
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interface structure, precursor rheology and dielectric properties of BaTiO3/PVDF–hfp nanocomposite films prepared from colloidal perovskite nanoparticles
Y. N. Hao a,
K. Bi*a,
S. O'Brien*b,
X. X. Wangc,
J. Lombardib,
F. Pearsallb,
W. L. Lib,
M. Lei*a,
Y. Wud and
L. T. Lic
a,
K. Bi*a,
S. O'Brien*b,
X. X. Wangc,
J. Lombardib,
F. Pearsallb,
W. L. Lib,
M. Lei*a,
Y. Wud and
L. T. Lic
aState Key Laboratory of Information Photonics and Optical Communications, School of Science, Beijing University of Posts and Telecommunications, Beijing 100876, China. E-mail: bike@bupt.edu.cn
bThe Department of Chemistry and Biochemistry, The City College of New York, The Ph.D. Program in Chemistry, The Graduate Center of the City University of New York, New York, NY 10016, USA. E-mail: sobrien@ccny.cuny.edu
cState Key Laboratory of New Ceramics and Fine Processing, School of Materials Science and Engineering, Tsinghua University, Beijing 100084, China
dSchool of Journalism and Communication, Beijing Normal University, Beijing 100875, China
First published on 27th June 2017
Abstract
Nanocomposite materials with uniform microstructure and high permittivity have attracted extensive interest in modern electronics. The compatibility between the filler phase and the polymer matrix is crucial in preparing high-performance composites. As an alternative to conventional surface modification treatment methodologies, hydroxylated colloidal BaTiO3 (BT) nanoparticles synthesized via a green and scalable process were directly used to fabricate high-permittivity nanocomposites. Interfacial interaction analyses between the BT nanoparticles and polymer matrix reveals that due to strong hydrogen bonding at the interface, transparent composite sols with excellent flow behavior can be observed. The sols are ideal formulations for the preparation of BT/PVDF–hfp 0–3 nanocomposite films. Owing to the unique interface structure, the composite films show a dense and uniform microstructure and superior dielectric properties. Parallel plate capacitor devices and the co-development of a sandwich architecture leads to the ability to prepare dielectric films with favorable performance characteristics. This method provides a novel and greatly simplified strategy for the fabrication of high-permittivity dielectric nanocomposites.
Introduction
Developing methods to prepare nanodielectric materials with high performance dielectric properties has great importance in academic research fields as well as for practical applications.1 A nanocomposite can combine the high permittivity of the ceramic filler phase and good machinability of the polymer matrix phase, making it a desirable approach for enhancing the capacitance,2 thermal stability,1a,3 energy density4 or field effect.5 With the ever growing trend towards integration, miniaturization and multi-functionalization of modern electronics, nanocomposite materials with high permittivity could find more widespread use in capacitors, field effect transistors, memory and energy storage devices.6 In such materials, the interactions between the filler phase and polymer matrix, as well as the interfacial structure of the two phases are crucial to the microstructure and dielectric properties of the composite.6d,7 Therefore, surface modification of the fillers has continued to be an active research area in improving the comprehensive performance of the nanocomposite.Due to the physical and chemical differences (surface tension, functional groups etc.) between the two phases, ceramic fillers are generally incompatible with the polymer matrix, which induces a poor combination at the interface and generates a third air phase, which causes a major problem:4a,8 the low permittivity and dielectric strength of the interstices greatly deteriorate the properties of the nanocomposite. The effective permittivity of the film is greatly suppressed, the loss increases usually by one to two orders of magnitude, and the dielectric strength (breakdown, voltage tolerance) as well as leakage become unsatisfactory. In addition, without appropriate surface coating, the exposed surface of the nanoparticles can readily absorb moisture, further worsening the dielectric properties of the nanocomposite over time.2a Another problem in composite materials is the aggregation of nanofillers, which commonly results in non-uniformity in both microstructure and property of the dielectrics. All these factors severely decrease the reliability of the nanocomposite for the applications of capacitors or energy storage devices.9 In order to improve the compatibility between the ceramic fillers and polymer matrix, great efforts have been paid to the surface modification and interface control of the nanoparticles. These surfactants can either promote the particle–polymer interfacial reaction, such as phosphonic acid,7,8 H2O2,9c,10 ammonia water,4a or improve the dispersion of the nanoparticles, for example, dopamine11 and ethylene diamine.12 However, with the decrease of particle size, the volume fraction of the low-permittivity organic coating layer increases sharply, which would have an increasingly dominant role in the properties and thus reduce the permittivity of the nanocomposite.13
In a different approach to conventional coating/mixing of commercially available nanopowders, in this study, we take advantage of the synthetic process of colloidal nanoparticles,14 which possess highly reactive hydroxyl ligand naturally, to fabricate 0–3 ceramic–organic nanocomposite films directly by mixing the as-synthesized BT nanoparticles with the polymer sol. In this study, poly(vinylidene fluoride-co-hexafluoro propylene), PVDF–hfp, which is a ferroelectric polymer that possesses a relative high electric displacement polarization, is chosen as the matrix phase to fabricate the nanocomposite films with comprehensive high dielectric constant and low dielectric mismatching between the two phases. Colloidal BT particles, which is a typical ferroelectric materials with very high dielectric constant, is used as the fillers phase. Based on the surface characteristics of the colloidal nanoparticles, transparent BT/PVDF–hfp nanocomposite sols with varied BT volume fractions are obtained, which greatly expands the fabrication method of composite materials. By spin-coating these sols, uniform BT/PVDF–hfp nanocomposite films are obtained. Fourier transform infrared spectroscopy (FTIR) and thermal analysis reveal that strong hydrogen bonding is likely generated between the ceramic particles and PVDF–hfp matrix. The interface interaction not only contributes to the compact combination of the two phases, but also promotes the uniform dispersion of BT nanoparticles in the matrix. The dielectric properties of the nanocomposite films are measured and investigated by several theoretical models. Based on the further analyses and hypothesis, a multilayer nanocomposite film with superior dielectric properties was developed and fabricated.
Experimental
The hydroxylated colloidal BT nanoparticles are synthesized by a simple gel-collection procedure that was reported earlier.2a,5,14 The obtained “gel rod” contains fully crystalline nanoparticles, which are readily stabilized in various organic solvent.1b In this study, transparent colloidal BT nanoparticle sol with a concentration of 100 mg mL−1 are prepared by sonication of the gel product in N,N-dimethylformamide (N,N-DMF) solvent. In the meantime, PVDF–hfp is dissolved in N,N-DMF solvent with a concentration of 100 mg mL−1 to form the polymer solvent. Then the two sols are mixed with certain BT volume fractions and then sonicated to form the nanocomposite sols. To fabricate the nanocomposite films, a silver bottom electrode with a dimension of 3 × 1 mm is first deposited on the clean glass substrate. The nanocomposite sols are spun-coat onto the substrate at 1000 rpm for 40 s and then dried at 80 °C to generate a thin film. Different composite sols are spun-coated onto the substrate sequentially to form the multilayer nanocomposite films. Then the dried nanocomposite films are baked on a hot plate at 80 °C for 4 hours to remove the residual solvent. Finally, top electrodes (3 × 1 mm) that are perpendicular to the bottom electrodes are deposited on the films for the following electronic performance tests.Transmission electron microscope (TEM) images of the nanoparticles and the nanocomposite were performed on a TEM (JEM-2100, JEOL Ltd., Tokyo, Japan) at an accelerating voltage of 200 kV. Thermal analyses were carried out on a TA instruments thermal analyser (New Castle, DE, USA) with a heating rate of 10 °C min−1 in nitrogen/air at a flow rate of 100 mL min−1. FTIR spectra were recorded in the 400–2000 cm−1 range using a FT-IR spectrometer (Nicolet 6700, Thermo Scientific, USA) with a 633 nm wavelength laser radiation at room temperature. Rheological properties of the nanocomposite sols are characterized using a rheometer (MCR-302, Anton Paar, Austria). Morphology of the nanocomposite films is obtained by a scanning electron microscope (SEM; Supra55VP, Zeiss, German). The dielectric properties of the single layer and multilayer nanocomposite films are measured using a precision impedance analyzer (4294A; Agilent, Palo Alto, CA).
Results and discussion
In a typical ceramic–organic polymer composite dielectric material, the ceramic fillers provide high electric displacement polarization and high permittivity of the composite. A uniform distribution of the filler phase in the polymer matrix, as well as the good interface combination is not only beneficial to the permittivity enhancement, but also important to the overall microstructure, electrical property and mechanical property of the composite.15 Here the gel-collection method is utilized to synthesize BT nanoparticle fillers. As shown in Fig. 1a, the nanoparticles are uniform both in size and morphology, with a particle size in the range of 7–11 nm. High-resolution (HR) TEM images reveal clear lattice fringes and confirm the single crystallinity of the individual nanoparticles. The aggregation-free feature of the nanoparticles is advantageous in forming a transparent nanocrystal sol. The surface chemistry of the nanoparticles was analyzed by TG-DTG (derivative thermogravimetry) curves measured in air. As shown in Fig. 1b, the weight loss below 200 °C can be ascribed to the desorption of water and ethanol molecules, which indicates that with a high surface free energy, the ultrafine nanoparticles retain solvent molecules on the surface and absorb moisture from the air. The ∼4.6% weight loss between 200–500 °C and the DTG peak at 275 °C and 329 °C can be assigned to the combustion of the residual –OC3H7 surface alkoxy groups. At higher temperature, the condensation of remaining hydroxyl groups induces the ∼1% weight loss above 500 °C. In consideration of the large specific surface area of such small particles, the surface organics are rather few and no other surfactant is coated on the particle surface.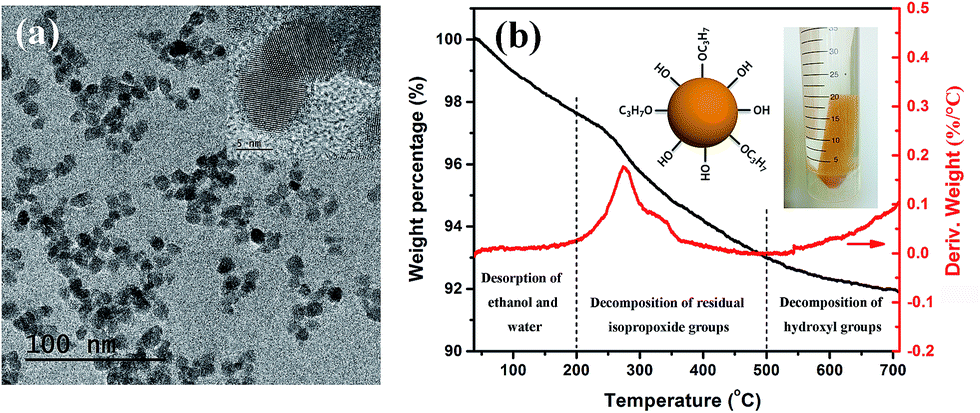 | ||
| Fig. 1 (a) TEM image (inset: HR-TEM image of the nanoparticles), and (b) TG-DTG curves of the BaTiO3 nanoparticles. The inserted photograph is the as-prepared “gel rod” in ethanol: a collected rod composed of fully crystallized nanoparticles as observed in (a). | ||
Our previous work has revealed that due to the abundant surface hydroxyl groups and some alkoxy groups, the nanoparticles have good dispersibility in organic polar solvents such as ethanol and furfuryl alcohol.14 As shown in Fig. 2a, the BT nanoparticles can also form stable and transparent sols with PVDF–hfp at different volume fractions in N,N-DMF, which makes them suitable for various solution-based processing methods such as spin-coating and printing. TEM observations on these sols indicate that although having rather good dispersibility, the pure BT nanoparticles are readily attracted to each other as a result of electrostatic or van der Waals interactions, and under certain conditions of solvent treatment (in this case N,N-DMF) can become loosely aggregated, mediated by interfacial attraction, as shown in Fig. 2b. While in the nanocomposite sol containing 40 vol% BT nanoparticles, the particles present very good monodispersity due to the interaction of the particle surface and the polymer (Fig. 2c), which appears to stabilize the nanoparticles and prevent aggregation, as predicted and observed in many systems. The individual particle in the HR-TEM image in Fig. 2d shows a fuzzy edge, which suggests that the particle surface is connected with PVDF–hfp molecules and thus a uniform particle–polymer mixture is formed. The appealing observation of such stable and transparent sols strongly suggested a chemical or H-bond interaction between polymer chain and nanoparticles surface, which warranted investigation by spectroscopy and thermal analysis.
 | ||
| Fig. 2 (a) Photograph of the PVDF–hfp sol, BT nanocrystal sol and the nanocomposite sols with BT volume fractions varying from 10% to 80%. (b) TEM image and schematic of the loosely interconnected BT nanoparticles. (c) TEM, and (d) HR-TEM images and schematic of the BT nanoparticles in the nanocomposite sol with 40 vol% BT loading. | ||
FTIR transmittance spectra and the thermal analysis data in Fig. 3 further confirm the interface interaction between BT nanoparticles and PVDF–hfp. The top curve in Fig. 3a is FTIR spectrum for pure PVDF–hfp. The vibrational peaks at wave numbers of 1413, 1190 and 1073 cm−1 are characteristic peaks for CH2 wagging, antisymmetric CF2 stretching and CF3 out-of-plane deformation, respectively.16 The CF3 rocking bands located at 987 and 773 cm−1 correspond to the α-phase of PVDF–hfp while the band at 855 cm−1 is assigned to the β-phase of PVDF–hfp. The CF2 rocking band at 888 cm−1 represents the amorphous band of PVDF–hfp.4 The band at 1190 cm−1 can be assigned to the β-phase of PVDF–hfp. The high intensity of 855 and 1190 cm−1 bands shows that PVDF–hfp in the nanocomposite films mainly possesses a high-performance ferroelectric β-phase. The bottom curve in Fig. 3a is FTIR spectrum for BT nanopowders. The absorption band at 1585 cm−1 is originated from the complex of alkoxy groups and the metal Ti centers of BT nanoparticles.17 Owing to the absorption of CO2 on the highly alkaline particle surfaces that have abundant hydroxyl groups, an obvious characteristic peak for CO32− is observed at 1442 cm−1.18 The band located at 1357 cm−1 is attributed to the C–H vibrations. When blended with PVDF–hfp, FTIR spectra of the nanocomposite films show obvious new features, presented in the curves between pure PVDF–hfp and BT nanoparticles. From top to bottom, the BT nanoparticle content increases from 10 to 80 vol% and the characteristic peak at 572 cm−1 for Ti–O vibration gradually increases. Meanwhile, the characteristic bands at 1073, 987, 888, 855 and 773 cm−1 also weaken as the polymer content decreases. However, although the strong antisymmetric CF2 stretching peak at 1190 cm−1 slightly decreases, it still maintains a relatively high intensity. As this peak is generally considered to be related to β-phase of PVDF–hfp, its variation reveals that the introduction of BT nanoparticles can enhance the polar β-phase in the polymer. The bands between 1600 and 1300 cm−1 for the nanocomposite films confirm the interaction between PVDF–hfp and BT nanoparticles. As illustrated in the inset in Fig. 3a, the particle surface contains electron pairs at the oxygen atoms of alkoxy groups and PVDF–hfp has electron pairs at fluorine atoms. Both of them can coordinate with the proton (H) atoms and form polymer–particle complexes. Therefore, the band at 1585 cm−1 for the complex of alkoxy groups and Ti centers is enhanced in the BT/PVDF–hfp nanocomposites. With decreasing particle content, the C–H vibrations initially at 1357 cm−1 slightly shift lower wavenumbers. Because formation of the hydrogen bond can promote further polarization of the original bond and therefore change the bond vibrational energy, the intensification of the FTIR absorbance and the peak position shift provide evidence of hydrogen bonding between F atoms in PVDF–hfp and H atoms belonging to the surface alkoxy groups. Similarly, the vibrational peak for CH2 vibrational “wagging” at 1413 cm−1 also shows an enhanced intensity and shifts to lower wavenumber with the increase of particle content, supporting a hypothesis that hydrogen bonding also occurs between H atoms in PVDF–hfp and O atoms in the surface alkoxy groups. The characteristic peak for CO32− at 1442 cm−1 disappears in all the nanocomposite films, confirming no barium carbonate impurity phase is present or evolving. This can be attributed to the occurrence of hydrogen bonds between hydroxyl groups and the F atoms in PVDF–hfp, which stabilized the highly alkaline surfaces. It is concluded that H-bonding at the interface, between polymer chains and surface hydroxyl groups, (as depicted in the inset in Fig. 3a) promotes favorable, interface mediated, compatibilization of the two entities in solution, increases the dispersibility of the nanoparticles, and serves to provide an advantageous formulation for preparing composite films.
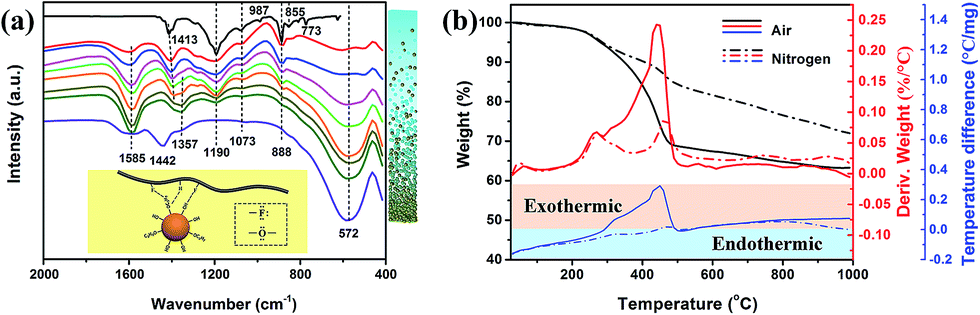 | ||
| Fig. 3 (a) FTIR spectra for PVDF–hfp (top curve); BT/PVDF–hfp nanocomposite films with BT volume fractions increasing from 10% (second to top curve) to 80% respectively in increments of 10%; and BT nanoparticles (bottom curve). The right bar schematic represents BT nanoparticle content increasing from top to bottom. (b) Thermal analysis results for the BT/PVDF–hfp nanocomposite film with 40 vol% BT loading in air and nitrogen, respectively (black lines are TG curves, red lines are DTG curves, and blue lines represent DTA curves). | ||
TG/DTG curves in air (black and red lines respectively) and nitrogen (dashed line) for the nanocomposite film with 40 vol% BT nanoparticles are presented in Fig. 3b. Similar to the as-obtained BT nanoparticles, the weight loss below 200 °C is attributed to the desorption of moisture and solvent molecules. A sharp weight loss on the TG curve along with a DTG peak between 200 and 400 °C is observed for the nanocomposite in both air and nitrogen atmosphere. Compared with the thermal analyses for the as-obtained BT nanoparticles, the two DTG peaks measured in air slightly shift to lower temperature due to the formation of hydrogen bonding. It is observed that the main DTG peak induced by decomposition of PVDF–hfp in air is located at 445 °C. However, the composite film persistently loses weight from 250 to 445 °C and the weight loss in 200–400 °C is obviously more than the surface organic content in the nanoparticles. From the DTA curve measured in air (blue line in solid), a continuous exothermic process from 300 to 500 °C is observed. This indicates that the hydrogen bonding between the surface of the BT nanoparticles and PVDF–hfp affect directly the thermostability of the polymer matrix at temperatures >250 °C. The reason for the consistent heat release is that the PVDF–hfp chains connected to the alkoxy groups by hydrogen bonding gradually decompose before the decomposition of the main chain. In the DTA curve recorded under nitrogen, only two exothermic peaks (lower than that in air, and located at the endothermic area) related to the decomposition of the surface organics on the nanoparticles and the PVDF–hfp main chains are observed, and no consistent exothermic process between them occurs. Under a nitrogen atmosphere, there is no combustion process available to the alkoxy groups, nor enough heat at this temperature to break the PVDF–hfp chains that connect with the alkoxy groups, PVDF–hfp therefore gradually starts to decompose until it reaches the decomposition temperature of ∼400 °C. The thermal analysis further confirm the interaction between BT nanoparticles and the polymer matrix and reveal that the hydrogen bonding not only promotes good dispersibility of the nanoparticles in the polymer matrix, but also influence the thermostability of the nanocomposite at high temperature.
The properties and stability of the BT/PVDF–hfp nanocomposite sol is crucial to the solution-based fabrication of uniform composite films. It also serves as a well-defined system for investigating the effect of rheological properties of the formulation, prior to nanocomposite film formation. To reveal how the interface interaction influence the processing of the nanocomposite films, the rheology was studied and the results are shown in Fig. 4. Fig. 4a shows the shear stress and viscosity variation with the increase of angular frequency (γ) for the nanocomposite sol that has been aged for several weeks. It is observed that the nanocomposite sol has a low viscosity (0–60 mPa, between the values for water and olive oil), confirming good fluidity of the sol. In the low-shear range, prior to the condition of a homogeneous flow, a transient viscosity peak is formed. The nanocomposite sol presents typical shear-thinning behavior since the shear stress increases, while the viscosity decreases with the increase of γ. This is mainly induced by the destruction of the internal net structure in the complex. It is observed that after several weeks' standing, the nanocomposite sol gradually transforms to a soft gel. However, this condition is reversible: the gel state can revert to a transparent sol by gentle shaking. From the low γ stage in the rheological curves in Fig. 4b, the storage modulus G′, which represents the elastic properties of the complex, is much higher than the loss modulus G′′, which represents the viscous portion of the complex shear stress, demonstrating typical gel-like behavior. With increasing γ, an apparent fluctuation in the rheological curve occurs between 1 and 5 rad s−1, showing that G′′ suddenly increases from ∼40 to 738, subsequently stabilizing at 450 for γ > 10. This fluctuation in the intermediate range of γ reveals the destruction of the internal net structure that formed by the polymer and the connected BT nanoparticles, as illustrated in the inset of Fig. 4b. Accordingly, the loss factor also fluctuated by nearly one order of magnitude. Although G′ is still larger than G′′, the complex turns into sol state and the reversible stable dispersion is obtained. This somewhat unique reversible property is attributed again to the formation of highly stabilized dispersions of the precursor nanocomposite sol formulation. With the observations of consistent fluidity and uniformity, the nanocrystal–polymer formulations (Fig. 2a) can be spun-coat to make uniform dielectric thin films.
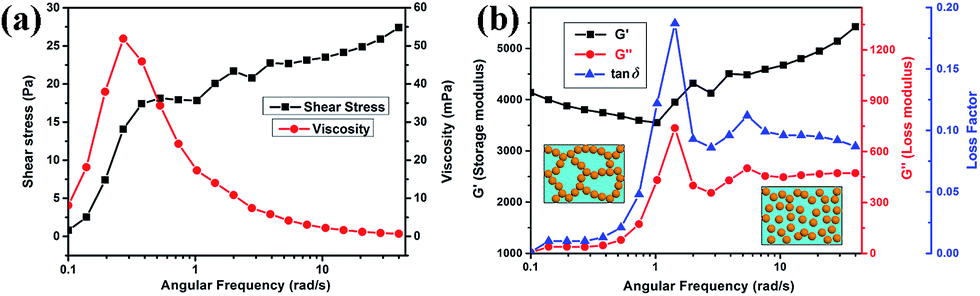 | ||
| Fig. 4 (a) Flow and viscosity curves, and (b) rheological curves for the nanocomposite sol. | ||
To assess dielectric performance, parallel plate capacitors were fabricated and tested, for which images, cross-sectional microstructure and the effective film permittivity, εr, derived from the frequency dependent impedance analysis are shown in Fig. 5. The effective film permittivity is taken to be the value of the real component of the complex permittivity, ![[small epsilon, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e107.gif) = ε′ + iε′′ (εr = ε′). The dissipation loss, tan
= ε′ + iε′′ (εr = ε′). The dissipation loss, tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ, is taken to be the imaginary part of the complex function, ε′′. At a given frequency, e.g. 104 Hz or 1 MHz, εr can be reported as the dielectric constant of the medium. It is important to report εr at a specified frequency, given the radical change in value of εr for nanocomposites as a function of frequency. The frequency dependence of the permittivity demonstrates relatively typical behaviour for a PVDF–hfp composite film. Overall, the introduction of the perovskite nanoparticles enhances εr. For example, at 104 Hz εr of pure PVDF–hfp is 12, with values increasing to 27.5, with increasing initial mixing volume fraction of BT nanoparticles, up to 60%. At higher estimated loadings εr decreases. This is expected, due to the predicted increase in void space that accompanies higher loadings. The frequency dependent behaviour of a dielectric is divided into frequency regimes. In the low frequency regime (<∼103 Hz) two phenomena known collectively as Maxwell–Sillars–Wagner polarization (also called interfacial polarization or space charge) occur, in which mobile charge at the interface between electrode and nanocomposite, and charge within the polymer–nanoparticle interface contribute to an apparent increase in εr. There is a concurrent increase in dissipation loss due to these polarization relaxation mechanisms. The charges at these interfaces can be associated with under coordinated atomic sites or ionic species, expected of a polymer–inorganic interface, especially in the case where the predominant bonding mechanism is intermolecular in nature. Overall, for device applications minimization of this behaviour is desirable. It is interesting to note that the nanoparticles serve to significantly lower the dissipation (tanδ) at higher frequencies with an inflection point at ∼104 Hz for which the contribution to the dissipation loss of the PVDF–hfp polymer matrix is known to increase. The decrease in εr and increase in tanδ > 104 Hz in this regime can be attributed to dipolar/molecular orientational polarization relaxation. The response frequency range (MHz to GHz) depends on the species, local environment, viscosity and the mobility of the electron clouds. The addition of nanoparticles to the PVDF–hfp and subsequent H-bonding, restricts motion of the polymer chains and dipolar groups, thereby inhibiting this dipolar relaxation effect. Such a phenomenon highlights the advantage of multicomponent nanocomposite systems, in which the polymer and nanoparticle act synergistically to produce a dielectric medium with more appealing characteristics.
δ, is taken to be the imaginary part of the complex function, ε′′. At a given frequency, e.g. 104 Hz or 1 MHz, εr can be reported as the dielectric constant of the medium. It is important to report εr at a specified frequency, given the radical change in value of εr for nanocomposites as a function of frequency. The frequency dependence of the permittivity demonstrates relatively typical behaviour for a PVDF–hfp composite film. Overall, the introduction of the perovskite nanoparticles enhances εr. For example, at 104 Hz εr of pure PVDF–hfp is 12, with values increasing to 27.5, with increasing initial mixing volume fraction of BT nanoparticles, up to 60%. At higher estimated loadings εr decreases. This is expected, due to the predicted increase in void space that accompanies higher loadings. The frequency dependent behaviour of a dielectric is divided into frequency regimes. In the low frequency regime (<∼103 Hz) two phenomena known collectively as Maxwell–Sillars–Wagner polarization (also called interfacial polarization or space charge) occur, in which mobile charge at the interface between electrode and nanocomposite, and charge within the polymer–nanoparticle interface contribute to an apparent increase in εr. There is a concurrent increase in dissipation loss due to these polarization relaxation mechanisms. The charges at these interfaces can be associated with under coordinated atomic sites or ionic species, expected of a polymer–inorganic interface, especially in the case where the predominant bonding mechanism is intermolecular in nature. Overall, for device applications minimization of this behaviour is desirable. It is interesting to note that the nanoparticles serve to significantly lower the dissipation (tanδ) at higher frequencies with an inflection point at ∼104 Hz for which the contribution to the dissipation loss of the PVDF–hfp polymer matrix is known to increase. The decrease in εr and increase in tanδ > 104 Hz in this regime can be attributed to dipolar/molecular orientational polarization relaxation. The response frequency range (MHz to GHz) depends on the species, local environment, viscosity and the mobility of the electron clouds. The addition of nanoparticles to the PVDF–hfp and subsequent H-bonding, restricts motion of the polymer chains and dipolar groups, thereby inhibiting this dipolar relaxation effect. Such a phenomenon highlights the advantage of multicomponent nanocomposite systems, in which the polymer and nanoparticle act synergistically to produce a dielectric medium with more appealing characteristics.
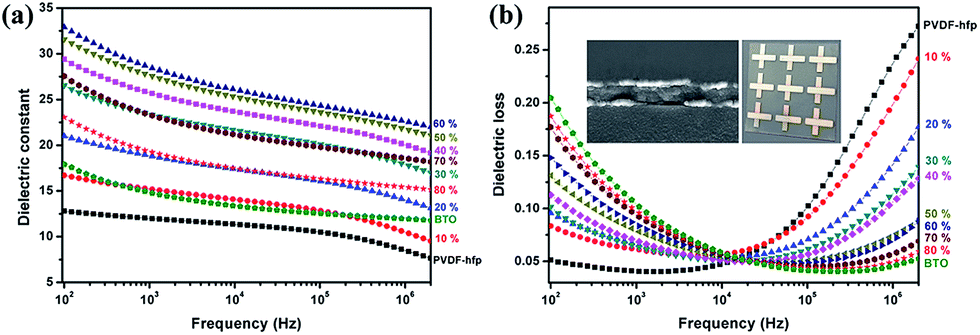 | ||
| Fig. 5 (a) Frequency dependent dielectric constant and (b) dielectric loss of the PVDF–hfp film, nanocomposite films with BT volume fraction varies from 10% to 80% and BT nanoparticle film. Inset: cross-sectional SEM image and photograph of the fabricated capacitor with the nanocomposite dielectric layer. | ||
With increasing particle volume fraction, the dielectric constant of the nanocomposite films gradually increases and reaches a maximum value for a loading of 60 vol%, then decreases sharply at higher estimated volume fractions. At these higher volume fractions, for which the amount of added PVDF–hfp is becoming sharply reduced, the volume fraction of nanoparticles is an estimate, based on the molar ratios devised in the precursor formulation (nanocomposite sol), which is then spun coat and dried at 80 °C. It can be anticipated that, without sufficient polymer to fill the space between the particles, the prevalence of voids during evaporative assembly and packing can occur. In the case of nanocomposite films at higher volume fractions of filler (>60%), it is well understood that packing of contiguous particles (expected for high volume fractions) can be irregular, creating interstitial space between the nanoparticles. Without the opportunity for the matrix polymer to fill this space, the interstitial void space (with a permittivity of εr ∼ 1) gradually increases to become one of the major components that will contribute to the overall dielectric properties of the nanocomposite. The decrease in permittivity at estimated volume fractions >60%, is observed from the experimental value (at 1 kHz and above). This is consistent with the prediction of void space increasing in the film. Theoretical predictions of the thin film effective permittivity, from several existing models at volume fractions above 40%, are shown in Fig. 6a. Although the Lichtenecker logarithmic law and Maxwell–Wagner equation can better describe the dielectric constant variation at estimated volume fractions <40%, the experimentally observed low permittivity at high volume fractions shows great discrepancy with the theoretical predictions. This can be attributed to an increasing amount of porosity in the nanocomposite film. Taking into account the interstitial porosity as a third phase in the 0–3 composite is one means to attempt to more accurately model the experimentally observed value of permittivity, using effective medium approximations. A three-phase model, adapted from the Lichtenecker logarithmic law is applied to analyse the dielectric behaviour of the nanocomposite films. Assuming that the volume fraction portion of BT nanoparticles and PVDF–hfp is fixed by the sol preparation process, the actual volume fractions of each component are calculated according to how well the measured dielectric constant at different designated BT fractions fit the Lichtenecker law and plotted in Fig. 6b. Although nanocomposite sols with higher BT volume fractions are obtained in the preparation, the actual BT fractions in the nanocomposite films can only be increased at limited values (below 60%). And the void phase is playing a more and more important role in determining the dielectric properties of the nanocomposite with the increase of the designated BT fractions. Therefore, the difference between the actual and designed BT volume fraction is the likeliest reason for the low permittivity and large discrepancy between the experimental and theoretical results at high particle fractions. The analysis indicates that a dense microstructure is a very important basis for the acquisition of high-permittivity dielectric nanocomposites, and that optimization of the surface chemistry of the nanoparticles with the polymer can lead to minimization of void space, surface absorbed moieties, and subsequent reduction in dissipation loss.
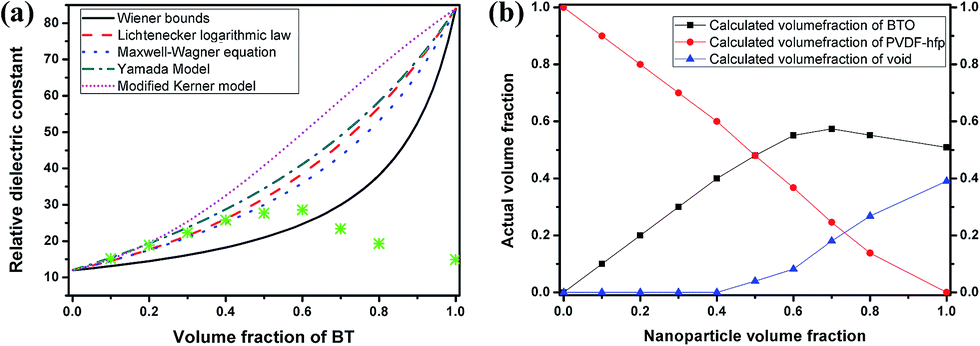 | ||
| Fig. 6 (a) Experimental and theoretical predicted relative dielectric constant of the nanocomposite films at different BT volume fractions at 1 kHz. (b) The calculated volume fractions of BT, PVDF–hfp and voids in the nanocomposite films in terms of the designed BT volume fractions by Lichtenecker logarithmic law. | ||
From Fig. 7a, the cross-sectional SEM image of the nanocomposite film with 30 vol% BT fractions, it can be observed that the film possesses a dense and uniform microstructure. Since it is assumed that moisture absorption is mainly occurring at the surface and internal voids of the nanocomposite film, a sandwich structure that formed by two dense, low particle loading outer layers and a high polarization but porous interlayer was proposed and constructed as an alternative, more favourable, configuration, as shown in the schematic and SEM image in Fig. 7b. The sandwich structure that combines the preferred properties of each layer is evolving as an effective way to further increase the comprehensive dielectric properties of composite materials.4b,19 Here, the outer high polymer fraction layer mainly serves as an inert encapsulation to prevent the hydrophilic particles from absorbing moisture at the surface of the interlayer. Fig. 7b shows that the each layer of the nanocomposite film is dense and homogeneous, with good combination at the interfaces of the layers.
 | ||
| Fig. 7 Cross-sectional SEM images of (a) the nanocomposite film with 30 vol% BT fractions, and (b) the multilayer nanocomposite film (outer layers: 20 vol% BT nanoparticles; interlayer: 40 vol% BT nanoparticles). | ||
In the fabricated sandwich-structure nanocomposite film, BT/PVDF–hfp nanocomposite films that contain 10 vol% and 20 vol% BT nanoparticles were chosen as the outer layers and the nanocomposite films containing 20–80 vol% BT as well as the pure BT nanoparticle film serve as the interlayer. As shown in Fig. 8, the low-frequency dielectric loss of the multilayer nanocomposite films is significantly reduced, and even the high-frequency dissipation is slightly lowered. Moreover, the dielectric constant is improved with proper interlayer, which demonstrates that by appropriate structure design, these hydroxylated colloidal perovskite nanoparticles can serve as high-permittivity fillers to improve the comprehensive property of the nanocomposite.
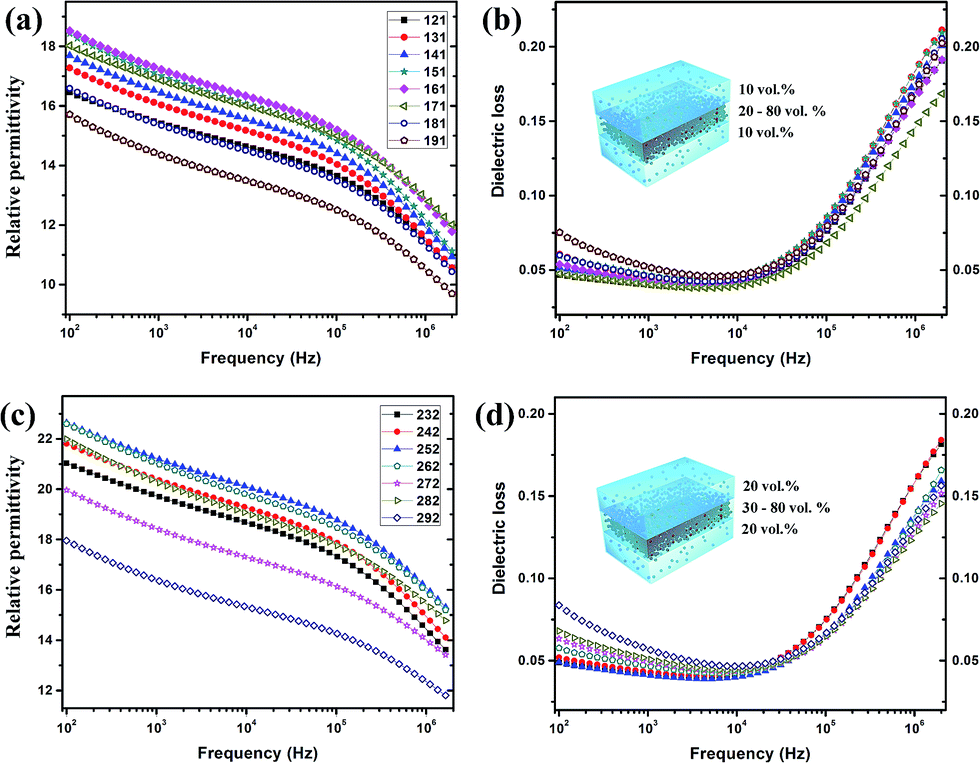 | ||
| Fig. 8 (a) Dielectric constant and (b) loss for the multilayer nanocomposite films with outer layers contain 10 vol% BT and the particle loading of interlayer varies from 20 to 80 vol%. (c) Dielectric constant and (d) loss for the multilayer nanocomposite films with outer layers contain 20 vol% BT and the particle loading of interlayer varies from 30 to 80 vol%. | ||
Conclusions
In summary, hydroxylated colloidal BT perovskite nanoparticles synthesized via a gel-collection method are directly used as fillers to fabricate high-permittivity nanocomposite films. Due to the surface hydroxyl groups and a few alkoxy groups, the nanoparticles are readily dispersed in N,N-DMF and, furthermore, form yellow transparent sols with PVDF–hfp. These transparent sols can be utilized as formulations to fabricate BT/PVDF–hfp nanocomposite films by spin-coating. A strong interface interaction, attributed to hydrogen bonding, is detected between the BT nanoparticles and the polymer matrix, essential to addressing issues of agglomeration of these highly active nanoparticles and promoting uniform dispersion of BT particles in a PVDF–hfp matrix. Based on the excellent rheological properties of the nanocomposite sols, the spun-coat nanocomposite films result in a uniform deposition that show dense microstructure (at low volume fraction) and favourable dielectric properties, with effectively improved dielectric constants and suppressed high-frequency dissipation. Moreover, a sandwich structure is designed to improve the low-frequency dissipation and multilayer nanocomposite films for which markedly improved comprehensive dielectric properties are obtained. This investigation reveals in detail the interaction between hydroxylated nanoparticles and polymer matrix, as well as its influence on the fabrication and dielectric performance of the nanocomposite, which not only offers a facile way to fabricate high-permittivity nanocomposite films, but also provides a reference for the deployment of ultrafine nanoparticles in composite materials.Acknowledgements
This work was supported by NSF DMR award #1461499, and NSF CREST# 1547830. The work was also supported by Ministry of Sciences and Technology of China through National Basic Research Program of China (973 Program 2015CB654604), National Natural Science Foundation of China for Creative Research Groups (Grant No. 51221291), Ministry of Science and Technology of China (Grant No. 2016YFA0301300), National Natural Science Foundation of China (Grant No. 51272123, 51402163, 61574020), and also supported by CBMI Construction Co., Ltd.Notes and references
- (a) Q. Li, L. Chen, M. R. Gadinski, S. H. Zhang, G. Z. Zhang, H. Y. Li, A. Haque, L. Q. Chen, T. N. Jackson and Q. Wang, Nature, 2015, 523, 576 CrossRef CAS PubMed; (b) B. Van Tassell, S. Yang, C. Le, L. Huang, S. Liu, P. Chando, X. Liu, A. Byro, D. L. Gerber, E. S. Leland, S. R. Sanders, P. R. Kinget, I. Kymissis, D. Steingart and S. O'Brien, IEEE T. Power Electr., 2016, 31, 2695 CrossRef; (c) Z. Wang, X. Ren, C. W. Leung, S. Shi and P. K. L. Chan, J. Mater. Chem. C, 2013, 1, 3825 RSC; (d) Z. M. Dang, J. K. Yuan, S. H. Yao and R. J. Liao, Adv. Mater., 2013, 25, 6334 CrossRef CAS PubMed.
- (a) L. Huang, S. Liu, B. J. Van Tassell, X. Liu, A. Byro, H. Zhang, E. S. Leland, D. L. Akins, D. A. Steingart, J. Li and S. O'Brien, Nanotechnology, 2013, 24, 415602 CrossRef PubMed; (b) C. Ehrhardt, C. Fettkenhauer, J. Glenneberg, W. Muenchgesang, H. S. Leipner, G. Wagner, M. Diestelhorst, C. Pientschke, H. Beige and S. G. Ebbinghaus, RSC Adv., 2014, 4, 40321 RSC.
- Z. M. Dang, Y. Q. Lin, H. P. Xu, C. Y. Shi, S. T. Li and J. B. Bai, Adv. Funct. Mater., 2008, 18, 1509 CrossRef CAS.
- (a) B. C. Luo, X. H. Wang, Y. P. Wang and L. T. Li, J. Mater. Chem. A, 2014, 2, 510 RSC; (b) Y. N. Hao, X. H. Wang, S. O'Brien, J. Lombardi and L. T. Li, J. Mater. Chem. C, 2015, 3, 9740 RSC.
- L. Huang, Z. Jia, I. Kymissis and S. O'Brien, Adv. Funct. Mater., 2010, 20, 554 CrossRef CAS.
- (a) V. L. Pushparaj, M. M. Shaijumon, A. Kumar, S. Murugesan, L. Ci, R. Vajtai, R. J. Linhardt, O. Nalamasu and P. M. Ajayan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13574 CrossRef CAS PubMed; (b) Y. Rao, S. Ogitani, P. Kohl and C. P. Wong, J. Appl. Polym. Sci., 2002, 83, 1084 CrossRef CAS; (c) F. C. Chen, C. W. Chu, J. He, Y. Yang and J. L. Lin, Appl. Phys. Lett., 2004, 85, 3295 CrossRef CAS; (d) F. Li, T. W. Kim, W. Dong and Y.-H. Kim, Thin Solid Films, 2009, 517, 3916 CrossRef CAS.
- P. Kim, S. C. Jones, P. J. Hotchkiss, J. N. Haddock, B. Kippelen, S. R. Marder and J. W. Perry, Adv. Mater., 2007, 19, 1001 CrossRef CAS.
- (a) P. Kim, N. M. Doss, J. P. Tillotson, P. J. Hotchkiss, M. J. Pan, S. R. Marder, J. Y. Li, J. P. Calame and J. W. Perry, ACS Nano, 2009, 3, 2581 CrossRef CAS PubMed; (b) V. Tomer and C. A. Randall, J. Appl. Phys., 2008, 104, 074106 CrossRef; (c) S. Ramesh, B. A. Shutzberg, C. Huang, J. Gao and E. P. Giannelis, IEEE Trans. Adv. Packag., 2003, 26, 17 CrossRef CAS.
- (a) Y. Rao and C. P. Wong, J. Appl. Polym. Sci., 2004, 92, 2228 CrossRef CAS; (b) S. Ogitani, S. A. Bidstrup-Allen and P. A. Kohl, IEEE Trans. Adv. Packag., 2000, 23, 313 CrossRef CAS; (c) T. Zhou, J.-W. Zha, R.-Y. Cui, B.-H. Fan, J.-K. Yuan and Z.-M. Dang, ACS Appl. Mater. Interfaces, 2011, 3, 2184 CrossRef CAS PubMed.
- M. N. Almadhoun, U. S. Bhansali and H. N. Alshareef, J. Mater. Chem., 2012, 22, 11196 RSC.
- (a) Y. Song, Y. Shen, H. Liu, Y. Lin, M. Li and C.-W. Nan, J. Mater. Chem., 2012, 22, 8063 RSC; (b) H. Luo, C. Chen, K. Zhou, X. Zhou, Z. Wu and D. Zhang, RSC Adv., 2015, 5, 68515 RSC.
- J. Li, J. Claude, L. E. Norena-Franco, S. Il Seok and Q. Wang, Chem. Mater., 2008, 20, 6304 CrossRef CAS.
- (a) T. Hanemann and D. V. Szabo, Materials, 2010, 3, 3468 CrossRef CAS; (b) T. J. Lewis, J. Phys. D: Appl. Phys., 2005, 38, 202 CrossRef CAS.
- S. Liu, L. Huang, W. Li, X. Liu, S. Jing, J. Li and S. O'Brien, Nanoscale, 2015, 7, 11766 RSC.
- Y. Hao, X. Wang, K. Bi, J. Zhang, Y. Huang, L. Wu, P. Zhao, K. Xu, M. Lei and L. Li, Nano Energy, 2017, 31, 49 CrossRef CAS.
- (a) L. N. Sim, S. R. Majid and A. K. Arof, Vib. Spectrosc., 2012, 58, 576 CrossRef; (b) D. Olmos, E. V. Martin and J. Gonzalez-Benito, Phys. Chem. Chem. Phys., 2014, 16, 24339 RSC.
- K. Fujinami, K. Katagiri, J. Kamiya, T. Hamanaka and K. Koumoto, Nanoscale, 2010, 2, 2080 RSC.
- M. D. B. Lopez, G. Fourlaris, B. Rand and F. L. Riley, J. Am. Ceram. Soc., 1999, 82, 1777 CrossRef CAS.
- (a) Y. F. Wang, J. Cui, Q. B. Yuan, Y. J. Niu, Y. Y. Bai and H. Wang, Adv. Mater., 2015, 27, 6658 CrossRef CAS PubMed; (b) P. H. Hu, Y. Shen, Y. H. Guan, X. H. Zhang, Y. H. Lin, Q. M. Zhang and C. W. Nan, Adv. Funct. Mater., 2014, 24, 3172 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |