
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of α-CaSO4·0.5H2O from flue gas desulfurization gypsum regulated by C4H4O4Na2·6H2O and NaCl in glycerol-water solution
Qing-Jun Guan,
Wei Sun *,
Yue-Hua Hu,
Zhi-Gang Yin and
Chang-Ping Guan
*,
Yue-Hua Hu,
Zhi-Gang Yin and
Chang-Ping Guan
Central South University, No. 932 South Lushan Road, Changsha 410083, China. E-mail: sunmenghu1@163.com
First published on 25th May 2017
Abstract
A brand new method to transform flue gas desulfurization gypsum (FGD gypsum) into α-calcium sulfate hemihydrate (α-HH) with short hexagonal prisms mediated by succinic acid disodium salt hexahydrate (C4H4O4Na2·6H2O) and NaCl in glycerol-water solution is studied, in which the appropriate reaction temperature is 90 °C. The addition of NaCl facilitates the dissolution of calcium sulfate dihydrate (DH) and creates much higher supersaturation which is a greater driving force for the phase transformation from DH to α-HH. C4H4O4Na2·6H2O as the crystal modifier effectively suppresses the α-HH crystal growth along the c axis, and the products generally change from needle-like particles to fat and short hexagonal prisms, which is attributed to the preferential adsorption of C4H4O42− on the top facets of the crystals by chelating Ca2+.
1. Introduction
Flue gas desulfurization gypsum (FGD gypsum), which is mainly composed of CaSO4·2H2O (DH), is the industrial waste residue produced by the limestone-gypsum wet process of flue gas desulfurization technology. The bulk deposition of FGD gypsum not only takes up plenty space, but also results in land and water pollution. Methods to rationally utilize FGD gypsum have drawn much attention in recent years. One of the most important methods to reuse FGD gypsum is the production of calcium sulfate hemihydrate (HH), including α-calcium sulfate hemihydrate (α-HH) and β-calcium sulfate hemihydrate (β-HH). Indisputably, α-HH is superior to β-HH due to its better workability and higher strength. It has been widely applied in moldings, special binder systems, dental materials, and in the construction industry.Numerous researchers have explored methods for the production of α-HH reusing FGD gypsum. One method is the autoclaving method,1 which involves heating DH in an autoclave at elevated temperatures and pressure under an atmosphere of saturated vapor. Obviously, this is an expensive procedure. Another method is the salt solution process under atmospheric pressure and at mild temperature,2–5 which requires high-concentration salt solutions, but evidently results in serious corrosion to equipment.
Recent scientific research has suggested that the transformation from DH to α-HH can be realized in alcohol water solution under mild conditions.6,7 Compared with the autoclave and salt solution methods, this process has the advantages of mild reaction conditions and no corrosion to equipment, and is thus more favorable for continuous industrial production. Although the transition from DH to α-HH is kinetically unfavorable in alcohol water solution, the addition of a small amount of salt as a phase transformation accelerator can significantly shorten the transition time.8 However, few researchers have explored the preparation of α-HH with a specific morphology and size using FGD gypsum in alcohol water solution. The morphology and size of α-HH usually determine its properties and functionalities. For instance, α-HH particles with a low aspect ratio or spherical shape possess better injectability and mechanic strength, and are preferable for use as bone cements.9,10 Therefore, in order to prepare α-HH products with low aspect ratios, crystal modifiers, among which carboxylic acids are highly effective,5,11,12 tend to be added during the transformation process. However, in alcohol water solutions, the influence of crystal modifiers on the morphology of α-HH crystals and transformation time from DH to α-HH has been rarely reported.
Herein, we introduce a brand new process to prepare α-HH with short hexagonal prisms by reusing FGD gypsum in glycerol-water solution. In the procedure, succinic acid disodium salt hexahydrate (C4H4O4Na2·6H2O) and NaCl are used as the crystal modifier and phase transformation accelerator, respectively, and the influence of the crystal modifier on the morphology of the α-HH crystals and the transformation time in glycerol-water solution is studied.
2. Experimental
2.1 Materials
FGD gypsum, the chemical composition of which is given in Table 1, was received from Panzhihua Iron & Steel Co., Ltd., Sichuan Province, China. Glycerol, NaCl and succinic acid disodium salt hexahydrate (C4H4O4Na2·6H2O) were purchased from Sinopharm Chemical Reagent Co., Ltd., Shanghai, China.| CaO | SO3 | H2O | SiO2 | Fe2O3 | Al2O3 | MgO | PbO | K2O | Total |
|---|---|---|---|---|---|---|---|---|---|
| 36.87 | 42.63 | 14.95 | 0.73 | 0.31 | 0.31 | 0.11 | 0.06 | 0.04 | 96.01 |
2.2 Experimental procedure
Glycerol (65 wt%)-water solutions with a certain amount of NaCl (by weight of FGD gypsum) and succinic acid disodium salt hexahydrate (C4H4O4Na2·6H2O) (by weight of FGD gypsum) were first added to a three-necked flask equipped with a reflux condenser on top of it, and the solution was stirred with a magnetic stirrer at a constant rate of 150 rpm and preheated to 90 °C in an oil bath. Then 60 g pretreated FGD gypsum (20 wt% solid content) was added to the reactor. During the reaction, 20 mL slurry was withdrawn at certain time intervals. Half of the sample was filtrated immediately, washed three times with boiling water and then rinsed once with ethanol before it was dried at 60 °C for 2 hours in an oven. The other 10 mL of slurry was filtered through a syringe filter with a 0.2 μm cellulose membrane for Ca2+ concentration determination.2.3 Characterization
The chemical compositions of FGD gypsum were investigated using X-ray fluorescence spectroscopy (XRF, Axios mAX, PANalytical B.V., Netherlands). The structures of the samples were determined by X-ray diffraction (XRD D8 Advanced, Bruker, Germany) using Cu Kα radiation (λ = 1.54178 Å) at a scanning rate of 5° min−1 in the 2θ range of 5° to 70°. The morphology of the samples was characterized with an optical microscope (DFC 480, Leica Microsystems Ltd., Germany). The surfaces of the crystal products were analyzed by X-ray photoelectron spectroscopy (XPS, ESCALAB 250Xi, Thermo Fisher, USA) with an.Al Kα photon energy of 1486.6 eV. The C 1s peak at 284.8 eV, which is related to the carbon adsorbed on the surface during the exposure of the samples to ambient atmosphere, was used as the reference for all spectra. The interaction between the crystal modifier and the crystal surfaces was analyzed by Fourier transform infrared spectroscopy (FT-IR, IRAffinity-1, Shimadzu, Japan) at a resolution of 4 cm−1 over the frequency range of 400−4000 cm−1. The concentration of Ca2+ in the filtrate was determined via inductively coupled plasma-atomic emission spectrometry (ICP-AES, PS-6, Baird, USA). The average diameters and average aspect ratios of the α-HH products for each sample were determined from about 100 crystal products using the optical images and the Image-Pro plus 6.0 software.
3. Results and discussion
3.1 Influence of reaction temperature on the phase transition kinetics
In order to explore the influence of reaction temperature on the kinetics of phase transformation from FGD gypsum to α-HH and determine the appropriate reaction temperature, we investigated the complete transformation time at different reaction temperatures using XRD, as shown in Fig. 1. The diffraction peaks of α-HH appear at 2θ = 14.72°, 25.64°, 29.70°, 31.86° and FGD gypsum (DH) at 2θ = 11.61°, 20.69°, 23.35° and 29.08°.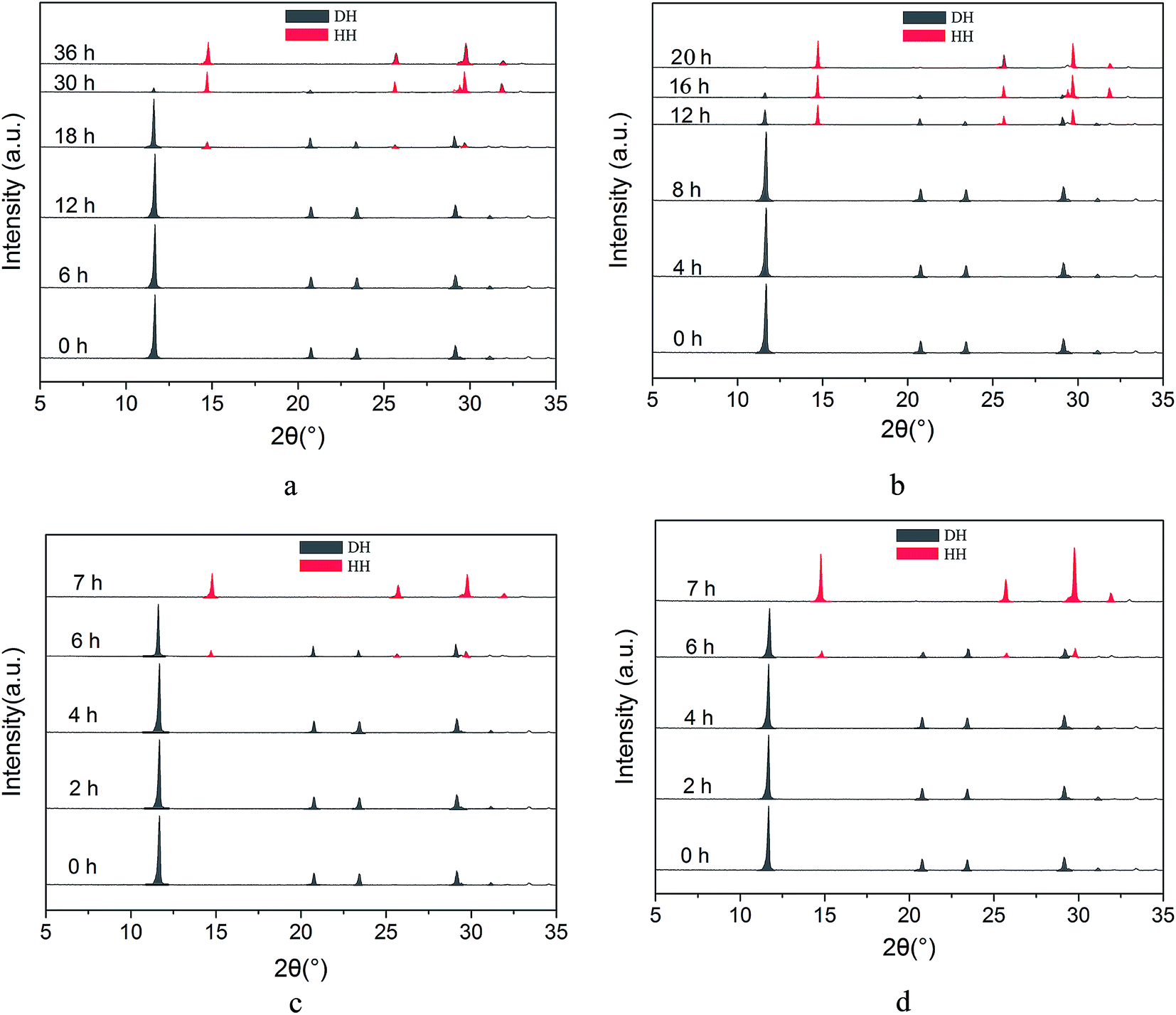 | ||
| Fig. 1 XRD patterns of the crystal products at different time intervals during the phase transformation in glycerol (65 wt%)-water solution without NaCl at different reaction temperatures ((a) 80 °C; (b) 85 °C; (c) 90 °C and (d) 95 °C). | ||
From Fig. 1a, it can be seen that at 80 °C the diffraction peaks of α-HH appear at 18 hours, which suggests that α-HH crystals were formed. After that, the intensity of the DH peaks generally weakened and the α-HH peaks gradually strengthened due to the gradual phase transformation from FGD gypsum to α-HH. At 36 hours, the FGD gypsum was completely transformed into α-HH. When the reaction temperature was increased to 85 °C, the complete transformation time reduced sharply to 20 hours (Fig. 1b), and with an increase in the temperature to 90 °C, the FGD gypsum was completely converted into α-HH within 7 hours (Fig. 1c). Upon further increasing the temperature to 95 °C, the complete transformation time did not significantly reduce, as shown in Fig. 1d. Therefore, considering efficiency and saving energy, the appropriate reaction temperature should be 90 °C.
3.2 Influence of NaCl on the rate of phase transformation from FGD gypsum to α-HH
In the preparation of α-HH whiskers using FGD gypsum under facile conditions, Na+ was used to accelerate the phase transformation rate.8 As shown in Fig. 2, the XRD patterns of the crystal products withdrawn at different interval times were used to track the phase transformation process. | ||
| Fig. 2 XRD patterns of the crystal products at different interval times during the phase transformation in the presence of different NaCl concentrations in glycerol (65 wt%)-water solution at 90 °C ((a) 0%; (b) 1%; (c) 3%; (d) 5% and (e) 7%). | ||
Without NaCl (Fig. 2a), the transformation from FGD gypsum to α-HH began at 360 min and was completed within 420 min. When 1% NaCl (by weight of FGD gypsum) (Fig. 2b) was added to the glycerol (65 wt%)-water solutions, the transformation started at 45 min and at 90 min the FGD gypsum was completely converted into α-HH. When the NaCl concentration was increased to 3% (Fig. 2c), the gypsum began to dehydrate at 15 min and the phase transformation was completed within 1 h. Upon further increasing the NaCl concentration to 5% (Fig. 2d) and 7% (Fig. 2e), the time of complete transformation from FGD gypsum to α-HH decreased to 45 min. As the phase transition reaction proceeded, α-HH generated by the FGD gypsum continued to dehydrate and was gradually transformed into anhydrous calcium sulfate (AH).
The promoting effect of NaCl on the transformation rate is attributed to the supersaturation (SHH) of α-HH in the solution. A higher supersaturation provided a greater driving force for α-HH nucleus formation, resulting in a shorter phase transformation time from FGD gypsum to α-HH. SHH is defined as:
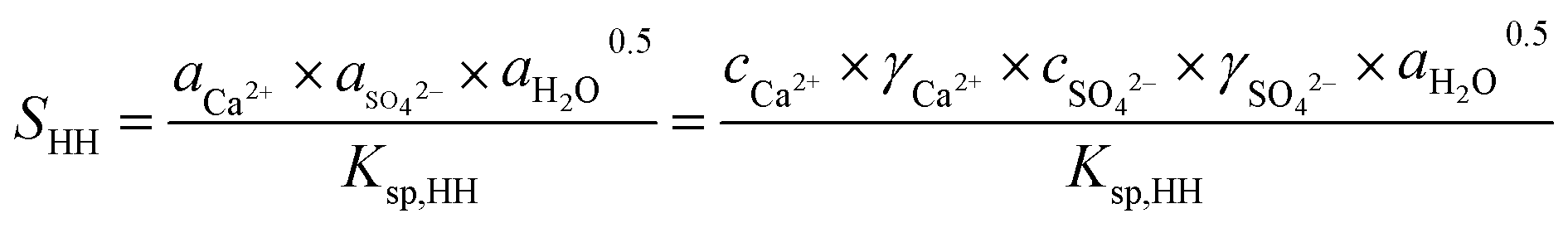 | (1) |
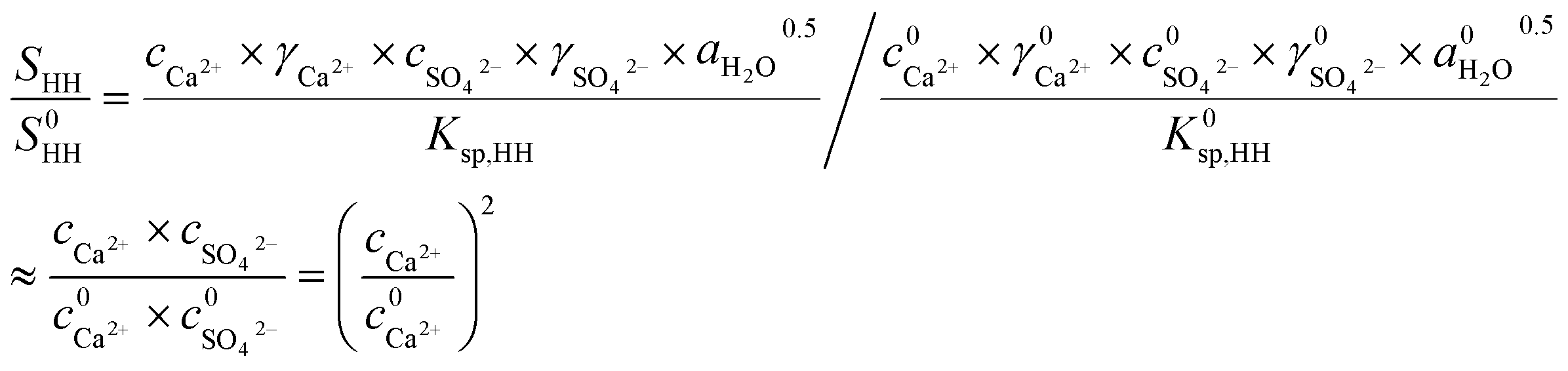 | (2) |
As depicted in Fig. 3, as the NaCl concentration increased, the Ca2+ concentration and SHH/S0HH experienced an upward trend, and when the NaCl concentration increased from 0 to 7%, the concentration of Ca2+ increased from 0.24 to 0.90 mg L−1 and SHH/S0HH from 1.00 to 14.06. It is clear that the addition of NaCl facilitated the dissolution of FGD gypsum and provided more available lattice ions, which created a much higher supersaturation and thus was a greater driving force for the phase transformation.
 | ||
| Fig. 3 Influence of NaCl on Ca2+ concentration and SHH/S0HH in glycerol (65 wt%)-water solution at 90 °C. | ||
3.3 Influence of C4H4 O4Na2·6H2O on the morphology of the crystal products
Fig. 4 and 5 show the morphological images and crystal size distribution of crystal products prepared with different concentrations of C4H4O4Na2·6H2O. Without the crystal modifier, the α-HH products tended to be acicular in shape owing to their preferential 1-D growth along the c axis.13–15 As shown in Fig. 4a, acicular crystals with an average aspect ratio of 28.60 were formed in the absence of the crystal modifier. When 0.03% C4H4O4Na2·6H2O (by weight of FGD gypsum) was added to the crystallization reaction system, the average length of the α-HH crystals decreased sharply from 53.79 to 32.23 μm and the average diameter increased from 1.89 to 7.84 μm, which resulted in a significant decline in the average aspect ratio to 4.15. With an increase in C4H4O4Na2·6H2O concentration from 0.03% to 0.06%, the average length of the crystals remained nearly unchanged, but their average diameter underwent a remarkable increase from 7.84 to 11.85 μm, leading to a decrease in the average aspect ratio from 4.15 to 2.72. From Fig. 4b and c, it can be seen that the α-HH products were transformed gradually from rod-like crystals into fat hexagonal prismatic crystals. With a further increase in the crystal modifier concentration to 0.09% (Fig. 4d), the crystal products appeared as shorter columns with an average diameter of 8.84 μm and aspect ratio of 1.96. As the concentration of C4H4O4Na2·6H2O increased to 0.12% (Fig. 4e), fat and short hexagonal prisms with a 7.36 μm average length, 5.70 μm average diameter and 1.32 average aspect ratio were acquired. From Fig. 4, 5 and the above discussion, it can be concluded that C4H4O4Na2·6H2O effectively tuned the size and morphology of the α-HH crystals. | ||
| Fig. 4 Influence of C4H4O4Na2·6H2O on the morphology of the α-HH products prepared in glycerol (65 wt%)-water solution with 5% NaCl (by weight of FGD gypsum) at 90 °C ((a) 0%; (b) 0.03%; (c) 0.06%; (d) 0.09% and (e) 0.12%). | ||
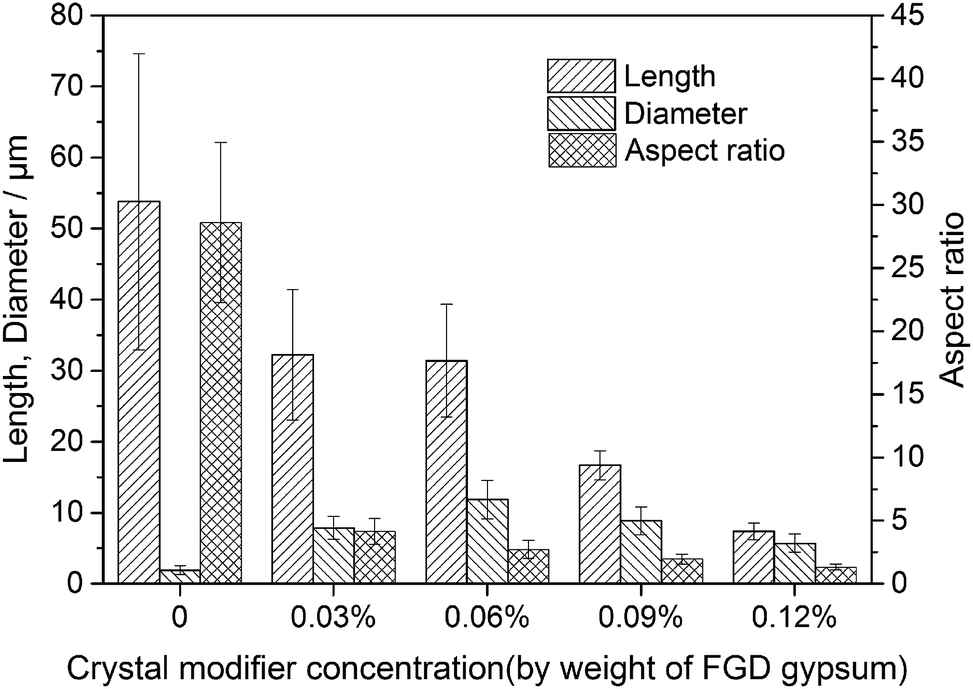 | ||
| Fig. 5 Average lengths, diameters and aspect ratios of the α-HH products prepared with different C4H4O4Na2·6H2O concentrations in glycerol (65 wt%)-water solution with 5% NaCl (by weight of FGD gypsum) at 90 °C. | ||
To confirm the interactions between the crystal modifier and the crystal surfaces of the α-HH crystals, FT-IR spectra were explored, as shown in Fig. 6. As shown in spectrum a, the peaks at 3612, 3564 and 1620 cm−1 are assigned to the O–H vibration of the crystal water molecules in the α-HH crystals. The three peaks at 1156, 1115, and 1097 cm−1 are assigned to the asymmetric stretching vibration of υ3 SO42−, and the peak at 1007 cm−1 is assigned to the distorted symmetric stretching vibration of υ1 SO42−. Also, the peaks at 660 and 602 cm−1 are indexed to the υ4 SO42− stretching.16,17
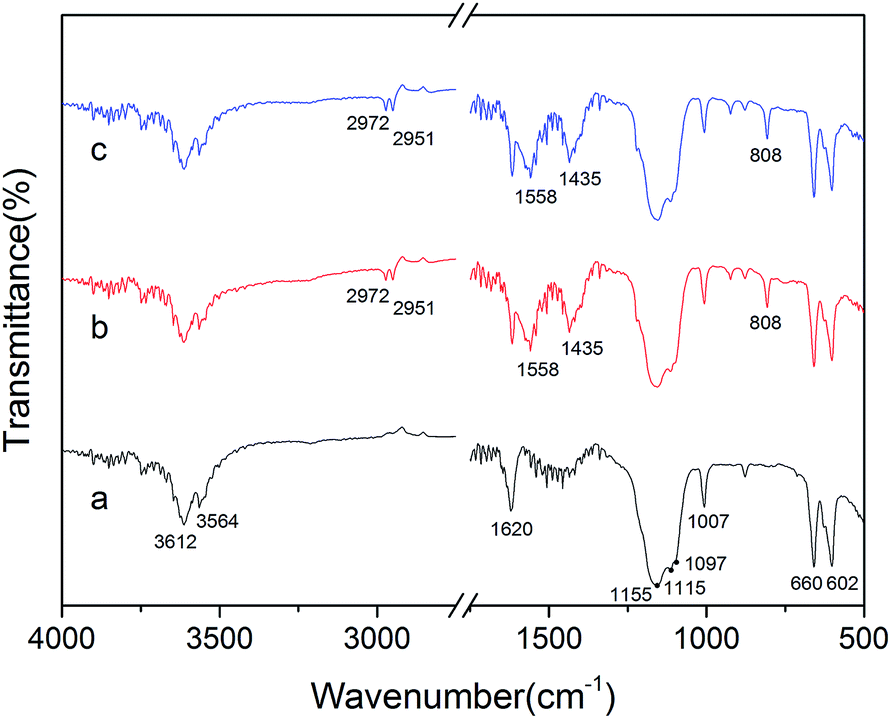 | ||
| Fig. 6 FT-IR spectra of the α-HH crystals prepared with different C4H4Na2·6H2O concentrations in glycerol (65 wt%)-water solution with 5% NaCl (by weight of FGD gypsum) at 90 °C ((a) 0%; (b) 0.06% and (c) 0.12%). | ||
As depicted in spectra b and c, the peaks at 2972 and 2951 cm−1 are attributed to the asymmetric and symmetric stretching vibrations of methylene (–CH2–), and the peaks at 1558 and 1435 cm−1 are derived from the asymmetric and symmetric stretching vibrations of the ester group (–COO−), which all demonstrate the adsorption of C4H4O4Na2·6H2O on the α-HH crystal surfaces.18 Additionally, the peak at 808 cm−1 might be due to the distorted vibration of methylene (–CH2–).
Fig. 7 depicts the C 1s XPS spectra of the crystal products formed in the presence of different C4H4O4Na2·6H2O concentrations. As shown in the C 1s spectra (Fig. 7), the most intense C 1s peak observed in all the samples is at 284.8 eV, which is associated with carbon impurity19 or the C–(C, H) species in the crystal modifier.20,21 Without the crystal modifier (Fig. 7a), the C 1s spectra of the α-HH crystals prepared could be fitted with three components. Besides the major component at 284.8 eV, the component located at 286.5 eV is associated with the C–O in glycerol,19,20,22,23 which implies the adsorption of a trace amount of glycerol on the crystal surfaces.24–26 The high binding energy component of C 1s at 288.9 eV is derived from the carbonate species (CO32−) of the limestone existing in FGD gypsum.27,28 As depicted in Fig. 7b and c, when a certain amount of crystal modifier was added to the crystallization reaction system, another C 1s component appeared at 288.2 eV, which is assigned to the carboxylic or carboxylate group (COO−),29–31 and with an increase in the crystal modifier concentration, the intensity of the peak at 288.2 eV improved gradually, implying the adsorption of C4H4Na2·6H2O on the surfaces of the crystal products.
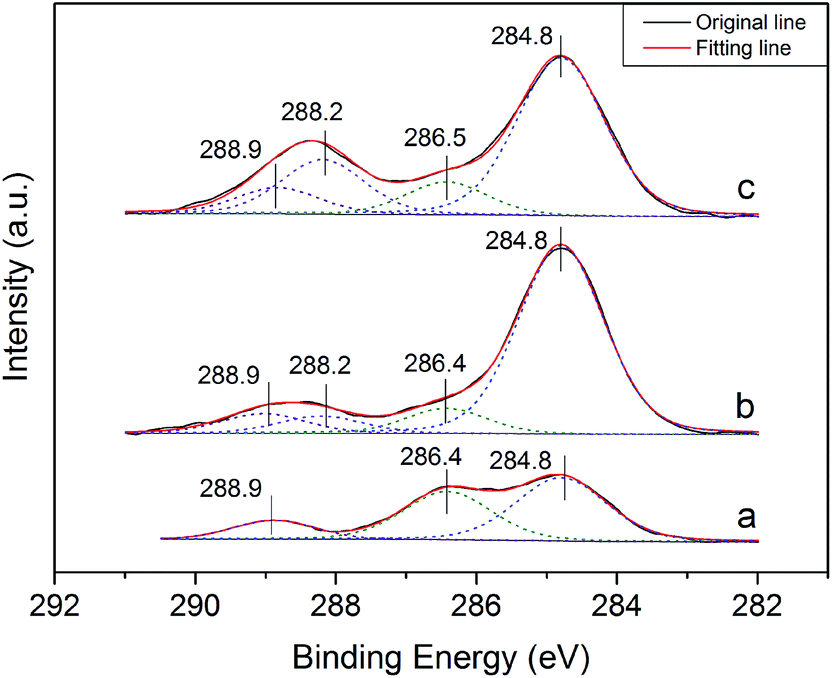 | ||
| Fig. 7 C 1s XPS spectra of α-HH crystals prepared with different concentrations of C4H4Na2·6H2O in glycerol (65 wt%)-water solution with 5% NaCl (by weight of FGD gypsum) ((a) 0%; (b) 0.06% and (c) 0.12%). | ||
Fig. 8 shows the Ca 2p XPS spectra of the α-HH crystals prepared in the presence of different C4H4O4Na2·6H2O concentrations. As shown in Fig. 8a, in the absence of the crystal modifier, the Ca 2p spectra of α-HH could be fitted with two components. The component at 347.2 eV is attributed to the calcium of limestone existing in FGD gypsum, and the other is derived from the calcium of α-HH. When the crystal modifier was added to the reaction system, a third peak appeared at 347.7 eV, which is associated with the calcium chelated by the carboxyl group of the crystal modifier, as shown in Fig. 8b and c.32 With an increase in the modifier concentration, the strength of the peak at 347.7 eV enhanced correspondingly. These results demonstrate the chelating reaction between the calcium on the crystal surface of α-HH and the carboxyl group of the crystal modifier.
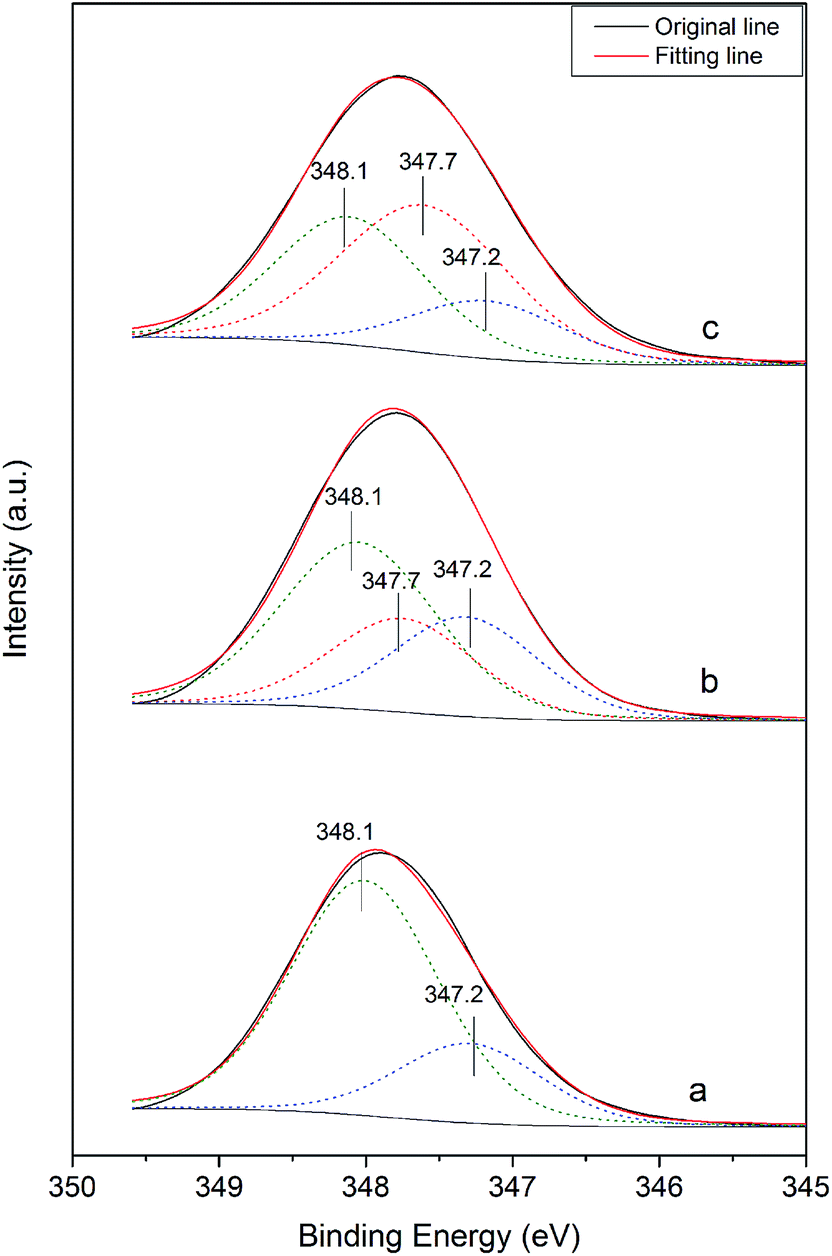 | ||
| Fig. 8 Ca 2p XPS spectra of the α-HH crystals prepared with different concentrations of C4H4Na2·6H2O in glycerol (65 wt%)-water solution with 5% NaCl (by weight of FGD gypsum) ((a) 0%; (b) 0.06% and (c) 0.12%). | ||
Table 2 shows the surface atomic distribution of the α-HH crystals formed in the presence of different concentrations of the crystal modifier. The surface carbon concentration of the α-HH products prepared without the crystal modifier might be mainly derived from carbon impurities or glycerol adsorbed on the crystal surfaces. From Table 2, it can be seen that with an increase in C4H4O4Na2·6H2O concentration, the surface oxygen and carbon concentrations of the α-HH products were generally enhanced, which implies the adsorption of C4H4O4Na2·6H2O on the α-HH crystal surfaces. However, the adsorption of the crystal modifier on the α-HH surfaces was much less than the amount added, and a certain amount of carboxylate ions (from the crystal modifier) could reduce water-gypsum ratio and improve the product strength to some extent. Therefore, there was no need to remove the crystal modifier from the produced α-HH crystals.
| C4H4Na2·6H2O concentration (by weight of FGD) | Ca/% | S/% | O/% | C/% |
|---|---|---|---|---|
| 0 | 15.30 | 16.80 | 64.92 | 2.98 |
| 0.06% | 14.31 | 15.48 | 65.91 | 4.30 |
| 0.12% | 11.56 | 12.72 | 66.32 | 9.40 |
As shown in Fig. 9, the α-HH lattices are composed of repeating, ionically bonded Ca and SO4 atoms in chains of –Ca–SO4–Ca–SO4–, and SO4 is a tetrahedral structure in which each S atom is covalently bonded to four O atoms.14,15 The structure of the chains may account for the fact that α-HH tended to grow in a 1D shape. These chains are hexagonally and symmetrically arranged and form a framework parallel to the c axis, and along the c axis continuous channels exist with a diameter of about 4.5 Å, where one water molecule is attached to every two calcium sulfate molecules.33 This crystal structure causes the distribution of Ca2+ to be denser on the 111 top facets and the distribution of SO42− denser on the 110 and 100 side facets, which makes the 111 facets positively charged and the 110 and 100 facets negatively charged.14,33 Therefore, when C4H4O4Na2·6H2O was added to the reaction system, C4H4O42− preferentially absorbed on the top facets by chelating Ca2+ and inhibited the α-HH crystal growth along the c axis, as shown in Fig. 10.34
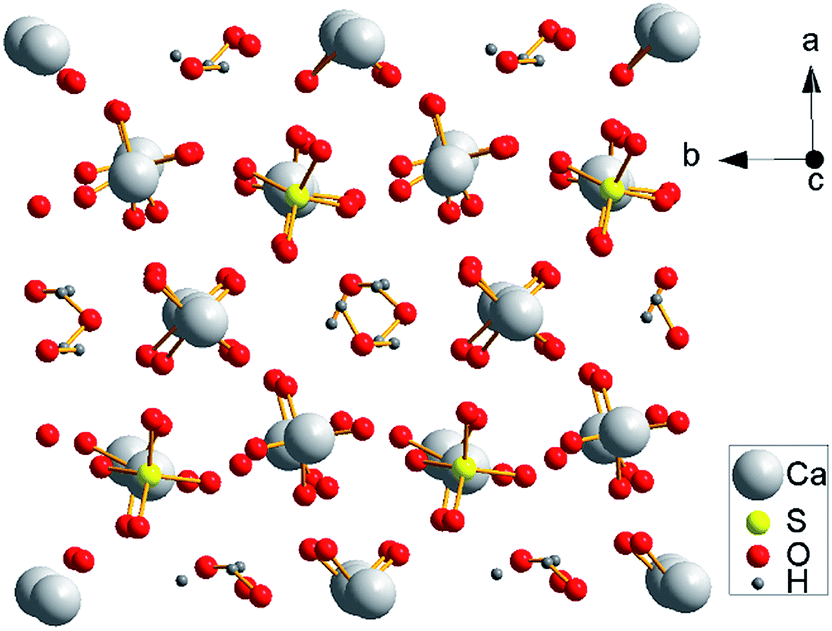 | ||
| Fig. 9 Crystal structure of the α-CaSO4·0.5H2O lattice along the c axis. | ||
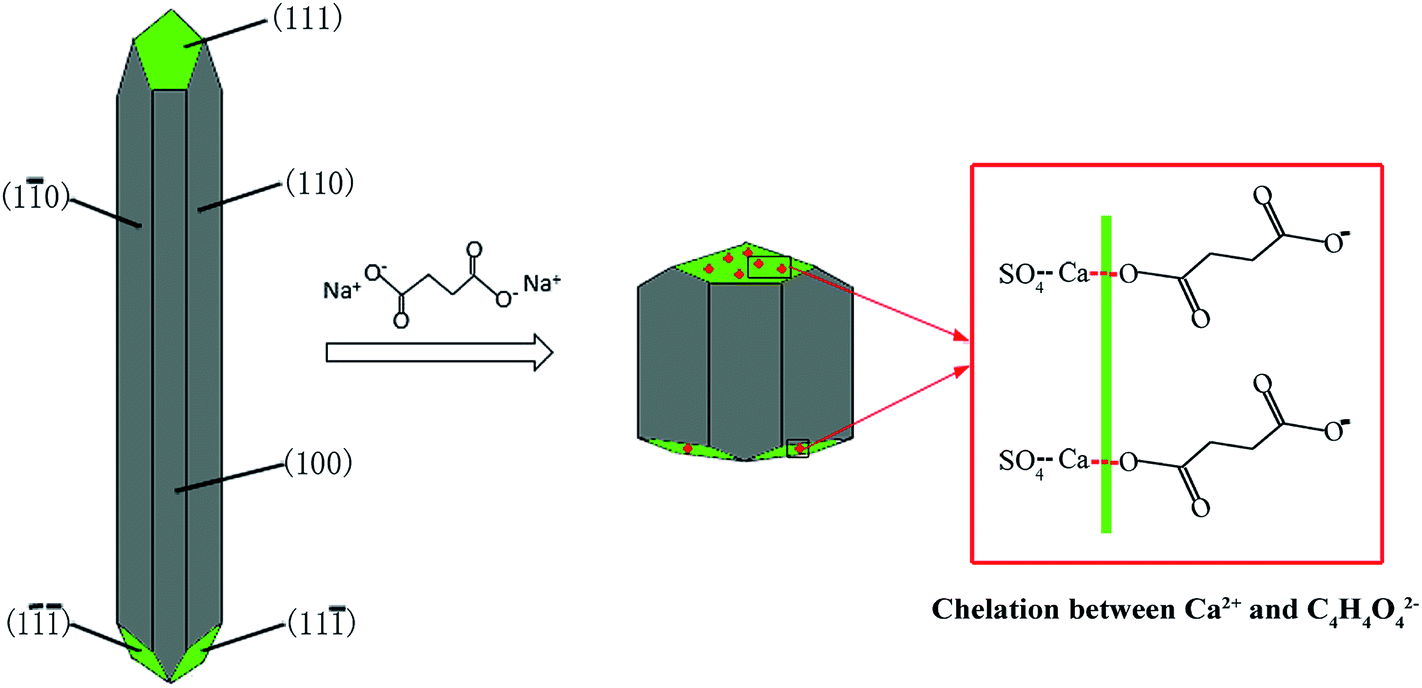 | ||
| Fig. 10 Schematic illustration of the adsorption of C4H4O4Na2·6H2O on the α-HH crystal surfaces. | ||
Although succinic acid disodium salt hexahydrate (C4H4O4Na2·6H2O) as the crystal modifier could effectively tune the morphology of the α-HH products, it suppressed the nucleation and crystal growth of α-HH and prolonged the time for the complete transformation from FGD gypsum to α-HH. As shown in Fig. 11, with an increase in the crystal modifier concentration from 0 to 0.12%, the complete transformation time was extended from 25 min to 17 h. The formation of α-HH can be divided into three processes: dissolution of DH, α-HH nucleation and α-HH growth, of which nucleation is the rate-determining step.35,36 When the crystal modifier was added to the reaction system, C4H4O42− chelated free Ca2+ in the solution and lower the supersaturation for α-HH, which restrained the nucleation of α-HH, and thus prolonged the transformation time.12
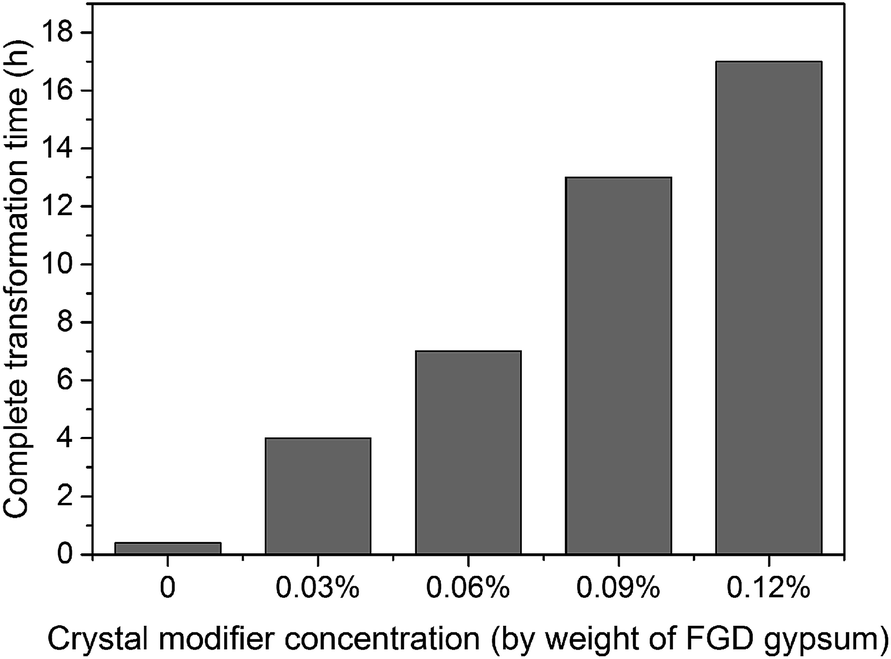 | ||
| Fig. 11 Time for the complete transformation from FGD gypsum to α-HH products with different C4H4O4Na2·6H2O concentrations and 5% NaCl (by weight of FGD gypsum) in glycerol (65 wt%)-water solution at 90 °C. | ||
The substitution of sodium ions in the modifier with calcium ions might have a considerable impact on the formation and properties of the α-HH products. Succinate calcium does not fully dissociate, and the succinate ions dissociated from succinate calcium in the solution would much less than that from succinate sodium, which would reduce the transformation time, but increase the aspect ratio of the α-HH products, in comparison with succinate sodium. However, the substitution of sodium ions in the modifier with other ions, such as potassium and magnesium, would not have a significant impact on the preparation of α-HH crystals because all of them are fully dissociated.
4. Conclusions
A new method was created for reusing FGD gypsum by preparing α-HH with short hexagonal prisms in glycerol-water solution with a small amount of succinic acid disodium salt hexahydrate (C4H4O4Na2·6H2O) and NaCl. Considering efficiency and saving energy, the appropriate reaction temperature was 90 °C. NaCl played an important role in accelerating the transformation from DH to α-HH. With an increase in NaCl concentration from 0 to 5%, the concentration of Ca2+ increased from 0.24 to 0.73 g L−1 and the corresponding SHH/S0HH increased from 1.00 to 9.25, which led to a sharp decrease in the complete transformation time from 7 h to 45 min. C4H4O4Na2·6H2O acted as a crystal modifier to regulate the crystal morphology, and as the concentration of C4H4O4Na2·6H2O increased from 0 to 0.12%, the crystal morphology was tuned from acicular to fat and short prismatic and the corresponding average aspect ratio decreased remarkably from 28.60 to 1.32. The suppression of the α-HH crystal growth along the c axis by C4H4O4Na2·6H2O is due to the preferential adsorption of C4H4O42− on the top facets of the crystal by chelating Ca2+. However, C4H4O42− chelates free Ca2+ in the solution and lowers the supersaturation of α-HH, thus restraining the nucleation of α-HH, which prolongs the transformation time.Acknowledgements
This work is financially supported by the Fundamental Research Funds for the Central Universities of Central South University (No. 2016zzts104); by the project of Sublimation Scholar's Distinguished Professor of Central South University; by the National 111 Project (No. B14034) and Collaborative Innovation Center for Clean and Efficient utilization of Strategic Metal Mineral Resources.References
- E. C. Combe and D. C. Smith, J. Appl. Chem., 2007, 18, 307–312 CrossRef.
- A. Kostic-Pulek, S. Marinkovic, S. Popov, M. Djuricic and J. Djinovic, Ceram.-Silik., 2005, 49, 115–119 CAS.
- S. Marinković, A. Kostić-Pulek, S. Durić, V. Logar and M. Logar, J. Therm. Anal. Calorim., 1999, 57, 559–567 CrossRef.
- Z. Alfred, O. Ivan, T. Felicia and B. Katarina, J. Am. Ceram. Soc., 1991, 74, 1117–1124 CrossRef.
- Z. Shen, B. Guan, H. Fu and L. Yang, J. Am. Ceram. Soc., 2009, 92, 2894–2899 CrossRef CAS.
- B. Guan, G. Jiang, H. Fu, L. Yang and Z. Wu, Ind. Eng. Chem. Res., 2011, 50, 13561–13567 CrossRef CAS.
- B. Guan, G. Jiang, Z. Wu, J. Mao and K. Bao, J. Am. Ceram. Soc., 2011, 94, 3261–3266 CrossRef CAS.
- G. Jiang, H. Fu, K. Savino, J. Qian, Z. Wu and B. Guan, Cryst. Growth Des., 2013, 13, 5128–5134 CAS.
- T. Feldmann and G. P. Demopoulos, J. Cryst. Growth, 2012, 351, 9–18 CrossRef CAS.
- P. Wang, E. J. Lee, C. S. Park, B. H. Yoon, D. S. Shin, H. E. Kim, Y. H. Koh and S. H. Park, J. Am. Ceram. Soc., 2008, 91, 2039–2042 CrossRef CAS.
- J. Peng, M. Chen, J. Zhang, J. Qu and C. Zou, J. Sichuan Univ., Eng. Sci. Ed., 2012, 44, 166–172 CAS.
- C. Jia, Q. Chen, X. Zhou, H. Wang, G. Jiang and B. Guan, Ind. Eng. Chem. Res., 2016, 55, 9189–9194 CrossRef CAS.
- S. Hou, J. Wang, X. Wang, H. Chen and L. Xiang, Langmuir, 2014, 30, 9804–9810 CrossRef CAS PubMed.
- P. Ballirano, A. Maras, S. Meloni and R. Caminiti, Eur. J. Mineral., 2001, 13, 985–993 CrossRef CAS.
- C. Bezou, A. Nonat, J. C. Mutin, A. N. Christensen and M. S. Lehmann, J. Solid State Chem., 1995, 117, 165–176 CrossRef CAS.
- X. Mao, X. Song, G. Lu, Y. Sun, Y. Xu and J. Yu, Ind. Eng. Chem. Res., 2015, 54, 4781–4787 CrossRef CAS.
- B. Kong, B. Guan, M. Z. Yates and Z. Wu, Langmuir, 2012, 28, 14137–14142 CrossRef CAS PubMed.
- M. D. Morris, A. Berger and A. Mahadevan-Jansen, Infrared and Raman spectroscopy, Marcel Dekker, Inc, 2001 Search PubMed.
- J. A. Mielczarski, J. M. Cases, A. M. Alnot and J. J. Ehrhardt, Langmuir, 1996, 12, 455–479 Search PubMed.
- F. Ikumapayi, M. Makitalo, B. Johansson and K. H. Rao, Miner. Eng., 2012, 39, 77–88 CrossRef CAS.
- Y. Mikhlin, A. Karacharov, Y. Tomashevich and A. Shchukarev, Vacuum, 2016, 125, 98–105 CrossRef CAS.
- M. Deng, D. Karpuzov, Q. Liu and Z. Xu, Surf. Interface Anal., 2013, 45, 805–810 CrossRef CAS.
- G. Fairthorne, D. Fornasiero, J. Ralston, G. Fairthorne, D. Fornasiero and J. Ralston, Anal. Chim. Acta, 1997, 346, 237–248 CrossRef CAS.
- Q. Chen, G. Jiang, C. Jia, H. Wang and B. Guan, CrystEngComm, 2015, 17, 8549–8554 RSC.
- Z. Pan, G. Yang, L. Yi, E. Xue, H. Xu, X. Miao, J. Liu, C. Hu and Q. Huang, Int. J. Appl. Ceram. Technol., 2013, 10, E219–E225 CrossRef CAS.
- W. Zhao, Y. Wu, J. Xu and C. Gao, RSC Adv., 2015, 5, 50544–50548 RSC.
- M. Ni and B. D. Ratner, Surf. Interface Anal., 2008, 40, 1356–1361 CrossRef CAS PubMed.
- M. Engelhard and D. Baer, Surf. Sci. Spectra, 1999, 6, 126–135 CrossRef.
- J. S. Hammond, J. W. Holubka, J. E. Devries and R. A. Dickie, Corros. Sci., 1981, 21, 239–253 CrossRef CAS.
- A. S. Tselesh, Thin Solid Films, 2008, 516, 6253–6260 CrossRef CAS.
- S. J. Huo, J. M. He, L. H. Chen and J. H. Fang, Spectrochim. Acta, Part A, 2015, 156, 123–130 CrossRef PubMed.
- B. Demri and D. Muster, J. Mater. Process. Technol., 1995, 55, 311–314 CrossRef.
- D. Freyer and W. Voigt, Monatsh. Chem., 2003, 134, 693–719 CrossRef CAS.
- M. Mathew, S. Takagi, B. O. Fowler and M. Markovic, J. Chem. Crystallogr., 1994, 24, 437–440 CrossRef CAS.
- A. Maher, D. M. Croker, Å. C. Rasmuson and B. K. Hodnett, Cryst. Growth Des., 2014, 14, 3967–3974 CAS.
- Z. Alfred, O. Ivan, T. Felicia and B. Katarina, J. Am. Ceram. Soc., 2005, 74, 1117–1124 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |