, 40–45 CrossRef.
- P. T. P. Aryanti, R. Yustiana, R. E. D. Purnama and I. G. Wenten, Membrane Water Treatment, 2015, 6, 379–392 CrossRef.
- I. G. Wenten and I. N. Widiasa, Desalination, 2002, 149, 279–285 CrossRef CAS.
- A. K. Wardani, A. N. Hakim, K. Khoiruddin and I. G. Wenten, Water Sci. Technol., 2017 DOI:10.2166/wst.2017.173.
- I. G. Wenten, H. Julian and N. T. Panjaitan, Desalination, 2012, 306, 29–34 CrossRef CAS.
- K. Khoiruddin, A. N. Hakim and I. G. Wenten, Membrane Water Treatment, 2014, 5, 87–108 CrossRef.
- E. Drioli and E. Curcio, J. Chem. Technol. Biotechnol., 2007, 82, 223–227 CrossRef CAS.
- I. G. Wenten, K. Khoiruddin, F. Arfianto and Z. Zudiharto, Desalination, 2013, 314, 109–114 CrossRef CAS.
- N. Kosinov, J. Gascon, F. Kapteijn and E. J. M. Hensen, J. Membr. Sci., 2016, 499, 65–79 CrossRef CAS.
- K. P. Lee, T. C. Arnot and D. Mattia, J. Membr. Sci., 2011, 370, 1–22 CrossRef CAS.
- L. Eykens, K. De Sitter, C. Dotremont, L. Pinoy and B. Van Der Bruggen, Ind. Eng. Chem. Res., 2016, 55, 9333–9343 CrossRef CAS.
- S. Qiu, M. Xue and G. Zhu, Chem. Soc. Rev., 2014, 43, 6116–6140 RSC.
- H. Vinh-Thang and S. Kaliaguine, Chem. Rev., 2013, 113, 4980–5028 CrossRef CAS PubMed.
- J. Sunarso, S. S. Hashim, N. Zhu and W. Zhou, Progress in Energy and Combustion Science, 2017, 61, 57–77 CrossRef.
- F. Meng, S.-R. Chae, A. Drews, M. Kraume, H.-S. Shin and F. Yang, Water Res., 2009, 43, 1489–1512 CrossRef CAS PubMed.
- N. F. Himma, S. Anisah, N. Prasetya and I. G. Wenten, J. Polym. Eng., 2016, 36, 329–362 Search PubMed.
- I. G. Wenten and K. Khoiruddin, J. Eng. Sci. Technol., 2016, 11, 916–934 Search PubMed.
- D. Ariono, K. Khoiruddin, Subagjo and I. G. Wenten, Mater. Res. Express, 2017, 4, 24006 CrossRef.
- N. F. Himma, A. K. Wardani and I. G. Wenten, Polym.-Plast. Technol. Eng., 2017, 56, 184–194 CrossRef CAS.
- N. F. Himma, A. K. Wardani and I. G. Wenten, Mater. Res. Express, 2017, 4, 54001 CrossRef.
- R. Mallada and M. Menéndez, Inorganic membranes: synthesis, characterization and applications, Elsevier, 2008, vol. 13 Search PubMed.
- S. L. Wee, C. T. Tye and S. Bhatia, Sep. Purif. Technol., 2008, 63, 500–516 CrossRef CAS.
- M. Pera-Titus, R. Mallada, J. Llorens, F. Cunill and J. Santamaría, J. Membr. Sci., 2006, 278, 401–409 CrossRef CAS.
- A. Huang, N. Wang and J. Caro, Microporous Mesoporous Mater., 2012, 164, 294–301 CrossRef CAS.
- Z. Xue, J. Ma, W. Hao, X. Bai, Y. Kang, J. Liu and R. Li, J. Mater. Chem., 2012, 22, 2532–2538 RSC.
- T. C. Bowen, R. D. Noble and J. L. Falconer, J. Membr. Sci., 2004, 245, 1–33 CrossRef CAS.
- Y. Morigami, M. Kondo, J. Abe, H. Kita and K. Okamoto, Sep. Purif. Technol., 2001, 25, 251–260 CrossRef CAS.
- J. Caro, M. Noack, P. Kölsch and R. Schäfer, Microporous Mesoporous Mater., 2000, 38, 3–24 CrossRef CAS.
- J. Caro, M. Noack and P. Kölsch, Adsorption, 2005, 11, 215–227 CrossRef CAS.
- J. Caro and M. Noack, Microporous Mesoporous Mater., 2008, 115, 215–233 CrossRef CAS.
- J. Caro and M. Noack, Adv. Nanoporous Mater., 2010, 1, 1–96 Search PubMed.
- J. Gascon, F. Kapteijn, B. Zornoza, V. Sebastián, C. Casado and J. Coronas, Chem. Mater., 2012, 24, 2829–2844 CrossRef CAS.
- K. L. Yeung and W. Han, Catal. Today, 2014, 236, 182–205 CrossRef CAS.
- E. E. McLeary, J. C. Jansen and F. Kapteijn, Microporous Mesoporous Mater., 2006, 90, 198–220 CrossRef CAS.
- M. P. Pina, R. Mallada, M. Arruebo, M. Urbiztondo, N. Navascués, O. De La Iglesia and J. Santamaria, Microporous Mesoporous Mater., 2011, 144, 19–27 CrossRef CAS.
- J. Coronas and J. Santamaria, Chem. Eng. Sci., 2004, 59, 4879–4885 CrossRef CAS.
- M. Mulder, Basic Principle of Membrane Technology, Kluwer Academic Publisher, Netherlands, 1996 Search PubMed.
- H. O. E. Karlsson and G. Tragardh, Trends Food Sci. Technol., 1996, 7, 78–83 CrossRef CAS.
- S. Sridhar, B. Smitha and A. Shaik, Sep. Purif. Rev., 2005, 34, 1–33 CrossRef CAS.
- H. Matuschewski and U. Schedler, Desalination, 2008, 224, 124–131 CrossRef CAS.
- H. R. Mortaheb, F. Ghaemmaghami and B. Mokhtarani, Chem. Eng. Res. Des., 2012, 90, 409–432 CrossRef CAS.
- B. Van der Bruggen and P. Luis, Curr. Opin. Chem. Eng., 2014, 4, 47–53 CrossRef.
- P. D. Chapman, T. Oliveira, A. G. Livingston and K. Li, J. Membr. Sci., 2008, 318, 5–37 CrossRef CAS.
- M. Kazemimoghadam and T. Mohammadi, Desalination, 2010, 262, 273–279 CrossRef CAS.
- L. M. Vane, J. Chem. Technol. Biotechnol., 2005, 80, 603–629 CrossRef CAS.
- P. Shao and R. Y. M. Huang, J. Membr. Sci., 2007, 287, 162–179 CrossRef CAS.
- L. Y. Jiang, Y. Wang, T.-S. Chung, X. Y. Qiao and J.-Y. Lai, Prog. Polym. Sci., 2009, 34, 1135–1160 CrossRef CAS.
- G. Liu, W. Wei and W. Jin, ACS Sustainable Chem. Eng., 2014, 2, 546–560 CrossRef CAS.
- K. C. Khulbe, T. Matsuura, C. Y. Feng and A. F. Ismail, RSC Adv., 2016, 6, 42943–42961 RSC.
- H. Maghsoudi, Sep. Purif. Rev., 2016, 45, 169–192 CrossRef CAS.
- T. Tago, Y. Nakasaka and T. Masuda, J. Jpn. Pet. Inst., 2012, 55, 149–159 CrossRef CAS.
- Y. Y. Fong, A. Z. Abdullah, A. L. Ahmad and S. Bhatia, Chem. Eng. J., 2008, 139, 172–193 CrossRef CAS.
- M. Pera-Titus, J. Llorens, J. Tejero and F. Cunill, Catal. Today, 2006, 118, 73–84 CrossRef CAS.
- B. Smitha, D. Suhanya, S. Sridhar and M. Ramakrishna, J. Membr. Sci., 2004, 241, 1–21 CrossRef CAS.
- K. Okamoto, H. Kita and K. Horii, Ind. Eng. Chem. Res., 2001, 40, 163–175 CrossRef CAS.
- M. Pera-Titus, J. Llorens and F. Cunill, Chem. Eng. Sci., 2008, 63, 2367–2377 CrossRef CAS.
- R. Krishna and J. A. Wesselingh, Chem. Eng. Sci., 1997, 52, 861–911 CrossRef CAS.
- M. Pera-Titus, C. Fité, V. Sebastián, E. Lorente, J. Llorens and F. Cunill, Ind. Eng. Chem. Res., 2008, 47, 3213–3224 CrossRef CAS.
- D. Shah, K. Kissick, A. Ghorpade, R. Hannah and D. Bhattacharyya, J. Membr. Sci., 2000, 179, 185–205 CrossRef CAS.
- M. Kondo and H. Kita, J. Membr. Sci., 2010, 361, 223–231 CrossRef CAS.
- M. E. Davis and R. F. Lobo, Chem. Mater., 1992, 4, 756–768 CrossRef CAS.
- A. R. Loiola, J. C. R. A. Andrade, J. M. Sasaki and L. R. D. da Silva, J. Colloid Interface Sci., 2012, 367, 34–39 CrossRef CAS PubMed.
- H. Yan, N. Ma, Z. Zhan and Z. Wang, Microporous Mesoporous Mater., 2015, 215, 244–248 CrossRef CAS.
- C. A. Ríos, C. D. Williams and M. A. Fullen, Appl. Clay Sci., 2009, 42, 446–454 CrossRef.
- Z. Gao and L. V. C. Rees, Zeolites, 1982, 2, 79–86 CrossRef CAS.
- B. Biškup and B. Subotić, Sep. Purif. Technol., 2004, 37, 17–31 CrossRef.
- T. M. Salama, I. O. Ali, A. I. Hanafy and W. M. Al-Meligy, Mater. Chem. Phys., 2009, 113, 159–165 CrossRef CAS.
- P. Demontis, J. Gulín-González, H. Jobic, M. Masia, R. Sale and G. B. Suffritti, ACS Nano, 2008, 2, 1603–1614 CrossRef CAS PubMed.
- A. Kulak, Y. Lee, Y. S. Park and K. B. Yoon, Angew. Chem., 2000, 112, 980–983 CrossRef.
- X. Liu, Y. Wang, X. Cui, Y. He and J. Mao, Powder Technol., 2013, 243, 184–193 CrossRef CAS.
- A. Huang, W. Yang and J. Liu, Sep. Purif. Technol., 2007, 56, 158–167 CrossRef CAS.
- S. Basak, D. Kundu and M. K. Naskar, Ceram. Int., 2014, 40, 12923–12930 CrossRef CAS.
- S. Aguado, J. Gascon, D. Farrusseng, J. C. Jansen and F. Kapteijn, Microporous Mesoporous Mater., 2011, 146, 69–75 CrossRef CAS.
- H. Li, J. Xu, J. Wang, J. Yang, K. Bai, J. Lu, Y. Zhang and D. Yin, J. Membr. Sci., 2014, 471, 84–93 CrossRef CAS.
- A. W. C. Van Den Berg, L. Gora, J. C. Jansen, M. Makkee and T. Maschmeyer, J. Membr. Sci., 2003, 224, 29–37 CrossRef CAS.
- S. Aguado, J. Gascón, J. C. Jansen and F. Kapteijn, Microporous Mesoporous Mater., 2009, 120, 170–176 CrossRef CAS.
- Y. Li, H. Chen, J. Liu and W. Yang, J. Membr. Sci., 2006, 277, 230–239 CrossRef CAS.
- A. Huang and W. Yang, Mater. Lett., 2007, 61, 5129–5132 CrossRef CAS.
- A. Huang and J. Caro, Chem. Mater., 2010, 22, 4353–4355 CrossRef CAS.
- A. Huang and J. Caro, J. Mater. Chem., 2011, 21, 11424–11429 RSC.
- A. Huang, Q. Liu, N. Wang, X. Tong, B. Huang, M. Wang and J. Caro, J. Membr. Sci., 2013, 437, 57–64 CrossRef CAS.
- M. Moheb Shahrestani, A. Moheb and M. Ghiaci, Vacuum, 2013, 92, 70–76 CrossRef CAS.
- L. Lai, J. Shao, Q. Ge, Z. Wang and Y. Yan, J. Membr. Sci., 2012, 409–410, 318–328 CrossRef CAS.
- K. Sato and T. Nakane, J. Membr. Sci., 2007, 301, 151–161 CrossRef CAS.
- A. Huang, Y. S. Lin and W. Yang, J. Membr. Sci., 2004, 245, 41–51 CrossRef CAS.
- Z. Wang, Q. Ge, J. Gao, J. Shao, C. Liu and Y. Yan, ChemSusChem, 2011, 4, 1570–1573 CrossRef CAS PubMed.
- H. Li, J. Wang, J. Xu, X. Meng, B. Xu, J. Yang, S. Li, J. Lu, Y. Zhang, X. He and D. Yin, J. Membr. Sci., 2013, 444, 513–522 CrossRef CAS.
- W. Xiao, Z. Chen, L. Zhou, J. Yang, J. Lu and J. Wang, Microporous Mesoporous Mater., 2011, 142, 154–160 CrossRef CAS.
- X. Chen, J. Wang, D. Yin, J. Yang, J. Lu, Y. Zhang and Z. Chen, AIChE J., 2013, 59, 936–947 CrossRef CAS.
- J. Ma, J. Shao, Z. Wang and Y. Yan, Ind. Eng. Chem. Res., 2014, 53, 6121–6130 CrossRef CAS.
- C. H. Cho, K. Y. Oh, J. G. Yeo, S. K. Kim and Y. M. Lee, J. Membr. Sci., 2010, 364, 138–148 CrossRef CAS.
- Y. Liu, X. Wang, Y. Zhang, Y. He and X. Gu, Chin. J. Chem. Eng., 2015, 23, 1114–1122 CrossRef CAS.
- Y. Liu, Z. Yang, C. Yu, X. Gu and N. Xu, Microporous Mesoporous Mater., 2011, 143, 348–356 CrossRef CAS.
- Z. Yang, Y. Liu, C. Yu, X. Gu and N. Xu, J. Membr. Sci., 2012, 392–393, 18–28 CrossRef CAS.
- M. Jafari, A. Bayat, T. Mohammadi and M. Kazemimoghadam, Chem. Eng. Res. Des., 2013, 91, 2412–2419 CrossRef CAS.
- Z. Wang, Q. Ge, J. Shao and Y. Yan, J. Am. Chem. Soc., 2009, 131, 6910–6911 CrossRef CAS PubMed.
- J. Shao, Z. Zhan, J. Li, Z. Wang, K. Li and Y. Yan, J. Membr. Sci., 2014, 451, 10–17 CrossRef CAS.
- Y. Cao, M. Wang, Z. L. Xu, X. H. Ma and S. M. Xue, ACS Appl. Mater. Interfaces, 2016, 8, 25386–25395 CAS.
- S. M. Holmes, M. Schmitt, C. Markert, R. J. Plaisted, J. O. Forrest, P. N. Sharratt, A. A. Garforth, C. S. Cundy and J. Dwyer, Chem. Eng. Res. Des., 2000, 78, 1084–1088 CrossRef CAS.
- M. Pera-Titus, J. Llorens, F. Cunill, R. Mallada and J. Santamaría, Catal. Today, 2005, 104, 281–287 CrossRef CAS.
- M. P. Pina, M. Arruebo, M. Felipe, F. Fleta, M. P. Bernal, J. Coronas, M. Menéndez and J. Santamaría, J. Membr. Sci., 2004, 244, 141–150 CrossRef CAS.
- J. J. Jafar and P. M. Budd, Microporous Mater., 1997, 12, 305–311 CrossRef CAS.
- M. Pera-Titus, M. Bausach, J. Llorens and F. Cunill, Sep. Purif. Technol., 2008, 59, 141–150 CrossRef CAS.
- X. Xu, W. Yang, J. Liu and L. Lin, Microporous Mesoporous Mater., 2001, 43, 299–311 CrossRef CAS.
- N. Kuanchertchoo, R. Suwanpreedee, S. Kulprathipanja, P. Aungkavattana, D. Atong, K. Hemra, T. Rirksomboon and S. Wongkasemjit, Appl. Organomet. Chem., 2007, 21, 841–848 CrossRef CAS.
- J. Shao, Q. Ge, L. Shan, Z. Wang and Y. Yan, Ind. Eng. Chem. Res., 2011, 50, 9718–9726 CrossRef CAS.
- S. Mintova, N. H. Olson, V. Valtchev and T. Bein, Science, 1999, 283, 958–960 CrossRef CAS PubMed.
- O. Larlus, S. Mintova and T. Bein, Microporous Mesoporous Mater., 2006, 96, 405–412 CrossRef CAS.
- B. J. Schoeman, J. Sterte and J.-E. Otterstedt, Zeolites, 1994, 14, 110–116 CrossRef CAS.
- H. Yang, H. Chen, H. Du, R. Hawkins, F. Craig, Z. Ring, O. Omotoso, V. Munoz and R. Mikula, Microporous Mesoporous Mater., 2009, 117, 33–40 CrossRef CAS.
- W. Liu, J. Zhang, N. Canfield and L. Saraf, Ind. Eng. Chem. Res., 2011, 50, 11677–11689 CrossRef CAS.
- M. Noack, P. Kölsch, A. Dittmar, M. Stöhr, G. Georgi, M. Schneider, U. Dingerdissen, A. Feldhoff and J. Caro, Microporous Mesoporous Mater., 2007, 102, 1–20 CrossRef CAS.
- L. C. Boudreau and M. Tsapatsis, Chem. Mater., 1997, 9, 1705–1709 CrossRef CAS.
- J. H. Koegler, H. van Bekkum and J. C. Jansen, Zeolites, 1997, 19, 262–269 CrossRef CAS.
- K. Aoki, K. Kusakabe and S. Morooka, AIChE J., 2000, 46, 221–224 CrossRef CAS.
- G. Li, E. Kikuchi and M. Matsukata, Microporous Mesoporous Mater., 2003, 62, 211–220 CrossRef CAS.
- L. C. Boudreau, J. A. Kuck and M. Tsapatsis, J. Membr. Sci., 1999, 152, 41–59 CrossRef CAS.
- P. Frontera, F. Crea, F. Testa and R. Aiello, J. Porous Mater., 2007, 14, 325–329 CrossRef CAS.
- M. K. Naskar, D. Kundu and M. Chatterjee, J. Porous Mater., 2011, 18, 319–327 CrossRef CAS.
- C. H. Cho, K. Y. Oh, S. K. Kim, J. G. Yeo and P. Sharma, J. Membr. Sci., 2011, 371, 226–238 CrossRef CAS.
- A. Malekpour, A. Samadi-Maybodi and M. R. Sadati, Braz. J. Chem. Eng., 2011, 28, 669–677 CrossRef CAS.
- A. Malekpour, M. R. Millani and M. Kheirkhah, Desalination, 2008, 225, 199–208 CrossRef CAS.
- E. Ameri, A. Moheb and S. Roodpeyma, Chem. Eng. J., 2010, 162, 355–363 CrossRef CAS.
- J. J. Jafar, P. M. Budd and R. Hughes, J. Membr. Sci., 2002, 199, 117–123 CrossRef CAS.
- W. Li, W. Liu, W. Xing and N. Xu, Ind. Eng. Chem. Res., 2013, 52, 6336–6342 CrossRef CAS.
- Ó. de la Iglesia, R. Mallada, M. Menéndez and J. Coronas, Chem. Eng. J., 2007, 131, 35–39 CrossRef.
- K. Aoki, K. Kusakabe and S. Morooka, J. Membr. Sci., 1998, 141, 197–205 CrossRef CAS.
- K. Aoki, K. Kusakabe and S. Morooka, Ind. Eng. Chem. Res., 2000, 39, 2245–2251 CrossRef CAS.
- X. Xu, W. Yang, J. Liu, X. Chen, L. Lin and H. Brunner, Chem. Commun., 2000, 603–604 RSC.
- X. Chen, W. Yang, J. Liu and L. Lin, J. Membr. Sci., 2005, 255, 201–211 CrossRef CAS.
- V. Van Hoof, C. Dotremont and A. Buekenhoudt, Sep. Purif. Technol., 2006, 48, 304–309 CrossRef CAS.
- L. H. Horsley, Azeotropic Data—III, AMERICAN CHEMICAL SOCIETY, 1973, vol. 116 Search PubMed.
- Y.-C. Wang, S.-C. Fan, K.-R. Lee, C.-L. Li, S.-H. Huang, H.-A. Tsai and J.-Y. Lai, J. Membr. Sci., 2004, 239, 219–226 CrossRef CAS.
- J.-M. Yeh, M.-Y. Yu and S.-J. Liou, J. Appl. Polym. Sci., 2003, 89, 3632–3638 CrossRef CAS.
- M. L. Gimenes, L. Liu and X. Feng, J. Membr. Sci., 2007, 295, 71–79 CrossRef CAS.
- J. Ge, Y. Cui, Y. Yan and W. Jiang, J. Membr. Sci., 2000, 165, 75–81 CrossRef CAS.
- Y. Q. Dong, L. Zhang, J. N. Shen, M. Y. Song and H. L. Chen, Desalination, 2006, 193, 202–210 CrossRef CAS.
- Y. C. Wang, Y. S. Tsai, K. R. Lee and J. Y. Lai, J. Appl. Polym. Sci., 2005, 96, 2046–2052 CrossRef CAS.
- S.-H. Chen, K.-C. Yu, S.-S. Lin, D.-J. Chang and R. M. Liou, J. Membr. Sci., 2001, 183, 29–36 CrossRef CAS.
- K.-R. Lee, R.-Y. Chen and J.-Y. Lai, J. Membr. Sci., 1992, 75, 171–180 CrossRef CAS.
- Z.-K. Xu, Q.-W. Dai, Z.-M. Liu, R.-Q. Kou and Y.-Y. Xu, J. Membr. Sci., 2003, 214, 71–81 CrossRef CAS.
- R. W. van Gemert and F. Petrus Cuperus, J. Membr. Sci., 1995, 105, 287–291 CrossRef CAS.
- L. Lai, J. Shao, Q. Ge, Z. Wang and Y. Yan, J. Membr. Sci., 2012, 409–410, 318–328 CrossRef CAS.
- C. Yu, Y. Liu, G. Chen, X. Gu and W. Xing, Chin. J. Chem. Eng., 2011, 19, 904–910 CrossRef CAS.
- J. Zhang and W. Liu, J. Membr. Sci., 2011, 371, 197–210 CrossRef CAS.
- D. Liu, Y. Zhang, J. Jiang, X. Wang, C. Zhang and X. Gu, RSC Adv., 2015, 5, 95866–95871 RSC.
- Z. Zhan, N. Ma, H. Yan, Y. Peng, Z. Wang and Y. Yan, J. Membr. Sci., 2015, 485, 94–102 CrossRef CAS.
- J. Li, J. Shao, Q. Ge, G. Wang, Z. Wang and Y. Yan, Microporous Mesoporous Mater., 2012, 160, 10–17 CrossRef CAS.
- Z. Zhan, J. Shao, Y. Peng, Z. Wang and Y. Yan, J. Membr. Sci., 2014, 471, 299–307 CrossRef CAS.
- M. Shahverdi, B. Baheri, M. Rezakazemi, E. Motaee and T. Mohammadi, Polym. Eng. Sci., 2013, 53, 1487–1493 CAS.
- M. Jafari, A. Bayat, T. Mohammadi and M. Kazemimoghadam, Chem. Eng. Res. Des., 2013, 91, 2412–2419 CrossRef CAS.
- B. Baheri, M. Shahverdi, M. Rezakazemi, E. Motaee and T. Mohammadi, Chem. Eng. Commun., 2015, 202, 316–321 CrossRef CAS.
- C. Yu, C. Zhong, Y. Liu, X. Gu, G. Yang, W. Xing and N. Xu, Chem. Eng. Res. Des., 2012, 90, 1372–1380 CrossRef CAS.
- H. Richter, I. Voigt and J.-T. Kühnert, Desalination, 2006, 199, 92–93 CrossRef CAS.
- W. Yuan, H. Chen, R. Chang and L. Li, Desalination, 2011, 273, 343–351 CrossRef CAS.
- G. W. Meindersma and A. B. de Haan, Desalination, 2002, 149, 29–34 CrossRef CAS.
- S. Tennison, Membr. Technol., 2000, 2000, 4–9 CrossRef.
- N. W. Ockwig and T. M. Nenoff, Chem. Rev., 2007, 107, 4078–4110 CrossRef CAS PubMed.
- J. Van den Bergh and T. U. Delft, DD3R zeolite membranes in separation and catalytic processes: modelling and application, Delft University of Technology, 2010 Search PubMed.
- D. Bastani, N. Esmaeili and M. Asadollahi, J. Ind. Eng. Chem., 2013, 19, 375–393 CrossRef CAS.
- H.-M. Guan, T.-S. Chung, Z. Huang, M. L. Chng and S. Kulprathipanja, J. Membr. Sci., 2006, 268, 113–122 CrossRef CAS.
- B. Louis, P. Reuse, L. Kiwi-Minsker and A. Renken, Appl. Catal., A, 2001, 210, 103–109 CrossRef CAS.
- E. Mateo, R. Lahoz, G. F. de la Fuente, A. Paniagua, J. Coronas and J. Santamaría, J. Membr. Sci., 2008, 316, 28–34 CrossRef CAS.
- M. Pera-Titus, J. Llorens and F. Cunill, Chem. Eng. Process. Process Intensif., 2009, 48, 1072–1079 CrossRef CAS.
- H. L. Jamieson, H. Yin, A. Waller, A. Khosravi and M. L. Lind, Microporous Mesoporous Mater., 2015, 201, 50–60 CrossRef CAS.
- M. T. Conato, M. D. Oleksiak, B. Peter McGrail, R. K. Motkuri and J. D. Rimer, Chem. Commun., 2015, 51, 269–272 RSC.
- I. Tiscornia, S. Valencia, A. Corma, C. Téllez, J. Coronas and J. Santamaría, Microporous Mesoporous Mater., 2008, 110, 303–309 CrossRef CAS.
- G. Li, E. Kikuchi and M. Matsukata, Sep. Purif. Technol., 2003, 32, 199–206 CrossRef CAS.
- Y. Kiyozumi, Y. Nemoto, T. Nishide, T. Nagase, Y. Hasegawa and F. Mizukami, Microporous Mesoporous Mater., 2008, 116, 485–490 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |