
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tunable Ag@SiO2 core–shell nanocomposites for broad spectrum antibacterial applications†
Mark A. Isaacs a,
Lee J. Durndella,
Anthony C. Hiltonb,
Luca Olivic,
Christopher M. A. Parletta,
Karen Wilson*a and
Adam F. Lee*a
a,
Lee J. Durndella,
Anthony C. Hiltonb,
Luca Olivic,
Christopher M. A. Parletta,
Karen Wilson*a and
Adam F. Lee*a
aEuropean Bioenergy Research Institute, Aston University, Birmingham, B4 7ET, UK. E-mail: a.f.lee@aston.ac.uk; k.wilson@aston.ac.uk
bLife and Health Sciences, Aston University, Birmingham, B4 7ET, UK
cSincrotrone Trieste, Trieste, 34149 Basovizza, Italy
First published on 27th April 2017
Abstract
Silica encapsulated silver nanoparticle core–shell nanocomposites of tunable dimensions were synthesised via a one-pot reverse microemulsion route to achieve controlled release of Ag+ ions for broad spectrum antibacterial application. Silver release rates and bactericidal efficacy against Gram-positive and Gram-negative bacteria, S. aureus and P. aeruginosa respectively, were inversely proportional to nanoparticle core diameter (3–8 nm) and silica shell (7–14 nm) thickness, and readily tuned through a facile hydrothermal etching protocol employing a PVP stabiliser to introduce mesoporosity.
Introduction
Healthcare associated infections (HCAI's) are illnesses resulting from a patient's stay within a hospital environment, and a matter of global concern, affecting up to 25% of patients in European clinics and costing billions of dollars.1,2 Around 4 million people per year acquire a HCAI within Europe, accounting for approximately 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths.3 Staphylococcus aureus has attracted particular attention due to its rapid developed resistance to conventional antibiotics; 10% of this bacterium exhibits methicillin-resistance (MRSA) across 19 of the 29 European economic area (EEA) countries.4
000 deaths.3 Staphylococcus aureus has attracted particular attention due to its rapid developed resistance to conventional antibiotics; 10% of this bacterium exhibits methicillin-resistance (MRSA) across 19 of the 29 European economic area (EEA) countries.4
Silver has a long history5–7 as an effective, broad spectrum antimicrobial in ionic, complex and bulk presentations.8–10 In contemporary times, advances in nanotechnology have led to a focus on the application of silver nanoparticles in combating drug-resistant bacteria.11–14 Systematic studies of silver nanoparticles deposited onto carefully selected supports suggest that tuning the properties of silver containing nanocomposites can lead to systems with variable dissolution rates, and in turn, effective antimicrobial lifetimes and strengths.12,15–17
Core–shell nanocomposites were developed in the 1990s18,19 and have found application within catalysis,20 spectroscopy21 and antimicrobials.22 In the latter case, there has been significant interest in the development of nanocomposites comprising silver nanoparticle cores encapsulated by inert oxides as a means to improve the thermal stability, sinter resistance and release kinetics of antimicrobial, soluble silver ions.23–25 Silver@silica materials have proven popular due to the relative simplicity of silica syntheses and low cost of associated alkoxide precursor compared with other metal oxides, and ease of introducing porosity via sacrificial organic templates.26,27 Silver has also been encapsulated within titania shells, adding photocatalytic degradation of pathogens (bacteria, moulds, viruses) and cancer cells28 to the mode of antimicrobial action offered by ionic Ag. However, the relationship between nanocomposite dimensions and silver release rates, critical to determining both the efficacy and longevity of associated antimicrobial performance, has never been quantified.
Here, two families of Ag@SiO2 core–shell nanocomposites were prepared possessing independently varying core diameter or shell thickness, to address structure–function and bioactivity relations, and the impact of post-synthetic etching protocols.
Experimental
Nanocomposite synthesis
Briefly, silver nanoparticles were formed and encapsulated in a one-pot reverse microemulsion method adapted from Li et al.29 as described in Fig. S1.† A microemulsion solution was prepared from cyclohexane (Sigma Aldrich ≥99%), Igepal co-520 (Sigma Aldrich) and deionised water in an aluminium foil-covered 250 ml round bottomed flask with vigorous stirring (750 rpm) at room temperature under flowing N2. Silver nitrate solution (0.1 M, Sigma Aldrich) was added and the solution left to equilibrate for 20 min. Hydrazine (Sigma Aldrich 50–60% vol) was added to reduce the silver. After 5 min stirring, ammonium hydroxide (Sigma Aldrich 35%) was added, followed by tetraethyl orthoxysilicate (TEOS, Sigma Aldrich 98%). The solution was stirred at 750 rpm for 24 h under flowing nitrogen (1 ml min−1) and the resulting microemulsion subsequently diluted with ethanol (Fisher 98%) to give a 1:1 molar ratio of ethanol:cyclohexane, then centrifuged at 6000 rpm for 8 min to separate the particulate phase and the solution decanted. Particulates were re-dispersed in a 50:50 vol% water:ethanol mixture by ultrasonication and the resulting sol again separated by centrifugation and washed twice more with a water/ethanol mixture to obtain a solid powder which was finally calcined at 500 °C for 4 h employing a ramp rate of 5 °C min−1.
Nanocomposite characterisation
XRD analysis was performed on a Bruker D8 advance diffractometer using Cu Kα X-ray radiation (0.15418 nm), with a LynxEYE high speed strip detector and calibrated against a corundum standard. Volume averaged crystallite diameters are reported. Diffraction patterns were recorded between 2θ = 10–80° in 0.01° steps. N2 porosimetry was performed on a Quantachrome Nova 4000 porosimeter, on samples degassed under vacuum at 120 °C for 2 h. Mesopore analysis was performed applying the BJH method to the desorption branch of the isotherm, and the t-plot method applied to calculate microporosity. Data was analysed using NovaWin version 11. UV-Vis measurements were performed in water using a Thermo evolution 220 UV-Vis spectrophotometer.XPS analysis was recorded using a Kratos Axis HSi X-ray photoelectron spectrometer utilising Al Kα (1486.6 eV) and Mg Kα (1253.6 eV) radiation and a magnetic charge neutraliser, with inelastic mean free paths obtained from the LA Surface database30,31 to determine silica shell thickness non-destructively. Spectra were energy referenced to adventitious carbon at 284.6 eV and analysed using CasaXPS version 2.3.15 with quantification using appropriate instrumental response factors. Ag 3d XP spectra were fitted to a Doniach–Sunjic peakshape, with a spin–orbit split doublet separation of 6 eV. Silica shell thickness was estimated from the relative intensities of Ag 3d photoelectrons excited by either Al or Mg Kα X-rays.
Bulk elemental analysis was undertaken using ICP-MS performed by MEDAC Ltd. HRTEM analysis was performed using a JEOL 2100 microscope with a LaB6 source and 180 kV acceleration. Samples were prepared using a drop casting method in ethanol onto continuous carbon grids. Particle sizing was performed using ImageJ 1.46r software. Particle size distributions were produced from counts of a minimum of 100 particles. Dissolution profiles were developed by stirring 10 mg of Ag@SiO2 core–shell nanocomposites in 25 ml of 0.5 M NaNO3 with periodic aliquots of the analyte solution measured for silver content using an Agilent 6130B single Quad (ESI) ICP-MS, calibrated against a range of silver concentrations made by serial dilution of a 1000 ppm Ag in 1% HNO3 standard (Sigma-Aldrich). Silver release rates were calculated using the initial rates of the dissolution profiles over the linear portion of the profiles (typically the first 30 min of immersion).
Nanocomposite etching
Ag@SiO2 core–shell nanocomposites were hydrothermal etched by mixing 100 mg of Ag@SiO2 core–shell material with 250 mg PVP (Sigma Aldrich, Mr = 40000) in 25 ml H2O and heated at 95 °C for 3 h in a 50 ml round bottom flask. The resulting etched material was subsequently filtered, washed three times with ethanol then dried overnight at 100 °C.32
Ag@SiO2 core–shell nanocomposites were also base etched by mixing 500 mg of Ag@SiO2 core–shell material with 2 g of PVP (Sigma Aldrich, Mr = 40000) in 150 ml H2O and stirred overnight. To this slurry, 20 ml of 0.1 M NaOH was added under continuous stirring, and solid samples periodically removed, filtered, washed three times with water and three times with ethanol prior to drying overnight at 100 °C.33
Antibacterial testing
The antibacterial performance of Ag@SiO2 core–shell nanocomposites was evaluated against Staphylococcus aureus ATCC 6538, MRSA ATCC 33591, Escherichia coli NCTC 10418, Bacillus subtilis NCTC 8236, Pseudomonas aeruginosa ATCC 15442 and Clostridium difficile ATCC 9689 which are representative Gram-positive and Gram-negative problematic organisms found in hospital environments. Zone of inhibition (ZoI) tests were performed by inoculating the surface of a nutrient agar plate (Oxoid, Basingstoke UK) with an excess volume (3 ml) of nutrient broth which had previously been inoculated and incubated to a cell density of ∼108 cfu ml−1 as determined spectrophotometrically using a Perkin-Elmer lambda 10 UV-Vis spectrophotometer. The liquid was manipulated by agitation to provide a confluent inoculum and the excess fluid removed to waste using a sterile pipette. Using a sterilised boring tool, 5 mm holes were then bored into the agar, and 100 μl of a solution of 10 mg of Ag@SiO2 core–shell nanocomposite in 5 ml of simulated body fluid (SBF, see Table S1†) dispensed into the borehole using a calibrated micropipette. SBF was prepared according to a method from Kokubo et al.34 750 ml of deionised water was stabilised at 37 °C with stirring, to this the following ions were added: NaCl (7.996 g, Sigma Aldrich >99%), NaHCO3 (0.35 g, Sigma Aldrich >99%), KCl (0.224 g, Sigma Aldrich >99%), K2HPO4·3H2O (0.228 g, Sigma Aldrich >99%), MgCl2 (0.305 g, Sigma Aldrich >99%), HCl (40 ml, 1 kmol l−1, Fisher scientific 37%), CaCl2 (0.278 g, Sigma Aldrich >99%), Na2SO4 (0.071 g, Sigma Aldrich >99%) and (CH2OH)3CNH2 (6.057 g, Sigma Aldrich 99%). Finally, the pH was adjusted to 7.35 using HCl solution (1 kmol l−1, Fisher scientific 37%). Plates were then incubated at 37 °C overnight, photographed, and calibrated zone areas determined using ImageJ software.Quantitative antimicrobial activity was determined by logarithmic reduction.15,16 Here, 5 mg of Ag@SiO2 core–shell nanocomposite material was added to an Eppendorf tube containing 1 ml of either S. aureus or P. aeruginosa in a nutrient broth at concentrations of 107 cfu ml−1. 100 μl aliquots of the resulting suspensions were subsequently removed at 0, 60, 240 min and 24 hours, and added to a 1 ml solution of Tween 20 (supplier, 1%), sodium dodecyl sulphate (supplier 0.4%) and sodium chloride (0.85%) to neutralise any soluble silver species.15,16 Each of the resulting neutralised solutions was serially diluted with phosphate buffered saline (PBS) prior to plating onto agar and incubation at 37 °C for 24 h. The experiments were all run with positive and negative controls of silver nitrate and without any nanocomposite respectively. After incubation, the number of colonies present on the agar was counted by visual inspection, and normalised relative to the initial colony count in the negative control at time t = 0 min to determine the logarithmic reduction of bacteria. All experiments were performed in triplicate, with mean values and standard deviations reported.
Results and discussion
Successful synthesis of the desired core–shell architecture was confirmed in all cases through HRTEM as illustrated in Fig. 1. The silver core size was controlled through varying the [water]:[surfactant] molar ratio (Wo); the diameter of the micelles and resulting silver nanoparticles was directly proportional to the water content (Fig. S2–S4†). The minimum core size of 3.5 ± 0.5 nm thus obtained is the smallest reported for such a core–shell composites. Control over the encapsulating silica shell thickness was achieved through varying the [water]:[TEOS] molar ratio (H); the silica shell thickness was directly proportional to the TEOS concentration (Fig. S5–S7†).
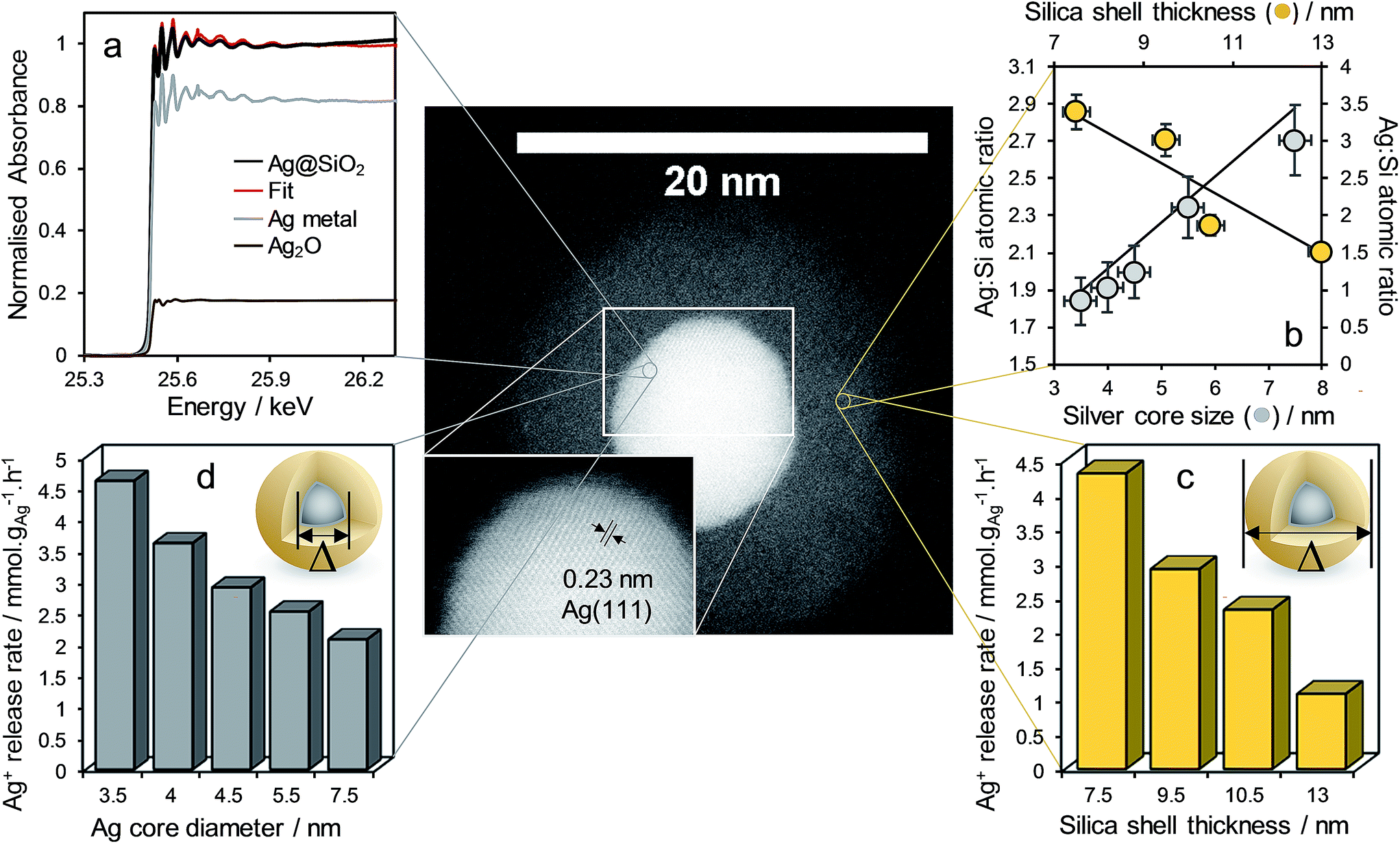 | ||
| Fig. 1 HR-STEM micrograph of representative Ag@SiO2 core–shell nanocomposite with silver lattice fringes shown inset (mean Ag core = 7.5 ± 0.5 nm, mean SiO2 shell thickness = 9.5 ± 1.2 nm, and clockwise from top left). (a) Fitted Ag K-edge XANES spectra of corresponding nanocomposite, and (b) Ag 3d:Si 2p XP surface atomic ratio and Ag+ release rate from nanocomposite as a function of (c) silica shell thickness (common mean Ag core diameter = 4.5 ± 0.4 nm) and (d) silver core diameter (common mean SiO2 shell = 9.5 ± 1.2 nm thick). | ||
Fig. 1 demonstrates complete encapsulation of, and the sharp interface between, silver core and silica shell for a Ag@SiO2 nanocomposite possessing a 7.5 ± 0.5 nm core and 9.5 ± 1.2 nm thick shell by HRTEM. UV-Vis spectroscopy revealed a characteristic feature at ∼410 nm arising from the surface plasmon resonance associated with silver nanoparticles (Fig. S8†). Powder XRD confirmed fcc silver metal (Fig. S9,† JCPDS no. 04-0783) as the only crystalline phase within the composite, in accordance with the observation of Ag(111) lattice fringes in Fig. 1. Corresponding volume averaged core diameters shown in Fig. 1, estimated from silver reflections using the Scherrer equation, were in good agreement with those from TEM (Fig. S9†). In contrast, Ag 3d XP spectra of all nanocomposites were dominated by a single spin–orbit doublet with 3d5/2 binding energy of 367.9 eV (Fig. S11†), consistent with Ag+ present in Ag2O, which exhibits an anomalous low binding energy shift with respect to silver metal.35 XPS was also employed to estimate the average silica shell thickness through comparing the Ag 3d intensity obtained under excitation by two different X-ray wavelengths (resulting in different photoelectron escape depths) as previously employed for Pt/PtOx36 and Pd/PdOx35,37 core–shell nanoparticles. The resulting shell thicknesses determined by XPS and reported in Fig. 1 were in good quantitative agreement with local HRTEM measurements (Fig. S12†). In order to understand the apparent discrepancy between the observation of predominantly ionic silver by surface sensitive XPS, and of metallic silver by XRD and HRTEM, Ag K-edge XANES were subsequently recorded to probe the chemical state of all silver species within a representative nanocomposite possessing a 4.5 ± 0.4 nm diameter silver core and 9.5 ± 1.2 nm thick SiO2 shell (Fig. 1 and S13†). Linear combination fitting of the XANES to silver metal, Ag2O and AgO standards revealed that while the majority of silver in the nanocomposite was present as the metal, approximately 20% was present as Ag+, in excellent quantitative agreement with the proportion predicted for a single layer of electronically perturbed silver atoms in the silver core at the interface with the silica shell (as described in the ESI†). This ionic silver likely serves as the initial reservoir for dissolved silver species upon immersion in aqueous media.
The impact of Ag@SiO2 nanocomposite structure on the kinetics of silver dissolution, expected to strongly influence their resulting antimicrobial performance, was subsequently investigated. Initial Ag+ release rates determined from the dissolution profiles in aqueous 0.5 M NaNO3 (Fig. S14†) are shown normalised to the mass of silver within each parent nanocomposite in Fig. 1. Previous studies on citrate and thiol functionalized methoxyl polyethylene glycol stabilised Ag nanoparticles report that the rate of oxidative silver dissolution is inversely proportional to particle diameter.27,38 These Ag@SiO2 core–shell nanocomposites exhibit a similar inverse relationship between silver core size and release rate, confirming that the geometric surface area of the core was rate-determining for Ag+ dissolution, with a rate constant of 14.664 μmol−1 h nmcore−1 gAg−1 (Fig. S15†). In contrast, the rate of Ag+ release exhibited an inverse linear dependence on silica shell thickness (Fig. S16†), suggesting a common diffusivity (and hence shell porosity) independent of shell thickness, such that dissolution is simply a function of the path length from the core to the shell exterior with a rate constant of 0.58 μmol−1 h nmshell−1 gAg−1. Extrapolating to a bare 4.5 ± 0.4 nm diameter silver core yields a predicted maximum release rate of 8.55 μmol h−1 gAg−1, almost identical to that of 8.9 μmol h−1 gAg−1 estimated from the literature for the same diameter (stabilised) silver nanoparticles.27,38
In light of the independence of Ag+ diffusivity on silica shell thickness, the 4.5 ± 0.4 nm diameter silver core and 9.5 ± 1.2 nm thick SiO2 shell nanocomposite was subsequently subjected to either hydrothermal32 or base etching pre-treatments33 (Fig. 2) in order to modify shell porosity and hence further control release kinetics. Neither pre-treatment influenced the silver core or silica shell dimensions (Fig. S16†), or the predominantly metallic nature of the crystalline core (Fig. S17†). However, base etching completely removed silver cores from a significant fraction of nanocomposite particles (up to 14%), proportional to the etching time (Fig. S18†); hydrothermal etching proved milder, removing only 3% of silver cores. The loss of silver from the nanocomposite through base etching can be rationalised in terms of the concomitant dramatic rise in surface area and pore diameter (Fig. S19†), accompanied by an apparent inhomogeneity in the appearance of silica shells (Fig. S16†), indicative of an open micro/mesoporous shell through which silver dissolution is facile. Hydrothermal etching had minimal impact on surface area, but increased the pore volume of the nanocomposite 10-fold (Fig. S19†).
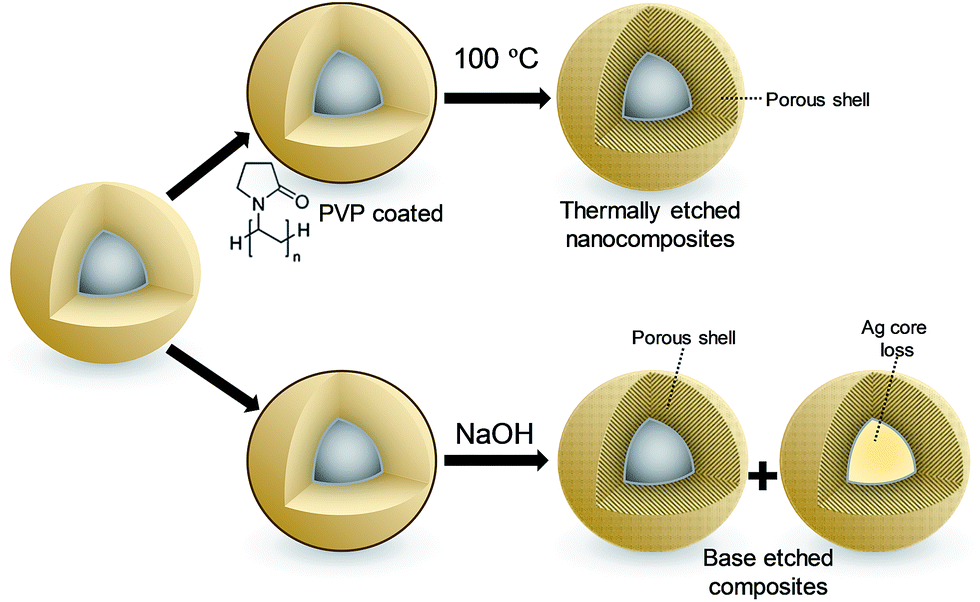 | ||
| Fig. 2 Schematic of post-synthetic treatment of Ag@SiO2 nanocomposites: (top) hydrothermal etching, (bottom) base etching. | ||
The loss of silver cores accompanying base etching significantly reduced the subsequent availability of soluble silver (Fig. S14 and S20†), an effect proportional to etching time, whereas hydrothermal pre-treatment enhanced total silver release by almost 50% after 24 h due to the improved mesoporosity.
Ag@SiO2 nanocomposites were subsequently quantitatively assayed against Staphylococcus aureus ATCC 6538 and Pseudomonas aeruginosa ATCC 15442, Gram-positive and Gram-negative bacteria respectively which are major pathogens in burns and dermal wounds,39–41 by the logarithmic reduction method in which one log reduction unit represents a 90% kill efficiency. All materials exhibited essentially complete bacterial death for both strains after 24 h incubation, hence performance was compared after 1 (S. aureus) and 4 h (P. aeruginosa) incubation. The longer assay time for the Gram-negative P. aeruginosa reflects the lower permeability of silver ions through the cell wall and membrane of this strain,42 increasing its antimicrobial resistance.26 Fig. 3, shows the impact of varying core diameter and shell thickness on the resulting log10 reductions, normalised to the mass of silver in each nano composite. Kill tests mirrored the silver release rates in Fig. 1, with the smallest cores enhancing bactericidal performance between 7- and 10-fold relative to the largest, and the thinnest shells offering between a 4- and 29-fold enhancement. Additional control over the efficacy of these Ag@SiO2 nanocomposites was achievable through etching of the silica shell. As anticipated from their Ag+ release behaviour (Fig. S20†), hydrothermal etching increased the antimicrobial performance 4-fold for S. aureus and 5-fold for P. aeruginosa, whereas base etching strongly suppressed this, by as much as 20-fold for P. aeruginosa (a reflection of the lower silver content following the latter aggressive pre-treatment).
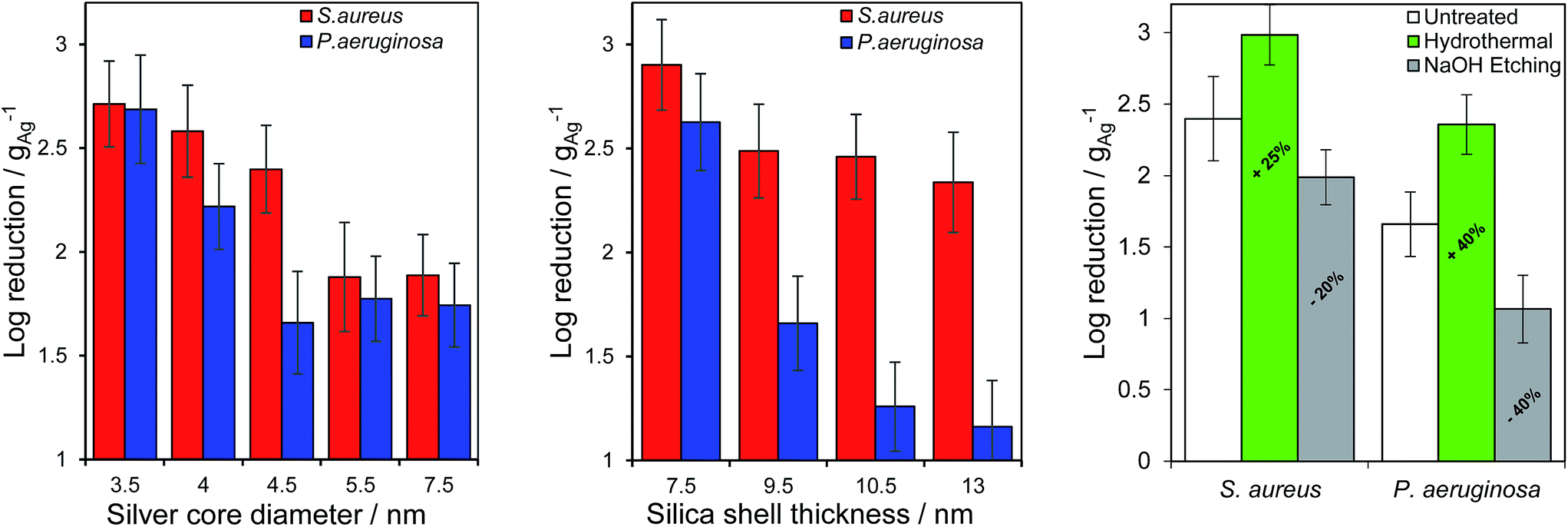 | ||
| Fig. 3 Mass-normalised bacterial kill tests as a function of (left) silver core diameter (common mean SiO2 shell thickness 9.5 ± 1.2 nm), (middle) SiO2 shell thickness (common mean silver core diameter = 4.5 ± 0.4 nm) against S. aureus ATCC 6538 and P. aeruginosa ATCC 15442, and (right) hydrothermal or base etching pretreatment (common mean silver core diameter = 4.5 ± 0.4 nm and SiO2 shell thickness = 9.5 ± 1.2 nm). Data after 1 and 4 h incubation of S. aureus ATCC 6538 and P. aeruginosa ATCC 15442 respectively. | ||
Broad spectrum antimicrobial efficacy of the 4.5 ± 0.4 nm diameter silver core and 9.5 ± 1.2 nm thick SiO2 shell nanocomposite was finally examined through semi-quantitative Zone of Inhibition experiments. Fig. 4 demonstrates that the Ag@SiO2 nanocomposite was potent against a variety of Gram-positive (S. aureus, MRSA, C. difficile, B. subtilis) and Gram-negative (P. aeruginosa, E. coli) bacteria in SBF.34
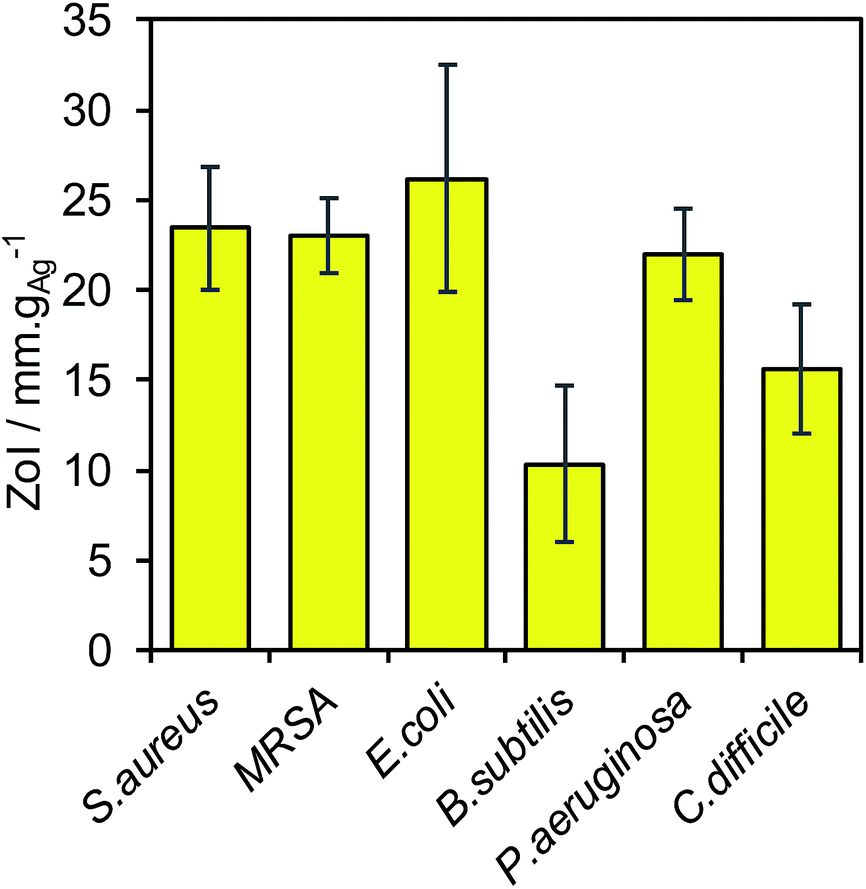 | ||
| Fig. 4 ZoI assays of a Ag@SiO2 nanocomposite (mean silver core 4.5 ± 0.4 nm and SiO2 shell thickness = 10.5 ± 1.2 nm) against Staphylococcus aureus ATCC 6538, MRSA ATCC 33591, Escherichia coli NCTC 10418, Bacillus subtilis NCTC 8236, Pseudomonas aeruginosa ATCC 15442 and Clostridium difficile ATCC 9689. | ||
The toxicity of our nanocomposites towards eukaryotic cells will be the subject of future studies. However, we note that at sizes comparable to those of the nanocomposites in this work (7 nm), silver nanoparticles are deemed non-toxic towards human skin cells at concentrations <6.25 μg ml−1, above which oxidative stress occurs through the generation of reactive oxygen species.43 Silica encapsulation in the present nanocomposites slows the release of soluble silver species by over an order of magnitude relative to naked silver nanoparticles, and hence is extremely unlikely to prove damaging in treating skin cells in wound care applications.
Ionic silver exhibits dramatically increased toxicity towards mammalian cells in comparison to its nanoparticulate form,44,45 although in vivo protein binding of free Ag+ greatly suppresses its toxicity.45 Silica nanoparticle toxicity is highly cell dependant, with half maximal inhibitory concentrations (IC50) towards macrophages (for similar sized particles to our nanocomposites) of around 73 μg ml−1: surface modification by facile silicon-alkoxide-based methodologies can drastically improve biocompatibility and cell viability.46
Conclusions
Systematically related families of Ag@SiO2 core–shell nanocomposites were synthesised through a reverse microemulsion route which enabled independent control over the silver core and silica shell dimensions. Bulks and surface sensitive X-ray spectroscopies indicate that metallic silver cores terminate in a Ag2O-like phase at the sharply delineated interface with the silica shell, which likely provides a reservoir for soluble ionic silver. Mass normalised rates of silver dissolution from the nanocomposites are inversely proportional to core diameter, i.e. a simple function of the core surface area:volume ratio, and display an inverse linear dependence on shell thickness consistent with a fixed porosity. Post-synthetic etching improves permeability of the silica shell, however base treatments enhance the shell porosity to such an extent that entire silver cores are lost from the nanocomposite resulting in poor subsequent antibacterial performance. In contrast, hydrothermal etching appears sufficiently mild to increase the shell porosity to permit faster silver dissolution while retaining a protective barrier against total core loss. Ionic silver dissolution kinetics mirrored their corresponding quantitative antibacterial efficacy against Gram-positive S. aureus ATCC 6538 and Gram-negative P. aeruginosa ATCC 15442, suggesting a facile route to the future design of nanocomposites possessing predicable activities and lifetimes. Future investigations will also explore the potential fungicidal properties of these Ag@SiO2 nanocomposites.
Acknowledgements
We would like to acknowledge the Knowledge Economic Skills Scholarship for their funding, along with Polymer Health Technology, Ebbw Vale, Wales for their contribution. We would also like to thank Dr Preena Mistry (Life and Health sciences, Aston University) for her guidance performing antimicrobial assays.Notes and references
- K. W. Lobdell, S. Stamou and J. A. Sanchez, Surg. Clin. North Am., 2012, 92, 65–77 CrossRef PubMed.
- V. D. Rosenthal and D. P. Phuong Anh, Am. J. Infect. Control, 2014, 42, 942–956 CrossRef PubMed.
- Healthcare associated infections (HAI), point prevalence survey, England, Public Health England, http://https://www.gov.uk/government/publications/healthcare-associated-infections-hcai-point-prevalence-survey-england, last updated 1/8/16.
- N. S. Leyland, J. Podporska-Carroll, J. Browne, S. J. Hinder, B. Quilty and S. C. Pillai, Sci. Rep., 2016, 6, 24770 CrossRef CAS PubMed.
- J. Duffy, The sanitarians: a history of American public health, University of Illinois Press, 1992 Search PubMed.
- J. Gibbard, Am. J. Public Health Nations Health, 1937, 27, 112–119 CrossRef CAS PubMed.
- W. Hill and D. Pillsbury, Argyria, the pharmacology of silver, Baltimore, 1939 Search PubMed.
- F. Almalioti, J. MacDougall, S. Hughes, M. M. Hasson, R. L. Jenkins, B. D. Ward, G. J. Tizzard, S. J. Coles, D. W. Williams and S. Bamford, Dalton Trans., 2013, 42, 12370–12380 RSC.
- H. H. Lara, E. N. Garza-Treviño, L. Ixtepan-Turrent and D. K. Singh, J. Nanobiotechnol., 2011, 9, 1 CrossRef PubMed.
- M. Rai, K. Kon, A. Ingle, N. Duran, S. Galdiero and M. Galdiero, Appl. Microbiol. Biotechnol., 2014, 98, 1951–1961 CrossRef CAS PubMed.
- J. S. Kim, E. Kuk, K. N. Yu, J.-H. Kim, S. J. Park, H. J. Lee, S. H. Kim, Y. K. Park, Y. H. Park and C.-Y. Hwang, Nanomed. Nanotech. Biol. Med., 2007, 3, 95–101 CrossRef CAS PubMed.
- S. Pal, Y. K. Tak and J. M. Song, Appl. Environ. Microbiol., 2007, 73, 1712–1720 CrossRef CAS PubMed.
- I. Sondi and B. Salopek-Sondi, J. Colloid Interface Sci., 2004, 275, 177–182 CrossRef CAS PubMed.
- V. K. Sharma, R. A. Yngard and Y. Lin, Adv. Colloid Interface Sci., 2009, 145, 83–96 CrossRef CAS PubMed.
- J. J. Buckley, P. L. Gai, A. F. Lee, L. Olivi and K. Wilson, Chem. Commun., 2008, 4013–4015 RSC.
- J. J. Buckley, A. F. Lee, L. Olivi and K. Wilson, J. Mater. Chem., 2010, 20, 8056–8063 RSC.
- G. Martinez-Castanon, N. Nino-Martinez, F. Martinez-Gutierrez, J. Martinez-Mendoza and F. Ruiz, J. Nanopart. Res., 2008, 10, 1343–1348 CrossRef CAS.
- C. F. Hoener, K. A. Allan, A. J. Bard, A. Campion, M. A. Fox, T. E. Mallouk, S. E. Webber and J. M. White, J. Phys. Chem., 1992, 96, 3812–3817 CrossRef CAS.
- I. Honma, T. Sano and H. Komiyama, J. Phys. Chem., 1993, 97, 6692–6695 CrossRef CAS.
- A. F. Lee, C. V. Ellis, K. Wilson and N. S. Hondow, Catal. Today, 2010, 157, 243–249 CrossRef CAS.
- G. A. Attard, J. Bennett, I. Mikheenko, P. Jenkins, S. Guan, L. Macaskie, J. Wood and A. Wain, Faraday Discuss., 2013, 162, 57–75 RSC.
- W. J. Chen, P. J. Tsai and Y. C. Chen, Small, 2008, 4, 485–491 CrossRef CAS PubMed.
- O. Niitsoo and A. Couzis, J. Colloid Interface Sci., 2011, 354, 887–890 CrossRef CAS PubMed.
- P. Saint-Cricq, J. Wang, A. Sugawara-Narutaki, A. Shimojima and T. Okubo, J. Mater. Chem. B, 2013, 1, 2451–2454 RSC.
- K. Xu, J.-X. Wang, X.-L. Kang and J.-F. Chen, Mater. Lett., 2009, 63, 31–33 CrossRef CAS.
- M. J. Brady, C. M. Lisay, A. V. Yurkovetskiy and S. P. Sawan, Am. J. Infect. Control, 2003, 31, 208–214 CrossRef PubMed.
- W. Zhang, Y. Yao, N. Sullivan and Y. Chen, Environ. Sci. Technol., 2011, 45, 4422–4428 CrossRef CAS PubMed.
- B. Cui, H. Peng, H. Xia, X. Guo and H. Guo, Sep. Purif. Technol., 2013, 103, 251–257 CrossRef CAS.
- T. Li, J. Moon, A. A. Morrone, J. J. Mecholsky, D. R. Talham and J. H. Adair, Langmuir, 1999, 15, 4328–4334 CrossRef CAS.
- M. P. Seah and W. Dench, Surf. Interface Anal., 1979, 1, 2–11 CrossRef CAS.
- P. J. Cumpson and M. P. Seah, Surf. Interface Anal., 1997, 25, 430–446 CrossRef CAS.
- Y. Hu, Q. Zhang, J. Goebl, T. Zhang and Y. Yin, Phys. Chem. Chem. Phys., 2010, 12, 11836–11842 RSC.
- Q. Zhang, T. Zhang, J. Ge and Y. Yin, Nano Lett., 2008, 8, 2867–2871 CrossRef CAS PubMed.
- T. Kokubo, H. Kushitani, S. Sakka, T. Kitsugi and T. Yamamuro, J. Biomed. Mater. Res., 1990, 24, 721–734 CrossRef CAS PubMed.
- C. M. Parlett, D. W. Bruce, N. S. Hondow, A. F. Lee and K. Wilson, ACS Catal., 2011, 1, 636–640 CrossRef CAS.
- A. F. Lee, K. Wilson, R. M. Lambert, C. P. Hubbard, R. G. Hurley, R. W. McCabe and H. S. Gandhi, J. Catal., 1999, 184, 491–498 CrossRef CAS.
- C. Parlett, D. W. Bruce, N. S. Hondow, M. A. Newton, A. F. Lee and K. Wilson, ChemCatChem, 2013, 5, 939–950 CrossRef CAS.
- T. S. Peretyazhko, Q. Zhang and V. L. Colvin, Environ. Sci. Technol., 2014, 48, 11954–11961 CrossRef CAS PubMed.
- A. Lansdown, A. Williams, S. Chandler and S. Benfield, J. Wound Care, 2005, 14, 155–160 CrossRef CAS PubMed.
- M. Fazli, T. Bjarnsholt, K. Kirketerp-Møller, B. Jørgensen, A. S. Andersen, K. A. Krogfelt, M. Givskov and T. Tolker-Nielsen, J. Clin. Microbiol., 2009, 47, 4084–4089 CrossRef PubMed.
- G. A. James, E. Swogger, R. Wolcott, P. Secor, J. Sestrich, J. W. Costerton and P. S. Stewart, Wound Repair Regen., 2008, 16, 37–44 CrossRef PubMed.
- K. I. Batarseh, J. Antimicrob. Chemother., 2004, 54, 546–548 CrossRef CAS PubMed.
- S. Arora, J. Jain, J. M. Rajwade and K. M. Paknikar, Toxicol. Lett., 2008, 179, 93–100 CrossRef CAS PubMed.
- S. Kittler, C. Greulich, M. Köller and M. Epple, Materialwiss. Werkst., 2009, 40, 258–264 CrossRef CAS.
- S. Kittler, C. Greulich, J. S. Gebauer, J. Diendorf, L. Treuel, L. Ruiz, J. M. Gonzalez-Calbet, M. Vallet-Regi, R. Zellner, M. Koller and M. Epple, J. Mater. Chem., 2010, 20, 512–518 RSC.
- T. Yu, A. Malugin and H. Ghandehari, ACS Nano, 2011, 5, 5717–5728 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Physicochemical characterisation, modification and antimicrobial testing. See DOI: 10.1039/c7ra03131a |
| This journal is © The Royal Society of Chemistry 2017 |