
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Four new phenylethanoid and flavonoid glycoside dimers from the fruits of Forsythia suspensa and their neuroprotective activities†
Fan Zhang‡
ab,
Ya-Nan Yang‡ a,
Zi-Ming Fenga,
Jian-Shuang Jianga and
Pei-Cheng Zhang*a
a,
Zi-Ming Fenga,
Jian-Shuang Jianga and
Pei-Cheng Zhang*a
aState Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, People's Republic of China. E-mail: pczhang@imm.ac.cn
bBeijing Changping Hospital of Integrated Chinese and Western Medicine, Beijing 102208, People's Republic of China
First published on 10th May 2017
Abstract
During our continuing study on the fruits of Forsythia suspensa, one new heterodimer (forsythoneoside E) containing a flavonoid and a phenethanoid glycoside formed through a pyran ring, one new phenylethanoid glycoside dimer (forsythoneoside F) with a C–O bond, along with a pair of flavonoid glycoside dimers (forsythobiflavone A and B) possessing different axial chirality, were obtained. Their chemical structures, including absolute configurations, were established by NMR, HRESIMS, UV, IR, and ECD spectroscopic data and comparison of experimental and calculated electronic circular dichroism (ECD) spectra. All of the compounds were evaluated for their neuroprotective effects against rotenone-induced neurotoxicity in PC12 cells at 1 μM.
Introduction
Forsythia suspensa (Thunb.) Vahl is widely distributed in China, Japan, and Korea as an ornamental plant for its lovely flowers in spring. But, more importantly, its fruit named Fructus Forsythiae is a famous traditional Chinese medicine, documented in every edition of the Chinese Pharmacopoeia and many classical medical books, e.g. Shen Nong's herbal Compendium of material medica and Mingyi Bielu as a remedy for inflammation, pyrexia, ulcer, and influenza. Phytochemical research of this plant revealed a number of chemical substances such as phenylethanoid glycosides,1,2 lignans,3–5 alkaloids,6 and triterpenoids.7 Among them, forsythoside A, belong to the phenylethanoid glycosides, is the major constituent of F. suspensa.8 A literature survey revealed that forsythoside A has shown a variety of pharmacological activities,9–14 which aroused us with great interests, especially the neuroprotective activity.15–17In our previous research, four neuroprotective adducts of phenylethanoid glycoside fused with flavonoid through pyran ring or carbon–carbon bond, named forsythoneoside A–D, were obtained from the fruits of F. suspensa.18 For the purpose of exploring novel neuroprotective compounds from this plant, a continuing study was performed, which resulted in the isolation of four compounds (Fig. 1), including one new heterodimer of flavonoid fused with phenylethanoid glycoside, named forsythoneoside E (1), one new phenylethanoid glycoside dimer, named forsythoneoside F (2), and a pair of asymmetric axial diastereoisomers of biflavone glycoside, named forsythobiflavones A (3) and B (4). Despite countless flavonoid and phenylethanoid glycosides from natural sources, the best of our knowledge, their dimers are isolated rarely. Given our previous neuroprotective screening results for the heterodimer of phenylethanoid glycoside and flavonoid,18 the neuroprotective activities of 1–4 were tested through in vitro assays. Herein, the isolation and structure elucidation of the compounds as well as their neuroprotective effects on rotenone-induced damage in PC12 cell line were described.
 | ||
| Fig. 1 Chemical structures of 1–4. | ||
Results and discussion
Forsythoneoside E (1) was obtained as brown amorphous powder with the distinctive UV absorption peaks at 337, 273, 253, and 200 nm. The molecular formula of C56H62O31 was established by the protonated molecular ion peak at m/z 1231.3342 [M + H]+ (calcd for C56H63O31, 1231.3348) observed in the HRESIMS, indicating 26 indices of hydrogen deficiency. The IR spectrum displayed typical absorption bands at 3361, 1694, and 1603 cm−1 assignable to hydroxyl, carbonyl, and aromatic functionalities, respectively.The analysis of 1H NMR spectrum (Table 1) showed two sets of ABX system aromatic protons at δH 7.91 (1H, d, J = 2.4 Hz), 6.99 (1H, d, J = 8.4 Hz), 7.76 (1H, dd, J = 8.4, 2.4 Hz), 7.12 (1H, d, J = 2.4 Hz), 6.84 (1H, d, J = 8.4 Hz), and 7.00 (1H, dd, J = 8.4, 2.4 Hz) for 3,4-dihydroxyphenyl; three single aromatic protons at δH 7.03 (1H, s), 6.77 (1H, s), and 6.67 (1H, s) for 1,3,4,5-tetrasubstituted or pentasubstituted benzene rings; one group of olefinic protons at δH 7.62 (1H, d, J = 15.6 Hz) and 6.31 (1H, d, J = 15.6 Hz). In addition, the aliphatic proton signals were present between δH 5.08 and 1.15, including one methene at δH 3.97 (1H, m) and δH 3.94 (1H, m), one methine at δH 4.42 (1H, m) and four anomeric proton signals at δH 5.08 (1H, d, J = 7.2 Hz), 4.47 (1H, d, J = 7.8 Hz), 4.74 (1H, brs), and 4.68 (1H, brs). The 13C NMR (Table 2) data, classified by the HSQC spectrum, revealed the presence of 56 carbon resonances, comprising two methyls, three methenes, 32 methines, and 19 quaternary carbon. Besides the four hexoses (24 carbons), the remaining 32 carbons could be attributed to a flavonol aglycon (15 carbons), a phenylethanoid unit (eight carbons), and a caffeoyl group (nine carbons).
| No. | 1a | 2b | 3a | 4a |
|---|---|---|---|---|
| a Data were measured in acetic acid-d4 at 600 MHz.b Data were measured in methanol-d4 at 500 MHz. | ||||
| 2 | 6.74 (d, 2.0) | |||
| 5 | 6.67 (d, 8.0) | |||
| 6 | 6.60 (dd, 8.0, 2.0) | 6.18 (d, 2.4) | 6.23 (d, 2.4) | |
| 7 | 4.75 (overlap) | |||
| 8 | 6.77 (s) | 4.02 (m, Ha) | 5.71 (d, 2.4) | 6.02 (s) |
| 3.57 (m, Hb) | ||||
| 10 | 7.00 (d, 2.0) | |||
| 12 | 7.91 (d, 2.4) | |||
| 13 | 6.72 (d, 8.0) | |||
| 14 | 6.90 (dd, 8.0, 2.0) | |||
| 15 | 6.99 (d, 8.4) | 7.52 (d, 16.0) | 7.20 (d, 8.4) | 7.17 (d, 8.4) |
| 16 | 7.76 (dd, 8.4, 2.4) | 6.22 (d, 16.0) | 7.37 (d, 8.4) | 7.48 (d, 8.4) |
| 17 | 5.08 (d, 7.2) | 4.56 (d, 7.8) | 4.69 (d, 7.8) | |
| 18 | 3.76 (m) | 4.39 (d, 8.0) | 3.51 (m) | 3.26 (m) |
| 19 | 3.73 (m) | 3.30 (m) | 3.58 (m) | 3.56 (m) |
| 20 | 3.61 (m) | 3.55 (m) | 3.58 (m) | 3.14 (m) |
| 21 | 3.47 (m) | 4.89 (t, 10.0) | 3.23 (m) | 3.41 (m) |
| 22 | 3.78 (m, Ha) | 3.59 (m) | 3.67 (m, Ha) | 3.80 (m, Ha) |
| 3.50 (m, Hb) | 3.41 (m, Hb) | 3.22 (m, Hb) | ||
| 23 | 4.68 (brs) | 3.69 (m, Ha) | 4.84 (brs) | 4.74 (brs) |
| 3.45 (m, Hb) | ||||
| 24 | 3.83 (m) | 4.59 (brs) | 4.10 (m) | 4.04 (m) |
| 25 | 3.76 (m) | 3.80 (m) | 3.71(m) | 3.84 (m) |
| 26 | 3.49 (m) | 3.62 (m) | 3.48 (m) | 3.54 (m) |
| 27 | 3.55 (m) | 3.30 (m) | 3.44 (m) | 3.62 (m) |
| 28 | 1.15 (overlap) | 3.54 (m) | 1.13 (d, 6.0) | 1.21 (d, 6.0) |
| 29 | 1.14 (d, 6.0) | |||
| 2′ | 7.03 (s) | 6.65 (d, 2.0) | ||
| 5′ | 6.67 (s) | 6.64 (d, 8.0) | ||
| 6′ | 6.53 (dd, 8.0, 2.0) | 6.43 (s) | 6.32 (s) | |
| 7′ | 4.42 (m) | 2.75 (m) | ||
| 8′ | 3.97 (m, Ha) | 3.94 (m, Ha) | ||
| 3.94 (m, Hb) | 3.67 (m, Hb) | |||
| 10′ | 7.12 (d, 2.4) | 7.00 (d, 2.0) | ||
| 12′ | 7.69 (d, 2.4) | 7.74 (d, 2.4) | ||
| 13′ | 6.84 (d, 8.4) | 6.74 (d, 8.0) | ||
| 14′ | 7.00 (dd, 8.4, 2.4) | 6.91 (dd, 8.0, 2.0) | ||
| 15′ | 7.62 (d, 15.6) | 7.55 (d, 16.0) | 6.88 (d, 8.4) | 6.86 (d, 8.4) |
| 16′ | 6.31 (d, 15.6) | 6.25 (d, 16.0) | 7.36 (dd, 8.4, 2.4) | 7.51 (dd, 8.4, 2.4) |
| 17′ | 4.99 (d, 7.8) | 4.90 (d, 7.8) | ||
| 18′ | 4.47 (d, 7.8) | 4.30 (d, 8.0) | 3.73 (m) | 3.71 (m) |
| 19′ | 3.54 (m) | 3.28 (m) | 3.70 (m) | 3.69 (m) |
| 20′ | 3.82 (m) | 3.55 (m) | 3.50 (m) | 3.63 (m) |
| 21′ | 4.98 (t, 10.0) | 4.86 (t, 10.0) | 3.43 (m) | 3.52 (m) |
| 22′ | 3.64 (m) | 3.59 (m) | 3.71 (m, Ha) | 3.72 (m, Ha) |
| 3.43 (m, Hb) | 3.53 (m, Hb) | |||
| 23′ | 3.69 (d, 8.5, Ha) | 3.64 (m, Ha) | 4.60 (brs) | 4.70 (brs) |
| 3.48 (m, Hb) | 3.43 (m, Hb) | |||
| 24′ | 4.74 (brs) | 4.53 (brs) | 3.74 (m) | 3.80 (m) |
| 25′ | 3.96 (m) | 3.80 (m) | 4.14 (m) | 3.82 (m) |
| 26′ | 3.83 (m) | 3.89 (t, 7.0) | 3.56 (m) | 3.53 (m) |
| 27′ | 3.47 (m) | 3.34 (m) | 3.78 (m) | 3.58 (m) |
| 28′ | 3.65 (m) | 3.51 (m) | 1.31 (d, 6.0) | 1.21 (d, 6.0) |
| 29′ | 1.15 (overlap) | 0.80 (d, 6.0) | ||
| No. | 1a | 2b | 3a | 4a |
|---|---|---|---|---|
| a Data were measured in acetic acid-d4 at 150 MHz.b Data were measured in methanol-d4 at 125 MHz. | ||||
| 1 | 132.1 | |||
| 2 | 161.4 | 115.9 | 163.1 | 163.5 |
| 3 | 137.9 | 146.3 | 139.3 | 139.3 |
| 4 | 181.5 | 146.4 | 181.0 | 181.1 |
| 5 | 161.8 | 116.6 | 164.4 | 164.5 |
| 6 | 108.3 | 120.5 | 102.3 | 102.3 |
| 7 | 161.8 | 83.7 | 167.2 | 167.3 |
| 8 | 97.8 | 75.1 | 96.8 | 96.9 |
| 9 | 158.2 | 127.7 | 160.6 | 160.4 |
| 10 | 109.1 | 115.3 | 107.7 | 107.8 |
| 11 | 125.0 | 146.8 | 127.9 | 126.9 |
| 12 | 120.1 | 149.7 | 121.2 | 121.5 |
| 13 | 146.8 | 116.4 | 146.4 | 146.6 |
| 14 | 151.2 | 123.2 | 150.8 | 151.3 |
| 15 | 118.2 | 147.6 | 118.4 | 118.2 |
| 16 | 126.2 | 114.7 | 126.0 | 127.2 |
| 17 | 107.2 | 168.3 | 108.7 | 107.6 |
| 18 | 77.3 | 104.2 | 77.1 | 77.3 |
| 19 | 79.6 | 74.8 | 78.8 | 79.5 |
| 20 | 72.3 | 75.8 | 71.0 | 72.0 |
| 21 | 78.2 | 72.2 | 78.1 | 78.3 |
| 22 | 70.4 | 75.2 | 68.2 | 70.2 |
| 23 | 103.6 | 67.6 | 103.9 | 103.1 |
| 24 | 73.7 | 102.4 | 73.5 | 73.8 |
| 25 | 74.3 | 72.3 | 74.2 | 74.4 |
| 26 | 75.8 | 72.5 | 75.7 | 75.8 |
| 27 | 71.2 | 74.0 | 71.4 | 71.3 |
| 28 | 19.6 | 70.0 | 19.6 | 19.8 |
| 29 | 18.2 | |||
| 1′ | 116.4 | 131.5 | ||
| 2′ | 118.7 | 117.2 | 160.5 | 161.3 |
| 3′ | 143.9 | 146.2 | 137.7 | 137.9 |
| 4′ | 147.6 | 144.7 | 181.2 | 181.8 |
| 5′ | 106.5 | 116.6 | 164.4 | 164.1 |
| 6′ | 147.5 | 121.4 | 103.3 | 102.3 |
| 7′ | 35.9 | 36.7 | 164.9 | 164.3 |
| 8′ | 75.8 | 72.5 | 105.3 | 105.1 |
| 9′ | 130.0 | 127.7 | 157.1 | 157.9 |
| 10′ | 117.5 | 115.3 | 108.2 | 108.0 |
| 11′ | 147.7 | 146.8 | 125.0 | 125.1 |
| 12′ | 150.5 | 149.7 | 120.3 | 120.3 |
| 13′ | 118.6 | 116.4 | 146.6 | 146.6 |
| 14′ | 125.6 | 123.2 | 150.9 | 150.3 |
| 15′ | 149.8 | 147.6 | 118.0 | 118.1 |
| 16′ | 116.9 | 114.7 | 126.0 | 126.5 |
| 17′ | 170.4 | 168.2 | 107.0 | 108.4 |
| 18′ | 105.1 | 104.6 | 77.4 | 77.3 |
| 19′ | 76.7 | 75.0 | 79.6 | 79.5 |
| 20′ | 77.4 | 75.9 | 72.5 | 72.5 |
| 21′ | 74.0 | 72.2 | 78.1 | 78.2 |
| 22′ | 76.6 | 75.2 | 71.0 | 70.6 |
| 23′ | 69.7 | 68.0 | 103.8 | 103.6 |
| 24′ | 103.2 | 102.3 | 73.7 | 73.9 |
| 25′ | 73.6 | 72.0 | 74.0 | 74.3 |
| 26′ | 74.2 | 73.1 | 76.1 | 75.8 |
| 27′ | 75.7 | 82.0 | 71.2 | 71.2 |
| 28′ | 71.3 | 68.9 | 19.9 | 19.6 |
| 29′ | 19.6 | 18.6 | ||
Based on the above analysis, compound 1 was assumed as the dimer of phenylethanoid glycoside and flavonoid glycoside which was similar to that of forsythoneoside A and B, a pair of heterodimers of flavonoid and phenylethanoid glycoside possess the pyran ring consisted by C7–C8–C7′–C1′–C6′–O. Through comparing NMR data of 1 with those of forsythoneoside A,18 the apparent differences between these two compounds were the chemical shifts of C-6 and C-8. According to the 13C NMR data, the chemical shift of C-6 (δC 108.3) in 1 was shift to downfield compared with that of C-6 (δC 102.6) in forsythoneoside A. By contrast, the chemical shift of C-8 (δC 97.8) in 1 was shift to upfield compared with that of C-8 (δC 103.6) in forsythoneoside A. From the above analysis, compound 1 was established as phenylethanoid–flavonoid heterodimer formed through a pyran ring, which consisted of [C7–C6–C7′–C1′–C6′–O]. This deduction was further corroborated by the key HMBC correlations (Fig. 2) from H-8 (δH 6.77) to C-6 (δC 108.3), C-9 (δC 158.2), and C-10 (δC 109.1).
 | ||
| Fig. 2 Key HMBC correlations of 1–3. | ||
After the planar structure had been established, the absolute configurations of compound 1 were determined by comparing the experimental and calculated ECD spectra. Conformational analysis of the whole molecule (1a) with 149 atoms was performed to locate the low-energy conformers using molecular mechanics force field (MMFF94). Two preferred conformers (Fig. S1, ESI†) were geometrically optimized by using time-dependent density functional theory (TD-DFT) method at the B3LYP/6-31G(d,p) level. Under the half-bandwidth as 0.35 eV, the calculated ECD spectrum of 1a was generated through Boltzmann-population weighting, which showed high similarity with the experimental ECD spectrum of 1 (Fig. 3). Therefore, the absolute configuration of 1 was defined as 7′R, and this compound was named forsythoneoside E.
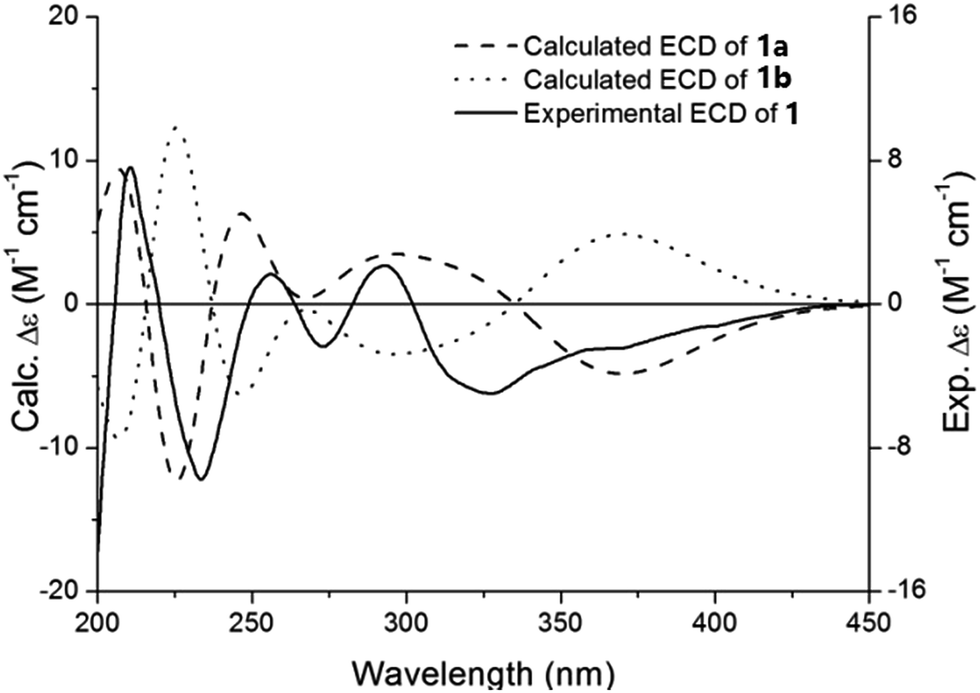 | ||
| Fig. 3 Experimental ECD spectrum of 1 and calculated ECD of 1a and 1b in MeOH. | ||
Forsythoneoside F (2) was isolated as greyish yellow amorphous powder with the max UV absorption at 330 and 218 nm. According to the 13C NMR data and a positive ion peak at m/z 1269.3842 [M + Na]+ (calcd for C58H70O30Na, 1269.3844) exhibited in the HRESIMS, its molecular formula was established as C58H70O30. The 1H NMR spectrum (Table 1) of 2 displayed resonances assignable to two caffeoyl groups at δH 7.00 (1H, d, 2.0), 6.72 (1H, d, 8.0), 6.90 (1H, dd, 8.0, 2.0), 7.00 (1H, d, 2.0), 6.74 (1H, d, 8.0), 6.91 (1H, dd, 8.0, 2.0), 7.52 (1H, d, 16.0), 7.55 (1H, d, 16.0), 6.25 (1H, d, 16.0), and 6.22 (1H, d, 16.0), two 3,4-dihydroxyphenyl groups at δH 6.74 (1H, d, 2.0), 6.67 (1H, d, 8.0), 6.60 (1H, dd, 8.0, 2.0), 6.65 (1H, d, 2.0), 6.64 (1H, d, 8.0), and 6.53 (1H, dd, 8.0, 2.0), together with four anomeric proton signals at δH 4.39 (1H, d, 8.0), 4.30 (1H, d, 8.0), 4.59 (1H, brs), and 4.53 (1H, brs). This result, combined with the 13C NMR data, suggested the presence of two molecules of forsythoside A in 2, which was also supported by comparison with the NMR data of forsythoside A.19 With the aid of HMBC spectrum, the linkage between these two units was characterized via three bond HMBC correlations of H-27′ (δH 3.34, 1H, m) to C-7 (δC 83.7) as shown in Fig. 2. From the above analysis, the planar structure was defined as a phenylethanoid glycoside dimer connected by a C7–O–C27′ ether bond, named forsythoneoside F. According to the structural characteristic, this compound could be regarded as 2-alkoxy phenylethanoid glycoside. Recently, we reported an approach for determining the absolute configuration of C-2 in 2-oxygenated phenylethanoid glycosides by 1H NMR spectroscopy.20 So, the absolute configuration of C-7 in 2 was determined to be R by a large chemical shift difference (Δδ = 0.45) of H-8a and H-8b.
Forsythobiflavone A (3) was isolated as a yellow amorphous powder. The molecular formula of C54H58O32 was assigned, based on the HRESIMS protonated molecular ion peak at m/z 1219.2976 [M + H]+ (calcd for C54H59O32, 1219.2984). IR spectrum indicated the presence of hydroxy (3402 cm−1), carbonyl (1652 cm−1), and aromatic (1604, 1504, and 1448 cm−1) groups.
The 1H NMR resonances displayed two typical proton signals at δH 6.18 (1H, d, J = 2.4 Hz), 5.71 (1H, d, J = 2.4 Hz) belonging to the H-6 and H-8 of flavonoid A-ring, a set of ABX spin system aromatic protons at δH 7.69 (1H, d, J = 2.4 Hz), 6.88 (1H, d, J = 8.4 Hz), 7.36 (1H, dd, J = 8.4, 2.4 Hz) belonging to the flavonoid B-ring, a pentasubstituted A-ring signal at δH 6.43 (1H, s), two ortho-coupled B-ring signals at δH 7.37 (1H, d, J = 8.4 Hz), 7.20 (1H, d, J = 8.4 Hz), and four anomeric proton signals at δH 4.56 (1H, d, J = 7.8 Hz), 4.99 (1H, d, J = 7.8 Hz), 4.84 (1H, br s), 4.60 (1H, br s). The 13C NMR spectrum (Table 2) exhibited 54 carbon signals, apart from two sets of rutinose carbon signals, the remaining 30 carbon signals could be assigned to two flavonoid units. This result, combined with the molecular formula, suggested that 3 was a dimeric rutin possessing a C–C linkage. The specific substitution pattern was disclosed by HMBC spectrum. The HMBC cross-peaks of the H-6′ (δH 6.43) with C-5′ (δC 164.4), C-7′ (δC 164.9), C-8′ (δC 105.3) and C-10′ (δC 108.2), together with the absence of H-12, confirmed that the two rutin units was connected by C12–C8′ bond. Thus, 3 was eventually characterized as shown, and named forsythobiflavone A.
Compound 4 (forsythobiflavone B) was determined as a diastereoisomer of 3 by the same molecular formula and the similar UV, IR, and NMR data. Owing to the totally opposite Cotton effect of 3 and 4, these two compounds were elucidated as a pair of asymmetric axial compounds with the opposite P/M configurations.
To determine the absolute configuration of 3 and 4, the ECD exciton chirality method was proposed.21,22 In the CD spectrum of 3, a positive exciton-split Cotton effect at 347 (Δε +13.88) and 308 (Δε −5.97) nm caused by the exciton coupling of the benzoyl and benzene chromophores illustrated M configuration of 3 (Fig. 4). While, a negative exciton-split Cotton effect at 347 nm (Δε −10.29) and 309 nm (Δε +1.16) in 4 established its P configuration.
 | ||
| Fig. 4 The determination of the absolute configuration for asymmetric axial by CD exciton chirality rule. | ||
Structurally, 1 was a heterodimer of rutin and forsythoside A, 2 was a dimer of forsythoside A, 3 and 4 were two dimers of rutin. In order to compare the neuroprotective effects between these compounds and their precursors, we introduced forsythoside A and rutin in the process of in vitro assay. As shown in Table 3, all of the samples exhibited neuroprotective effects against rotenone-induced neurotoxicity in PC12 cells at 1 μM. Notably, for compound 1, increasing the cell viability from 53.9 ± 7.1% for the model to 71.5 ± 1.7%, which was stronger than compounds 2–4, forsythoside A, and rutin. This result, combined with the neuroprotective activities of forsythoneosides A–D in our previous study, suggested that the pyran ring appear to contribute to their biological activities.
| Group | Rotenone (4 μM) |
|---|---|
| a ###p < 0.001 vs. control, ***p < 0.001 vs. model, **p < 0.01 vs. model, *p < 0.1 vs. model. | |
| Control | 100.0 ± 3.9 |
| Model | 53.9 ± 7.1### |
| 1 | 71.5 ± 1.7*** |
| 2 | 62.6 ± 4.5* |
| 3 | 68.8 ± 6.9 |
| 4 | 62.3 ± 0.9*** |
| Forsythoside A | 60.4 ± 5.5 |
| Rutin | 68.8 ± 3.3* |
Experimental
General experimental procedures
Optical rotations were measured using a JASCO P-2000 digital polarimeter (JASCO, Easton, MD, USA). UV spectra were recorded on a JASCO V650 spectrophotometer spectrometer (JASCO). IR spectra were obtained on a Nicolet 5700 spectrometer using an FT-IR microscope transmission method (Thermo Scientific, Waltham, MA, USA). ECD spectra were measured in the 500–215 nm spectral range on a JASCO J-815 spectropolarimeter (JASCO). NMR spectra were acquired on Bruker AVIII-500 and Bruker AVIIIHD-600 spectrometers (Bruker-Biospin, Billerica, MA, USA). HRESIMS were obtained by an Agilent 6520 HPLC-Q-TOF (Agilent Technologies, Waldbronn, Germany). Column chromatography was performed on macroporous resin (Diaion HP-20, Mitsubishi Chemical Corp., Tokyo, Japan), RP-18 (50 μm, YMC, Kyoto, Japan), and Sephadex LH-20 (Pharmacia Fine Chemicals, Uppsala, Sweden). Preparative HPLC separation was performed using a Shimadzu LC-10AT instrument (Shimadzu Corp., Tokyo, Japan) with an SPD-10A detector, using a YMC-Pack ODS-A column (250 mm × 20 mm, 5 μm; YMC Corp., Kyoto, Japan). HPLC-DAD analysis was performed using an Agilent 1200 series system (Agilent Technologies) with an Apollo C18 column (250 mm × 4.6 mm, 5 μm; Alltech Corp., Lexington, KY, USA).Plant material
The fruits of F. suspensa were collected in Yuncheng city, Shanxi Province, People's Republic of China, in Dec. 2011 and were identified by Prof. Lin Ma (Institute of Materia Medica, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing, 100050, P. R. China). A voucher specimen (ID-S-2597) was deposited at the Herbarium of the Department of Medicinal Plants, Institute of Materia Medica.Extraction and isolation
The air-dried fruits of F. suspensa (90.0 kg) were deseeded and reflux-extracted with 75% EtOH (600 L). And then, the extract was filtrated and evaporated under reduced pressure. The residue (12.6 kg) was suspended in water (10 L), and partitioned successively with petroleum ether, EtOAc and n-BuOH. After the solvent was removed, the n-BuOH-soluble portion (4.0 kg) was further suspended in water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) and filtrated to give an aqueous layer. The aqueous layer (1.5 kg) was concentrated and subjected to a macroporous adsorption resins (HP-20, 20 L) column chromatography, eluting with H2O, 15% ethanol, 30% ethanol, 50% ethanol, and 95% ethanol. Owing to the result of neuroprotective activity assay, the 30% EtOH fraction (460 g) was chromatographed over Sephadex LH-20 eluting with H2O–MeOH in gradient (a gradient from 100:0 to 0:100) to yield 5 fractions (Fr A–E). Fraction C (116 g) was separated by Sephadex LH-20 (MeOH–H2O from 10:90 to 40:60) to yield 14 fractions (Fr.C1–C14). Fr.C12 was purified by Prep-HPLC (MeOH–H2O, 25:75) to afford forsythobiflavone A (3, 20 mg) and forsythobiflavone B (4, 15 mg), respectively. Fraction E (45 g) was separated by Sephadex LH-20 eluting with MeOH–H2O (a gradient from 30% to 60%) to obtain 43 subfractions (Fr.E1–E43). Fr.E23 was purified by Prep-HPLC (MeOH–H2O, 30:70) to afford forsythoneuoside F (2, 25 mg). Forsythoneoside E (1, 16 mg) was isolated by Prep-HPLC (MeOH–H2O, 40:60) from Fr.E27.
:10) and filtrated to give an aqueous layer. The aqueous layer (1.5 kg) was concentrated and subjected to a macroporous adsorption resins (HP-20, 20 L) column chromatography, eluting with H2O, 15% ethanol, 30% ethanol, 50% ethanol, and 95% ethanol. Owing to the result of neuroprotective activity assay, the 30% EtOH fraction (460 g) was chromatographed over Sephadex LH-20 eluting with H2O–MeOH in gradient (a gradient from 100:0 to 0:100) to yield 5 fractions (Fr A–E). Fraction C (116 g) was separated by Sephadex LH-20 (MeOH–H2O from 10:90 to 40:60) to yield 14 fractions (Fr.C1–C14). Fr.C12 was purified by Prep-HPLC (MeOH–H2O, 25:75) to afford forsythobiflavone A (3, 20 mg) and forsythobiflavone B (4, 15 mg), respectively. Fraction E (45 g) was separated by Sephadex LH-20 eluting with MeOH–H2O (a gradient from 30% to 60%) to obtain 43 subfractions (Fr.E1–E43). Fr.E23 was purified by Prep-HPLC (MeOH–H2O, 30:70) to afford forsythoneuoside F (2, 25 mg). Forsythoneoside E (1, 16 mg) was isolated by Prep-HPLC (MeOH–H2O, 40:60) from Fr.E27.
Cell culture
PC12 cells were purchased from the American Type Culture Collection (ATCC). The cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen, Gibco, USA) supplemented with 5% heat-inactivated fetal bovine serum (Invitrogen, Gibco, USA), 5% horse serum (Thermo Scientific, Hyclone, USA), 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. The cultures were maintained in a humidified incubator at 37 °C in an atmosphere of 95% air and 5% CO2. The media were changed every two or three days.Structure characterization
ε): 337 (4.38), 273 (4.35), 253 (4.39), and 200 (4.96) nm. IR (KBr) νmax 3361, 1694, 1603, 1520, and 1453 cm−1; and (+)-HRESIMS m/z 1231.3342 [M + H]+ (calcd for C56H63O31, 1231.3348). For the NMR data, see Tables 1 and 2.:1); CD (MeOH–H2O 1:1) λmax (Δε): 344 (+0.53), 256 (+0.20), 230 (−0.52), and 209 (+3.87); UV (MeOH) λmax (logε): 330 (4.53), 290 (4.44), and 200 (5.06) nm. IR (KBr) νmax 3382, 1696, 1605, 1519, and 1447 cm−1; and (+)-HRESIMS m/z 1269.3842 [M + Na]+ (calcd for C58H70O30Na, 1269.3844). For the NMR data, see Tables 1 and 2.:1); CD (MeOH–H2O 1:1) λmax (Δε): 347 (+13.88), 308 (−5.97), 281.5 (+6.45), and 247.5 (−31.86); UV (MeOH) λmax (logε): 348 (4.39), 258 (4.62), and 204 (4.90) nm. IR (KBr) νmax 3402, 1652, 1604, 1504, and 1448 cm−1; and (+)-HRESIMS m/z 1219.2976 [M + H]+ (calcd for C54H59O32, 1219.2984). For the NMR data, see Tables 1 and 2.:1); CD (MeOH–H2O 1:1) λmax (Δε): 347 (−10.28), 308 (+1.16), 287 (−4.05), and 249 (+18.29); UV (MeOH) λmax (logε): 349 (4.28), 259 (4.53), and 204 (4.79) nm. IR (KBr) νmax 3379, 1653, 1607, 1512, and 1448 cm−1; and (+)-HRESIMS m/z 1219.2988 [M + H]+ (calcd for C54H59O32, 1219.2984). For the NMR data, see Tables 1 and 2.Determination of the absolute configuration of sugar
The D-glucose and L-rhamnose were determined by using the same method as described in the literature.18Neuroprotective activities of compounds 1–4
The neuroprotective effects of compounds (1–4) against rotenone-induced neurotoxicity in PC12 cells were examined following previously described procedures.18Conclusions
The objective of the present research was to conduct a further investigation of the novel neuroprotective compounds from the fruits of F. suspensa. Herein, we reported the structure elucidation of four dimer compounds and their neuroprotective effects against rotenone-induced neurotoxicity in PC12 cells. At 1 μM, each compound (1–4) exhibited neuroprotective effect with cell viability of 71.5 ± 1.7%, 62.6 ± 4.5%, 68.8 ± 6.9% and 62.3 ± 0.9%, respectively, compared with that of the model with cell viability of 53.9 ± 7.1%. Analyses of their structure features revealed that the unusual pyran ring formed by the fusion of forsythoside A, and rutin might have contributed to their pharmacological activities.Acknowledgements
The research described in this publication was supported by the National Natural Science Foundation of China (No. 81673313) and CAMS Innovation Fund for Medical Sciences (CIFMS) (No. 2016-I2M-1-007).References
- Y. Cui, Q. Wang, X. W. Shi, X. W. Zhang, X. N. Sheng and L. T. Zhang, Phytochem. Anal., 2010, 21, 253–260 CrossRef CAS PubMed.
- C. Li, Y. Dai, S. X. Zhang, Y. H. Duan, M. L. Liu, L. Y. Chen and X. S. Yao, Phytochemistry, 2014, 104, 105–113 CrossRef CAS PubMed.
- M. J. Chang, T. M. Hung, B. S. Min, J. C. Kim, M. H. Woo, J. S. Choi, H. K. Lee and K. Bae, Biosci., Biotechnol., Biochem., 2008, 72, 2750–2755 CrossRef CAS PubMed.
- X. L. Piao, M. H. Jang, J. Cui and X. Piao, Bioorg. Med. Chem. Lett., 2008, 18, 1980–1984 CrossRef CAS PubMed.
- X. J. Yan, X. Y. Bai, Q. B. Liu, S. Liu, P. Y. Gao, L. Z. Li and S. J. Song, J. Asian Nat. Prod. Res., 2014, 16, 374–382 Search PubMed.
- S. J. Dai, Y. Ren, L. Shen and D. W. Zhang, Planta Med., 2009, 75, 375–377 CrossRef CAS PubMed.
- Y. Ge, Y. Z. Wang, P. P. Chen, Y. F. Wang, C. C. Hou, Y. B. Wu, M. L. Zhang, L. G. Li, C. H. Huo, Q. W. Shi and H. X. Gao, J. Agric. Food Chem., 2016, 64, 125–131 CrossRef CAS PubMed.
- X. S. Fang, Y. Z. Wang, J. H. Wang, J. Zhang and X. Wang, J. Sep. Sci., 2013, 36, 2672–2679 CrossRef CAS PubMed.
- H. H. Qu, Y. M. Zhang, Y. Wang, B. X. Li and W. J. Sun, J. Pharm. Pharmacol., 2008, 60, 261–266 CrossRef CAS PubMed.
- H. W. Li, J. F. Wu, Z. W. Zhang, Y. Y. Ma, F. F. Liao, Y. Zhang and G. J. Wu, Phytother. Res., 2011, 25, 338–342 CAS.
- C. W. Pan, G. Y. Zhou, W. L. Chen, L. Zhuge, L. X. Jin, Y. Zheng, W. Lin and Z. Z. Pan, Int. Immunopharmacol., 2015, 26, 80–85 CrossRef CAS PubMed.
- H. S. Shin, S. Y. Park, H. G. Song, E. Hwang, D. G. Lee and T. H. Yi, Phytother. Res., 2015, 29, 870–876 CrossRef CAS PubMed.
- L. Cheng, F. Li, R. Ma and X. P. Hu, Int. Immunopharmacol., 2015, 28, 494–499 CrossRef CAS PubMed.
- Y. Wang, H. F. Zhao, C. X. Lin, J. Ren and S. Z. Zhang, Neurochem. Res., 2016, 41, 659–665 CrossRef CAS PubMed.
- C. K. Huang, Y. L. Lin, H. Su and D. Q. Ye, Neurochem. Res., 2015, 40, 27–35 CrossRef CAS PubMed.
- H. M. Wang, L. W. Wang, X. M. Liu, C. L. Li, S. P. Xu and A. D. Farooq, Pharmacol., Biochem. Behav., 2013, 105, 134–141 CrossRef CAS PubMed.
- J. M. Kim, S. Kim, D. H. Kim, C. H. Lee, S. J. Park, J. W. Jung, K. H. Ko, J. H. Cheong, S. H. Lee and J. H. Ryu, Eur. J. Pharmacol., 2011, 660, 326–333 CrossRef CAS PubMed.
- F. Zhang, Y. N. Yang, X. Y. Song, S. Y. Shao, Z. M. Feng, J. S. Jiang, L. Li, N. H. Chen and P. C. Zhang, J. Nat. Prod., 2015, 78, 2390–2397 CrossRef CAS PubMed.
- Q. L. Jin, H. G. Jin, J. E. Shin, J. K. Hong and E. R. Woo, Bull. Korean Chem. Soc., 2011, 32, 1721–1724 CrossRef CAS.
- S. Y. Shao, F. Zhang, Y. N. Yang, Z. M. Feng, J. S. Jiang and P. C. Zhang, Org. Lett., 2016, 18, 4084–4087 CrossRef CAS PubMed.
- W. Hüttel, M. Nieger and M. Müller, Synthesis, 2003, 12, 1803–1808 Search PubMed.
- G. Bringmann, K. P. Gulden, Y. F. Hallock, K. P. Manfredi, J. H. Cardellina II, M. R. Boyd, B. Kramer and J. Fleischhauer, Tetrahedron, 1994, 50, 7807–7814 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1D NMR, 2D NMR HRMS, IR, and ECD spectra. See DOI: 10.1039/c7ra04229a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |