
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceF127DA micelle cross-linked PAACA hydrogels with highly stretchable, puncture resistant and self-healing properties†
Yunfei Liua,
Wanfu Zhoub,
Quan Zhoub,
Kang Penga,
Akram Yasina and
Haiyang Yang *a
*a
aCAS Key Laboratory of Soft Matter Chemistry, School of Chemistry and Materials Science, University of Science and Technology of China, Hefei, 230026, P. R. China. E-mail: yhy@ustc.edu.cn
bOilfield Production Technology Institute, Daqing Oilfield Co. Ltd., Daqing, 163514, P. R. China
First published on 6th June 2017
Abstract
It is highly desirable to develop hydrogels that possess excellent mechanical properties and self-healing ability. In this study, a highly stretchable, puncture resistant and self-healing hydrogel is prepared, via in situ copolymerization of a functional monomer, N-acryloyl-6-aminocaproic acid (AACA), and Pluronic F127 diacrylate (F127DA) as macro-cross-linkers in aqueous solution at 50 °C. Owing to the toughness-enhancement mechanism induced by F127DA micelles, the elongation ratio and tensile strength at break of the optimal hydrogel reach up to 2500% and 69 kPa, respectively. Meanwhile, a stab test shows that the hydrogels can be stabbed by a steel needle without any damage. What's more, the hydrogels exhibit excellent self-healing ability, due to multiple hydrogen bonding between protonated PAACA groups. And the hydrogels show pH-responsive cyclic swelling and de-swelling behaviors because of the pH sensitivity of AACA. Thus, the present hydrogels would provide new opportunities with regard to the design and practical application of hydrogel systems.
Introduction
Polymer hydrogels, a class of “soft and wet” materials, have been widely researched in tissue engineering, drug delivery and actuators,1–3 because of their excellent biocompatibility and fascinating stimuli-responsive properties. However, they usually suffer from low mechanical strength and poor extensibility, due to their heterogeneous intrinsic structures and lack of efficient energy-dissipation mechanisms.4–6 To address this problem, researchers have developed various novel strategies to design high-strength hydrogels, including double network hydrogels,7,8 nanocomposite hydrogels,9,10 slide-ring hydrogels,11 tetra-PEG hydrogels,12 physical interaction enhanced hydrogels,13,14 hydrophobic association hydrogels15 and macromolecular microsphere composite hydrogels.16–18 The general principle is to implement mechanisms into hydrogels to dissipate large amounts of energy under deformation and to retain original configurations after deformation.5 For instance, Wang et al. reported macromolecular microsphere composite hydrogels for the first time.16 They prepared macromolecular microspheres from styrene as multifunctional cross-linkers, and then in situ chemically grafted poly(acrylic acid) chains in the surface of microspheres by using oxygen and γ-irradiation to initiate the polymerization of acrylic acid. These macromolecular microsphere composite hydrogels showed a unique microstructure and high mechanical strength. The covalent bonding, together with chain entanglements and hydrogen bonding between microspheres were suggested to account for the mechanical properties.16,18 Afterward, some other microspheroidal multifunctional cross-linkers, such as silica nanoparticles19,20 and surfactant micelles,21,22 were used to prepare mechanically strong hydrogels. Guo et al. fabricated a highly stretchable and resilient hydrogel based on polymer micelles.21 Although these hydrogels possessed good mechanical properties, most of them could not self-repair once damage, due to the lack of self-healing mechanisms.The self-healing ability is one of the most remarkable characters of living organisms, which can repair injury spontaneously to original state so as to increase the survivability and lifespan.23,24 In 2001, White et al. reported the early autonomic healing polymeric material as follow: a structural composite matrix incorporated a microencapsulated healing agent. And the healing agent was released and triggered polymerization by an embedded catalyst upon crack intrusion, bonding the crack faces.25 Since then, various strategies have been explored to generate self-healing hydrogels. The most utilized strategies are introducing reversible supramolecular interactions such as electrostatic interactions,26,27 host–guest recognitions,28 hydrophobic interactions29,30 or hydrogen bonding,31–33 etc. Among all these different kinds of reversible interactions, hydrogen bonding is one of the most well-known reversible interactions to design self-healing hydrogels. For instance, Phadke et al.31 demonstrated that the rapid self-healing ability can be achieved in chemically permanently cross-linked systems via strong hydrogen-bonding interactions.
In this study, we report a highly stretchable, puncture resistant and self-healing hydrogel via in situ copolymerization of a function monomer, N-acryloyl-6-aminocaproic acid (AACA), and Pluronic F127 diacrylate (F127DA). The reason of introducing F127DA is that F127DA can self-assemble to form polymeric micelles with vinyl groups on surface, and thus severing as multifunctional macro-cross-linkers34,35 evenly distributed in aqueous solution. The sparsely dispersed macro-cross-linkers, together with the extensive entanglements of the PAACA chains contribute to the excellent mechanical properties of hydrogels. Meanwhile, the flexible and long PAACA chains endow hydrogels with the self-healing ability. When PAACA chains of hydrogels are protonated, those chains cross the fracture surface to form abundant and multiple hydrogen bonding between the carboxyl groups of the other side chains and repair damaged hydrogels. What's more, the hydrogels show pH-responsive cyclic swelling-deswelling behavior because of the pH sensitivity of AACA. Thus, the present work not only provides a facile strategy to design mechanical strong and self-healing hydrogels but also broadens the practical applications of hydrogels.
Experimental
Materials
6-Aminocaproic acid and acryloyl chloride were obtained from Energy Chemical. Pluronic F127 was purchased from Sigma Aldrich. Potassium persulfate (KPS), sodium hydroxide (NaOH), trimethylamine (TEA), sodium sulfate (Na2SO4) and rhodamine B were supplied from Sinopharm Chemical Reagent Co. Ltd. All chemicals were used without further purification.Synthesis of N-acryloyl-6-aminocaproic acid (AACA)
AACA was synthesized according to literature.36 Briefly, 6-aminocaproic acid (10 g, 76.2 mmol) and NaOH (3.048 g, 76.2 mmol) were dissolved in 80 ml deionized water. Subsequently, 7.74 ml acryloyl chloride dissolved in 15 ml tetrahydrofuran was dropped slowly into the above mixture in an ice bath. The pH of the mixture was remained at 7.5–7.8 in the whole reaction process. And then the mixture was extracted with ethyl acetate. The aqueous layer was extracted with ethyl acetate again after its pH adjusted to 3. The organic layers were collected, combined and dried over with anhydrous Na2SO4. The solution was then filtered, concentrated, and precipitated in petroleum ether. The product was precipitated three times and the final product was dried in vacuum.Synthesis of Pluronic F127 diacrylate (F127DA)
F127DA was synthesized adopting the reported method.37 Briefly, 10 g F127 was dissolved in 100 ml dichloromethane in a three-neck flask, and subsequently 10-fold molar excess triethylamine and acryloyl chloride were added, dropwise, with dry nitrogen gas. The mixture was then stirred for 24 hours at room temperature under nitrogen gas and afterward filtered to any precipitates. The above filtrate was concentrated and precipitated three times into an excess of chilled diethyl ether. The white precipitate was dried in vacuum for 1 day at room temperature.Hydrogel preparation
Hydrogels were prepared via in situ free radical polymerization of AACA, using F127DA (6 mM) as a multifunctional cross-linker, and KPS (0.1 mol% of the monomer) as an initiator. First, F127DA was dissolved in deionized water in a vial, followed by the addition of AACA, equimolar NaOH and KPS. After stirring for 10 min, the reaction mixtures in the vial were poured into a plastic mold and the polymerization was carried out for 24 hours at 50 °C to obtain the macro-micelle cross-linked hydrogel (M-hydrogel). If no other description, the concentrations of AACA and F127DA were 4 M and 6 mM, respectively.Materials characterization
The size of F127DA micelles were characterized by a Malvern Nano-ZS Zetasizer Particle Analyser. The laser light scattering measurements were set at 90 °C. The samples were diluted with deionized water to 2 mg ml−1 before analysis. The test temperature was from 25 °C to 50 °C. Each sample was measured three times and taken as the average value. Fourier transform infrared spectroscopy (FT-IR) spectra of dry hydrogels were recorded on a Nicolet 8700 to confirm the successful polymerization of hydrogels.Characterization of mechanical properties
Rheological measurements were conducted on a TA AR-G2 rheometer at 25 °C. A parallel plate was used for testing with a diameter of 40 mm and a distance of 1000 µm. The strain sweep was recorded from 0.1% to 100% at a consistent frequency of 6.281 rad s−1. The frequency sweep was recorded from 0.1 to 100 rad s−1 at a consistent strain of 1%. Tensile tests were performed at room temperature, using a SunsUTM2502 universal testing machine with a 100 N load cell. The crosshead speed of 60 mm min−1 was chosen to perform the monotonic and cyclic tensile test. In cyclic tensile tests, the as-prepared samples were stretched to 1400% and then immediately unload. The loading–unloading cycle was kept for four times. The toughness was estimated using the area below the stress–strain curves.Characterization of self-healing ability
Two cylindrical hydrogels were cut into in half, respectively. For easy visualization, one of them was stained with rhodamine B. Subsequently, the two fresh fracture surfaces were contacted end to end. And then the connection part of hydrogels was soaked in diluted HCl solution with a slight press for 20 minutes. The self-healing efficiency was calculated by the tensile strength at the break before and after the self-healing process.Swelling and deswelling measurements
The as-prepared hydrogels were immersed into excess water to reach the swelling equilibrium. The Swelling Ratio (SR) was calculated as SR = (Wt − Wd)/Wd. Herein, Wt and Wd represent the weight of the swollen hydrogel at time t and dry hydrogel, respectively. The deswelling measurement was performed by immersing the completely swollen hydrogel into acid solution at pH = 3. Water retention (WR) was calculated as WR = (Wt − Wd)/Ws. Herein, Wt, Wd, and Ws represent the weight of the deswollen hydrogel at time t, dry hydrogel, and swollen hydrogel at equilibrium, respectively.Results and discussion
As shown in Fig. 1a, the M-hydrogels were prepared via in situ free radical copolymerization of AACA and F127DA with KPS as an initiator. The triblock copolymer F127DA, which could self-assembled as polymer micelles and evenly dispersed in deionized water, was served as multifunctional cross-linkers. The average diameter of F127DA micelles at different temperatures was shown in Fig. S1.† It should be noted that the AACA chains were grafted at the vinyl groups of the end of the polymer micelles. In addition, no chemical cross-linkers were used in the polymerization (F127DA micelles served as multifunctional cross-likers, as demonstrated in Fig. S2†). A series of M-hydrogels with varied concentration of AACA were successfully prepared (the FI-TR spectra were shown in Fig. S3†). Compared with conventional chemically cross-linked hydrogels, the as-prepared hydrogels showed a good extensibility and were able to withstand different high-level deformations. As shown in Fig. 1b and c, the cylinder shaped hydrogel could be stretched up to dozens times of original length and also could be knotted stretching without any damage.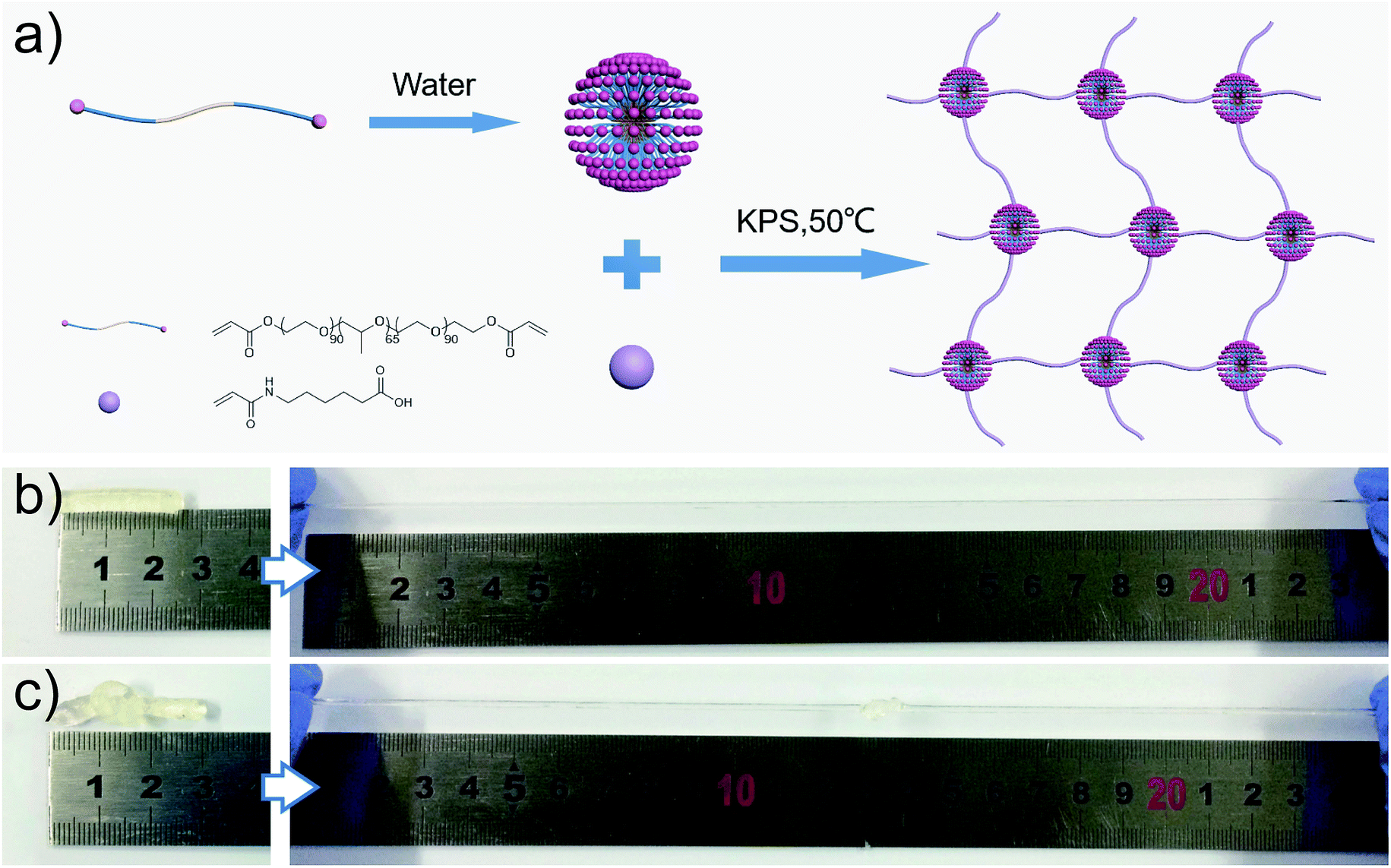 | ||
| Fig. 1 (a) Schematic illustration of the preparation of M-hydrogels. (b and c) The as-prepared hydrogels show a excellent extensibility, and can bear different high-level deformations such as elongation (b), and elongation after knotting (c). | ||
To investigate the mechanical properties of the M-hydrogels, rheology measurements were first performed with varied concentrations of AACA. Strain sweep experiments ensured the linear viscoelasticity region of the M-hydrogels. As shown in Fig. 2a, the elastic modulus G′ and loss modulus G″ were independent of the strain over the range from 0.1% to 20%. Then, the obvious frequency-dependent behaviors of G′ and G″ were observed in Fig. 2b, indicative of the viscoelastic nature of the hydrogels. The frequency dependence of G′ was possibly due to the physically interactions including the entanglements of PAACA chains and the hydrophobic association of F127DA micelles, which was consistent with other physically cross-linked hydrogels in previous reports.38,39 Meanwhile, over the entire frequency range, the plots of the storage modulus G′ shifted to higher values with increasing the concentration of AACA, attributing to the increment of the entanglements of PAACA chains.
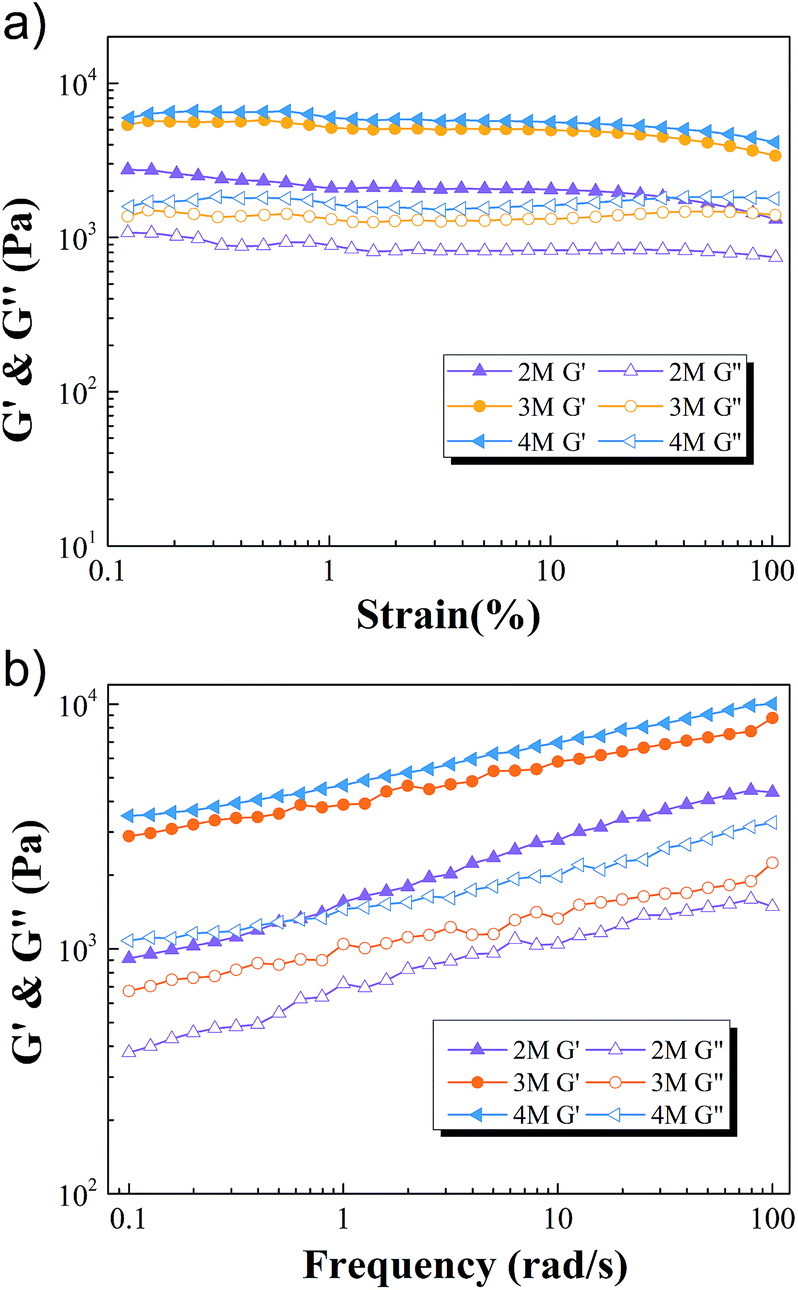 | ||
| Fig. 2 The elastic modulus G′ (full symbols) and loss modulus G″ (open symbols) as a function of strain (a) and frequency (b) of M-hydrogels with varied concentrations of AACA. | ||
The uniaxial tensile tests, which used a tensile machine with a 100 N loading cell, were then conducted by subjecting cylindrical samples at a constant speed of 60 mm min−1. The stress–strain curves of M-hydrogels with different concentrations of AACA were shown in Fig. 3a. As the concentration of AACA in hydrogel increased from 2 to 4 M, the fracture tensile strength increased from 10 kPa to 69 kPa. It was noted that these hydrogels were highly stretchable, of which with 4 M AACA the fracture elongation reached up to 2500%. The toughness of all hydrogels calculated from stress–strain curves were shown in Fig. 3b, the toughness monotonically increased from about 119 kJ m−3 to 316.35 kJ m−3 and 814 kJ m−3 with 2, 3 and 4 M AACA, respectively. The higher concentration of AACA resulted in more entanglements between PAACA chains, contributing to a denser and stronger network. Thus, it was reasonable that the hydrogels showed higher mechanical strength and toughness with higher concentration of AACA. This was also consistent with previous results of rheology tests.
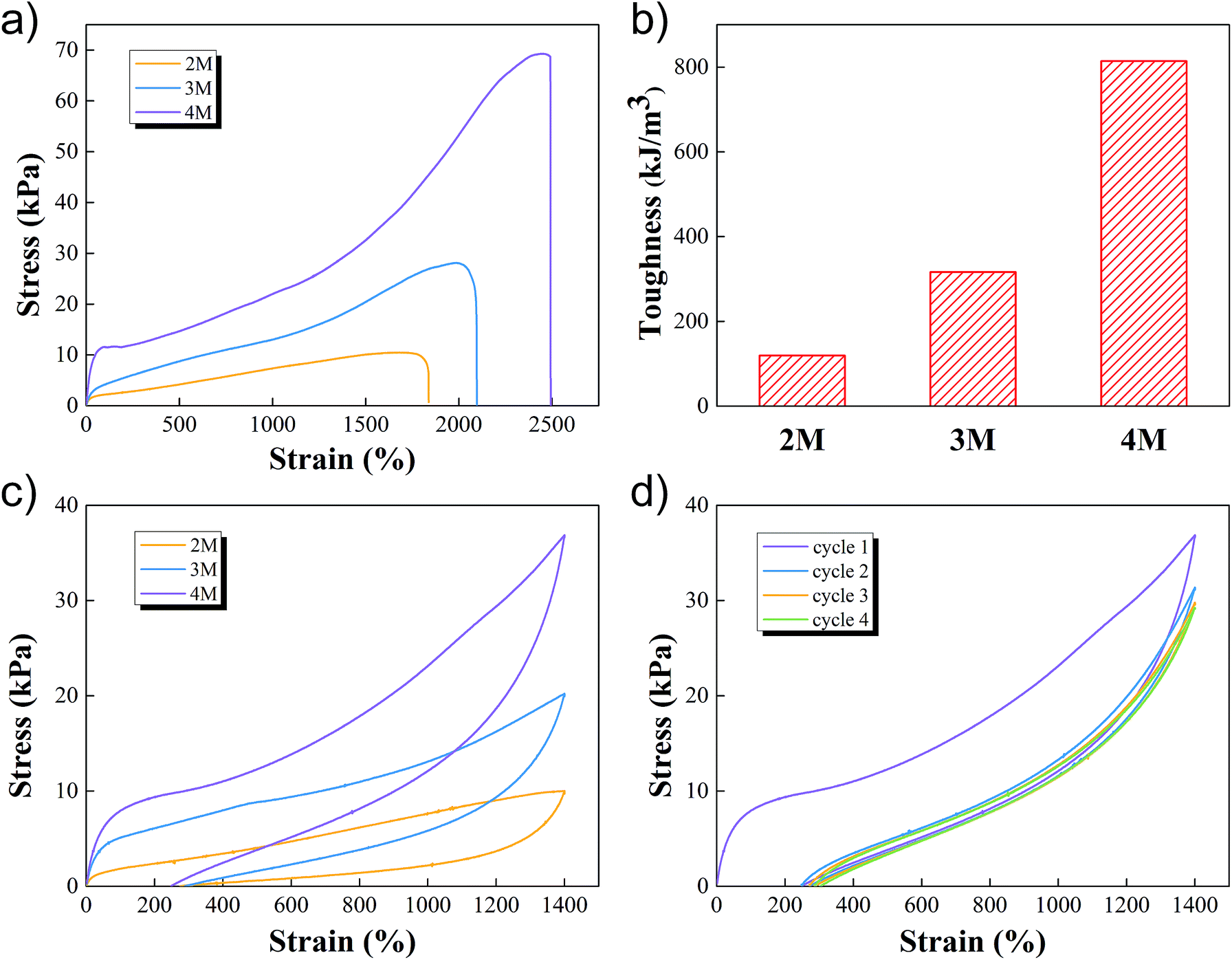 | ||
| Fig. 3 (a–c) Tensile stress–strain curves (a), toughness (b) and tensile loading–unloading curves (c) of M-hydrogels with varied concentrations of AACA. (d) Cyclic tensile loading-unloading curves of M-hydrogels with 4 M AACA. | ||
We conducted the loading-unloading tests at a constant speed of 60 mm min−1 to assess the energy dissipation of M-hydrogels, to further describe the mechanical properties. The results of the loading-unloading tests of M-hydrogels with varied concentration of AACA were shown in Fig. 3c. When the hydrogel was first loaded to 1400% strain and then unloaded immediately, clear hysteresis loops, which represented an energy dissipation in the process of stretching, were observed in all of the samples. And the loop area increased with the increasing of concentration of AACA. These results indicated that F127DA micelles together with PAACA chains could effectively dissipate energy under the large deformation, supporting the excellent mechanical properties such as the extensibility and toughness. Note that, the energy dissipation was not reversible. Under repeated loading–unloading cycles, the first cycle exhibited a pronounced hysteresis loop. However, the hysteresis became negligible in the next four cycle (Fig. 3d and S4†), probably resulting from the irreparable damage of F127DA micelles within the network.
In addition to the highly stretchable property, M-hydrogels exhibited excellent puncture resistant property, which was rarely reported in hydrogel field. This property could minimize the harm to hydrogels from sharp materials such as knife, needle, broken glass, etc.40,41 As shown in Fig. 4, we used a steel needle (diameter 1 mm) to puncture a sheet-shaped (size: 20 × 20 × 2 mm3) hydrogel which was stained by rhodamine B. Compared with origin thickness, the hydrogel was punctured as thin as possible. However, no damage appeared. Thus, the M-PAACA hydrogel showed a significant potential application as body armours.
 | ||
| Fig. 4 Photos show the puncture resistant property of M-hydrogels. A sheet sample can be stabbed without any damage. | ||
Once damaged, it was essential for M-hydrogels to rapidly self-repair. Then we investigated the self-healing behavior of hydrogels. As shown in Fig. 5a, two cylindrical hydrogels were cut in half, respectively. For better visualization, one of them was stained by rhodamine B. Then the fresh fracture surfaces were contacted end to end and the interface was immerged in diluted hydrochloric acid solution with a slight press for 20 minutes. The healed hydrogels could be stretched to a large deformation without breaking. The reason was that after treating the interface with hydrochloric acid, a small fraction of carboxyl groups on the surface was protonated and the hydrogen bonding formed between carboxyl groups in a face-on configuration (Fig. 5b). On the other hand, the side chains of hydrogels were long and flexible, and they possessed an optimal balance of hydrophobic and hydrophilic interactions which allowed the larger fraction of protonated carboxyl groups to cross one of interfaces and reach another interface, and finally interact with amide groups in an interleaved configuration (Fig. 5b).31 Those two kinds of strong hydrogen bonds as bridges could connect the interface to induce the hydrogels to self-heal. The FT-IR spectroscopy was used to confirm the hydrogen bonding. As shown in Fig. 5c, in contrast to the unhealed hydrogel, a prominent band at 1712 cm−1 in healed hydrogel FT-IR spectra refers to the hydrogen-bonded terminal carboxylic acid group. Note that the band at 1631 cm−1 was observed, which was attributed to amide I mode, indicating that the strong hydrogen bonds formed between carboxyl groups and amide of the opposing pendant side chain. It was noteworthy that putting the healed hydrogel into sodium hydroxide solution would separate the healed hydrogel again, indicating that the self-healing process was reversible.
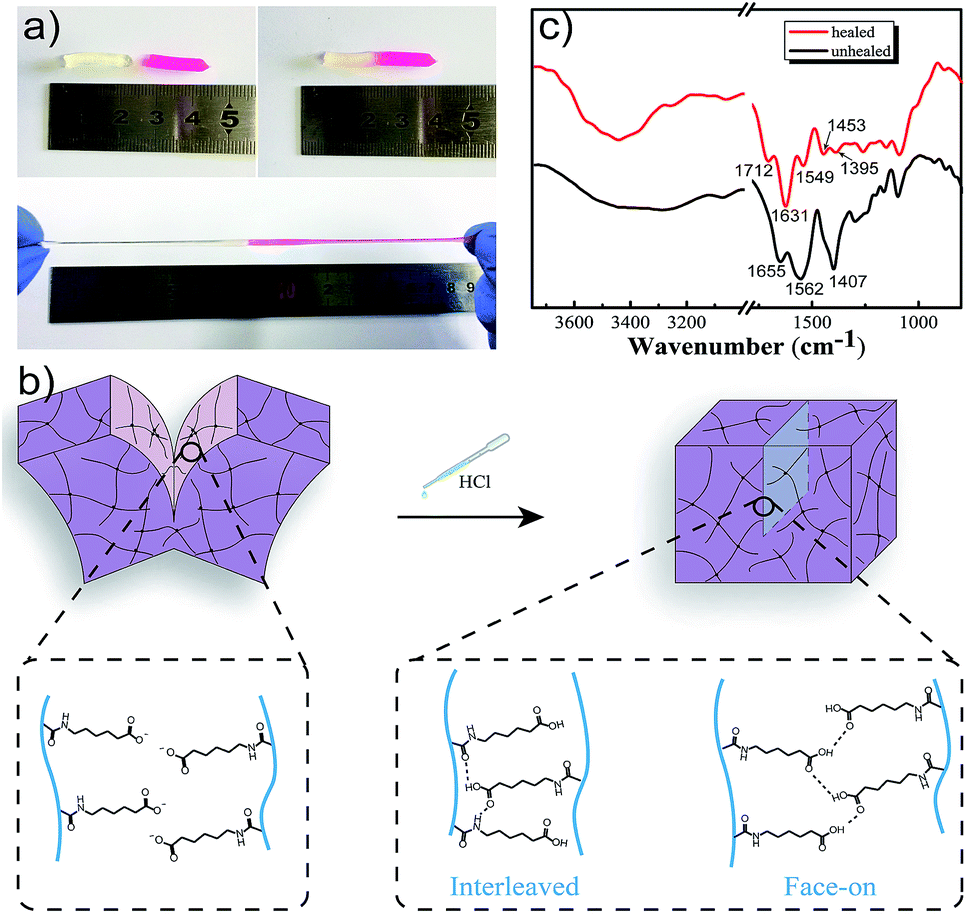 | ||
| Fig. 5 (a) Photos of the self-healing behavior of M-hydrogels: after treating hydrochloric acid into the interface of two pieces of hydrogels (one of them is stained with rhodamine B), the hydrogel is healed for 20 min and can be stretched to a large deformation. (b) Mechanism of the self-healing process in M-PAACA hydrogels. (c) FTIR spectrometry of healed and unhealed hydrogels. | ||
To further quantify self-healing efficiency, which was defined as the tensile strength ratio between healed gel and original gel, tensile tests were performed. Herein, the healed hydrogel prepared by merging two contact cylindrical hydrogel samples into acidic solution with pH = 3 for 2 h. The self-healing efficiency of M-hydrogels was able to reach 60%, as calculated from tensile stress–strain curves in Fig. S5.†
As the result of pH responsiveness of AACA, the M-hydrogels showed obvious swelling-deswelling dynamic behavior. Fig. 6a depicted the swelling ratios of M-hydrogels in solutions with different pH values. It was found that the swelling ratios were apparently different between pH < 4 and pH > 6. When pH value increased from 3 to 7, the swelling ratio increased sharply from 5 to 63. This phenomenon was attributed to that carboxyl groups of PAACA side chains were protonated and became hydrophobic when the pH value was under the ionization equilibrium constant (pKa = 4.4) of 6-aminocaproic acid. Meanwhile, hydrogen bonds formed between protonated AACA groups would cross-link polymer chains, further resulting in the decrease of the swelling ratio. When the pH value was higher than the pKa, the carboxyl groups became ionized. Due to the breakage of hydrogen bonds and the electrostatic repulsion between deprotonated (COO−) groups, the swelling ratio of hydrogel got a sharp increase.
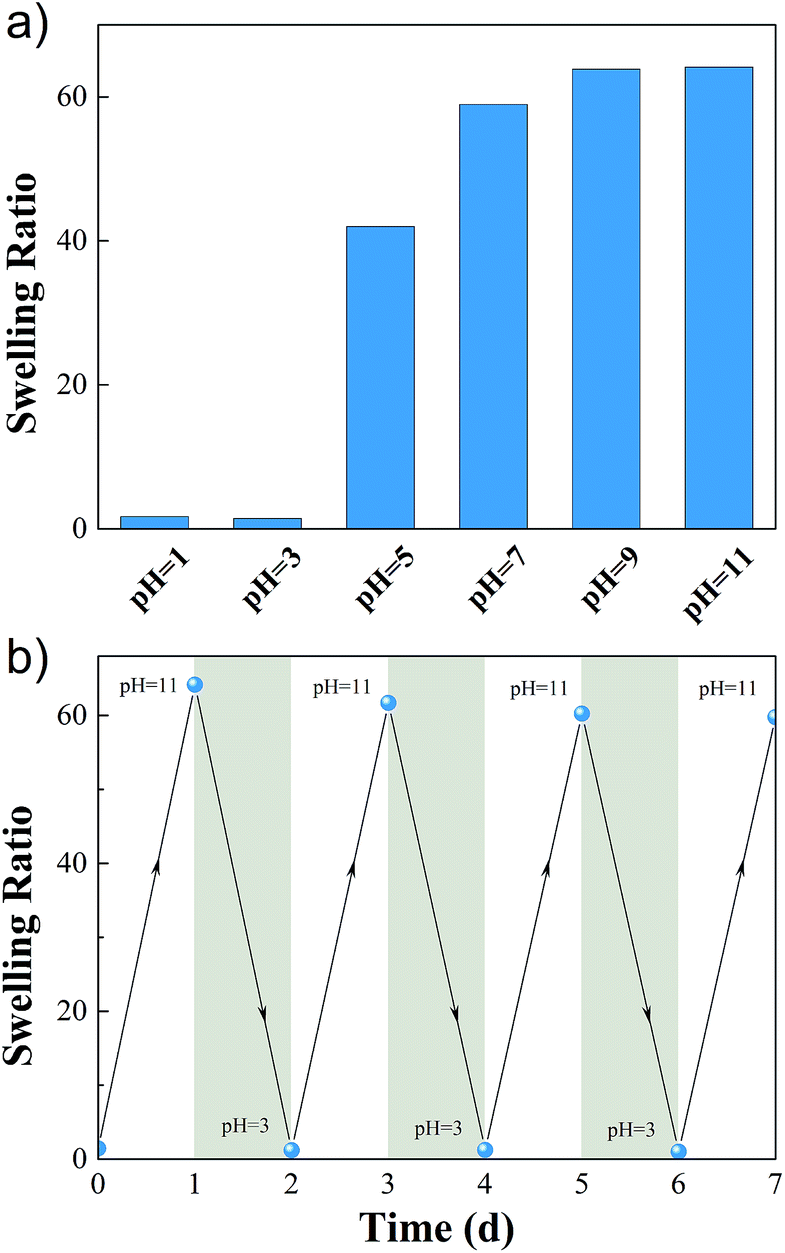 | ||
| Fig. 6 (a) Swelling ratio as a function of pH for M-hydrogels. (b) Cyclic swelling–deswelling behavior of M-hydrogels at pH = 3 and pH = 11. | ||
In addition, the swelling–deswelling kinetics of hydrogels were presented in Fig. S6.† The equilibrium swelling ratio of the hydrogel was achieved after 24 hours in basic solution, and then it was immersed in acidic solution at pH 3, it started to shrink, and the water retention reached a balance in 24 hours. The swelling–deswelling behavior could be recycled several times by changing the pH of solution (Fig. 6b), indicating that the hydrogels exhibited good reproducibility and the cyclic swelling and deswelling process did not destroy the structure of the hydrogels.
Conclusions
In summary, we have developed a kind of highly stretchable, puncture resistant and self-healing hydrogel via in situ copolymerization AACA with F127DA which serves as a multifunctional cross-linker. The homogenous network and effective energy dissipation mechanism allow the M-hydrogel to be stretched to more than 2000% and punctured by a steel needle without damage. Moreover, the flexible side PAACA chains of M-hydrogel endow the M-hydrogel with the self-healing ability. And the M-hydrogels show pH-responsive cyclic swelling–deswelling behavior because of the pH sensitivity of AACA. We believe that the present hydrogels with both excellent mechanical properties and self-healing ability could open a new pathway with regard to the design and practical applications of hydrogel systems.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant no. 51273189), the National Science and Technology Major Project of the Ministry of Science and Technology of China (2016ZX05016), and the National Science and Technology Major Project of the Ministry of Science and Technology of China (2016ZX05046).Notes and references
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS PubMed.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- Y. S. Kim, M. Liu, Y. Ishida, Y. Ebina, M. Osada, T. Sasaki, T. Hikima, M. Takata and T. Aida, Nat. Mater., 2015, 14, 1002–1007 CrossRef CAS PubMed.
- J. P. Gong, Soft Matter, 2010, 6, 2583–2590 RSC.
- X. Zhao, Soft Matter, 2014, 10, 672–687 RSC.
- Y. Tanaka, J. P. Gong and Y. Osada, Prog. Polym. Sci., 2005, 30, 1–9 CrossRef CAS.
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120–1124 CrossRef CAS.
- R. Liu, S. Liang, X.-Z. Tang, D. Yan, X. Li and Z.-Z. Yu, J. Mater. Chem., 2012, 22, 14160–14167 RSC.
- A. Bin Imran, K. Esaki, H. Gotoh, T. Seki, K. Ito, Y. Sakai and Y. Takeoka, Nat. Commun., 2014, 5, 5124 CrossRef PubMed.
- T. Sakai, T. Matsunaga, Y. Yamamoto, C. Ito, R. Yoshida, S. Suzuki, N. Sasaki, M. Shibayama and U.-i. Chung, Macromolecules, 2008, 41, 5379–5384 CrossRef CAS.
- X. Hu, M. Vatankhah-Varnoosfaderani, J. Zhou, Q. Li and S. S. Sheiko, Adv. Mater., 2015, 27, 6899–6905 CrossRef CAS PubMed.
- K. Peng, H. Yu, H. Yang, X. Hao, A. Yasin and X. Zhang, Soft Matter, 2017, 13, 2135–2140 RSC.
- D. C. Tuncaboylu, M. Sari, W. Oppermann and O. Okay, Macromolecules, 2011, 44, 4997–5005 CrossRef CAS.
- T. Huang, H. Xu, K. Jiao, L. Zhu, H. R. Brown and H. Wang, Adv. Mater., 2007, 19, 1622–1626 CrossRef CAS.
- J. Zhao, K. Jiao, J. Yang, C. He and H. Wang, Polymer, 2013, 54, 1596–1602 CrossRef CAS.
- F. Jiang, T. Huang, C. He, H. R. Brown and H. Wang, J. Phys. Chem. B, 2013, 117, 13679–13687 CrossRef CAS PubMed.
- J. Yang, X.-P. Wang and X.-M. Xie, Soft Matter, 2012, 8, 1058–1063 RSC.
- J. Yang, C.-R. Han, J.-F. Duan, F. Xu and R.-C. Sun, Nanoscale, 2013, 5, 10858–10863 RSC.
- M. Tan, T. Zhao, H. Huang and M. Guo, Polym. Chem., 2013, 4, 5570–5576 RSC.
- T. Zhao, M. Tan, Y. Cui, C. Deng, H. Huang and M. Guo, Polym. Chem., 2014, 5, 4965–4973 RSC.
- S. Burattini, B. W. Greenland, D. Chappell, H. M. Colquhoun and W. Hayes, Chem. Soc. Rev., 2010, 39, 1973–1985 RSC.
- Z. Wei, J. H. Yang, J. Zhou, F. Xu, M. Zrinyi, P. H. Dussault, Y. Osada and Y. M. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed.
- Z. Tao, K. Peng, Y. Fan, Y. Liu and H. Yang, Polym. Chem., 2016, 7, 1405–1412 RSC.
- A. Harada, R. Kobayashi, Y. Takashima, A. Hashidzume and H. Yamaguchi, Nat. Chem., 2011, 3, 34–37 CrossRef CAS PubMed.
- D. C. Tuncaboylu, M. Sahin, A. Argun, W. Oppermann and O. Okay, Macromolecules, 2012, 45, 1991–2000 CrossRef CAS.
- X. Hao, H. Liu, Z. Lu, Y. Xie and H. Yang, J. Mater. Chem. A, 2013, 1, 6920–6927 CAS.
- A. Phadke, C. Zhang, B. Arman, C.-C. Hsu, R. A. Mashelkar, A. K. Lele, M. J. Tauber, G. Arya and S. Varghese, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4383–4388 CrossRef CAS PubMed.
- J. Cui and A. d. Campo, Chem. Commun., 2012, 48, 9302–9304 RSC.
- H. Zhang, H. Xia and Y. Zhao, ACS Macro Lett., 2012, 1, 1233–1236 CrossRef CAS.
- Y.-n. Sun, G.-r. Gao, G.-l. Du, Y.-j. Cheng and J. Fu, ACS Macro Lett., 2014, 3, 496–500 CrossRef CAS.
- Y. Sun, S. Liu, G. Du, G. Gao and J. Fu, Chem. Commun., 2015, 51, 8512–8515 RSC.
- R. Ayala, C. Zhang, D. Yang, Y. Hwang, A. Aung, S. S. Shroff, F. T. Arce, R. Lal, G. Arya and S. Varghese, Biomaterials, 2011, 32, 3700–3711 CrossRef CAS PubMed.
- S.-Y. Lee and G. Tae, J. Controlled Release, 2007, 119, 313–319 CrossRef CAS PubMed.
- C.-J. Wu, A. K. Gaharwar, B. K. Chan and G. Schmidt, Macromolecules, 2011, 44, 8215–8224 CrossRef CAS.
- H. J. Kong, E. Wong and D. J. Mooney, Macromolecules, 2003, 36, 4582–4588 CrossRef CAS.
- T. J. Kang, K. H. Hong and M. R. Yoo, Fibers Polym., 2010, 11, 719–724 CrossRef CAS.
- L. L. Sun, D. S. Xiong and C. Y. Xu, J. Appl. Polym. Sci., 2013, 129, 1922–1928 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03137h |
| This journal is © The Royal Society of Chemistry 2017 |