
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Epoxide functionalization on cholane side chains in the identification of G-protein coupled bile acid receptor (GPBAR1) selective agonists†
Simona De Marino a,
Adriana Carinob,
Dario Masulloa,
Claudia Finamorea,
Valentina Sepea,
Silvia Marchianòb,
Francesco Saverio Di Levaa,
Vittorio Limongelliac,
Stefano Fioruccib and
Angela Zampella*a
a,
Adriana Carinob,
Dario Masulloa,
Claudia Finamorea,
Valentina Sepea,
Silvia Marchianòb,
Francesco Saverio Di Levaa,
Vittorio Limongelliac,
Stefano Fioruccib and
Angela Zampella*a
aDepartment of Pharmacy, University of Naples “Federico II”, Naples, Italy. E-mail: azampell@unina.it; Fax: +39-081676552; Tel: +39-081678525
bDepartment of Surgery and Biomedical Sciences, Nuova Facoltà di Medicina, Perugia, Italy
cUniversità della Svizzera Italiana (USI), Faculty of Informatics, Institute of Computational Science – Center for Computational Medicine in Cardiology, Lugano, Switzerland
First published on 27th June 2017
Abstract
The G-protein coupled receptor of bile acids GPBAR1 is a bile acid receptor that plays an important role in regulating innate immunity, glucose homeostasis and energy expenditure representing an interesting target for the treatment of metabolic and degenerative diseases. A selective control of its activity over the other bile acid-activated receptors is desirable to reduce unwanted effects due to activation of multiple downstream signals regulated by diverse receptors. Here, we report the design and the synthesis of a small series of bile acid derivatives with their side chains decorated with an epoxide ring. We demonstrate that all the compounds selectively activate GPBAR1 in cell-based assays and regulate the expression of pro-glucagon, a canonical GPBAR1 targeted gene in an intestinal endocrine cell line, while have no effect on FXR, LXRα and LXRβ. Finally, we elucidate the binding mode of the most potent compound of this family through molecular modeling studies. Our study contributes to increase the arsenal of bile acid derivatives serving as GPBAR1 modulators, widening the chemical space that can be exploited in drug design.
1. Introduction
GPBAR1 is the first example of the specific bile acid receptor subclass1,2 of human G-protein coupled receptors. GPBAR1 is expressed in several tissues, including non-parenchymal liver cells, gallbladder, ileum, colon, heart, spleen, kidney, placenta, lung, uterus, testis, mammary gland, prostate, skeletal muscle, brown adipose tissue, leukocytes, macrophages, endothelial cells and selected areas of the central nervous system. Activation by secondary bile acids, mainly the tauro conjugated form of lithocholic acid (TLCA, taurolithocholic acid in Fig. 1), increases intracellular levels of cAMP and protein kinase A, allowing signaling transduction. Responses to GPBAR1 activation are tissue-specific. In entero-endocrine L cells, GPBAR1 activation stimulates the release of glucagon-like peptide (GLP)-1, a well characterized incretin that improves insulin release, thus regulating glucose homeostasis,3 whereas in muscle and brown adipose tissue, GPBAR1 activation increases energy expenditure and oxygen consumption attenuating diet-induced obesity.4 Moreover, GPBAR1 reduces liver steatosis,5 regulates gallbladder filling and bile homeostasis,6–8 intestinal motility,9 itch, and analgesia.10 Finally, activation of the GPBAR1 signaling pathway has been reported to beneficially impact on multiple inflammatory diseases, such as atherosclerosis, steatohepatitis and colitis.11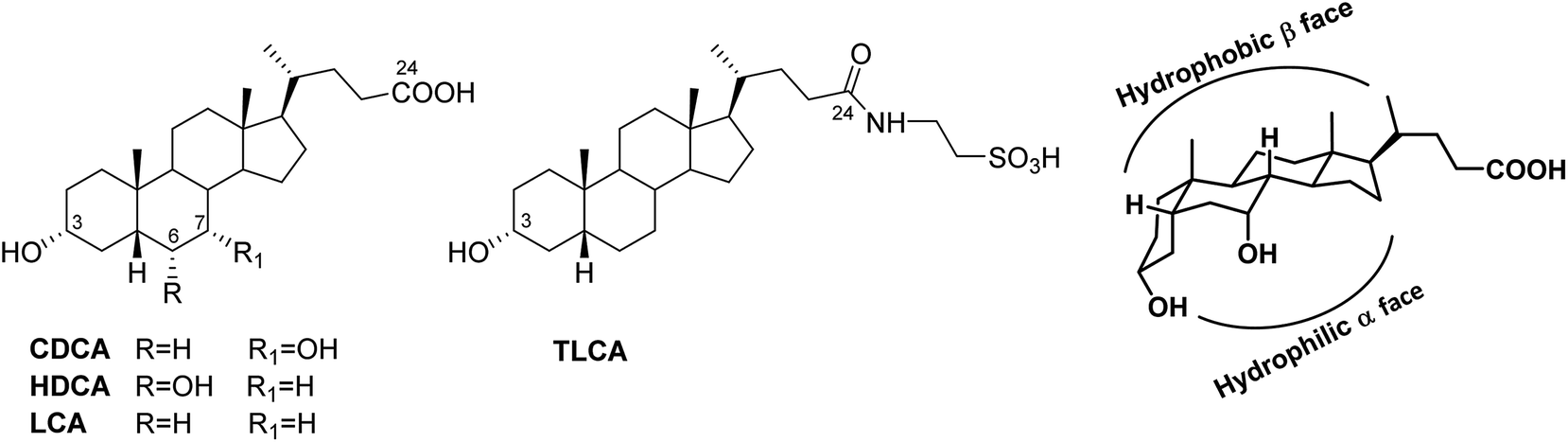 | ||
| Fig. 1 Endogenous bile acids with their bent shape. | ||
Consequently, modulation of GPBAR1 by small molecules has shown promise in the treatment of several human diseases ranging from type 2 diabetes, obesity and liver steatosis to inflammation and cancer.12
In the last ten years, chemical modification on bile acid and non-bile acid scaffolds from academia and industries have led to the identification of GPBAR1 modulators. In the context of bile acid mimetics, designing selective GPBAR1 agonists is challenging since bile acids are also agonists for several nuclear receptors, mainly farnesoid X receptor, FXR, the endogenous sensor of bile acid levels, and the two proteins share major structural requisites for ligand binding. Since FXR and GPBAR1 act on different downstream signals, targeting both receptors could be beneficial in a variety of clinical indications, making dual agonists attractive candidates as potential novel therapeutic options with applications across a range of chronic metabolic and liver diseases.13 On the other hand, dual FXR/GPBAR1 ligands might expose patients to multiple unwanted effects due to activation of the diverse downstream signals controlled by the two receptors.14,15
Bile acids are truncated side chain steroidal molecules with a carboxyl functional group at C-24 and with the steroidal nucleus adopting a bent shape due to the A/B cis ring juncture that forces ring A to lie outside of the plane of the BCD ring system (Fig. 1). In that manner, two well-differentiated surfaces are defined, the hydrophobic convex surface of the tetracyclic core and a hydrophilic surface where the hydroxyl groups lie.
The activity and selectivity of bile acids towards their receptors is related to the index of hydrophilicity that in turn depends on the number, position and configuration of OH groups. Thus, among human bile acid pool, chenodeoxycholic acid (CDCA) with two hydroxyl group α-oriented at C-3 and C-7 is the most potent endogenous FXR agonist, whereas lithocholic acid (LCA) and its tauro-conjugated form (TLCA) are privileged activators of GPBAR1 (Fig. 1). Of interest, the introduction of a hydroxyl group at C-6 as in hyodeoxycholic acid (HDCA), a secondary bile acid generated in human small intestine,16 completely abolishes FXR activity whereas retains GPBAR1 modulation showing a weak agonistic profile.17
We have recently investigated the chemical space of HDCA,18 introducing various apolar side chains and making specific chemical modifications on its steroidal scaffold. This study led to the identification of the first example of LXRα/GPBAR1 dual modulator useful in treatment of metabolic disorders. Here, we have designed and synthesized a new series of HDCA derivatives with the aim of discover novel selective agonists of GBPAR1. Specifically, we have introduced side chains of different length and size containing an epoxide group on the HDCA scaffold, which are endowed of amphipathic properties and thus able to establish both polar and hydrophobic contacts with the receptor counterpart. The rationale of the design is based on our previous computational studies showing that the side chain of GPBAR1 ligands is placed in proximity of an amphipathic region of the receptor.13b,18,19,20 The novel ligands described in this study were expected to preserve or even enhance affinity towards GPBAR1. Furthermore, since FXR agonists typically have polar side chains, mainly carboxylic or alcoholic end groups, the epoxide compounds should show a preferred activity towards GPBAR1 over FXR.13d
In addition to the side chain of HDCA, we have also modified the substituent at the C-6 position of the steroidal scaffold and investigated the ability of the newly synthesized HDCA analogues (1–6, Fig. 2) to activate GPBAR1 in cell-based assays and to regulate the expression of GPBAR1 targeted genes. The selectivity profile of the new compounds towards GPBAR1 over other pharmacologically relevant bile acid receptors such as FXR, LXRα and LXRβ is also reported along with molecular modeling studies on GPBAR1.
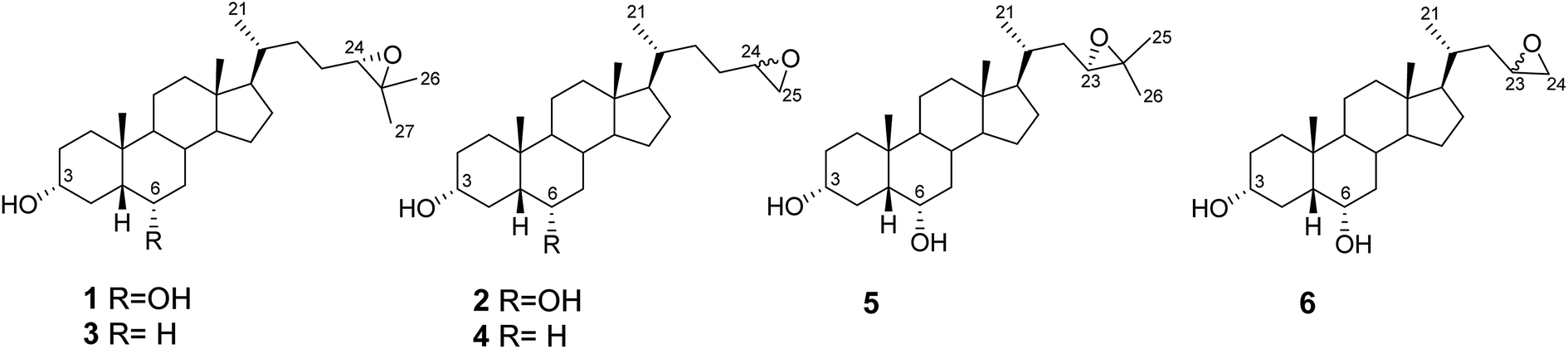 | ||
| Fig. 2 HDCA derivatives prepared in this study. | ||
2. Results and discussion
2.1 Chemistry
Aldehyde 7 was used as intermediate in the preparation of compounds 1 and 2. As previously described,18 a four-steps reaction sequence on HDCA methyl ester, including protection of alcoholic functions at C-3 and C-6 (2,6-lutidine, t-butyldimethylsilyl trifluoromethanesulfonate, CH2Cl2), reduction of the side chain methyl ester (LiBH4, MeOH/THF), and subsequent one pot Swern oxidation furnished aldehyde 7 in 76% chemical yield (Scheme 1).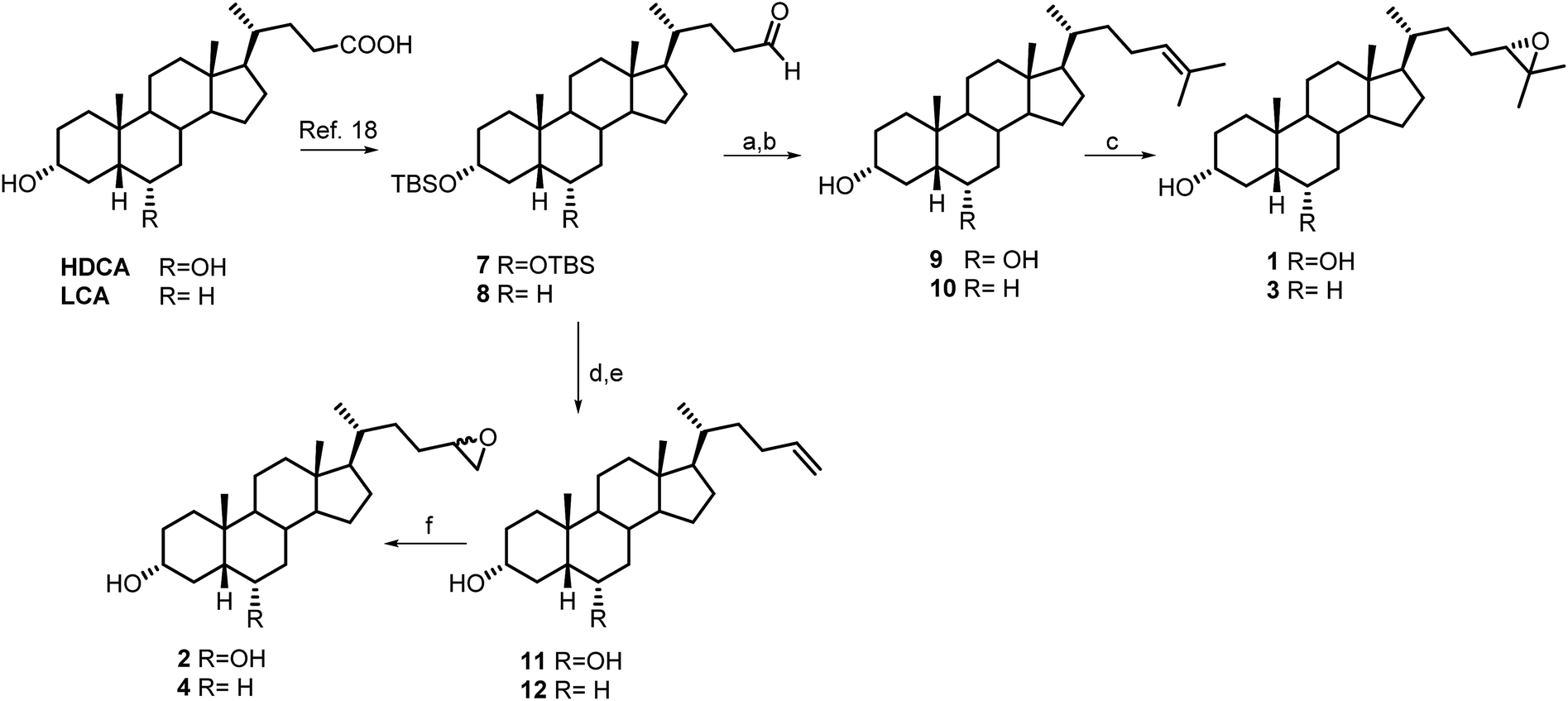 | ||
| Scheme 1 Synthesis of compounds 1–4. Reagents and conditions: (a) n-BuLi, isopropyl triphenylphosponium iodide, THF dry, r.t.; (b) HCl 37%, MeOH; (c) MCPBA, CHCl3 dry; (d) n-BuLi, methyl triphenylphosponium iodide, THF dry, r.t.; (e) HCl 37%, MeOH; (f) MCPBA, CHCl3 dry. | ||
Wittig olefination with isopropyl triphenylphosphonium iodide followed by the removal of 3α,6α-dihydroxy protective groups gave the unsaturated compound 9 that was also used as starting material in double bound epoxidation with meta-chloroperbenzoic acid (MCPBA) affording the epoxy derivative 1 in 87% yield. The 24(S) configuration of 1 was deduced by the complete agreement of the chemical shifts values of the Me-26 and Me-27 in the side chain with those of 24(S),25-epoxycholesterol.21
Compound 2 (Scheme 1) was prepared as an inseparable mixture of the two diasteroisomers at C-24 position following the same synthetic protocol described for compound 1, using methyl triphenylphosponium iodide in Wittig olefination and meta-chloroperbenzoic acid (MCPBA) in epoxidation.
To obtain the corresponding 6-deoxy derivatives 3 and 4, LCA methyl ester was subjected to the same four-step sequence depicted for HDCA (Scheme 1). Witting olefination with isopropyl triphenylphosphonium iodide and methyl triphenylphosponium iodide followed by the removal of 3α-hydroxy protective group gave the unsaturated compounds 10 and 12, respectively, that were used as starting material in double bound epoxidation with meta-chloroperbenzoic acid (MCPBA) affording the epoxy derivatives 3 (68% yield) and 4, respectively.
Scheme 2 illustrates the synthetic protocols for the preparation of HDCA derivatives 5 and 6 with nor epoxy side chains. HDCA was transformed in the C-23 protected aldehyde 13 following our previously published procedure.18,22
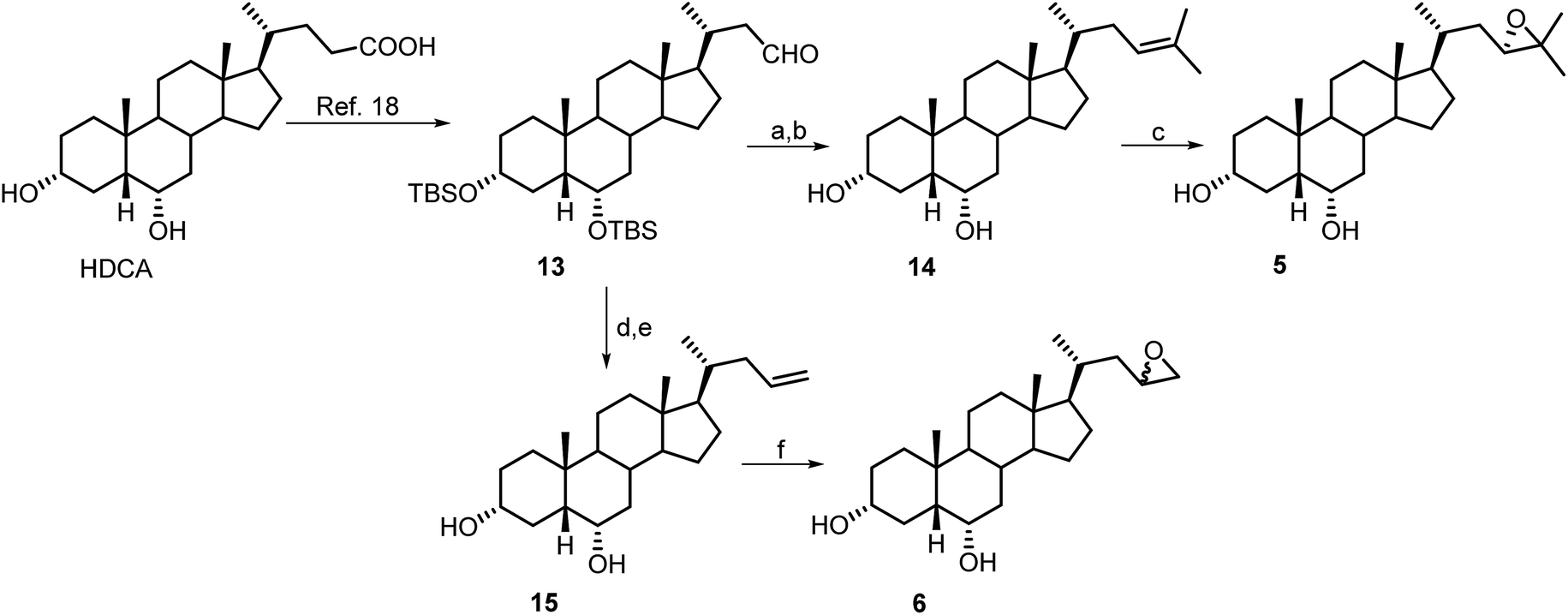 | ||
| Scheme 2 Synthesis of compounds 5 and 6. Reagents and conditions: (a) n-BuLi, isopropyl triphenylphosponium iodide, THF dry, r.t.; (b) HCl 37%, MeOH; (c) MCPBA, CHCl3 dry; (d) n-BuLi, methyl triphenylphosponium iodide, THF dry, r.t.; (e) HCl 37%, MeOH; (f) MCPBA, CHCl3 dry. | ||
Wittig olefination and double bond epoxidation in the same operative conditions described for the preparation of derivatives 1 and 2, gave the corresponding nor derivatives 5 and 6.
2.2 Pharmacological evaluation
Compounds 1–6 were evaluated in a luciferase reporter assay on HEK-293T cells transiently transfected with human GPBAR1. Cells were stimulated with compounds 1–6 using TLCA, the most potent endogenous activator of GPBAR1 (10 μM), as reference compound. As shown in Fig. 3, all derivatives generated in this study were able to induce cAMP-luciferase reporter gene, with an efficacy at 10 μM concentration comparable to that of TLCA.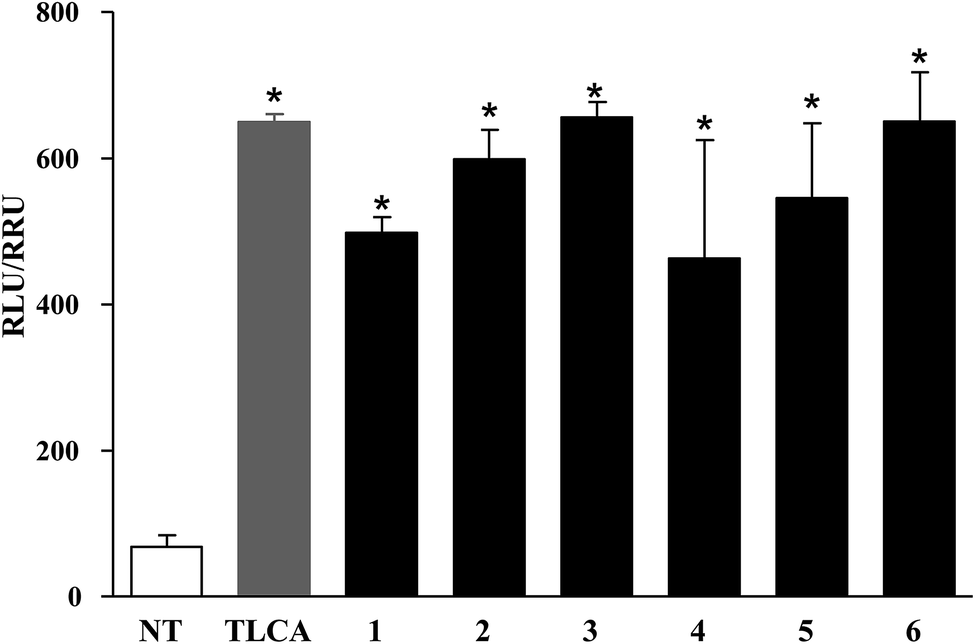 | ||
| Fig. 3 Agonism on GPBAR1 by transactivation assay. Luciferase reporter assay was performed on HEK-293T cells co-transfected with GPBAR1 and a reporter gene containing a cAMP responsive element in front of the luciferase gene. Cells were stimulated with 1–6 (10 μM). TLCA (10 μM) was used as a positive control. Luciferase activity served as a measure of the rise in intracellular cAMP following activation of GPBAR1. Value was expressed as RLU/RRU (Relative Luciferase Unit/Relative Renilla Unit). Results are expressed as mean ± standard error. *p < 0.05 versus not treated cells (NT). | ||
The relative potency was then investigated by a detailed measurement of the concentration–response curves with 1–6 transactivating GPBAR1 with EC50 values in the range of 1.49–5.80 μM (Fig. 4).
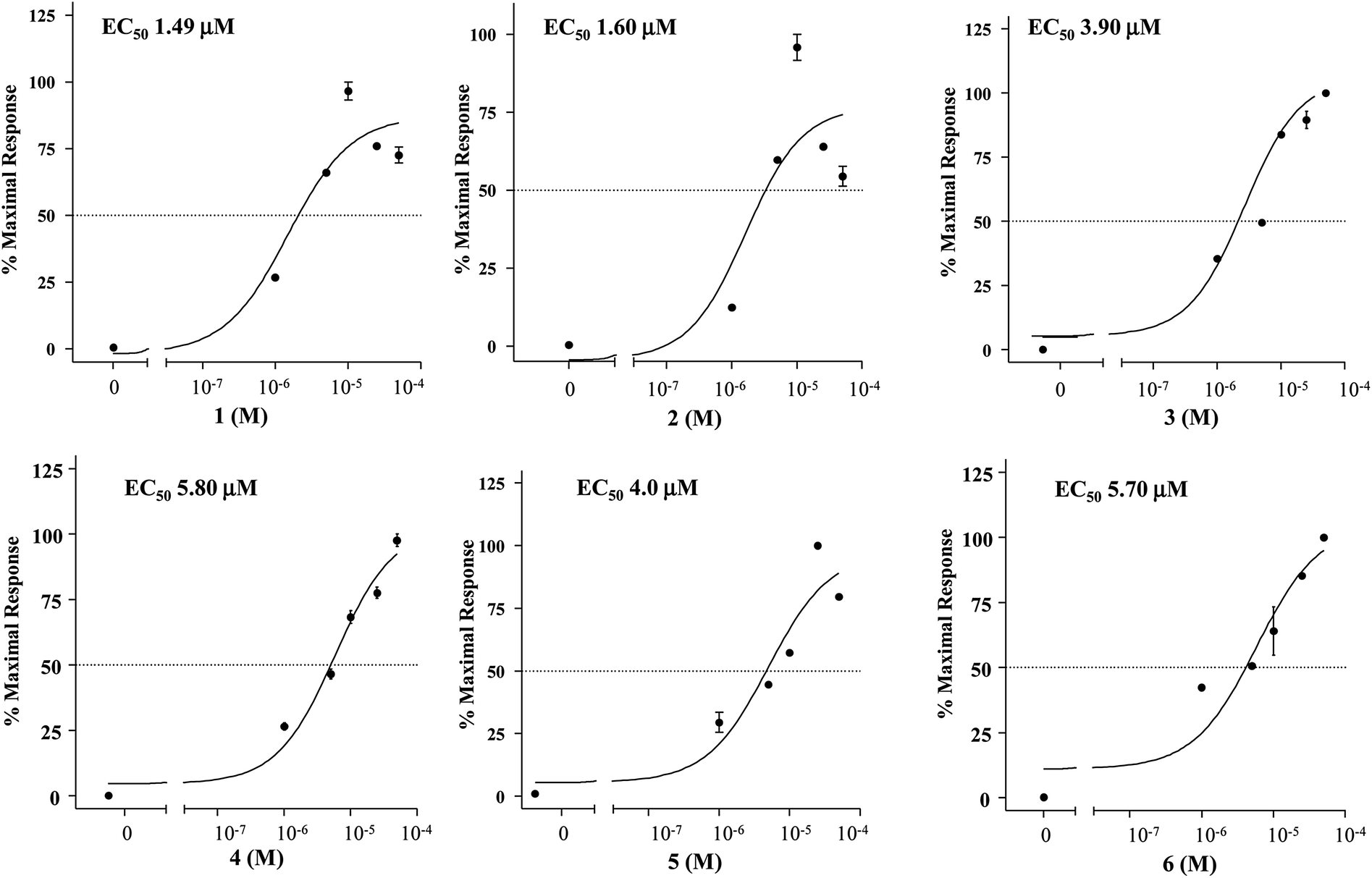 | ||
Fig. 4 Concentration–response curve of compounds 1–6 on GPBAR1. GPBAR1 activity was measured in HEK-293T cells co-transfected with GPBAR1 and a reporter gene containing a cAMP responsive element in front of the luciferase gene (CRE). Twenty-four hour post transfection cells were stimulated with increasing concentrations of each agent: range from 1 μM to 50 μM. Results are expressed as mean ± ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) standard error. standard error. | ||
Furthermore, compounds 1–6 were evaluated in vitro on common off-targets, such as FXR and LXRα and β. As showed in Fig. 5, none compound, when tested at 10 μM concentration, induced FXR as well as LXRα and LXRβ transactivation on HepG2 cells, thus demonstrating that the introduction of an epoxy functionality on HDCA and LCA scaffold represents a good strategy to obtain potent and selective GPBAR1 agonists.
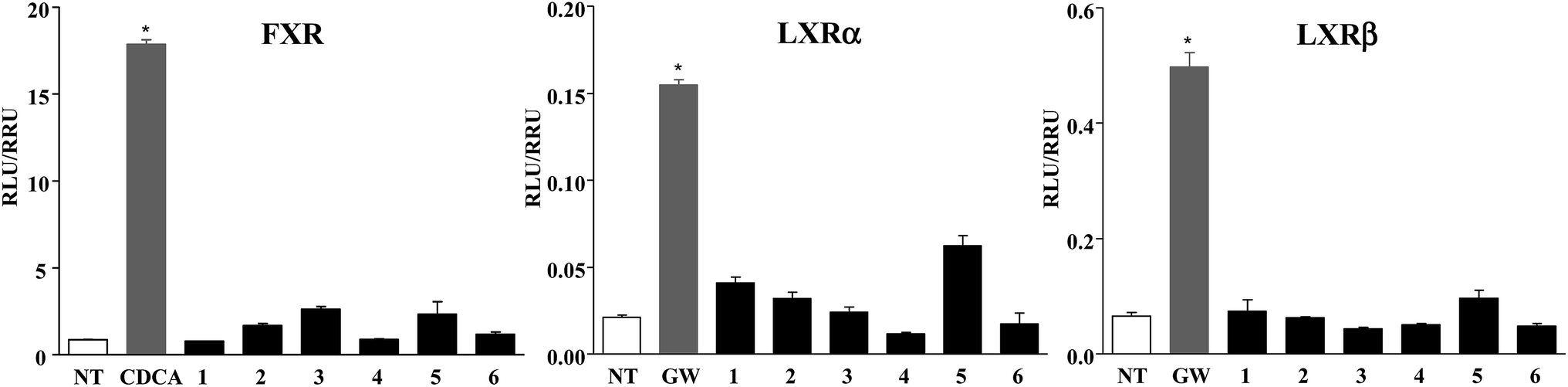 | ||
| Fig. 5 Specificity of compounds 1–6 on nuclear receptors. (FXR) HepG2 cells were co-transfected with pSG5-FXR, pSG5-RXR, and with the reporter vector phsp27TKLuc and then stimulated 18 h with CDCA (10 μM), a FXR agonist, or with 1–6 (10 μM); (LXRα and LXRβ) HepG2 cells were co-transfected with the Gal4 luciferase reporter vector and with a chimera in which the Gal4 DNA binding domain is fused to the LBD of LXRα or LXRβ, and stimulated 18 h with GW3965 (10 μM), a LXR agonist, or with 1–6 (10 μM). Data are the mean ± SE of three experiments. *p < 0.05 versus not treated cells (NT). | ||
Considering the reactivity of epoxide compounds, we have analyzed the presence in the GPBAR1 binding site and along the ligand-binding pathway of nucleophilic amino acids that can typically react with epoxide functions (e.g., Cys, Asp).23 The analysis has revealed that none of these amino acids is present either in the binding site or along the ligand-binding pathway. However, within our family of GPBAR1 agonists, we have investigated the stability of compounds 1 and 2 as prototypes of a branched and a linear epoxide, respectively, in HEK-293T cell culture medium (D-MEM additioned with 10% FBS) and in HEK-293T cell culture medium additioned with serine, in order to mimic the chemical environment of the GPBAR1 binding site in which three serine residues are present. HPLC analysis revealed the presence of intact compounds 1 and 2 after incubation for 4 h at 37 °C in cell medium and in cell medium/serine (Fig. S2 and S3 in the ESI†), thus demonstrating the stability of the above compounds in physiological conditions and their non-reactivity within GPBAR1.
Finally, the pharmacological potential of derivatives 1–6 is well demonstrated through RT-PCR data showed in Fig. 6. When administered in vitro on GLUTAg cells, all compounds generated in this study caused a robust increase of the mRNA of proglucagon-1, a GPBAR1 targeted gene. GLUTAg cells are an intestinal enteroendocrine cell line, releasing glucagon-like peptide 1 (GLP-1) by differential processing of proglucagon-1, the gene which is expressed in these cells, in response to GPBAR1 agonists,24 therefore are considered a suitable model in evaluating the interactions between GPBAR1 and its ligands. Furthermore, GLP-1 is an appealing pharmacological target in the treatment of diabetes and GLP-1 receptor agonists are currently used in the treatment of type 1 diabetes.25
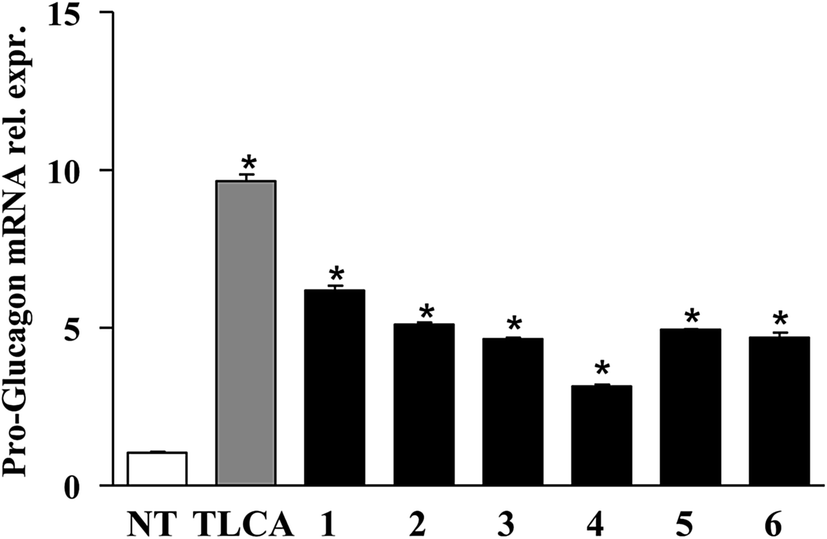 | ||
| Fig. 6 Effect of compounds 1–6 on pro-glucagon mRNA expression. Real-time PCR analysis of mRNA expression of GPBAR1 target gene pro-glucagon in GLUTAg cells stimulated with 10 μM TLCA or compounds 1–6. Values are normalized relative to GAPDH mRNA and are expressed relative to those of not treated cells (NT), which are arbitrarily set to 1. The relative mRNA expression is expressed as 2 − (ΔΔCt). *p < 0.05 versus NT (non treated cells). | ||
2.3 Molecular modeling
In order to investigate the binding mode of the newly synthesized HDCA derivatives to GPBAR1, we performed docking simulation that is a widely used computational technique to generate and rank ligand/protein complexes based on empirical scoring functions.26 In the present case, we performed docking calculations on the most potent compound of the series 1 in the receptor homology model of GPBAR1 that has been successfully used in our previous drug design studies.13b,18,19,20 The docking results showed that 1 binds to GPBAR1 similarly to other BA compounds previously investigated by us as agonists of this receptor (Fig. 7).13b,18,19,20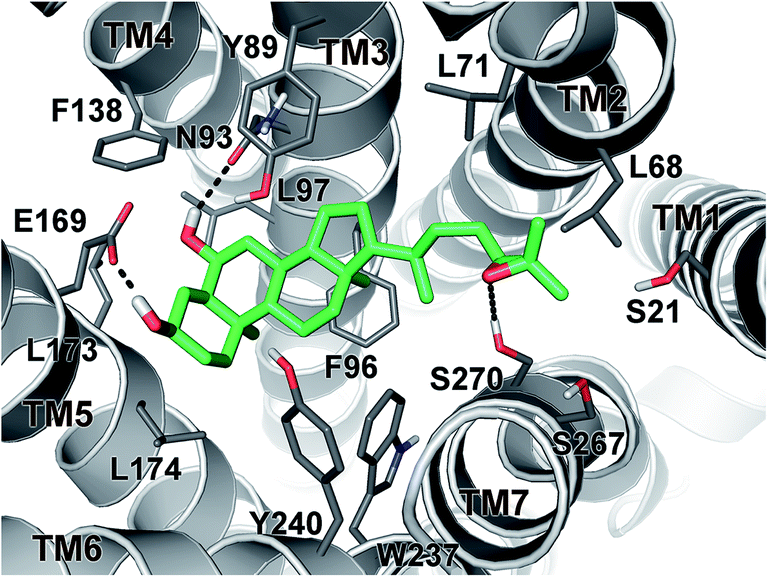 | ||
| Fig. 7 Binding mode of 1 (green sticks) in hGPBAR1 (gray cartoon). Receptor amino acids important for ligand binding are displayed as sticks. Receptor extracellular loops and all nonpolar hydrogens are omitted for clarity. | ||
In particular, the ligand steroidal scaffold establishes a number of hydrophobic interactions with the side chains of Tyr89, Phe96, Leu174 and Trp237 and Tyr240, while the 3α- and 6α-hydroxyl groups engage H-bond interactions with Glu169 on the transmembrane helix-5 (TM-5) and with Asn93 on TM3, respectively. The latter interaction is however not fundamental to activate the receptor, in fact, compounds 3 and 4, which do not present the 6α-OH group and thus cannot interact with Asn93, display EC50 values comparable to their 6α-hydroxylated analogues. On the other side of the binding site, the epoxide side chain of 1 can reach and contact the serine residues on TM-1 and TM-7 (Fig. 7). In the same region, the ligand epoxide side chain can even form hydrophobic interactions with the side chains of Leu68 and Leu71 and Leu263. The amphipathic nature and the plasticity of this receptor region allow bile acids to accommodate side chains with different steric and electrostatic properties, thus explaining the similar agonist profiles shown by all the epoxide analogues presented in this study.
3. Experimental
3.1 Chemistry
All chemicals were obtained from Sigma-Aldrich, Inc. Solvents and reagents were used as supplied from commercial sources with the following exceptions. Dichloromethane, tetrahydrofuran and trimethylamine were distilled from calcium hydride immediately prior to use. Methanol was dried from magnesium methoxide as follow. Magnesium turnings (5 g) and iodine (0.5 g) were refluxed in a small (50–100 mL) quantity of methanol until all of the magnesium has reacted. The mixture was diluted (up to 1 L) with reagent grade methanol, refluxed for 2–3 h then distilled under nitrogen. All reactions were carried out under argon atmosphere using flame-dried glassware. Reaction progress was monitored via thin-layer chromatography (TLC) on Alugram silica gel G/UV254 plates. Silica gel MN Kieselgel 60 (70–230 mesh) from Macherey-Nagel Company was used for column chromatography. Tested samples were obtained by HPLC purification on a Waters Model 510 pump equipped with a Waters Rheodine injector and a differential refractometer, model 401 and the purities of compounds were determined to be greater than 95%.High-resolution ESI-MS spectra were performed with a Micromass Q-TOF mass spectrometer. NMR spectra were obtained on Varian Inova 400, 500 and 700 NMR spectrometer (1H at 400, 500 and 700 MHz, 13C at 100, 125 and 175 MHz, respectively) equipped with a SUN microsystem ultra 5 hardware and recorded in CDCl3 (δH = 7.26 and δC = 77.0 ppm). All of the detected signals were in accordance with the proposed structures. Coupling constants (J values) are given in hertz (Hz), and chemical shifts (δ) are reported in ppm and referred to CHCl3 as internal standards. Spin multiplicities are given as s (singlet), dt (doublet triplet), d (doublet), or m (multiplet).
3.1.1.1 3α,6α-Di(tert-butyldimethylsilyloxy)-5β-cholan-24-al (7). Aldehyde 7 was prepared starting from HDCA as previously described.18
3.1.1.2 5β-Cholest-24-en-3α,6α-diol (9). To a solution of isopropyl triphenylphosphonium iodide (5.4 g, 12.5 mmol) in THF (2 mL), n-BuLi (5 mL, 12.5 mmol) was added dropwise at room temperature until the solution reached a red color. After 30 min, a solution of aldehyde 7 (1.5 g, 2.5 mmol) in THF (5 mL) was added. After 1 h, the mixture was quenched by addition of saturated aqueous NaHCO3 (50 mL) and extracted with EtOAc (3 × 50 mL). The organic phase was dried (Na2SO4) and concentrated. Purification on silica gel (hexane) gave the protected intermediate in 84% yield. To a solution of this intermediate (1.3 g, 2.1 mmol) in MeOH, 1 mL of HCl 37% v/v was added. After 1 h, silver carbonate was added, the reaction mixture was centrifuged, and the supernatant was concentrated in vacuo to give compound 9 as a colorless amorphous solid (850 mg, quantitative yield). An analytic sample was purified by HPLC on a Nucleodur 100-5 C18 (5 μm; 10 mm i.d. × 250 mm) with MeOH/H2O (96
:4) as eluent (flow rate 3 mL min−1, tR = 12.0 min). Selected 1H NMR (400 MHz, CDCl3): δH 5.10 (1H, t, J = 6.7 Hz, H-24), 4.08 (1H, dt, J = 11.9, 4.6 Hz, H-6β), 3.64 (1H, m, H-3β), 1.68 (3H, s, Me-26), 1.60 (3H, s, Me-27), 0.92 (3H, d, J = 7.0 Hz, Me-21), 0.91 (3H, s, Me-19), 0.65 (3H, s, Me-18); 13C NMR (100 MHz, CDCl3): δC 130.9, 125.3, 71.6, 68.2, 56.2 (2 × C), 48.4, 42.9, 40.0, 39.9, 36.4, 36.2, 35.6, 35.3, 35.0, 34.9, 30.3, 29.3, 28.4, 25.7, 24.7, 24.3, 23.3, 20.9, 18.4, 17.7, 12.1. HR ESIMS m/z 403.3576 [M + H]+, C27H46O2 requires 403.3578.
3.1.1.3 24(S),25-Epoxy-5β-cholestan-3α,6α-diol (1). To a solution of compound 9 (500 mg, 1.24 mmol) in CHCl3 dry, meta-chloroperbenzoic acid was added (643 mg, 3.73 mmol) at room temperature. The reaction was stirred overnight and then extracted with an aqueous solution of Na2SO3 (5%) and CH2Cl2 (3 × 50 mL) to give compound 1 (450 mg, 87%). Tested sample was obtained through HPLC (Fig. S1, Panel A in ESI†) on Phenomenex Luna C18 (5 μm; 4.6 mm i.d. × 250 mm) with MeOH/H2O (85
:15) as eluent (flow rate 1 mL min−1, tR = 15.0 min). The S configuration at C24 was determined in comparison with 1H-NMR data reported in literature for 24(S),25-epoxycholesterol.21 1H NMR (700 MHz, CDCl3) and 13C NMR (175 MHz, CDCl3) in Tables 1 and 2, ESI;† HR ESIMS m/z 419.3525 [M + H]+, C27H47O3 requires 419.3528.
3.1.1.4 25-Homo-5β-chol-24-en-3α,6α-diol (11). Compound 11 (370 mg, 0.99 mmol, 60%) was synthesized, starting from compound 7 (1 g, 1.65 mmol) by an analogous procedure to that detailed above for compound 9, using a solution of methyl triphenylphosphonium iodide. An analytic sample was purified by HPLC on a Nucleodur 100-5 C18 (5 μm; 10 mm i.d. × 250 mm) with MeOH/H2O (96
:4) as eluent (flow rate 3 mL min−1) giving pure compound 11 (tR = 11.2 min). Selected 1H NMR (400 MHz, CDCl3): δH 5.80 (1H, m, H-24), 4.99 (1H, d, J = 17.1 Hz, H-25), 4.91 (1H, d, J = 9.8 Hz, H-25), 4.05 (1H, dt, J = 12.0, 4.5 Hz, H-6β), 3.62 (1H, m, H-3β), 0.91 (3H, s, Me-19), 0.92 (3H, d, J = 7.0 Hz, Me-21), 0.64 (3H, s, Me-18);13C NMR (100 MHz, CDCl3): δC 139.6, 113.9, 71.6, 68.1, 56.2, 56.1, 48.4, 42.8, 39.9, 39.8, 36.0, 35.5, 35.3, 35.2, 35.1, 34.8, 30.5, 30.2, 29.2, 28.2, 24.2, 23.5, 20.7, 18.4, 12.0. HR ESIMS m/z 375.3263 [M + H]+, C25H43O2 requires 375.3266.
3.1.1.5 24,25-Epoxy-25-homo-5β-cholan-3α,6α-diol (2). The epoxidation on compound 11 (125 mg, 0.33 mmol) has been performed in the same conditions described for compound 1 obtaining, crude compound 2. HPLC purification (Fig. S1, Panel B in the ESI†) on a Phenomenex Luna C18 (5 μm; 4.6 mm i.d. × 250 mm) with MeOH/H2O (85
:15) as eluent (flow rate 1 mL min−1) gave pure 2 (tR = 11.2 min) as a mixture of two diastereoisomers at C-24 position. 1H NMR (700 MHz, CDCl3) and 13C NMR (175 MHz, CDCl3) in Tables 1 and 2, ESI;† HR ESIMS m/z 391.3212 [M + H]+, C25H43O3 requires 391.3214.
3.1.1.6 3α-tert-Butyldimethylsilyloxy-5β-cholan-24-al (8). Aldehyde 8 (1.8 g, 3.8 mmol, 72%), was synthesized starting from LCA (2 g, 5.3 mmol) as previously described.18
3.1.1.7 5β-Cholest-24-en-3α-ol (10). Compound 10 (367 mg, 0.95 mmol, 40% over two steps) was prepared from 8 (900 mg, 1.9 mmol) as detailed for compound 9. An analytic sample was purified by HPLC on a Nucleodur 100-5 C18 (5 μm; 4.6 mm i.d. × 250 mm) with MeOH/H2O (99
:1) as eluent (flow rate 1 mL min−1, tR = 12.4 min). Selected 1H NMR (400 MHz, CDCl3): δH 5.08 (1H, t, J = 6.6 Hz, H-24), 3.62 (1H, m, H-3β), 1.68 (3H, s, Me-26), 1.60 (3H, s, Me-27), 0.92 (3H, ovl, Me-21), 0.92 (3H, s, Me-19), 0.64 (3H, s, Me-18); 13C NMR (100 MHz, CDCl3): δC 130.9, 125.2, 71.9, 56.5, 56.2, 42.7, 42.1, 40.4, 40.2, 36.5, 36.1, 35.8, 35.6, 35.3, 34.6, 30.6, 28.3, 27.2, 26.4, 25.7, 24.7, 24.2, 23.4, 20.8, 18.6, 17.6, 12.0. HR ESIMS m/z 387.3620 [M + H]+, C27H47O requires 387.3623.
3.1.1.8 24(S),25-Epoxy-5β-cholestan-3α-ol (3). Compound 3 (213 mg, 68%) was prepared from 10 (300 mg, 0.78 mmol) by the procedure detailed for 1. Tested sample was obtained by HPLC purification on Nucleodur 100-5 C18 (5 μm; 4.6 mm i.d. × 250 mm) with MeOH/H2O (92
:8) as eluent (flow rate 1 mL min−1, tR = 16.0 min). 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) in Tables 1 and 3, ESI;† HR ESIMS m/z 403.3576 [M + H]+, C27H47O2 requires 403.3579.
3.1.1.9 25-Homo-5β-chol-24-en-3α-ol (12). Compound 12 (653 mg, 1.8 mmol, quantitative yield over two steps) was prepared from 8 (900 mg, 1.9 mmol) as described for 9, using a solution of methyl triphenylphosphonium iodide. An analytic sample was purified by HPLC on a Nucleodur 100-5 C18 (5 μm; 4.6 mm i.d. × 250 mm) with MeOH/H2O (99
:1) as eluent (flow rate 1 mL min−1, tR = 10.2 min). Selected 1H NMR (400 MHz, CDCl3): δH 5.80 (1H, m, H-24), 4.99 (1H, d, J = 17.1 Hz, H-25), 4.91 (1H, d, J = 9.8 Hz, H-25), 4.05 (1H, dt, J = 12.0, 4.5 Hz, H-6β), 3.62 (1H, m, H-3β), 0.91 (3H, s, Me-19), 0.92 (3H, d, J = 7.0 Hz, Me-21), 0.64 (3H, s, Me-18); 13C NMR (100 MHz, CDCl3): δC 139.6, 113.9, 71.6, 56.5, 56.2, 42.7, 42.2, 40.5, 40.2, 36.5, 35.8, 35.3, 35.2, 34.8, 34.6, 30.5, 30.2, 28.3, 27.2, 26.4, 24.2, 23.5, 20.8, 18.4, 12.0. HR ESIMS m/z 359.3314 [M + H]+, C25H43O requires 359.3317.
3.1.1.10 24,25-Epoxy-25-homo-5β-cholan-3α-ol (4). Crude compound 4 (521 mg, 1.39 mmol) was prepared from 12 (500 mg, 1.39 mmol) in the same operative conditions previously described for 2. Tested sample was obtained as a mixture of two diastereoisomers at C-24 position by HPLC purification on Nucleodur 100-5 C18 (5 μm; 4.6 mm i.d. × 250 mm) with MeOH/H2O (92
:8) as eluent (flow rate 1 mL min−1, tR = 15.1 min). 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) in Tables 1 and 3, ESI;† HR ESIMS m/z 375.3263 [M + H]+, C25H43O2 requires 375.3265.
3.1.1.11 3α,6α-Di(tert-butyldimethylsilyloxy)-24-nor-5β-cholan-23-al (13). Aldehyde 13 was prepared from HDCA as previously described.18
3.1.1.12 25-nor-5β-Cholest-23-en-3α,6α-diol (14). Compound 14 (264 mg, 0.7 mmol, 80% over two steps) was prepared from 13 (500 mg, 0.85 mmol) by the same procedure described for 9. An analytic sample was purified by HPLC on a Nucleodur 100-5 C18 (5 μm; 10 mm i.d. × 250 mm) with MeOH/H2O (96
:4) as eluent (flow rate 3 mL min−1, tR = 16.0 min) giving pure compound 14. Selected 1H NMR (400 MHz, CD3OD): δH 5.12 (1H, br t, J = 6.8 Hz, H-23), 4.01 (1H, dt, J = 11.9, 4.6 Hz, H-6β), 3.50 (1H, m, H-3β), 1.70 (3H, s, Me-26), 1.59 (3H, s, Me-25), 0.93 (3H, s, Me-19), 0.90 (3H, d, J = 6.7 Hz, Me-21), 0.69 (3H, s, Me-18).13C NMR (100 MHz, CD3OD): δC 132.8, 124.2, 72.4, 68.6, 57.6 (2 × C), 49.9, 44.1, 41.3 (2 × C), 38.2, 37.0, 36.8, 36.2, 35.6, 35.5, 31.1, 29.9, 29.5, 26.1, 25.5, 24.1, 21.9, 19.2, 18.0, 12.4. HR ESIMS m/z 389.3420 [M + H]+, C26H45O2 requires 389.3424.
3.1.1.13 23(S),24-Epoxy-25-nor-5β-cholestan-3α,6α-diol (5). Epoxidation on compound 14 (100 mg, 0.3 mmol) was performed in the same operative conditions described for 1, furnishing compound 5 (105 mg, 0.26 mmol, 87%). Tested sample was obtained by HPLC on a Nucleodur 100-5 C18 (5 μm; 10 mm i.d. × 250 mm) with MeOH/H2O (90
:10) as eluent (flow rate 3 mL min−1), giving pure 5 (tR = 10.4 min). The S configuration was determined in comparison with 1H NMR data reported in literature for compound 24(S),25-epoxycholesterol.21 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) in Tables 1 and 2, ESI;† HR ESIMS m/z 405.3369 [M + H]+, C26H45O3 requires 405.3371.
3.1.1.14 5β-Chol-24-en-3α,6α-diol (15). Wittig olefination on aldehyde 13 (500 mg, 0.85 mmol), as described for compound 14, using a solution of methyl triphenylphosphonium iodide, gave compound 15 (290 mg, 0.8 mmol, 95% over two steps). An analytic sample was purified by HPLC on Nucleodur 100-5 C18 (5 μm; 10 mm i.d. × 250 mm) with MeOH/H2O (96
:4) as eluent (flow rate 3 mL min−1), giving pure compound 15 (tR = 11.0 min). Selected 1H NMR (400 MHz, CDCl3): δH 5.77 (1H, m, H-23), 5.01 (1H, ovl, H-24), 4.99 (1H, ovl, H-24), 4.06 (1H, dt, J = 12.1, 4.6 Hz, H-6β), 3.63(1H, m, H-3β), 0.92 (3H, d, J = 6.6 Hz, Me-21), 0.91 (3H, s, Me-19), 0.66 (3H, s, Me-18). 13C NMR (100 MHz, CDCl3): δC 137.3, 115.7, 71.6, 68.1, 56.1, 55.8, 48.4, 42.8, 40.5, 39.9, 39.8, 35.9, 35.8, 35.5, 35.1, 34.8, 30.3, 29.2, 28.2, 24.2, 23.5, 20.7, 18.6, 12.0. HR ESIMS m/z 361.3101 [M + H]+, C24H41O2 requires 361.3103.
3.1.1.15 23,24-Epoxy-5β-cholan-3α,6α-diol (6). Epoxidation on 15 (100 mg, 0.3 mmol) in the same operative conditions described for 2 furnished crude 6 (108 mg, 0.3 mmol). Tested sample was obtained as a mixture of two diastereoisomers at C-23 position by HPLC purification on Nucleodur 100-5 C18 (5 μm; 4.6 mm i.d. × 250 mm) with MeOH/H2O (80
:20) as eluent (flow rate 1 mL min−1, tR = 37.5 min). 1H NMR (500 MHz, CD3OD and 400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) in Tables 1 and 2, ESI;† HR ESIMS m/z 377.3056 [M + H]+, C24H41O3 requires 377.3058.
3.2 Biological assay
3.3 Stability of compound 1 and compound 2 in cell medium and toward serine amino acid
One microliter of individual 60 μM solutions in DMSO of the compound 1 and compound 2 was diluted either in 499 μL of cell medium (D-MEM additioned with 10% FBS) or in 499 μL of cell medium additioned with 5 equivalent of serine in a 1.5 mL Eppendorf. The Eppendorf were gently shaken 4 h at 37 °C and injected on an HPLC apparatus equipped with a differential refractometer, model 401. Analysis was performed on Phenomenex Luna C18 (5 μm; 4.6 mm i.d. × 250 mm) column with MeOH/H2O (85:15) as eluent (flow rate 1 mL min−1). One microliter of individual DMSO solutions of compound 1 and compound 2 was also diluted in 499 μL of MeOH/H2O (85:15). For each sample, the stability was determined comparing HPLC peak area of the sample in the standard solution with HPLC peak area in cell medium and in cell medium additioned with serine.
3.4 Molecular docking
The Glide (version 7.1) software package27 was used to perform molecular docking calculations in the three-dimensional model of hGPBAR1.13b Ligand and receptor structures were prepared as described in a previous paper.19 A box centered on the GPBAR1 binding cavity was created; the Cartesian coordinates of the box, X, Y, and Z length were all set to 24.74, 26.74, 22.74 Å, respectively. The standard precision (SP) mode of the GlideScore function was used to score the predicted binding poses. All the residue labels were taken from the wild-type amino acidic sequence of human GPBAR1. All figures were rendered using PyMOL (http://www.pymol.org).4. Conclusions
With the aim at developing selective agonists of GPBAR1, we have described a novel family of HDCA-derivatives. All of these compounds activate the receptor in cell-based assay displaying EC50 values in the low micromolar range and increase the release of GLP-1, a well validated GPBAR1 target, from GLUTAg cells, a murine cell line of intestinal endocrine L cells. Remarkably, the novel derivatives result inactive on common off-targets of GPBAR1 ligands, such as LXRs and FXR and this finding leads to the conclusion that the introduction of an epoxide group on the bile acid side chain can shift ligand selectivity towards GPBAR1 over FXR and other nuclear receptors. These aspects have been investigated through molecular docking studies disclosing the binding mode to GPBAR1 of the newly synthesized BA derivatives and providing new valuable hints for the selective modulation of the membrane bile acid receptor.The present results contribute to increase the arsenal of bile acids derivatives serving as GPBAR1 modulators, widening the chemical space that can be exploited in drug design and paving the way for testing the novel derivatives in rodent models of common metabolic disorders including obesity and diabetes.
Conflict of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by grants from MIUR-ITALY PRIN2015 “Top-down and Bottom-up approach in the development of new bioactive chemical entities inspired on natural products scaffolds” (Project No. 2015MSCKCE_003) and the Swiss National Science Foundation (Project No. 200021_163281). The authors thank European Cooperation in Science and Technology (COST Action CA15135; multi-target paradigm for innovative ligand identification in the drug discovery process) for the support.Notes and references
- Y. Kawamata, R. Fujii, M. Hosoya, M. Harada, H. Yoshida, M. Miwa, S. Fukusumi, Y. Habata, T. Itoh, Y. Shintani, S. Hinuma, Y. Fujisawa and M. Fujino, J. Biol. Chem., 2003, 278, 9435–9440 CrossRef CAS PubMed.
- T. Maruyama, Y. Miyamoto, T. Nakamura, Y. Tamai, H. Okada, E. Sugiyama, T. Nakamura, H. Itadani and K. Tanaka, Biochem. Biophys. Res. Commun., 2002, 298, 714–719 CrossRef CAS PubMed.
- D. P. Kumar, S. Rajagopal, S. Mahavadi, F. Mirshahi, J. R. Grider, K. S. Murthy and A. J. Sanyal, Biochem. Biophys. Res. Commun., 2012, 427, 600–605 CrossRef CAS PubMed.
- M. Watanabe, S. M. Houten, C. Mataki, M. A. Christoffolete, B. W. Kim, H. Sato, N. Messaddeq, J. W. Harney, O. Ezaki, T. Kodama, K. Schoonjans, A. C. Bianco and J. Auwerx, Nature, 2006, 439, 484–489 CrossRef CAS PubMed.
- G. Vassileva, W. Hu, L. Hoos, G. Tetzloff, S. Yang, L. Liu, L. Kang, H. R. Davis, J. A. Hedrick, H. Lan, T. Kowalski and E. L. Gustafson, J. Endocrinol., 2010, 205, 225–232 CrossRef CAS PubMed.
- V. Keitel, K. Cupisti, C. Ullmer, W. T. Knoefel, R. Kubitz and D. Häussinger, Hepatology, 2009, 50, 861–870 CrossRef CAS PubMed.
- T. Li, S. R. Holmstrom, S. Kir, M. Umetani, D. R. Schmidt, S. A. Kliewer and D. J. Mangelsdorf, Mol. Endocrinol., 2011, 25, 1066–1071 CrossRef CAS PubMed.
- N. Pean, I. Doignon, I. Garcin, A. Besnard, B. Julien, B. Liu, S. Branchereau, A. Spraul, C. Guettier, L. Humbert, K. Schoonjans, D. Rainteau and T. Tordjmann, Hepatology, 2013, 58, 1451–1460 CrossRef CAS PubMed.
- F. Alemi, D. P. Poole, J. Chiu, K. Schoonjans, F. Cattaruzza, J. R. Grider, N. W. Bunnett and C. U. Corvera, Gastroenterology, 2013, 144, 145–154 CrossRef CAS PubMed.
- F. Alemi, E. Kwon, D. P. Poole, T. Lieu, V. Lyo, F. Cattaruzza, F. Cevikbas, M. Steinhoff, R. Nassini, S. Materazzi, R. Guerrero-Alba, E. Valdez-Morales, G. S. Cottrell, K. Schoonjans, P. Geppetti, S. J. Vanner, N. W. Bunnett and C. U. Corvera, J. Clin. Invest., 2013, 123, 1513–1530 CAS.
- V. Z. Rocha and P. Libby, Nat. Rev. Cardiol., 2009, 6, 399–409 CrossRef CAS PubMed.
- (a) S. Fiorucci, A. Mencarelli, G. Palladino and S. Cipriani, Trends Pharmacol. Sci., 2009, 30, 570–580 CrossRef CAS PubMed; (b) A. Tiwari and P. Maiti, Drug Discovery Today, 2009, 14, 523–530 CrossRef CAS PubMed; (c) S. Fiorucci, S. Cipriani, F. Baldelli and A. Mencarelli, Prog. Lipid Res., 2010, 49, 171–185 CrossRef CAS PubMed.
- (a) S. Fiorucci, A. Mencarelli, E. Distrutti, G. Palladino and S. Cipriani, Curr. Med. Chem., 2010, 17, 139–159 CrossRef CAS PubMed; (b) C. D'Amore, F. S. Di Leva, V. Sepe, B. Renga, C. Del Gaudio, M. V. D'Auria, A. Zampella, S. Fiorucci and V. Limongelli, J. Med. Chem., 2014, 57, 937–954 CrossRef PubMed; (c) C. Festa, B. Renga, C. D'Amore, V. Sepe, C. Finamore, S. De Marino, A. Carino, S. Cipriani, M. C. Monti, A. Zampella and S. Fiorucci, J. Med. Chem., 2014, 57, 8477–8495 CrossRef CAS PubMed; (d) V. Sepe, E. Distrutti, V. Limongelli, S. Fiorucci and A. Zampella, Future Med. Chem., 2015, 7, 1109–1135 CrossRef CAS PubMed; (e) S. Fiorucci and E. Distrutti, Trends Mol. Med., 2015, 21, 702–714 CrossRef CAS PubMed; (f) C. Finamore, C. Festa, B. Renga, V. Sepe, A. Carino, D. Masullo, M. Biagioli, S. Marchianò, A. Capolupo, M. C. Monti, S. Fiorucci and A. Zampella, Sci. Rep., 2016, 6, 29320 CrossRef CAS PubMed; (g) A. Carino, S. Cipriani, S. Marchianò, M. Biagioli, C. Santorelli, A. Donini, A. Zampella, M. C. Monti and S. Fiorucci, Sci. Rep., 2017, 7, 42801 CrossRef CAS PubMed.
- F. G. Schaap, M. Trauner and P. L. Jansen, Nat. Rev. Gastroenterol. Hepatol., 2014, 11, 55–67 CrossRef CAS PubMed.
- S. Cipriani, B. Renga, C. D'Amore, M. Simonetti, A. A. De Tursi, A. Carino, M. C. Monti, V. Sepe, A. Zampella and S. Fiorucci, PLoS One, 2015, 10, e0129866 Search PubMed.
- H. J. Eyssen, G. De Pauw and J. Van Eldere, Appl. Environ. Microbiol., 1999, 65, 3158–3163 CAS.
- H. Sato, A. Macchiarulo, C. Thomas, A. Gioiello, M. Une, A. F. Hofmann, R. Saladin, K. Schoonjans, R. Pellicciari and J. Auwerx, J. Med. Chem., 2008, 51, 1831–1841 CrossRef CAS PubMed.
- S. De Marino, A. Carino, D. Masullo, C. Finamore, S. Marchianò, S. Cipriani, F. Saverio Di Leva, E. Novellino, B. Catalanotti, V. Limongelli, S. Fiorucci and A. Zampella, Sci. Rep., 2017, 7, 43290 CrossRef PubMed.
- V. Sepe, B. Renga, C. Festa, C. D'Amore, D. Masullo, S. Cipriani, F. S. Di Leva, M. C. Monti, E. Novellino, V. Limongelli, A. Zampella and S. Fiorucci, J. Med. Chem., 2014, 57, 7687–7701 CrossRef CAS PubMed.
- F. S. Di Leva, C. Festa, B. Renga, V. Sepe, E. Novellino, S. Fiorucci, A. Zampella and V. Limongelli, Sci. Rep., 2015, 5, 16605 CrossRef CAS PubMed.
- N. C. O. Tomkinson, T. M. Willson, J. S. Russel and T. A. Spencer, J. Org. Chem., 1998, 63, 9919–9923 CrossRef CAS.
- V. Sepe, R. Ummarino, M. V. D'Auria, B. Renga, S. Fiorucci and A. Zampella, Eur. J. Org. Chem., 2012, 5187–5194 CrossRef CAS.
- (a) M. A. Argiriadi, C. Morisseau, B. D. Hammock and D. W. Christianson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10637–10642 CrossRef CAS PubMed; (b) G. A. Gomez, C. Morisseau, B. D. Hammock and D. W. Christianson, Biochemistry, 2004, 43, 4716–4723 CrossRef CAS PubMed.
- H. E. Parker, K. Wallis, C. W. le Roux, K. Y. Wong, F. Reimann and F. M. Gribble, Br. J. Pharmacol., 2012, 165, 414–423 CrossRef CAS PubMed.
- A. B. Petersen and M. Christensen, Diabetes, Metab. Syndr. Obes.: Targets Ther., 2013, 6, 217–231 CrossRef CAS PubMed.
- (a) M. Anzini, C. Braile, S. Valenti, A. Cappelli, S. Vomero, L. Marinelli, V. Limongelli, E. Novellino, L. Betti, G. Giannaccini, A. Lucacchini, C. Ghelardini, M. Norcini, F. Makovec, G. Giorgi and R. Ian Fryer, J. Med. Chem., 2008, 51, 4730–4743 CrossRef CAS PubMed; (b) D. Heckmann, B. Laufer, L. Marinelli, V. Limongelli, E. Novellino, G. Zahn, R. Stragies and H. Kessler, Angew. Chem., Int. Ed., 2009, 48, 4436–4440 CrossRef CAS PubMed; (c) M. Anzini, S. Valenti, C. Braile, A. Cappelli, S. Vomero, S. Alcaro, F. Ortuso, L. Marinelli, V. Limongelli, E. Novellino, L. Betti, G. Giannaccini, A. Lucacchini, S. Daniele, C. Martini, C. Ghelardini, L. Di Cesare Mannelli, G. Giorgi, M. P. Mascia and G. Biggio, J. Med. Chem., 2011, 54, 5694–5711 CrossRef CAS PubMed.
- Glide, version 7.1, Schrödinger, LLC, New York, NY, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra for all products. See DOI: 10.1039/c7ra04922f |
| This journal is © The Royal Society of Chemistry 2017 |