
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpirolactam capped cyanine dyes for designing NIR probes to target multiple metal ions†
Chirantan Kara,
Yutaka Shindob,
Kotaro Okab,
Shigeru Nishiyamac,
Koji Suzukia and
Daniel Citterio *a
*a
aDepartment of Applied Chemistry, Faculty of Science and Technology, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan. E-mail: citterio@applc.keio.ac.jp
bDepartment of Biosciences and Informatics, Faculty of Science and Technology, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan
cDepartment of Chemistry, Faculty of Science and Technology, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan
First published on 10th May 2017
Abstract
A cyanine based spirocyclic metal ion probe has been reported, which can be tuned easily for targeting various metal ions. The mechanism of selective sensing was verified by designing two novel compounds IR-NCS and IR-PYR, which can selectively sense Hg2+ and Zn2+/Cd2+ ions respectively in a mixed aqueous medium and physiological pH. The spectral changes in both the cases are in the near infrared (NIR) region. Both chemodosimetric and coordination based approaches were used for sensing the cations. The complete mechanism of the sensing events was studied using UV-Vis and fluorescence spectroscopy. Additional insight about the experimental findings was corroborated by theoretical studies using DFT calculations. The confocal microscopic study revealed that the probe IR-NCS can be effectively used for detecting intracellular levels of Hg2+ ions without showing any toxic effect to the model human cells.
Introduction
The physiological effects of heavy metal pollution have been well established through several experiments in natural as well as laboratory environments. Malformation in various organisms is now routinely used as a potent tool to identify toxic metal contamination of fresh and marine waters. According to the recent research metal-induced malformations in early Palaeozoic plankton can be an indicator of mass extinction.1 Apart from there toxic effect metals are also essential for life, playing significant parts in various biological processes (e.g., the transcription and translation of nucleic acids and synthesis of proteins). The biological importance of metal ions is remarkably versatile inside living organism; along with providing permanent and transient structural reinforcement metal ions can also facilitate electron transfer, transport small-molecules, and act as catalysts in cellular processes.2 Redox-inactive metals including alkali metals such as sodium, potassium, alkaline earth metals like calcium, and transition metals like zinc are commonly recognized as dynamic signals, on the other hand redox-active metals like iron and copper are traditionally known for being concealed by protein ligands and static enzyme cofactors. Due to the major role of metal ions in environmental and biological systems a better understanding of transition metals in their labile form is necessary. Accordingly, significant interest has been observed in developing selective and sensitive signalling units for different important metal ions.3Recently, various groups have reported different chemosensors for selective sensing of metal ions, with spectral response in the visual region.4–14 Although optical probes showing visual changes have considerable roles in various research areas, lately chemosensors with absorption and emission at the near-infrared region (NIR, 650–1000 nm) are gaining special interest.15 The amount of scattering of any electromagnetic radiation is inversely proportional to its wavelength, thus for long wavelength NIR radiation the possibility of scattering is less. Due to minimized scattering NIR radiation provides high sample penetration compared to ultra violet or visible radiation. Moreover, the interference generated due to autofluorescence from the chromophores and macromolecules present in the analytic sample is reduced, which enables the study of molecular and physiological events in several layers deep inside the analyte samples and tissues.16 Although there have been a few reports on NIR sensing of cations, but newer strategies of designing versatile NIR chemosensors for targeting various metal ions are still required.
To date, various NIR dyes have been applied as signal reporters of chemosensors, such as 1,8-naphthalimide,17,18 xanthenes,19 boron dipyrromethene difluoride (BODIPY),20,21 etc. Among them cyanine based platforms have attracted substantial interest from researchers around the world because these dyes show favourable spectral properties such as high fluorescence quantum yield, high absorption coefficient with narrow absorption band and spectral response (absorbance and fluorescence) in relatively longer wavelength.22 Additionally, low toxicity and high degree of biocompatibility to living cells and organisms further make cyanine dyes an attractive platform for biological and medicinal application. Although most of the chemosensors designed on cyanine platforms are based on photoinduced electron transfer (PET) based switch OFF–ON, due to their high lying occupied molecular orbital (HOMO) energy, quenching the fluorescence by the PET process alone is difficult.23
General strategies which are employed to generate metal dependent optical response from a chemosensor, are either reversible sensing based on coordination of the sensor molecule with the metal ions or irreversible sensing based on chemical reaction of the chemosensor (chemodosimeter) with specific metal ions. Most of the classical metal ion sensors are based on coordination approach, this kind of chemosensors are generally reversible in nature and can be used for several cycles. In some cases where the sensor-analyte coordination has optimum bond strength, they can even be used for extracting the target metal ions from a mixture.24 As one of the emerging sensing strategies, chemodosimetric detection of metal ions has attracted increasing attention because of irreversible chemical change of the dosimeter molecule in presence of target species, causing specific spectral response in the emission or absorption spectra. Thus, these kinds of sensors have additional advantages in terms of accuracy and selectivity of detection compared to those based on complexation or coordination effect.25,26 Taking the above needs into consideration various chemosensors for environmentally hazardous and benign metal ions have been reported. Among various hazardous metal ions mercury is one of the most harmful and abundant pollutant, its contamination is extensive and can be generated from various natural and anthropogenic sources.27,28 Once mercury makes its way into the food chains, it causes serious damage to the human health and ecology.29–31 Furthermore, introduction of a small quantity of mercury can be concentrated to harmful levels due to the natural process of bioaccumulation. Similar to mercury, cadmium is also a notorious contaminant and carcinogenic metal.32 Route of intake for cadmium is also widespread, although inhaling cadmium containing dust is the major way of intake but it may also take place through smoking and food. Cadmium is found mostly in electroplated steel, pigments in plastics, electric batteries, etc.33 A high exposure to cadmium can damage liver and kidneys, increase the risks of cardiovascular diseases and cancer. Owing to these health hazards, cadmium is listed as number 7 on ATSDR's “CERCLA Priority List of Hazardous Substances”34 which can cause acute and chronic toxicity. Apart from hazardous mercury and cadmium, zinc is an essential element involved in various crucial biological processes, like cellular metabolism, neurotransmission, and apoptosis.35 Zinc is also known to exist as a structural component of proteins or in the catalytic site of enzymes.36 As an essential micronutrient, deficiency of zinc can lead to immune dysfunction, impaired cognition, diarrhea, and death, primarily in children under the age of 5 years.37 The human genome is known to encode two dozen Zn2+ specific transporters and many metal-buffering proteins, expressed in a tissue specific manner.38 However, unlike other essential transition metal ions (viz., Fe2+, Cu2+, Mn2+), Zn2+ ion is spectroscopically and magnetically inert. Therefore, common analytical techniques, like Mossbauer, NMR, and EPR spectroscopy cannot detect a typical zinc ion in complex biological or environmental systems. Considering the environmental importance and physiological implications of Hg2+, Cd2+, and Zn2+ there is significant interest in developing selective and sensitive sensors for these metal ions.
Herein, we described a new method for designing and synthesizing cyanine based NIR fluorescence probes which can be easily tuned for targeting various metal ions. We have verified the potential of our strategy by the synthesis of two new hydrazine functionalized spirolactam capped cyanine probes where we have demonstrated the chemodosimetric approach to detect Hg2+ ions and coordination based approach for sensing Zn2+/Cd2+ metal ions. We have also conducted quantum mechanical calculation to rationalize our experimental findings.
Experimental section
General information and materials
All of the materials for synthesis were purchased from commercial suppliers and used without further purification. The absorption spectra were recorded on a UV-1800 (Shimadzu co. Kyoto, Japan) spectrophotometer using 10 mm path length quartz cuvettes in the range of 300–900 nm wavelengths, and the fluorescence measurements were carried on a SREX Fluorolog-3 (Model FL-3-11, Horiba Jobin Yvon, Kyoto, Japan) spectrofluorometer using 10 mm path length quartz cuvettes with a slit width of 5 nm at 298 K. The High-Resolution MS spectra (HR-MS) of IR-NCS, IR-HGP, IR-PYR and the corresponding metal complex were obtained on a Waters LCT Premier XE with MeOH as the solvent. NMR spectra were recorded on a JEOL ECA 500 MHz instrument. The chemical shifts were recorded in parts per million (ppm) on the scale. The following abbreviations are used to describe spin multiplicities in 1H NMR spectra: s = singlet; d = doublet; t = triplet; m = multiplet. Tetramethylsilane (TMS) was used as an internal standard for NMR spectroscopy.Synthesis of I1
IR-780 (200.0 mg, 0.3 mmol), 2-carboxyphenylboric acid (100.0 mg, 0.6 mmol), and K3PO4·7H2O (75.0 mg, 0.35 mmol) dissolved in 6 mL DMF/H2O (v/v = 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were heated at 90 °C in the presence of Pd(PPh3)4 (catalytic amount) for overnight under argon atmosphere. The reaction mixture was cooled to room temperature, and poured in 100 mL ice water. The mixture was extracted thrice using CH2Cl2/C2H5OH (v/v = 10:1). The organic phase was separated, washed with brine, and dried with anhydrous Na2SO4. The solvent was removed under reduced pressure to give the crude product, which was purified by column chromatography (using Merck Aluminium oxide 90 active basic) using CH2Cl2 to CH2Cl2/C2H5OH (v/v 100:1 to 95:5) as eluent to afford the compound I1 as a green solid (78 mg, 33.3% yield) 1H NMR [500 MHz, CD3OD, SiMe4, J (Hz), δ (ppm)]: 8.10–8.12 (1H, m), 7.52–7.54 (2H, m), 7.32 (1H, d, J = 5.0 Hz), 7.28 (4H, t, J = 6.5 Hz), 7.13 (4H, quartet, J = 16.25 Hz), 7.05–7.07 (2H, m), 6.08 (2H, d, J = 13.5 Hz), 3.98 (4H, t, J = 7.0 Hz), 2.70–2.75 (2H, m), 2.61–2.67 (2H, m), 2.15 (1H, broad s), 1.96 (1H, broad s), 1.75–1.82 (4H, m), 1.22 (6H, s), 1.12 (6H, s), 0.99 (6H, t, J = 7.5 Hz). 13C NMR [125 MHz, CDCl3, SiMe4, δ (ppm)]: 173.7, 172.8, 168.5, 149.8, 144.1, 142.3, 141.6, 139.8, 132.9, 131.6, 131.5, 130.1, 129.6, 129.1, 125.5, 123.4, 111.5, 100.5, 49.9, 46.2, 28.7, 28.3, 25.9, 22.5, 21.7, 11.8. ESI-MS (positive mode, m/z). Calcd for C43H49N2O2: 625.3794 (M). Found: m/z 625.3878. Melting point: 146–154 °C.
:1) were heated at 90 °C in the presence of Pd(PPh3)4 (catalytic amount) for overnight under argon atmosphere. The reaction mixture was cooled to room temperature, and poured in 100 mL ice water. The mixture was extracted thrice using CH2Cl2/C2H5OH (v/v = 10:1). The organic phase was separated, washed with brine, and dried with anhydrous Na2SO4. The solvent was removed under reduced pressure to give the crude product, which was purified by column chromatography (using Merck Aluminium oxide 90 active basic) using CH2Cl2 to CH2Cl2/C2H5OH (v/v 100:1 to 95:5) as eluent to afford the compound I1 as a green solid (78 mg, 33.3% yield) 1H NMR [500 MHz, CD3OD, SiMe4, J (Hz), δ (ppm)]: 8.10–8.12 (1H, m), 7.52–7.54 (2H, m), 7.32 (1H, d, J = 5.0 Hz), 7.28 (4H, t, J = 6.5 Hz), 7.13 (4H, quartet, J = 16.25 Hz), 7.05–7.07 (2H, m), 6.08 (2H, d, J = 13.5 Hz), 3.98 (4H, t, J = 7.0 Hz), 2.70–2.75 (2H, m), 2.61–2.67 (2H, m), 2.15 (1H, broad s), 1.96 (1H, broad s), 1.75–1.82 (4H, m), 1.22 (6H, s), 1.12 (6H, s), 0.99 (6H, t, J = 7.5 Hz). 13C NMR [125 MHz, CDCl3, SiMe4, δ (ppm)]: 173.7, 172.8, 168.5, 149.8, 144.1, 142.3, 141.6, 139.8, 132.9, 131.6, 131.5, 130.1, 129.6, 129.1, 125.5, 123.4, 111.5, 100.5, 49.9, 46.2, 28.7, 28.3, 25.9, 22.5, 21.7, 11.8. ESI-MS (positive mode, m/z). Calcd for C43H49N2O2: 625.3794 (M). Found: m/z 625.3878. Melting point: 146–154 °C.
Synthesis of I2
I1 (78.0 mg, 0.1 mmol), N-hydroxysuccinimide (23.5 mg, 0.2 mmol), and N,N′-dicyclohexyl carbodiimide (58.0 mg, 0.3 mmol) were dissolved in 3 mL dichloromethane, and the reaction mixture was stirred at room temperature for 1 h. The solvent was removed under reduced pressure to give the intermediate, which was dissolved in 2 mL DMF and 0.5 mL of hydrazine hydrate was added, the solution was stirred at room temperature for another 12 h. The colour of the reaction mixture changed from green to orange. The mixture was poured to 50 mL cold water and extracted three times using CH2Cl2. The organic phase was collected, dried using Na2SO4 and concentrated under reduced pressure to get the crude mixture. The reaction mixture was concentrated under reduced pressure to give the crude product, which was purified by column chromatography (using basic Alumina) using CH2Cl2 to CH2Cl2/C2H5OH (v/v 100:0 to 97:3) as eluent to afford the compound I2 as a dark orange solid, the compound has a tendency to degrade during column chromatography (50 mg, 0.08 mmol, 80% yield). 1H NMR [500 MHz, CDCl3, SiMe4, J (Hz), δ (ppm)]: 7.91 (1H, d, J = 8.0 Hz), 7.43–7.50 (2H, m), 7.38 (1H, t, J = 8.0 Hz), 7.08 (2H, t, J = 7.0 Hz), 7.0 (2H, d, J = 7.0 Hz), 6.75 (2H, t, J = 7.5 Hz), 6.5 (2H, d, J = 7.5 Hz), 6.16 (2H, d, J = 12 Hz), 5.27 (2H, d, J = 12 Hz), 3.97 (2H, s), 3.47 (4H, t, J = 7.5 Hz), 2.88–2.95 (2H, m), 2.58–2.64 (2H, m), 2.00–2.06 (1H, m), 1.83–1.90 (1H, m), 1.66 (4H, dd, J = 14.4, J = 7.5 Hz), 1.38 (6H, s), 1.14 (6H, s), 0.95 (6H, t, J = 8.0 Hz). 13C NMR [125 MHz, CDCl3, SiMe4, δ (ppm)]: 167.3, 156.1, 152.2, 145.3, 138.8, 131.8, 128.6, 127.7, 127.4, 126.2, 123.9, 121.9, 121.7, 120.9, 119.0, 105.8, 91.0, 75.2, 45.2, 44.1, 28.5, 26.1, 23.6, 19.8, 11.9. ESI-MS (positive mode, m/z). Calcd for C43H51N4O: 639.4063 (M + H). Found: m/z 639.4087. Melting point: 72 °C (decomposed).
Synthesis of IR-NCS
The hydrazide I2 (30 mg, 0.046 mmol) and phenyl isothiocyanate (18.16 mg, 0.13 mmol) were dissolved in 1 mL of DMF and the resulting solution was stirred for 10 h in room temperature. The reaction mixture was concentrated under reduced pressure, and the solid was collected. The crude mixture was purified with column chromatography (using basic Alumina) using CH2Cl2 to CH2Cl2/C2H5OH (v/v 100:0 to 97:3) as eluent to afford the compound IR-NCS as greenish solid (11 mg, 30% yield). 1H NMR [500 MHz, CDCl3, SiMe4, J (Hz), δ (ppm)]: 8.39 (1H, s), 8.04 (1H, d, J = 7.5), 7.72 (1H, t, J = 7.5 Hz), 7.59 (1H, t, J = 7.5 Hz), 7.53 (2H, d, J = 8 Hz), 7.41 (2H, d, J = 8 Hz), 7.18 (2H, t, J = 7.5 Hz), 7.11 (2H, t, J = 7.5 Hz), 7.07 (1H, t, J = 7.5 Hz), 6.96 (2H, d, J = 7.0 Hz), 6.78 (2H, t, J = 7.0 Hz), 6.56 (2H, d, J = 8 Hz), 6.35 (2H, d, J = 11 Hz), 5.23 (2H, d, J = 11 Hz), 3.486 (4H, broad peak), 2.79–2.83 (2H, m), 2.57–2.61 (2H, m), 2.07–2.12 (1H, m), 1.77–1.83 (1H, m), 1.62 (4H, broad peak), 0.95 (6H, t, J = 6.5 Hz), 1.01 (6H, s), 1.08 (6H, s). 13C NMR [125 MHz, CDCl3, SiMe4, δ (ppm)]: 159.4, 144.9, 138.7, 138.1, 133.2, 131.2, 128.6, 127.9, 125.8, 124.6, 123.9, 121.7, 119.9, 106.4, 91.3, 57.3, 45.3, 39.5, 35.5, 29.9, 28.0, 27.4, 26.4, 22.9, 19.9, 15.7, 11.9. ESI-MS (positive mode, m/z). Calcd for C50H56N5O: 774.4206 (M + H). Found: m/z 774.4205. Melting point: 133 °C (decomposed).
Synthesis of IR-PYR
The hydrazide I2 (25.0 mg, 0.04 mmol) and 2-pyridine carboxaldehyde (8.38 mg, 0.08 mmol) were dissolved in 1 mL of DMF and the resulting solution was stirred for 18 h in room temperature (1 drop of acetic acid was added as catalyst). The reaction mixture was concentrated under reduced pressure, and the solid was collected. The crude mixture was separated using column chromatography (using basic Alumina) using hexane/ethyl acetate (75:25) mixture. The separated product was further purified using basic alumina preparative layer chromatographic plates, using CH2Cl2/C2H5OH (v/v 99:1) as eluent to afford the compound IR-PYR as yellowish-green solid (5.5 mg, 20% yield). 1H NMR [500 MHz, CDCl3, SiMe4, J (Hz), δ (ppm)]: 8.5 (1H, d, J = 5 Hz), 8.3 (1H, d, J = 8 Hz), 8.02–8.03 (2H, m), 7.69 (2H, m), 7.56 (1H, t, J = 8 Hz), 7.42 (1H, t, J = 7 Hz), 7.13–7.16 (1H, m), 7.03 (2H, t, J = 7.5 Hz), 6.94 (2H, d, J = 7.5 Hz), 6.7 (2H, t, J = 7.5 Hz), 6.45 (2H, d, J = 8 Hz), 6.19 (2H, d, J = 12 Hz), 5.20 (2H, d, J = 12 Hz), 3.40–3.43 (4H, m), 3.10–3.14 (2H, m), 2.62–2.67 (2H, m), 2.16–2.20 (1H, m), 1.86–1.94 (1H, m), 1.60–1.66 (4H, m), 1.2 (6H, s), 1.15 (6H, s), 0.93 (6H, t, J = 7.5 Hz). 13C NMR [125 MHz, CDCl3, SiMe4, δ (ppm)]: 156.47, 149.22, 148.76, 145.25, 139.02, 136.45, 133.11, 127.55, 126.33, 125.39, 123.83, 122.41, 121.62, 121.26, 120.61, 118.95, 105.72, 91.06, 75.21, 45.22, 44.09, 28.76, 28.28, 26.21, 23.69, 19.83, 11.93. ESI-MS (positive mode, m/z). Calcd for C49H54N5O: 728.4328 (M + H). Found: m/z 728.4323. Melting point: 92–97 °C (decomposed).
UV-Vis and fluorescence spectral studies
Stock solutions of various ions (5.0 × 10 mM) were prepared in deionized water. The stock solutions of ligands (5.0 × 10 mM) were prepared in DMF. The solution of IR-NCS was then diluted to 10.0 μM with methanol/aqueous 1× PBS buffer (pH 7.4) and the solution of IR-PYR was diluted to 10 μM with ethanol/aqueous 1× PBS buffer (pH 7.4; 8:2 v/v). In titration experiments for IR-PYR, each time a 1 mL solution of the probes (10 μM) was filled in a quartz optical cell of 1 cm optical path length, and the metal ion stock solution was added into the quartz optical cell gradually by using a micropipette. Spectral data were recorded 1 min after the addition of the metal ion. In titration experiments for IR-NCS, separate sample solutions were prepared by addition of increasing amount of Hg2+ ions to the probe solution (10 μM) and spectral data were recorded within 5 min after the addition of metal ion. In selectivity experiments, the test samples were prepared by placing appropriate amounts of the corresponding cation stock solution into 1 mL solution of the probes IR-NCS and IR-PYR (10 μM). For fluorescence measurements, excitation was provided at 695 nm, and emission was collected from 725 to 900 nm.
Evaluation of the binding constant for the formation of IR-PYR–Zn complex
Receptor IR-PYR with an effective concentration of 10 μM in an ethanol/aqueous 1× PBS buffer (pH 7.4; 8:2 v/v) was used for the emission titration studies with a Zn2+ metal ion solution. A stock solution of zinc nitrate, having a concentration of 0.5 mM ethanol/aqueous 1× PBS buffer (pH 7.4; 8:2 v/v) solution was used. The effective metal ion concentration was varied between 0 and 30 μM.
The binding constant for the formation of the respective complexes were evaluated using the Benesi–Hildebrand (B–H) plot (eqn (1)).39
| 1/(I − I0) = 1/{K(Imax − I0)C} + 1/(Imax − I0) | (1) |
Finding the detection limit
The detection limit for Hg2+ was not calculated because the initial changes in fluorescence intensity were not linear (due to intermediate complex formation). The detection limit for Zn2+ and Cd2+ were calculated using the fluorescence titration data. The fluorescence emission spectra of IR-PYR were measured for five times and the standard deviation of blank measurement were achieved. To gain the slope, the ratio of the fluorescence emission at 780 nm was plotted as a concentration of Zn2+ and Cd2+.The detection limit was calculated with the following equation:
| Detection limit = 3σ/k | (2) |
Computational methods
Full geometry optimizations were carried out using the density functional theory (DFT) method with Becke-3-Lee-Yang-Parr (B3LYP) exchange functional40 for all the molecules. The 6-31G* basis set was assigned for all C, N, S, O and H atoms whereas lanl2dz basis set was assigned for Zn atom. All calculations were performed with Gaussian 09 program with the help of the GaussView visualization program.41Result and discussion
Design and synthesis of the NIR probes IR-NCS and IR-PYR
Controlling the emissive response of a cyanine moiety by spirocyclization is a very recent approach.42 Our effort is to modify the spirocyclization approach to design chemosensors for targeting different metal ions by minor tuning of the design. Similar phenomena are also known in rhodamine based spirolactam systems where simple change in the metal binding site of the moiety can completely change the selectivity of the probes.43 To the best of our knowledge, this is the first report of a hydrazine functionalized spirolactam capped cyanine probe for sensing multiple metal ions. Hence, this study enables a new strategy for synthesizing NIR probes for metal ions.In this article we have reported two new molecules IR-NCS and IR-PYR (Scheme 1). In case of IR-NCS we have planned to couple the process of spirolactam ring opening with the stoichiometric and irreversible Hg2+-induced conversion of thiosemicarbazides to 1,3,4-oxadiazoles (Scheme 2).44 We have expected that this would work as the foundation for a novel Hg2+ selective chemodosimeter. The cyanine derivative IR-NCS seemed to be perfect for this application since the thiosemicarbazide moiety of IR-NCS can be easily liberated to form the oxadiazole in presence of Hg2+ ions, which in terms will induce the ring opening of the spirolactam unit.
 | ||
| Scheme 1 Synthetic scheme for the probes IR-NCS and IR-PYR. | ||
 | ||
| Scheme 2 Hg2+ induced oxadiazole formation. | ||
In case of IR-PYR we have designed a pyridine functionalised metal binding site, which along with the pyridine nitrogen, the lactam oxygen and the sp2 hybridized imine nitrogen provides the necessary support for binding metal ions. This kind of metal binding site constituting three donor atoms is well known for binding transition metal ions like Cu2+/Zn2+/Cd2+ etc.45 We have expected that the binding of specific metal ions with IR-PYR will trigger the opening of spirolactam ring of the cyanine derivative, which can be easily detected through changes in absorbance and fluorescence property of the solution.
The synthesis of compound I1, I2, IR-NCS and IR-PYR is shown in Scheme 1. The overall synthesis of the probes is very simple involving three easy steps. The first step is the synthesis of I1 by the Suzuki–Miyaura coupling, where we have attached benzoic acid moiety to the cyanine unit by refluxing cyanine chloride (IR-780) with carboxyphenylboric acid in the presence of Pd(PPh3)4 in DMF/H2O. The second step is the formation of the hydrazide, where I1 was first activated with N-hydroxysuccinimide (NHS) followed by reacting with hydrazine hydrate. The highly reactive amine group present in the hydrazine gives extra flexibility to the system and in the third step the metal selective sites were connected with the fluorophore through the free amine group of the hydrazide. The structure of all the compounds were fully characterised by 1H NMR, 13C NMR and MS-ESI spectroscopic studies.
UV-Vis spectroscopy of IR-NCS and IR-PYR
The photo-physical data of the compounds in methanol (MeOH) are compiled in Table S1.†Our aim was to study the metal–ligand binding induced opening of the spirolactam ring of IR-NCS and generation of the cyanine form. This structural change can be manifested in the electronic and fluorescence spectral patterns. UV-Vis spectra of IR-NCS recorded in methanol/aqueous PBS buffer indicated an absorption hump around 770–825 nm (Fig. 1a), absorbance around 780 nm is indicative of the presence of conjugated cyanine moiety in the solution, which might be due to the existence of a reversible equilibrium between the non-conjugated IR-NCS and the ring opened IR-NCS with conjugated cyanine backbone (Scheme 3). The selectivity of IR-NCS was checked in presence of various metal ions (Na+, Mg2+, Cr3+, Hg2+, Cu2+, Zn2+, Fe3+, Fe2+, Co2+, Ni2+, Cd2+, and Al3+). A significant change in the UV-Vis spectral pattern of IR-NCS was observed only in presence of Hg2+ ions, among all the other metal ions used (Fig. S1a†). During sequential titration with increasing concentration of Hg2+ ions (0 equiv. to 3 equiv. of Hg2+) a sharp absorption peak appeared around 769 nm (Fig. 1a). Additionally it is also observed that the initial absorbance peak of IR-NCS is broad, indicative of multiple species present in the solution. Possibility of the existence of more than one species in the solution of IR-NCS was further discussed in the Theoretical study section.
 | ||
| Fig. 1 (a) UV-Vis titration spectra of IR-NCS (10 μM) upon incremental addition of Hg2+ ions in CH3OH/aqueous PBS buffer (1×, pH 7.4; 3:7 v/v). (b) UV-Vis titration spectra of IR-PYR (10 μM) upon incremental addition of Zn2+ ions in C2H5OH/aqueous PBS buffer (1×, pH 7.4; 8:2 v/v). (c) UV-Vis titration spectra of IR-PYR (10 μM) upon incremental addition of Cd2+ ions in C2H5OH/aqueous PBS buffer (1×, pH 7.4; 8:2 v/v). | ||
 | ||
| Scheme 3 Equilibrium between the closed and open form of IR-NCS or IR-PYR. | ||
We have also performed metal dependent UV-Vis studies with IR-PYR, in this case we have found that the ligand itself shows some absorbance around 770 nm, which is expected due to the reversible equilibrium between the closed and open forms of IR-PYR (Scheme 3), similar to our observation in IR-NCS. The selectivity of the ligand is checked in presence of various metal ions (Na+, Mg2+, Mn2+, Hg2+, Cu2+, Zn2+, Fe3+, Fe2+, Co2+, Ni2+, Cd2+, and Al3+). A significant change in the UV-Vis spectral pattern was observed selectively in presence of Cd2+ and Zn2+ ions (Fig. S1b†).
Upon continuous addition of Zn2+/Cd2+ metal ions absorbance at 770 nm increases sharply forming an absorption band characteristic of a conjugated cyanine moiety (Fig. 1b and c).
Fluorescence spectroscopic studies
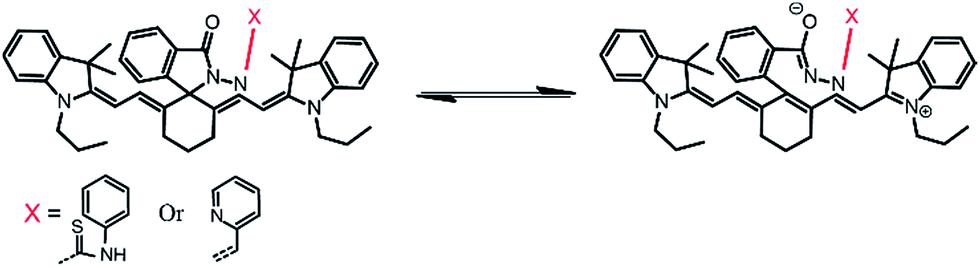
The selectivity of the synthesized probes IR-NCS and IR-PYR towards different metal ions (Na+, Mg2+, Cr3+, Hg2+, Cu2+, Zn2+, Fe3+, Fe2+, Co2+, Ni2+, Cd2+, and Al3+in CH3OH/aqueous PBS buffer) were also studied by fluorescence emission spectroscopy. As shown in Fig. 2a, a solution of IR-NCS (10 μM) displayed a low intensity emission maxima near 785 nm when excited at 695 nm. Addition of Hg2+ ions to this probe solution induced a dramatic increase in the fluorescence intensity. It can also be witnessed from Fig. 2a that the metal dependent switch-ON response of IR-NCS is entirely selective towards Hg2+ ions, and no significant change in emission intensity occurred even in the presence of excess of other metal ions. To gain an insight of the properties of IR-NCS as a sensor for Hg2+ ions, a titration of the chemosensor was performed with sequential addition of Hg2+ ions. As shown in Fig. 2b the fluorescence intensity of the solution containing 10 mM of IR-NCS was enhanced (with an initial decrease) with increasing concentration of Hg2+ ions, the initial decrease in fluorescence intensity was maybe due to the formation of an intermediate complex between the fluorophore and the heavy metal ion (Hg2+) (Scheme 4).46 The fluorescence titration also confirmed that IR-NCS exhibits high sensitivity toward Hg2+. The saturation behavior of the emission intensity of IR-NCS after addition of 1 equiv. of Hg2+ reveals that the Hg2+ chemosensor/chemodosimeter has a 1:1 stoichiometry.
 | ||
| Fig. 2 (a) Fluorescence spectra of IR-NCS in CH3OH/aqueous PBS buffer (1×, pH 7.4; 4:6 v/v) in presence of various metal ions (10 equivalent). (b) Fluorescence titration spectra of IR-NCS (10 μM) upon incremental addition (0 to 1.4 equivalent) of Hg2+ ions in CH3OH/aqueous PBS buffer (1×, pH 7.4; 4:6 v/v). | ||
 | ||
| Scheme 4 Metal sensing mechanism of IR-NCS and IR-PYR. | ||
The mechanism of chemodosimetric response of IR-NCS is further confirmed by ESI-MS studies of the sensor solution in presence of Hg2+ ions (ESI Fig. S14†), the molecular ion peak at m/z 740.4316 proves the presence of desired product IR-HGP in the solution.
The change in emission spectra of IR-NCS in presence of Hg2+ can be explained by the chemical reaction shown in Scheme 4. Reaction of Hg2+ ions with the chemodosimeter breaks the spirolactam ring with the formation of IR-HGP as a product. As shown in Scheme 4, formation of the product IR-HGP also caused the reappearance of the positive charge on the nitrogen atom resulting in the generation of a highly conjugated cyanine backbone. Thus, with increasing concentration of Hg2+ ions the concentration of cyanine moiety in the solution also increases, consequently a sharp change in the absorption as well as emission spectra is observed.
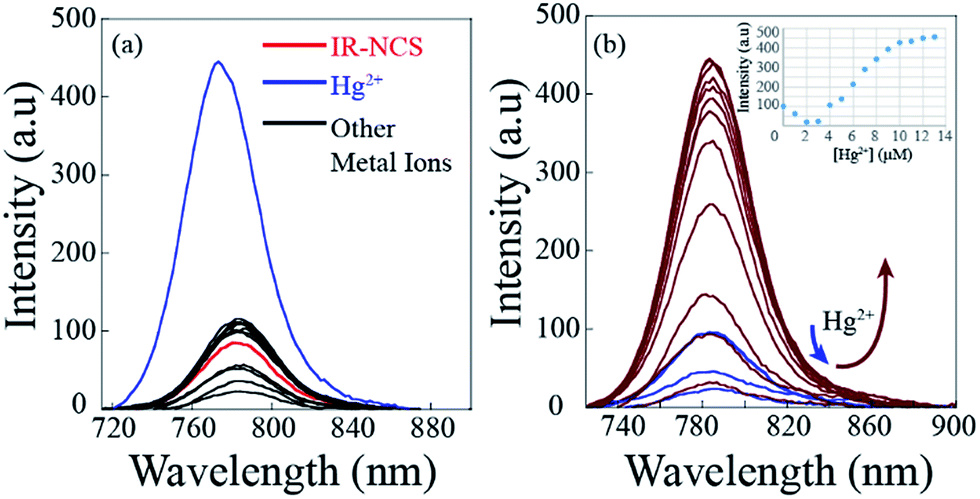
In case of IR-PYR the excitation of the initial solution at 695 nm wavelength shows an emission peak near 780 nm. The low intensity of the peak supports the fact that in absence of metal ions the molecule remains predominantly in the spirolactam form. Addition of Zn2+/Cd2+ ions to the solution containing IR-PYR induces a sharp increase in fluorescence intensity near 780 nm. Increase in the absorption spectral band near 770 nm and the emission band near 780 nm on binding to Zn2+/Cd2+ suggested opening of the spirolactam ring in IR-PYR on metal ion coordination. As shown in Fig. 3a the metal–chemosensor binding induced ring opening of IR-PYR and the formation of conjugated cyanine moiety is selective towards Zn2+ and Cd2+ ions and shows very weak response for other tested metal ions (Na+, Mg2+, Mn2+, Hg2+, Cu2+, Zn2+, Fe3+, Fe2+, Co2+, Ni2+, Cd2+, and Al3+). To further understand the metal sensing mechanism of IR-PYR as a sensor for Zn2+ and Cd2+ ions, a titration of the solution containing IR-PYR was performed with increasing concentration of Zn2+ and Cd2+ ions respectively. As shown in Fig. 3b and c the emission intensity of a 10 μM solution of IR-PYR was enhanced with sequential addition of Zn2+ and Cd2+ ions, in both the cases the ligand exhibited a high sensitivity towards the corresponding metal ions, with near about 6 folds increase of its fluorescence intensity upon addition of 1.2 equivalent of metal ions.
 | ||
| Fig. 3 (a) Fluorescence spectra of IR-PYR in C2H5OH/aqueous PBS buffer (1×, pH 7.4; 8:2 v/v) in presence of various metal ions (10 equivalent). (b) Fluorescence titration spectra of IR-PYR (10 μM) upon incremental addition (0 to 1.2 equivalent) of Zn2+ ions in C2H5OH/aqueous PBS buffer (1×, pH 7.4; 8:2 v/v). (c) Fluorescence titration spectra of IR-PYR (10 μM) upon incremental addition (0 to 1.2 equivalent) of Cd2+ ions in C2H5OH/aqueous PBS buffer (1×, pH 7.4; 8:2 v/v). | ||
The stoichiometry of the complex formed between IR-PYR and metal ions was established with the help of Job's plot. The complex formed between Zn2+ and IR-PYR is found to be 1:1 in stoichiometry (Fig. S6†), whereas the complex formed between Cd2+ and IR-PYR is initially 1:2 in nature, but tends to form a 1:1 complex in presence of excess metal ions in the solution (Fig. S7†). The 1:1 stoichiometry of both complexes was further proved from the ESI-MS studies. The molecular-ion peak obtained at m/z 853.3376 in the mass spectrum is related to the mass of [IR-PYR + Zn2+ + NO3] (ESI Fig. S15†) and the molecular ion peak at m/z 903.3183 is related to the mass of [IR-PYR + Cd2+ + NO3] (ESI Fig. S16†). Binding constant for the Zn-complex was calculated using Benesi–Hildebrand method, which was found to be 1.9 × 106 M−1. The detection limit of IR-PYR for Zn2+ and Cd2+ ions was found to be 3.3 × 10−8 M (33 nM) and 4.7 × 10−8 M (47 nM) respectively.
Theoretical studies
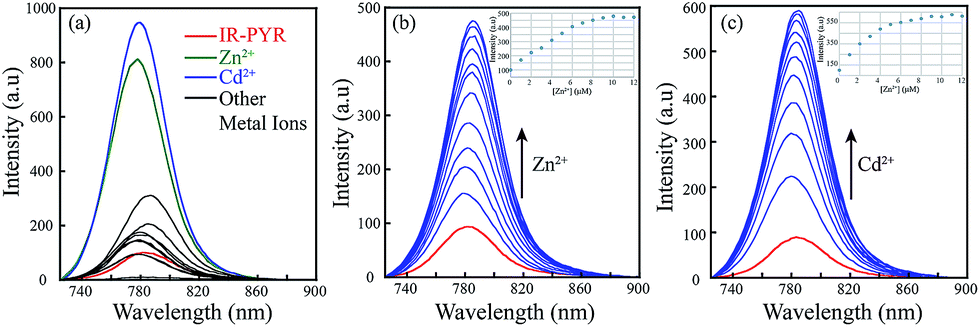
For a theoretical assessment of the optical response of IR-NCS and IR-PYR toward specific metal ions Density Functional Theory (DFT) based calculations were performed using Gaussian 09 package. DFT calculations of the probes also gave us a clear idea about their geometries. The findings from the optical spectroscopic and mass spectrometric studies indicated that IR-NCS undergoes a cyclization reaction in presence of Hg2+ ions, which induced breaking of the spirolactam ring to form a new compound IR-HGP (Scheme 4). Thus, the geometry of both IR-NCS and IR-HGP was optimized. The DFT optimization of IR-NCS revealed that the spirolactam ring of IR-NCS stays perpendicular to the heptamethine cyanine backbone and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S double bond prefers to align in the opposite direction to the carbonyl CO bond (Fig. 4).
S double bond prefers to align in the opposite direction to the carbonyl CO bond (Fig. 4).
 | ||
| Fig. 4 Theoretically optimized structures of IR-NCS and IR-HGP and energy diagrams of HOMO and LUMO orbitals of IR-NCS and IR-HGP calculated at the DFT level using a B3LYP/6-31G*. All protons are removed from the image for clarity. | ||
It was also noticed that the planarity of the cyanine backbone in IR-NCS is decreased compared to that of the free acid (I1), which may be attributed to the lack of conjugation in the former. On the other hand in case of IR-HGP the newly formed oxadiazole ring stays completely perpendicular to the cyanine backbone and due to the formation of the conjugated structure the planarity in the system is regained (Fig. S3†).
As the electronic spectra of both IR-NCS and IR-HGP are mainly generated from the electronic transition from HOMO to LUMO, we have also calculated the corresponding HOMO and LUMO energies. The HOMO–LUMO energy difference of IR-NCS is 3.38455381 eV whereas the respective energy difference in case of IR-HGP is 2.04548087 eV. The steep decrease in energy difference values explains the increase in absorbance of the NIR band (770 nm) in presence of Hg2+ ions.
We have also used theoretical calculations to understand the equilibrium between the ring-opened and ring-closed form of IR-NCS. As shown in Fig. 5a we have assumed that the O-atom in ring-opened form of IR-NCS can either be in a protonated state or in a deprotonated state in aqueous solution. The broadness of the UV-Vis peak over a wide range also indicated that multiple species were present in the solution. Thus to validate our experimental results we have calculated the HOMO and LUMO energies of the protonated and deprotonated species viz. IR-P and IR-D. For IR-P the HOMO–LUMO energy difference of the geometrically optimized structure is found to be 2.0585412 eV (Fig. 5b), which is similar to the HOMO–LUMO energy gap of IR-HGP.
 | ||
| Fig. 5 (a) Equilibrium between the probable species present in the solution of IR-NCS. (b) Theoretically optimized structures of IR-D and IR-P and the corresponding energy diagrams of HOMO and LUMO orbitals calculated at the DFT level using a B3LYP/6-31G*. All protons are removed from the image for clarity. | ||
The similarity between the corresponding energy gaps of IR-P and IR-HGP confirms that the hump near 770 nm in an aqueous solution of IR-NCS is due to the presence of IR-P in the solution. On the other hand the corresponding energy difference of IR-D was found to be 1.34233758 eV (Fig. 5b). The relatively small HOMO–LUMO energy gap of IR-D indicates that the hump in the longer wavelength region (over 800 nm) of the absorbance spectra is due to the presence of IR-D in the aqueous solution of IR-NCS. Furthermore we have confirmed these facts again by recording absorbance of IR-NCS respectively in an acidic and a basic medium. In a basic medium of pH 9 most of the ring opened IR-NCS molecules are in IR-D form and the absorbance spectra show a broad peak with a prominent hump in the longer wavelength region (over 800 nm), whereas in acidic medium of pH 4 most of the ring opened IR-NCS molecules are in IR-P form and the absorbance spectra show a sharp peak at relatively shorter wavelength (near 770 nm) (ESI, Fig. S5†).
In case of IR-PYR, we have confirmed from the Job's plot analysis and mass spectrometric studies that a 1:1 complex is formed in presence of Zn2+ ions, whereas both 1:2 and 1:1 complexes were possible in presence of Cd2+ ions. Thus, for the sake of simplicity we have pursued the geometry optimization of IR-PYR and its Zn complex (IR-PYR–Zn).
Similar to IR-NCS, IR-PYR also showed lesser planarity of the non-conjugated cyanine backbone and in both the cases the spirolactam unit is perpendicular to the heptamethine chain. In presence of Zn2+ ions the electron pairs from carbonyl-O, imine-N and pyridine-N participate in the binding event (Fig. 6).
 | ||
| Fig. 6 Theoretically optimized structures of IR-PYR and IR-PYR–Zn (nitrate as a counter anion) and the corresponding energy diagrams of HOMO and LUMO orbitals calculated at the DFT level using a B3LYP/6-31G*. All protons are removed from the image for clarity. | ||
After chelation with the Zn2+ ion, the newly formed IR-PYR–Zn complex retained a more planar geometry (cyanine backbone) than its precursor IR-PYR (Fig. S4†). The HOMO–LUMO energy difference of IR-PYR is 3.13774642 eV whereas the respective energy difference in case of IR-PYR–Zn is 2.04711354. Similar to the probe IR-NCS in this case also sharp decrease in the energy difference value is consistent with the result obtained from UV-Vis spectral analysis. Furthermore in both of the cases we have noticed that after the formation of IR-HGP and IR-PYR–Zn atomic contribution to the HOMO/LUMO is solely from the cyanine backbone, which again verifies our assumption that the increase in UV-Vis absorbance after metal recognition is observed due to the presence of highly conjugated cyanine unit in the system.
Metal-ion competition studies
The absorption and emission response of IR-PYR and IR-NCS in presence of individual metal ions had shown a notable selectivity for Zn2+/Cd2+ and Hg2+ metal ions. However, the most significant criterion of a chemosensor is its ability to recognize the target analyte in a complex environment of other competing analytes. This hypothesis has been validated by testing the selectivity of IR-NCS and IR-PYR in presence of various cations, which can interfere with the sensing of Zn2+/Cd2+ and Hg2+ metal ions. The fluorescence response of IR-NCS in presence of Hg2+ metal ions was nearly unchanged even after addition of 5 equivalents of competing metal ions (Mg2+, Cu2+, Mn2+, Zn2+, Fe2+, Co2+, Ni2+ and Al3+) Fig. 7a. On the other hand IR-PYR shows significant interference only in presence of Cu2+ ions, which might have a better binding compatibility with the ligand, thus in presence of paramagnetic Cu2+ ions the emission intensity is completely quenched46 and did not re-appear with the addition of Zn2+/Cd2+ ions (Fig. 7b).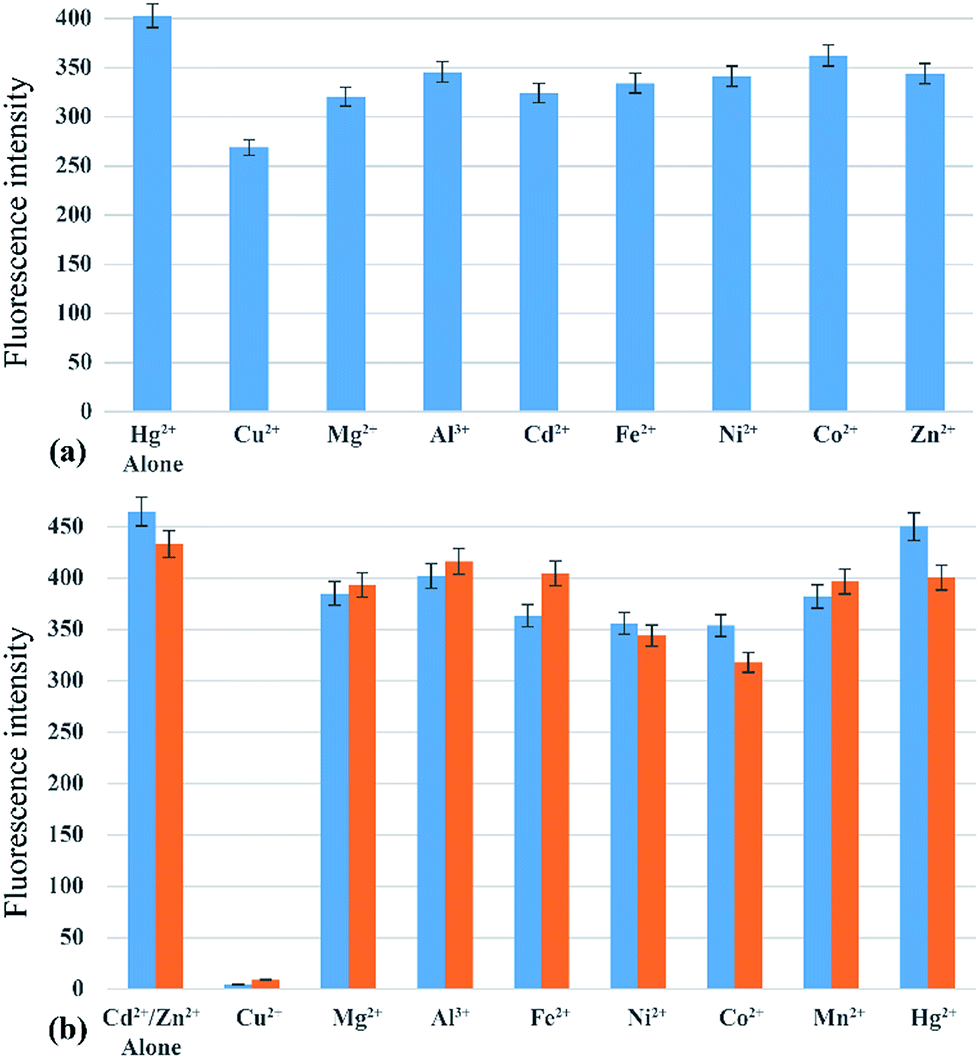 | ||
| Fig. 7 (a) Normalized fluorescence responses of IR-NCS (10 μM) in presence of 5 equivalent of various metal ions and after subsequent addition of 1 equivalent Hg2+ ions. (b) Normalized fluorescence responses of IR-PYR (10 μM) in presence of 5 equivalent of various metal ions and after subsequent addition of 1 equivalent Cd2+ (blue bars) or Zn2+ (orange bars) metal ions. | ||
Cell imaging studies
The importance of any chemosensing probe is evaluated by its potential to detect the analyte inside living systems like cells or organism. The effective detection of analyte inside living cells proves the ability of the probe to work within a complex environment. As evident cellular intake of any chemosensor crucially depends on the solubility of the probe. The plasma membrane of cells is lipophilic in nature whereas the intracellular environment is of opposite nature, thus molecular probe must have an optimum solubility which allow it to get pass the lipophilic membrane. Along with the solubility, the charge character of a probe also plays an important role. A neutral molecule can easily diffuse through the membrane whereas a charged molecule may not readily diffuse through membranes, instead they show a tendency to accumulate in subcellular compartments.47In this study both of our ligands were primarily uncharged but form a charged species when reacted with selective metal ions. Given the suitable water solubility of IR-NCS and significant physiological implications of cellular mercury, we were motivated to investigate the ability of IR-NCS for detecting intracellular levels of mercury. As evident from our solution based spectroscopic studies, IR-NCS undergoes a chemical reaction in presence of Hg2+ ions to form a new molecule with increase in fluorescence intensity. Our aim was to determine if the chemical reaction and the corresponding change in fluorescence intensity is prominent enough to be determined inside living human cells by confocal microscopy (Fig. 8). When the HeLa cells were primarily treated with 20 μM of IR-NCS, the cells show very mild fluorescence intensity. In the next step we have gradually increased the concentration of Hg2+ ions from 5 μM to 100 μM. As shown in Fig. 8 the cells start to show significant increment in fluorescence intensity even when the mercury concentration is as low as 5 μM, indicating selective sensing of Hg2+ ions in complex cellular environment.
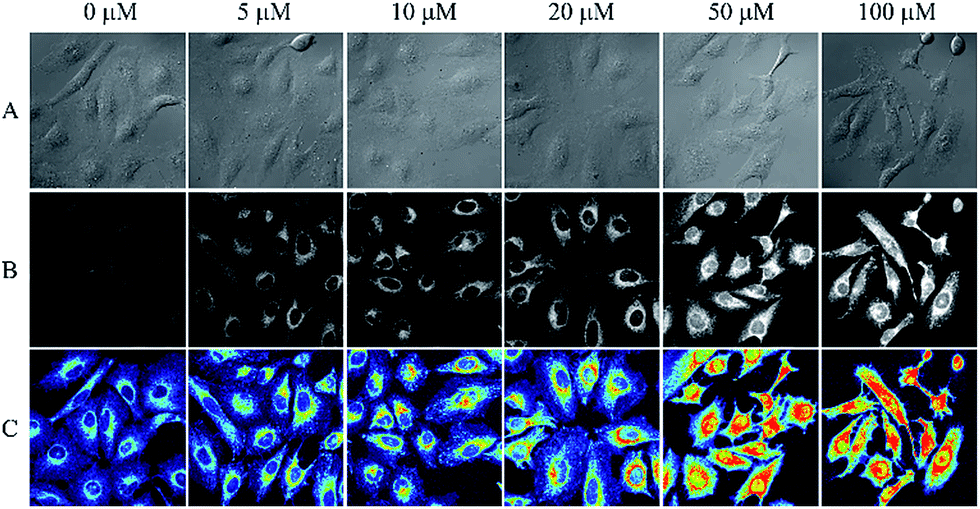 | ||
| Fig. 8 Each column from left to right represents the confocal microscopic images of HeLa cells (pre-treated with 20 μM IR-NCS) after addition of 0–100 μM Hg2+ ions. Row (A) represents the differential interference contrast (DIC) images. Row (B) represent the fluorescence images. Row (C) represents the pseudo-colored images, blue to red color is assigned to fluorescence intensity from 0 to 1500. | ||
Interestingly, the increase in fluorescence intensity inside the cells is consistent with the increase in Hg2+ metal ion concentration, thus along with the sensing of mercury ions inside cells a prediction about the extent of contamination can also be done. The uniform distribution of fluorescence across the intracellular region suggests that IR-NCS can pass through the cell membrane and permeate the cells and upon further addition of Hg2+ ions the probe IR-NCS undergoes a ring opening reaction to produce IR-HGP as a product. As the concentration of IR-HGP inside cells increases the corresponding fluorescence intensity also increases. It is also noteworthy that the typical morphological trait of HeLa cells were retained throughout the experiment, which indicates that the probe IR-NCS and the product IR-HGP are not cytotoxic to the HeLa cells.
Conclusion
In summary, we have developed a new technique to design chemosensors for metal ions which are easily modifiable for targeting different metals ions. To verify our system we have synthesised two compounds IR-NCS and IR-PYR which show significant changes in their fluorescence and absorbance spectra in presence of selective metal ions. We found that in the case of IR-NCS, the Hg2+ ion selective response was due to a chemodosimetric reaction of the probe with Hg2+ ions to form a new compound IR-HGP, which triggers the opening of the spirolactam ring in IR-NCS to form a conjugated cyanine dye. On the other hand, the Zn2+/Cd2+ metal ion selective response of IR-PYR was found to be completely due to the formation of a coordination complex between the probe and the metal ion, which induces the ring opening of the spirolactam cycle and subsequently increases the emission intensity in the NIR spectral region. We have also used DFT based calculation techniques as supporting evidences of our experimental results. As the probe IR-NCS has an optimum solubility in aqueous medium so we have used it for intracellular detection of Hg2+ ions, we found that the cells pre-treated with the probe display significant increase in emission intensity in presence of Hg2+ ions under the confocal microscope. We expect that this new technique can be further applied for developing NIR sensors for various environmentally and biologically important metal ions.Acknowledgements
CK acknowledge JSPS for the research grant and his alma mater IIT Guwahati for the Gaussian 09 package.References
- T. R. A. Vandenbroucke, P. Emsbo, A. Munnecke, N. Nuns, L. Duponchel, K. Lepot, M. Quijada, F. Paris, T. Servais and W. Kiessling, Nat. Commun., 2015, 6, 7966, DOI:10.1038/ncomms8966.
- S. J. Lippard and J. M. Berg, Principles of Bioinorganic Chemistry, University Science Books, Mill Valley, CA, 1994 Search PubMed.
- A. T. Aron, K. M. Ramos-Torres, J. A. Cotruvo Jr and C. J. Chang, Acc. Chem. Res., 2015, 48, 2434–2442 CrossRef CAS PubMed.
- N. Shao, Y. Zhang, S. M. Cheung, R. H. Yang, W. H. Chan, T. Mo, K. A. Li and F. Liu, Anal. Chem., 2005, 77, 7294–7303 CrossRef CAS PubMed; D. W. Domaille, E. L. Que and C. J. Chang, Nat. Chem. Biol., 2008, 4, 168–175 CrossRef PubMed; K. M. K. Swamy, S. K. Ko, S. K. Kwon, H. N. Lee, C. Mao, J. M. Kim, K. H. Lee, J. Kim, I. Shin and J. Yoon, Chem. Commun., 2008, 5915–5917 RSC.
- E. Kimura, Pure Appl. Chem., 1986, 58, 1461–1466 CrossRef CAS; Z. Zhou and C. J. Fahrni, J. Am. Chem. Soc., 2004, 126, 8862–8863 CrossRef PubMed; Y. Zhou, F. Wang, Y. Kim, S. J. Kim and J. Yoon, Org. Lett., 2009, 11, 4442–4445 CrossRef PubMed.
- L. Fabbrizzi, M. Licchelli, P. Pallavicini, A. Perotti and D. Sacchi, Angew. Chem., 1994, 106, 2051–2053 CrossRef CAS; E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517–1549 CrossRef PubMed.
- J. Cody and C. J. Fahrni, Tetrahedron, 2004, 60, 11099–11107 CrossRef CAS; X. Qi, E. J. Jun, L. Xu, S. J. Kim, J. S. J. Hong, Y. J. Yoon and J. Y. Yoon, J. Org. Chem., 2006, 71, 2881–2884 CrossRef PubMed; Y. Xiang, A. J. Tong and Y. Ju, Org. Lett., 2006, 8, 2863–2866 CrossRef PubMed; G. K. Li, Z. X. Xu, C. F. Chen and Z. T. Huang, Chem. Commun., 2008, 1774–1776 RSC; M. X. Yu, M. Shi, Z. G. Chen, F. Y. Li, X. X. Li, Y. H. Gao, J. Xu, H. Yang, Z. G. Zhou, T. Yi and C. H. Huang, Chem.–Eur. J., 2008, 14, 6892–6900 CrossRef PubMed.
- S. Goswami, D. Sen and N. K. Das, Org. Lett., 2010, 12, 856–859 CrossRef CAS PubMed; Z. Xu, J. Yoon and D. R. Spring, Chem. Commun., 2010, 46, 2563–2565 RSC; K. Kaur and S. Kumar, Dalton Trans., 2011, 40, 2451–2458 RSC; K. C. Ko, J. S. Wu, H. J. Kim, P. S. Kwon, J. W. Kim, R. A. Bartsch, J. Y. Lee and J. S. Kim, Chem. Commun., 2011, 47, 3165–3167 RSC.
- R. Martinez, F. Zapata, A. Caballero, A. Espinosa, A. Tarraga and P. Molina, Org. Lett., 2006, 8, 3235–3238 CrossRef CAS PubMed; E. Sanna, L. Martinez, C. Rotger, S. Blasco, J. Gonzalez, E. Garcia-Espana and A. Costa, Org. Lett., 2010, 12, 3840–3843 CrossRef PubMed; A. F. Chaudhry, M. Verma, M. T. Morgan, M. M. Henary, N. Siegel, J. M. Hales, J. W. Perry and C. J. Fahrni, J. Am. Chem. Soc., 2010, 132, 737–747 CrossRef PubMed; M. Verma, A. F. Chaudhry and C. J. Fahrni, Org. Biomol. Chem., 2010, 8, 363–370 Search PubMed.
- Y. Zheng, Q. Huo, P. Kele, F. M. Andreopoulos, S. M. Pham and R. M. Leblanc, Org. Lett., 2001, 3, 3277–3280 CrossRef CAS PubMed; L. Gao, J. Q. Wang, L. Huang, X. X. Fan, J. H. Zhu, Y. Wang and Z. G. Zou, Inorg. Chem., 2007, 46, 10287–10293 CrossRef PubMed; V. S. Jisha, A. J. Thomas and D. Ramaiah, J. Org. Chem., 2009, 74, 6667–6673 CrossRef PubMed; H. H. Wang, L. Xue, Z. J. Fang, G. P. Li and H. Jiang, New J. Chem., 2010, 34, 1239–1242 RSC.
- T. Gunnlaugsson, J. P. Leonard and N. S. Murray, Org. Lett., 2004, 6, 1557–1560 CrossRef CAS PubMed; R. Martinez, A. Espinosa, A. Tarraga and P. Molina, Org. Lett., 2005, 7, 5869–5872 CrossRef PubMed; Z. Xu, K. Baek, H. N. Kim, J. Cui, X. Qian, D. R. Spring, I. Shin and J. Yoon, J. Am. Chem. Soc., 2010, 132, 601–610 CrossRef PubMed.
- L. Yang, R. McRae, M. M. Henary, R. Patel, B. Lai, S. Vogt and C. J. Fahrni, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11179–11184 CrossRef CAS PubMed; E. W. Miller, L. Zeng, D. W. Domaille and C. J. Chang, Nat. Protoc., 2006, 1, 824–827 CrossRef PubMed; L. Zeng, E. W. Miller, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 10–11 CrossRef PubMed; Z. Xu, S. J. Han, C. Lee, J. Yoon and D. R. Spring, Chem. Commun., 2010, 46, 1679–1681 RSC; D. W. Domaille, L. Zeng and C. J. Chang, J. Am. Chem. Soc., 2010, 132, 1194–1195 CrossRef PubMed.
- H. D. Axelrod, J. H. Cary, J. E. Bonelli and J. P. Lodge Jr, Anal. Chem., 1969, 41, 1856–1858 CrossRef CAS PubMed; M. F. Choi and P. Hawkins, Anal. Chim. Acta, 1997, 344, 105–110 CrossRef; M. A. Spaziani, J. L. Davis, M. Tinani and M. K. Carroll, Analyst, 1997, 122, 1555–1557 RSC; J. Rodriguez-Fernandez, J. M. Costa, R. Pereiro and A. Sanz-Medel, Anal. Chim. Acta, 1999, 398, 23–31 CrossRef.
- M. G. Choi, S. Cha, H. Lee, H. L. Jeon and S.-K. Chang, Chem. Commun., 2009, 7390–7392 RSC; X. F. Yang, L. Wang, H. Xu and M. Zhao, Anal. Chim. Acta, 2009, 631, 91–95 CrossRef CAS PubMed; L. Zhang, X. Lou, Y. Yu, J. Qin and Z. Li, Macromolecules, 2011, 44, 5186–5193 CrossRef.
- G. Patonay and M. D. Antoine, Anal. Chem., 1991, 63, 321A–327A CrossRef CAS; R. B. Thompson, Top. Fluoresc. Spectrosc., 1994, 4, 151–181 Search PubMed; S. Stoyanov, Pract. Spectrosc., 2001, 25, 35–93 Search PubMed; W. M. Leevy, S. T. Gammon, H. Jiang, J. R. Johnson, D. J. Maxwell, M. Marquez, D. Piwnica-Worms and B. D. Smith, J. Am. Chem. Soc., 2006, 128, 16476–16477 CrossRef PubMed; W. M. Leevy, S. T. Gammon, J. R. Johnson, A. J. Lampkins, H. Jiang, M. Marquez, D. Piwinica-Worms and B. D. Smith, Bioconjugate Chem., 2008, 19, 686–692 CrossRef PubMed.
- J. Fabian, H. Nakazumi and M. Matsuoka, Chem. Rev., 1992, 92, 1197–1226 CrossRef CAS; J. V. Frangioni, Curr. Opin. Chem. Biol., 2003, 7, 626–634 CrossRef PubMed; B. A. Smith, W. J. Akers, W. M. Leevy, A. J. Lampkins, S. Z. Xiao, W. Wolter, M. A. Suckow, S. Achilefu and B. D. Smith, J. Am. Chem. Soc., 2010, 132, 67–69 CrossRef PubMed; J. M. Baumes, J. J. Gassensmith, J. Giblin, J.-J. Lee, A. G. White, W. J. Culligan, W. M. Leevy, M. Kuno and B. D. Smith, Nat. Chem., 2010, 2, 1025–1030 CrossRef PubMed.
- R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936–3953 RSC.
- S. Banerjee, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2013, 42, 1601–1618 RSC.
- X. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Chem. Rev., 2012, 112, 1910–1956 CrossRef CAS PubMed.
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC.
- T. Kowada, H. Maeda and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953–4972 RSC.
- A. Mishra, R. K. Behera, P. K. Behera, B. K. Mishra and G. B. Behera, Chem. Rev., 2000, 100, 1973–2011 CrossRef CAS PubMed.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259 CrossRef CAS; T. Egawa, K. Hanaoka, Y. Koide, S. Ujita, N. Takahashi, Y. Ikegaya, N. Matsuki, T. Terai, T. Ueno, T. Komatsu and T. Nagano, J. Am. Chem. Soc., 2011, 133, 14157 CrossRef PubMed.
- C. Kar, M. D. Adhikari, A. Ramesh and G. Das, RSC Adv., 2012, 2, 9201–9206 RSC.
- Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2013, 113, 192–270 CrossRef CAS PubMed.
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed.
- D. W. Boening, Chemosphere, 2000, 40, 1335–1340 CrossRef CAS PubMed.
- H. H. Harris, I. Pickering and G. N. George, Science, 2003, 301, 1203 CrossRef CAS PubMed.
- T. W. Clarkson, L. Magos and G. J. N. Myers, N. Engl. J. Med., 2003, 349, 1731–1737 CrossRef CAS PubMed.
- C. M. L. Carvalho, E.-H. Chew, S. I. Hashemy and J. Lu, J. Biol. Chem., 2008, 283, 11913–11923 CrossRef CAS PubMed.
- G. Guzzi and C. A. M. La Porta, Toxicology, 2008, 244, 1–12 CrossRef CAS PubMed.
- R. R. Lauwerys, A. M. Bernard, H. A. Reels and J.-P. Buchet, Clin. Chem., 1994, 40, 1391 CAS.
- G. F. Nordberg, R. F. M. Herber and L. Alessio, Cadmium in the Human Environment, Oxford University Press, Oxford, UK, 1992 Search PubMed.
- Agency for Toxic Substances and Disease Registry, 4770 Buford Hwy NE, Atlanta, GA 30341, http://www.atsdr.cdc.gov/cercla/07list.html.
- E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517–1549 CrossRef CAS PubMed; C. J. Frederickson, J. Y. Koh and A. I. Bush, Nat. Rev. Neurosci., 2005, 6, 449–462 CrossRef PubMed; A. G. Scrimgeoura, C. H. Stahl, J. P. McClung, L. J. Marchitelli and A. J. Young, J. Nutr. Biochem., 2007, 18, 813–819 CrossRef PubMed; J. M. Berg and Y. Shi, Science, 1996, 271, 1081–1085 Search PubMed.
- B. L. Vallee and K. H. Falchuk, Physiol. Rev., 1993, 73, 79–118 CAS.
- M. Hambidge, J. Nutr., 2000, 130, 1344S–1349S CAS.
- L. A. Lichten and R. Cousins, Annu. Rev. Nutr., 2009, 29, 153–176 CrossRef PubMed.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef; D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- L. He, W. Lin, Q. Xu, M. Ren, H. Wei and J.-Y. Wang, Chem. Sci., 2015, 6, 4530–4536 RSC.
- J. Huang, Y. Xu and X. Qian, J. Org. Chem., 2009, 74, 2167–2170 CrossRef CAS PubMed; H. Yang, Z. Zhou, K. Huang, M. Yu, F. Li, T. Yi and C. Huang, Org. Lett., 2007, 9, 4729–4732 CrossRef PubMed; J. F. Zhang, Y. Zhou, J. Yoon, Y. Kim, S. J. Kim and J. S. Kim, Org. Lett., 2010, 12, 3852–3855 CrossRef PubMed; M. Vendrell, D. Zhai, J. C. Er and Y. T. Chang, Chem. Rev., 2012, 112, 4391–4420 CrossRef PubMed; C. Kar, M. Adhikari, A. Ramesh and G. Das, Inorg. Chem., 2013, 52, 743 CrossRef PubMed.
- Y.-K. Yang, K.-J. Yook and J. Tae, J. Am. Chem. Soc., 2005, 127, 16760–16761 CrossRef CAS PubMed.
- K. Li, Y. Xiang, X. Wang, J. Li, R. Hu, A. Tong and B. Z. Tang, J. Am. Chem. Soc., 2014, 136, 1643–1649 CrossRef CAS PubMed; F.-J. Huo, C.-X. Yin, Y.-T. Yang, J. Su, J.-B. Chao and D.-S. Liu, Anal. Chem., 2012, 84, 2219–2223 CrossRef PubMed; S. Chaudhuri, S. Kundu, M. K. Biswas, T. Weyhermuller and P. Ghosh, Inorg. Chim. Acta, 2015, 430, 199–207 CrossRef.
- R. Krämer, Angew. Chem., 1998, 110, 804–806 (Angew. Chem., Int. Ed., 1998, 37, 772–773) CrossRef CAS; J. Yoon, N. E. Ohler, D. H. Vance, W. D. Aumiller and A. W. Czarnik, Tetrahedron Lett., 1997, 38, 3845–3848 CrossRef; A. Torrado, G. K. Walkup and B. Imperiali, J. Am. Chem. Soc., 1998, 120, 609–610 CrossRef; M. A. Bernardo, F. Pina, B. Escuder, E. Garcia-España, M. L. Godino-Salido, J. LaTorre, S. V. Luis, J. A. Ramirez and C. Soriano, J. Chem. Soc., Dalton Trans., 1999, 915–921 RSC.
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization spectra and supporting data along with peripheral findings. See DOI: 10.1039/c7ra03246c |
| This journal is © The Royal Society of Chemistry 2017 |