
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInertial microfluidics combined with selective cell lysis for high throughput separation of nucleated cells from whole blood†
Harisha Ramachandraiah,
Helene A. Svahn and
Aman Russom*
and
Aman Russom*
Division of Proteomics and Nanobiotechnology, Science for Life Laboratory, KTH Royal Institute of Technology, Stockholm, Sweden. E-mail: aman.russom@scilifelab.se
First published on 6th June 2017
Abstract
The ability to rapidly analyze and extract information from peripheral blood cells has the potential of providing a wealth of new information about immune function and general health of the patient. In spite of the tremendous progress achieved in the field of leukocyte analysis, one of the major impediments for routine analysis is the enrichment of cell populations from heterogeneous sources such as blood, as the currently used techniques tend to be laborious. Moreover, the isolation of small and transient cell populations in blood, like circulating tumor cells during cancer metastasis, is even more challenging. Here, we report an integrated device for label-free continuous flow separation of nucleated cells from unprocessed whole blood at high throughput. The method utilizes exposure to hypotonic buffer to completely remove red blood cells and at the same time a size increase of nucleated cells for inertial focusing and separation in spiral microchannel. Using an integrated device with two outlets, we isolated total leukocytes at a high yield of 99%. Furthermore cancer cells spiked into whole blood could be separated at a yield of 88% while 80% of leukocyte could be depleted into separate outlet by simply changing the resistance between the two outlets. Finally, using a three-outlet integrated device, we demonstrate fractionation of leukocyte into subpopulation. The device continuously separates granulocytes at a purity of 86%, monocyte at a purity of 43% and lymphocytes at a purity of 91% simultaneously. Finally, a cell activation study of the immune system using blood from healthy subjects, stimulated ex vivo with lipopolysaccharides (LPS), confirmed that the high operational flow rate of the device does not alter the activation levels of leukocytes or introduce artifacts. Hence, the simple, high-throughput and low-cost integrated device requiring neither external force fields nor mechanical parts to operate should readily be applicable to sort nucleated cells as stand-alone and/or as integrated lab-on-a-chip devices with high-throughput requirements.
1. Introduction
Leukocytes comprise only ∼1% of the total number of cells in blood and are important components of the immune system. Their population varies in the presence of infection, autoimmune reactions and malignancies.1 Therefore, enumeration of leukocytes subpopulations is one of the most commonly performed tests in clinical laboratories to diagnose and monitor many diseases.2 Moreover, important diagnostic and prognostic information can be gained from capturing cells transiently present in small numbers in blood, like circulating tumor cells (CTC), a subpopulation of tumor cells that spread and subsequently give rise to secondary tumors.Isolation of nucleated cells from blood without loss or activation is a challenging task. Red blood cells (RBCs) make up the majority of the cellular fraction and subsequent molecular and cellular analysis of leukocytes often requires depletion or removal of RBCs. The conventional cell separation techniques rely on size, density and differential expression of surface antigens to isolate desired cell populations, including density gradient centrifugation,3 preferential lysis of RBCs, Ficoll-Hypaque density gradient centrifugation,4 porous filters, and cell filtration.5 These macro scale methods are labor-intensive, non-standardized, and require large samples. For isolation of pure subpopulations, immunomagnetic (MACS) or immunofluorescent (FACS) separation methods based on antibodies against specific cell surface markers are routinely used for blood fractionation. However, there are several limitations with these methods, including low sample throughput, cell death, limited quantitation capability or high cost. In addition, antibody-based techniques can lead to activation of cells using certain antibodies, as well as the need for multiple markers for identification of certain cell types, or large quantities of depletion antibodies as in the case of RosetteSep (from StemCell Technology). Fueled by recent developments in microfluidics and microfabrication, a number of miniaturized systems have been developed with the aim of separating nucleated cells from whole blood. By controlling the microenvironment, microfluidics offers unique capabilities in processing multiple serial or parallel tasks and is promising for fundamental and applied immunological studies.6 Similar to macroscale methods, these microfluidic devices separate cells based on their physical, chemical and functional properties. Cell isolation methods can be roughly divided into active and passive systems. Active separation systems require external forces for separation. Dielectrophoretic field-flow-fractionation (DEP-FFF) isolates leukocytes through balancing between DEP and sedimentation forces.7 Magnetophoresis uses the different magnetic properties of blood cells for separation.8 Immunomagnetic cell separation has been used to sort leukocytes by selectively attaching magnetic beads to the cells.9 Shear-mediated cell adhesion within functionalized microfluidic channels has been used cells from whole blood and has been successfully applied in cancer diagnostics.10–15 Passive microfluidic systems achieve separation by filtering particles through sieving structures or by exploiting the differential interaction of particles with local flow profiles.16–18 These methods have proven to be simple because they do not require an external force field to separate particles based on size, shape, and deformability, but here also the relatively low throughput has restricted their adaptation into clinical settings. The highest yield of nucleated cells can be accomplished using a microfluidic lysis protocol subjecting blood to a controlled exposure to deionized water.19,20 This exposure preferentially causes rapid lysis of RBCs and has a minimal effect on other nucleated cells as long as the cells are returned to isotonic conditions within 10 seconds. Though this technique ensures the highest yield of nucleated cells with minimal activation, the need for subsequent off-chip centrifugation step to get rid of RBC debris limits the overall throughput and is a major source of cell loss and variations due to human factors. In this paper, we address these limitations by combining selective RBC lysis with size-based fractionation WBCs into total and subpopulation by employing inertial microfluidics.
Inertial microfluidics, a passive and size dependent separation technique, has gained considerable interest in recent years for cell separation.21,22 In inertial microfluidics, suspended particles can migrate across the streamline and focus at the equilibrium position as a result of a lift force (FL), which is a balance between a shear-gradient-induced force (FLS) that is directed toward the wall and a wall-induced force (FLW) directed away from a stationary wall pushing particles to an equilibrium position.23–27 The net lift force (FL) on particle is derived by combination of both and FLS and FLW: FL = ρU2a4/Dh2. fc (Re, Xc). Here, ρ is the density, U is the mean velocity and μ is dynamic viscosity of the fluid. Particle diameter is denoted by a, and Dh (=2wh/(w + h)) is the hydraulic diameter of microchannel, where “w” and “h” are the width and height of microchannel respectively. The coefficient of inertial lift force fc (Re, Xc) is dependent on position of particle in the microchannel (Xc) and Reynold's number (Re = ρUDh/μ), which defines the scale of these net forces within channel. In flows through curved channels, secondary flows (i.e. Dean flow) due to the curvature of the channel, alternate the equilibrium position by recirculating the velocity profile.28 The magnitude of the secondary flow is measured by Dean number, De (defined as De = Re(Dh/2r)1/2); where r is the radius of the curvature. The Dean flow results in a drag force (FD) that scales with De2 (from Stokes drag (FD ∼ ρUm2aDh2/r)). The Dean flow results in a drag force (FD) acting in the direction of vortex flow upon particles entrained in the vortices. The balance between dominant FL and FD dictates particle focusing in flow through curved channels. Unlike focusing in straight channels, which is typically limited to the channel cross-section in the range of the particle size to a create single focusing point,29,30 flow through curved channels enable particle focusing in a large cross-sectional area. We previously reported particle focusing using a channel aspect ratio of up to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10.31 Subsequently, most recent work dealing with blood processing, such as the separation of white blood cells32–38 and CTCs35,39–46 from blood, have used different spiral devices.47–50 For CTC isolation, the method exploits the fact that cancer cells are larger than most types of blood cells.42,51–54 Although the flow rates in inertial microfluidics are very high, typically in the mL min−1 range, an inherent limitation to processing large blood volumes is the breakdown of focusing behaviors at high volume fractions. This is due to the fact that particle–particle interactions compromise the focusing behavior, and typically require the solid content to be below 1% for single stream focusing. In blood, RBCs outnumber other nucleated cells by approximately 5000:1. This necessitates a significant dilution to process blood, which increases the processing time, or affects the purity of fractionated nucleated cells.
:10.31 Subsequently, most recent work dealing with blood processing, such as the separation of white blood cells32–38 and CTCs35,39–46 from blood, have used different spiral devices.47–50 For CTC isolation, the method exploits the fact that cancer cells are larger than most types of blood cells.42,51–54 Although the flow rates in inertial microfluidics are very high, typically in the mL min−1 range, an inherent limitation to processing large blood volumes is the breakdown of focusing behaviors at high volume fractions. This is due to the fact that particle–particle interactions compromise the focusing behavior, and typically require the solid content to be below 1% for single stream focusing. In blood, RBCs outnumber other nucleated cells by approximately 5000:1. This necessitates a significant dilution to process blood, which increases the processing time, or affects the purity of fractionated nucleated cells.
In this work, we combined inertial microfluidics with selective RBC lysis to separate nucleated cells from unprocessed whole blood, and report for the first time the fractionation of WBCs into sub-populations. We first analyzed the effect of solid content on yield using only a spiral device to focus and separate nucleated cells. Based on these results, we designed an integrated device that enables complete removal of RBCs via osmotic lysis and at the same time concentrates nucleated cells by size. An important and enabling feature of this protocol is the fact that the focusing occurs when blood is exposed to the hypotonic buffer, at which time nucleated cells increase in size enabling differential focusing from RBC debris. This device therefore accomplishes simultaneous RBC depletion, nucleated cell size amplification for focusing and separation and finally return to isotonic conditions for final delivery of the sample in concentrations suitable for subsequent analysis. We show three applications of the integrated device: separation of total leukocytes and the use of cell swelling as a novel parameter for enrichment of cancer cells and fractionation of leukocytes into subpopulations, all using unprocessed whole blood as input sample.
2. Material and methods
Microfluidic devices were fabricated in polydimethylsiloxane (PDMS) using standard soft lithography techniques.55 Briefly, channel replicas were produced using a negative photoresist SU-8 (MicroChem) onto the silicon wafer using standard microelectromechanical systems (MEMs) technology. The height of the SU-8 pattern on the master was measured using a surface profilometer. Microfluidics devices were produced by casting PDMS onto the patterned silicon wafers. The elastomeric PDMS (Dow Corning) was mixed with a cross-linker with a ratio of 10:1(wt/wt), and poured onto the master used as a mold, degassed and cured at 65 °C for 6 hours. The curved PDMS with replicated channels was peeled off from the silicon wafer and channel holes were punched with a Harris Uni-CoreTM, Tip ID 0.75 mm. The PDMS replica was bonded to a glass slide (70 mm × 30 mm) after brief oxygen plasma treatment. Access tubing (Tygon; Saint-Globain PPL corp) of slightly larger diameter than the access holes was press-fitted into the holes.
2.1 Particle and cancer cell line suspensions
Internally dyed green and red fluorescent polystyrene microspheres (Fluoro-Max™) with a diameter of 10 μm and 15 μm respectively, were purchased from Thermo Scientific. Bead suspensions were prepared by diluting to 0.5% vol with Milli-Q (MQ) water (Millipore) containing 0.1% Tween 20 (Merck, Germany). Bead mixtures were prepared by mixing beads in a ratio of 1:1 (v/v). Cancer cell lines A549, SH-SY5Y and MCF7 were cultured in Dulbecco's modified Eagle's medium (DMEM), containing 10% fetal bovine serum, and 0.1% non-essential amino acids (Sigma-Aldrich). Cells were cultured in a 5% CO2 humidified atmosphere at 37 °C. Cells were grown to 80–90% confluence before harvesting and media was changed every 2–3 days. Prior to sample introduction the devices were primed with 1× PBS. Cells and particle suspension were pumped through the device inlets by syringe pump (Harvard Apparatus PHD 2000), connected to the inlet by tubing. Two outlets (outlet 1 and outlet 2) were connected to the device outlet ports and collection tubes. The device was mounted onto the stage of an inverted fluorescent microscope (Zeiss Axiovert 135 TV). Images were captured with a ProgRes® CCD SpeedXT core 3 camera (Jenoptik) and analyzed using Icy bioimage analysis software.
2.2 Blood sampling and processing
Blood was drawn from anonymous healthy donors and collected in venous blood collection tubes containing EDTA (BD Vacutainer, BD Biosciences) at the Skanstull Blood Centre (Blodcentralen Skanstull, Stockholm, Sweden). All samples were processed on the day of sample collection. Blood samples were gently rocked back and forth before use. The experimental setup for whole blood experiments consists of the microfluidics devices and syringe pumps. For the spiral device whole blood was diluted with 1× PBS for control experiments, or with MQ water for 10 s to selectively lyse the RBCs. Lysis was terminated by adding an equal volume of 2× PBS in a ratio of 1:1. One syringe pump drove the blood through the inlet and the enriched leukocyte population was collected at outlet 1, while the leukocyte-depleted fraction was collected at outlet 2. A Coulter counter (Beckman Coulter) was used to quantify the blood components in the unprocessed and collected fractions. Briefly, for Coulter counter analysis, known volumes of sample were added to the Coulter Isoton II solution (Beckman Coulter) and 3 drops of Zapoglobin II lytic reagent (Beckman Coulter) were added only to the control samples to lyse the RBCs for white blood cells (WBC) count. For the integrated chip, MQ water was pumped through inlet 1 using a syringe pump (Harvard Apparatus PHD 2000), while whole blood was pumped through inlet 2 using another syringe pump (neMESYS, Cetoni GmbH, Germany). A third pump (neMESYS, Cetoni GmbH, Germany) drove 2× PBS through inlet 3 to terminate blood lysis and adjust the flow rate for inertial focusing. The ratio between whole blood and water was adjusted, while the ratio of lysed solution to 2× PBS was kept constant, to give the desired total concentration. For instance, 1 mL min−1 total flow rate for a 2.5% blood concentration consists of 25 μL whole blood: 475 μL water: 500 μL 2× PBS, for a 5% blood concentration of 50 μL whole blood: 450 μL water: 500 μL 2× PBS and for a 10% blood concentration of 100 μL whole blood: 400 μL water: 500 μL 2× PBS. The enriched leukocyte population was collected at outlet 1 and the leukocyte depleted fraction at outlet 2. For differential focusing, the channel resistance was varied by connecting the tubing from outlet 1 to a straight channel. For fractionation of leukocyte into sub-population, a three-outlet device was used. Effect of cell-specific expansion was studies for separation of leukocytes into sub-population. The Coulter counter was used to quantify the particles and blood components.
2.3 Spiking of cancer cells
Cancer cell lines A549, SH-SY5Y and MCF7 were purchased from the American Type Culture Collection (http://www.atcc.org). Cancer cell lines were harvested and re-suspended in cultured media. Cells were counted and viability assessed using a TC20 automated cell counter (Bio-Rad). About 100 μL of cancer cell suspension, typically containing 10000–30000 cells was spiked into 4 mL of whole blood diluted as indicated, after which the blood samples were gently rocked back and forth for 1 minute before use. A volume of 100 μL was removed as a control before loading the sample into the syringe.
2.4 Cell swelling
Trypsinized A549 cells were re-suspended in 1× PBS and allowed to sediment on a WillCo-dish Glass Bottom dish. 1× PBS was replaced with hypotonic buffer and real time images were taken and analyzed using Icy bio image analysis software.2.5 LPS stimulation ex vivo studies
LPS of 1 mg mL−1 was added to one tube of freshly drawn peripheral blood from healthy subjects and incubated for 2 hours at 37 °C. Another blood tube same subject was divided into three parts: one processed through integrated spiral device, the second through P. Setu chip and third kept unprocessed for control and the one processed through the device. Samples collected from the both the device and controls were stained for CD11b, CD18 and CD69 and analyzed using FACS.2.6 FACS analysis
For the control sample, 100 μL of whole blood spiked with cancer cells was lysed with 500 μL of MQ water, the lysis was terminated with 500 μL of 2× PBS and centrifuged at 1500 rpm for 5 min at 21 °C, the supernatant was discarded and the pellet was re-suspended in 100 μL of 1× PBS. For microfluidic experiments, samples were processed through the chip and the samples collected at the outlet were centrifuged and re-suspended in 1× PBS. For flow cytometry analysis, cells were incubated for 15 minutes with primary antibodies. Blood cells were stained with the Multicolor Human T cell marker panel (CD3-FITC, CD8-APC and CD45-PE/Cy7®) (Abcam) and cancer cells were stained with anti-EpCAM-PE monoclonal antibodies (Thermo Scientific). A Beckman Coulter flow cytometer was used for FACS analysis and data obtained from flow cytometry was analyzed with the analysis software Kaluza. For cell counts, 20 μL of flow count beads (Beckman Coulter) containing 937 beads per μL were added to the samples and diluted to 1 mL.3. Results and discussion
In this work, we have combined selective RBC lysis with focusing and separation of nucleated cells based on differential inertial focusing in flow through spiral devices. First, we designed a spiral microchannel and optimized the flow and geometry parameters to focus and sort live cells. Fig. 1 shows a schematic of the method. First, whole blood is exposed to water for RBC lysis. During this time the nucleated cells start to swell and in the subsequent step PBS is used to terminate the lysis followed by inertial based separation of the cells into subpopulation. In the following sections, we will first briefly describe and discuss cell focusing and separation in flow through spiral channels, after which we present our results on sorting and separating nucleated cells including total and sub-population from unprocessed whole blood using an integrated device.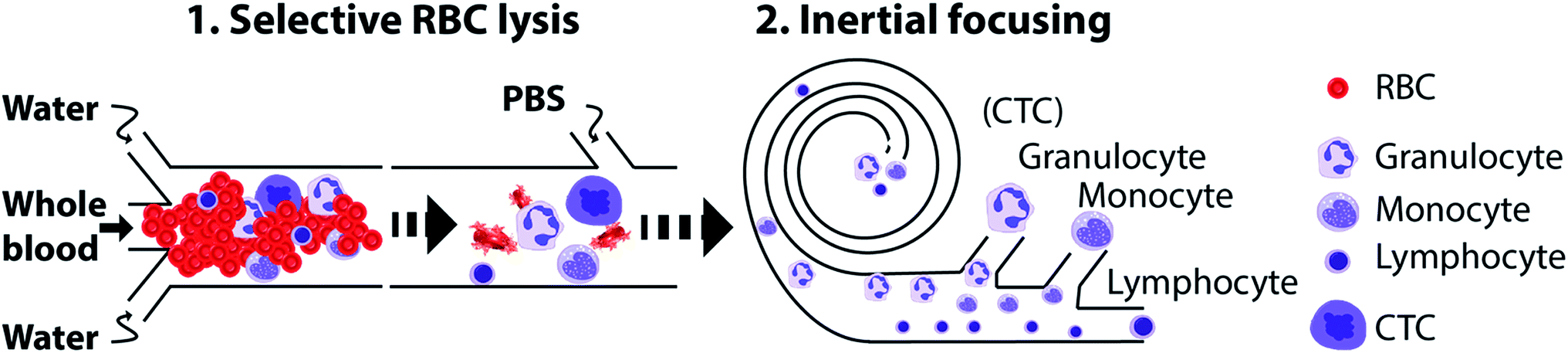 | ||
| Fig. 1 Schematic of selective RBC lysis combined with inertial microfluidic based separation of nucleated cells. Using an integrated device, whole blood is exposed to water for osmtic lysis of RBCs and at the same time the nucleated cells swell and can be separated by size-based inertial focusing using a spiral. | ||
3.1 Inertial focusing and separation of cells in spiral microdevices
Particles and cells flowing through curved spiral microchannels experience two major forces; namely, lift forces (FL) due to inertial flow, and Dean forces (FD) resulting from Dean flow due to curvature. Depending on the relative magnitude of FL and FD acting on a particle, focusing close to the inner wall (dominant lift) or mixing (dominant Dean force) can occur.20 While we focus on blood cell separation here, we describe the microspheres based experiments and observations in ESI† (Fig. S1 and S2). Based on the initial experiments using microspheres, we designed a 10-loop spiral channel device for blood cell experiments. Specifically the spiral device is a 10-loop 500 μm × 115 μm (width × height) channel with spiral geometry consisting of a 5-loop anti-clock wise turn followed by a 5-loop clock wise turn from the center to focus and sort cells (Fig. 2A). Adjacent channels are separated by 500 μm and the width of the last loop increases gradually to 1600 μm with the radius of curvature. The in-and-out spiral geometry is designed to allow integration of the inertial focusing with the upstream processing.We first evaluated the device by flowing 10 μm microspheres at various flow rates and found an optimized flow rate at 1 mL min−1 for focusing and extraction through outlet 1 (Fig. 2D). For clarity, unless specified, the Dean number given henceforward is for the last row of the 10-loop channel (5-loop anti-clock wise turn and 5-loop clock wise turn). To find the optimum flow conditions for leukocyte separation, 1%, 5% and 10% blood samples (diluted in 1× PBS) were initially passed through the device at different flow rates (Fig. 3). The fractionated cells (outlet 1 and outlet 2) were then analyzed using Coulter counter. Fig. 3 shows the recovery of leukocytes for the 1%, 5% and 10% blood concentrations at outlet 1, with a separation efficiency of 93%, 89% and 80% respectively (for De = 6.2).
 | ||
| Fig. 2 Inertial particle focusing in flow through spiral channels. (A) 10 loop spiral device with one inlet and two outlets. (B–D) 10 μm particles shown at different position as they flow through the spiral device. The particles, initially unfocused are focused as they flow through the channel (B) and pass through the center point, s-turn (C), and can be separated through the inner outlet 1 (D). Flow rate: 1 mL min−1. | ||
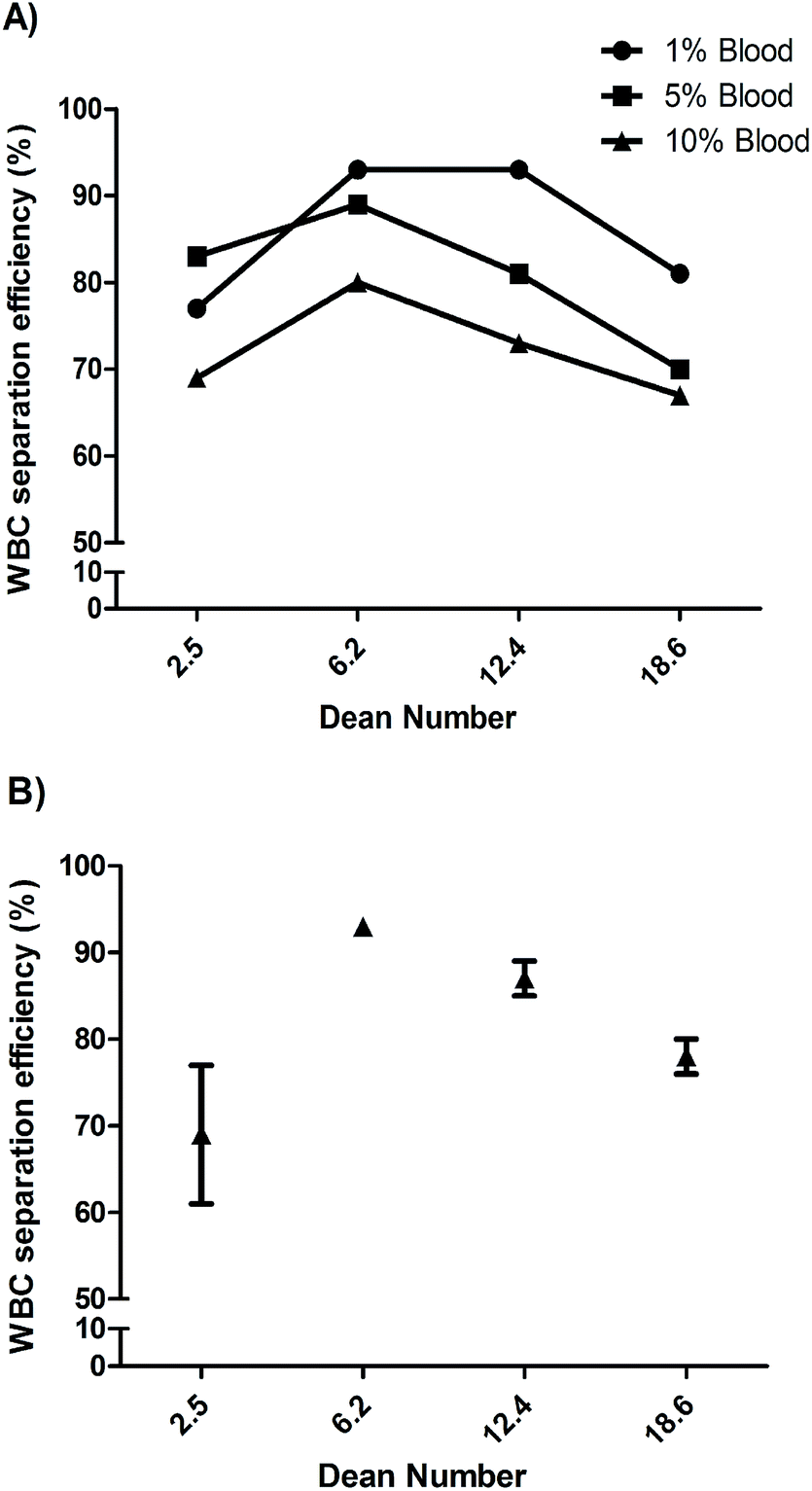 | ||
| Fig. 3 (A) Separation of WBC at different dilution (1, 5 and 10%) of whole blood, at different flow rates respectively. At De = 6.2, the separation efficiency was 93%, 89% and 80% for the different diluted sample. (B) Separation of WBCs after processing lysed 10% blood sample at different flow rates. A separation efficiency of 95% was achieved for De = 6.2. | ||
The decreased separation efficiency at higher solid content is due to collision between cells (primarily RBCs) that disrupts the equilibrium positions. A way to reduce the solid content of whole blood is by dilution prior to flowing through the spiral device. Although relatively high volumetric flow rates are obtained in inertial microfluidics, the need to dilute (often below 1% solid content) is a major limitation for applications that require processing of large (mL) blood volumes. Most inertial microfluidic devices used for blood processing rely on off-chip dilution.35,42,54,56–59 In this work, we explored selective RBC lysis in combination with inertial microfluidics. We selectively lysed the RBC by exposing them to a hypotonic solution for 10 seconds, while keeping the WBC cells intact as previously described.60,61 When we processed lysed blood from the 10% dilution through the device (Fig. 3B), the separation efficiency was immediately increased 1.7 fold to 93%, and was now equal to that of 1% diluted blood. Hence, while reducing the solid content in blood to less than 1% dramatically reduces throughput, here we show that selective RBC lysis has the potential of improving performance while maintaining high throughput.
3.2 Integrated device for blood processing
Following the promising results in applying a selective RBC lysis protocol upstream to the spiral, we developed an integrated device for automated whole blood processing (Fig. 4A). The integrated device uses unprocessed whole blood as input and performs the following functions: (i) uses a hypotonic buffer to eliminate RBC, (ii) uses inertial focusing to sort nucleated cells based on size, and finally (iii) returns cells to isotonic conditions to their original size following sorting. The integrated device consists of three regions: a mixing region, an incubation chamber and an inertial separation region (Fig. 4A). The width and height of the channel were 500 μm and 100 μm for both mixing and spiral regions, while the height of the incubation chamber was 150 μm. In the mixing region, whole blood is continuously mixed with deionized water. At the inlet, water branches into 2 streams, 1 on either side of the entry stream of whole blood in the channel. This focuses the whole blood into a narrow stream flanked on both sides by deionized water. In addition, the mixing channel contains herringbone structures on the roof to facilitate chaotic mixing. The incubation chamber enables controlled contact time of the lysis solution. After the incubation chamber, the solution is mixed with a lysis termination buffer (2× PBS) before reaching the spiral section for size-based separation of the leukocytes.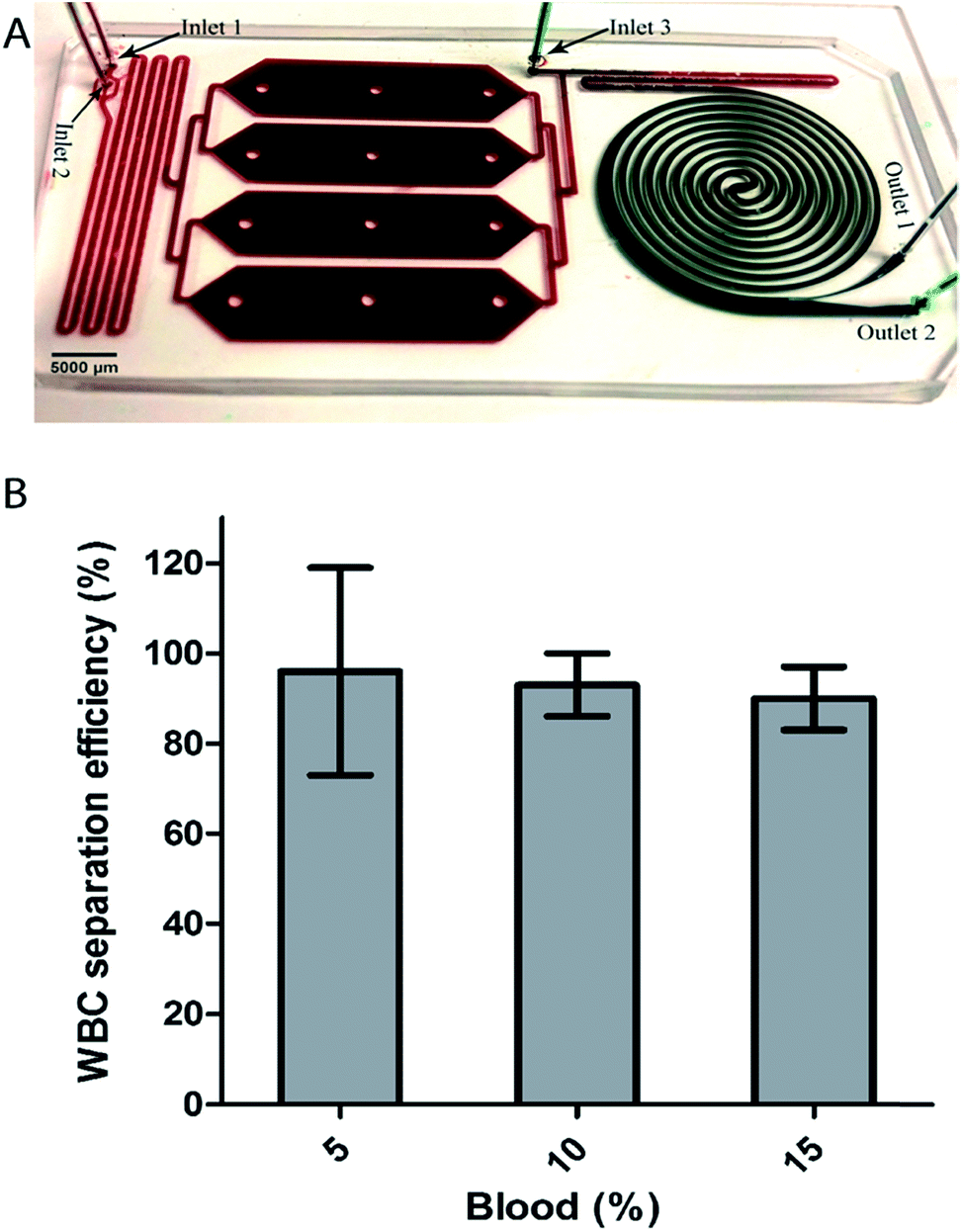 | ||
| Fig. 4 Integrated devices for leukocyte separation from whole blood. (A) The device design. The integrated device has three regions: in the first region, deionized water is mixed with whole blood in the channel with herringbone structure for chaotic mixing. In the second region, the pre-mixed solution is incubated and in the third region PBS is added to terminate the RBC lysis and focus the remaining leukocyte for size based separation in the spiral. The fractionated sample from outlet 1 is readily available for downstream analysis. (B) WBCs separation from whole blood, collected at outlet 1. Whole blood was introduced and on-chip diluted to a final dilution of 5%, 10% and 15%. The separation efficiency, collected at outlet 1, was 98%, 99% and 92% for the on-chip diluted samples. Flow rate: 1 mL min−1. | ||
Experimentally, unprocessed whole blood was introduced and diluted on-chip to a final dilution of 5%, 10% and 15% as described in the Materials and methods section. The total flow rate (1 mL min−1) was maintained constant and the samples were collected at the outlets and analyzed using Coulter counter and/or flow cytometer. As can be seen in Fig. 4B, the leukocytes were separated with a separation efficiency of 98%, 99% and 92% for the on-chip 5%, 10% and 15% diluted blood respectively. Compared to off-chip lysis followed by spiral device separation, the integrated device increased separation efficiency, presumably due the difference in the osmotic behavior of nucleated cells that causes the cells to increase in size, thereby enhancing their inertial separation. For instance, 10% blood processed after off-chip lysis achieved an efficiency of 95% (see Fig. 3B), while the integrated device achieved an efficiency of 99%. Hence, an important and enabling feature of the protocol in the integrated device is the fact that the focusing occurs while the blood is exposed to the hypotonic buffer. During this time nucleated cells increase in size enabling differential focusing from RBC debris. This device therefore accomplishes simultaneous RBC depletion, nucleated cell size amplification for focusing, and final delivery of sample in concentrations suitable for subsequent downstream analysis. Using the integrated device, the fact that cells swell at a different rate can be exploited for separation. In the next section, we report and discuss the use of cell swelling as a parameter to improve separation resolution.
3.3 Cell swelling as a parameter for cell separation
It has been shown previously that WBC sub types swell at different rates upon exposure to hypotonic solution.62 The mechanism of cell swelling depends on factors like cytoplasm to nucleus ratio, cell membrane proteins like aquaporin, membrane properties and F-actin to G-actin ratio.63 To exploit the possibility for controlled exposure of cells to hypotonic solutions for cell sorting into different subpopulations and other applications such as separation of cancer cells based on size, we first tested cell swelling using cancer cell line. MCF7 cancer cell line was used to measure the effect of hypotonic solution and lysis. These cells expanded very rapidly and could withstand more than 2 min exposure to hypotonic solution without lysing (Fig. 5A).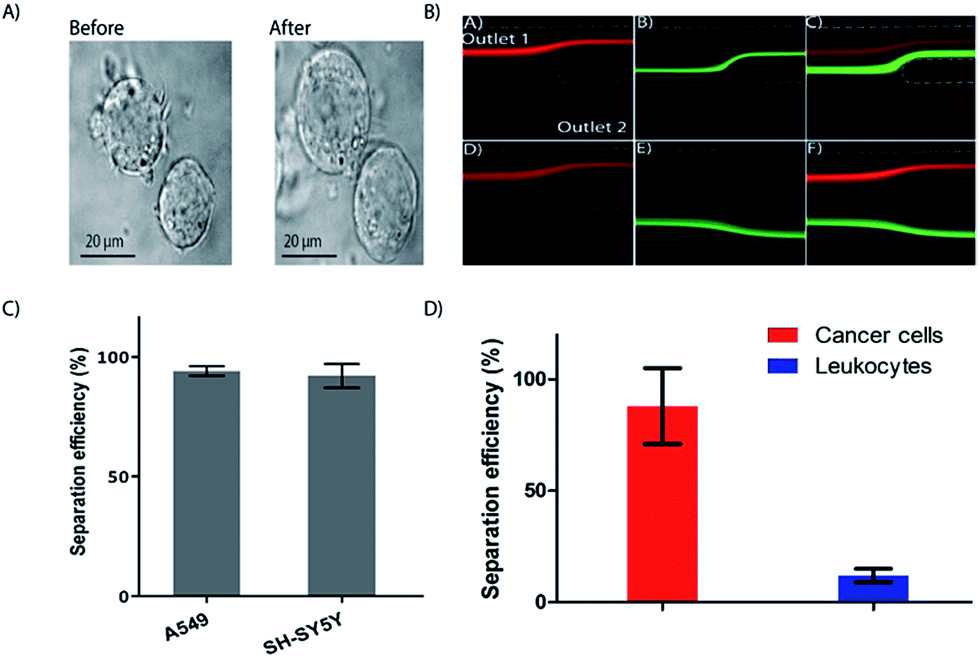 | ||
| Fig. 5 Cell swelling for enhanced inertial separation. (A) Cancer cell lines before and after a 2 minute exposure to a hypotonic solution. (B) Integrated device characterization using microspheres. The 15 μm (red) and 10 μm (green) particles are differentially focused and exiting either through outlet one (upper panel, equal resistance) or through the two separate outlets (lower panel, higher resistance at outlet 1). Flow rate was 1 mL min−1 (corresponding De = 5.9). (C) Separation of cancer cell lines. Two cancer cell lines in PBS processed through the device are collected through outlet 1 at an efficiency of 97% or higher (n = 3). (D) Separation of spiked cancer cells (A549) from whole blood diluted to 5% in the integrated device. A yield of 88% was achieved (n = 3) for the cancer cells while 80% of the leukocytes could be depleted to outlet two. | ||
While the focus of the current study is on isolation of total and subpopulation leukocyte separation, we also tested the possibility to separate cancer cells from whole blood without changing the design of the integrated device. Size based CTC separation using spiral devices have been covered extensively in recent years.64 Here, we are testing the use of osmosis as a parameter to separate CTCs from other nucleated cells. We have previously reported on differential inertial focusing in flow through spiral channels, where particles can be differentially focused into separate lateral positions based on size for separation.20
Using the current integrated device, differential inertial focusing can be exploited by simply changing the resistance at the outlets (i.e. making the tubing at outlet 1 longer than outlet 2). First, without the added resistance, we obtained the conditions to differentially focus both particles but such that both 10 and 15 μm microspheres exiting through outlet 1 (Fig. 5B, upper panel). Next, we varied the flow resistance of the device at outlet 1 to selectively separate the 10 μm particles at the fixed flow rate of 1 mL min−1 without defocusing, while 15 μm particles retained their position and could be collected at outlet 1 (Fig. 5B, lower panel). The particles were differentially separated with an efficiency of 87% for 15 μm beads at outlet 1 and 95% for 10 μm beads at outlet 2 (data not shown). The volume fraction at the outlets 1 and 2 was 1:10 using the resistance. We kept this constant and tested the processing of two different cancer cell lines in PBS and obtained high (>97%) separation efficiency (Fig. 5C). The higher separation efficiency of cancer cell lines compared to the 15 μm microspheres is likely due cell swelling, resulting in increased cell size. Finally, as a proof of principle, we spiked cancer cell lines into whole blood and processed the sample through the integrated device, keeping the total flow rate at 1 mL min−1 (Fig. 5D). The spiked whole blood was exposed to the hypertonic solution for RBC lysis to a final concentration of 5%, and the enriched cancer cells were collected though outlet 1 at a yield of 88% (Fig. 5D), while the WBCs were depleted by 80%. Further improvement of the yield is possible by reducing the whole blood fraction. For instance, reducing the blood fraction to 2.5% and changing the flow rate improved the cancer cell yield improved to 95% (De = 5.8) (Fig. S3†).
3.4 Fractionation of leukocytes into sub-population
The focus of the current study was to develop a simple method to separate leukocyte from unprocessed whole blood into total and subpopulation. For fractionation of the leukocytes into subpopulation, we used the fact that exposure to hypotonic solution results in cell-specific swelling. By simply adding one more outlet to the integrated device (Fig. 6A), we were able to separate the cells into subpopulations. The integrated device uses unprocessed whole blood as its input and performs the following functions: (a) uses a hypotonic buffer to simultaneously eliminate RBCs and increase the size of other nucleated cells, (b) uses inertial focusing to sort inflated nucleated cells based on size, and finally (c) returns cells to isotonic conditions and to their original size following sorting. By optimizing the total contact time of the hypotonic buffer, it was possible to continuously separate granulocytes at a purity of 86% and lymphocytes at a purity of 91%, while the purity for the monocyte population was 43% (Fig. 6B).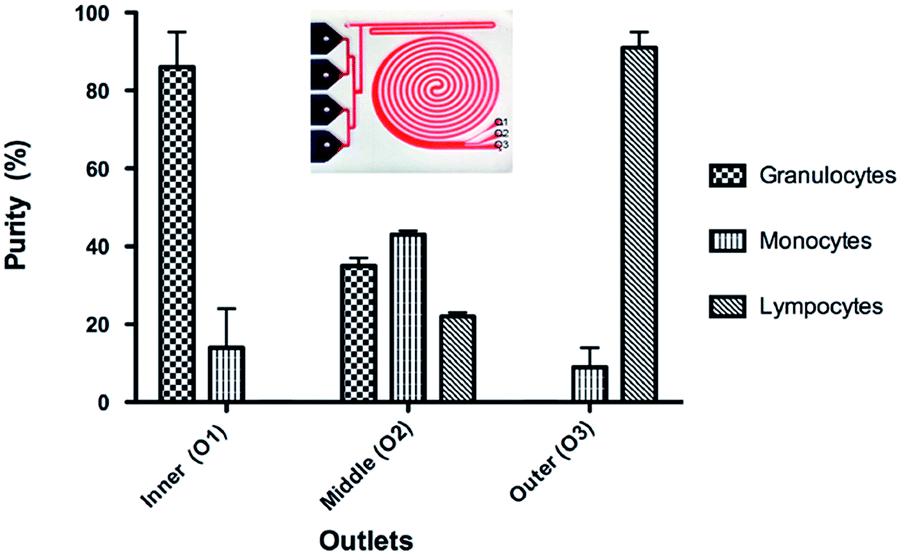 | ||
| Fig. 6 Integrated device for leukocyte fractionation into subpopulation. (A) The integrated device showing the three outlets. (B) Fractionation of leukocyte sub population at outlets one to three. A purity of 86%, 41% and 91% was obtained for granulocyte (outlet 1), monocyte (outlet 2) and lymphocyte (outlet 3) respectively for a contact time of 13 s. | ||
The summary of the integrated device performance results for different contact times is presented in Table 1. The purity of the granulocyte and lymphocyte subtypes is comparable to macroscale methods. The purity of the monocyte population, collected at outlet 2, is much lower. The highest purity was obtained for a contact time of 13 seconds (86%, 43% and 91% respective for granulocyte, monocyte and lymphocyte collected at outlets 1, 2 and 3). When the contact time is increased to 25 seconds the purity of granulocyte is decreased while the other cell subtypes are maintained relatively unchanged. Additional increased contact time to 30 seconds resulted in significantly decrease purity for all subtypes. As it can be seen from the graph in Fig. 6, the monocyte subpopulation is the only contaminating one at outlet one and three. One simple way of improving purity for all sub populations would be to increase the number of outlets. For instance, a six-outlet device would provide higher purity for all three sub-populations.
| Blood cells | Exposure time (s) | Granulocytes | Monocytes | Lymphocytes | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Outlet 1 | Outlet 2 | Outlet 3 | Outlet 1 | Outlet 2 | Outlet 3 | Outlet 1 | Outlet 2 | Outlet 3 | ||
| Yield (%) | 11 | 23 ± 1 | 73 ± 1 | 1 ± 0 | 5 ± 0 | 90 ± 7 | 5 ± 4 | 0 | 56 ± 2 | 44 ± 2 |
| 13 | 27 ± 7 | 73 ± 7 | 0 | 5 ± 4 | 90 ± 7 | 5 ± 3 | 0 | 47 ± 2 | 53 ± 2 | |
| 26 | 39 ± 10 | 60 ± 10 | 1 ± 1 | 12 ± 3 | 86 ± 2 | 2 ± 1 | 0 | 72 ± 20 | 28 ± 20 | |
| 30 | 43 ± 4 | 57 ± 5 | 0 | 18 ± 4 | 74 ± 6 | 8 ± 2 | 0 | 75 ± 9 | 25 ± 9 | |
| Purity (%) | 11 | 85 ± 1 | 33 ± 1 | 1 ± 0 | 15 ± 1 | 41 ± 2 | 10 ± 8 | 0 | 26 ± 1 | 89 ± 8 |
| 13 | 86 ± 9 | 35 ± 2 | 0 | 14 ± 10 | 43 ± 1 | 9 ± 5 | 0 | 22 ± 1 | 91 ± 4 | |
| 26 | 75 ± 4 | 28 ± 6 | 3 ± 1 | 24 ± 4 | 40 ± 2 | 7 ± 4 | 0 | 33 ± 8 | 90 ± 4 | |
| 30 | 70 ± 3 | 28 ± 1 | 1 ± 1 | 30 ± 3 | 36 ± 1 | 24 ± 3 | 0 | 36 ± 1 | 75 ± 2 | |
Overall, the method is very robust, requires only 2 pumps and the integrated, stand alone, device to continuously focus and fractionate cells into sub-population. While we don't anticipate fluctuation in efficiency due to external parameters such as temperature and relative humidity, additional properties such as cell stiffness might be used as parameter for separation. Other solution, such as detergents might be used. For instance, we have recently used saponin to lyse all blood cells while keeping bacteria viable.65 For immunological studies, the isolated cells from a heterogeneous population of cells in blood should ideally not be activated due to the isolation method itself.
3.5 Activation study of the leukocytes
To explore the effect of the integrated spiral device on the activation of cells, the activation status of blood cells processed through the device was compared against unprocessed blood cells by studying the activation of the immune system. We compared the differential expression surface activation markers of native leukocytes in whole blood versus leukocytes stimulated ex vivo with lipopolysaccharide (LPS). Using flow cytometry, we used known early activation markers to score activation of the three main leukocytes subpopulations neutrophils, lymphocytes and monocytes. As can be seen in Fig. 7, we observed no difference between the unprocessed (control) and processed neutrophils in both the stimulated and unstimulated blood, indicating that our integrated microfluidic system does not alter the activation levels of the cells. Similar results were obtained using activation markers for lymphocytes and monocytes.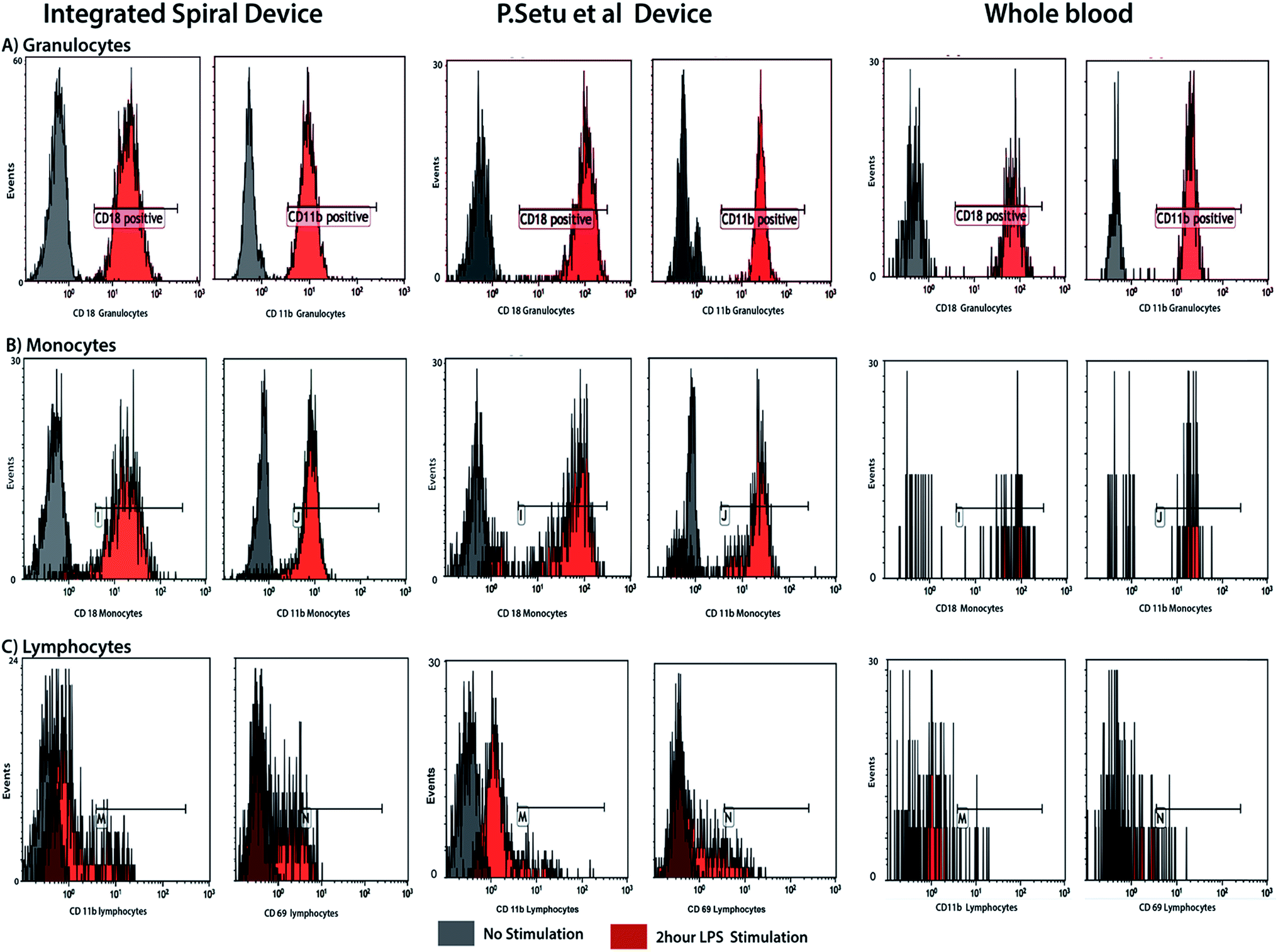 | ||
| Fig. 7 Cell activation study and comparison with RBC lysis device by Sethu et al.60 FACS results of the activation level of neutrophils prior and following ex vivo LPS stimulation of whole blood. There is no significant difference in pattern between unprocessed and processed whole blood for both stimulated and unstimulated neutrophils, indicating that the device does not activate or introduce artifacts. | ||
The aim of this study was to develop a rapid and efficient method for separating nucleated cells from whole blood. By employing inertial microfluidics, the continuous isolation of nucleated cells offers label free isolation and minimizes the time of blood processing compared to other methods. This integrated device enables large volume processing: 6 mL for leukocyte isolation from whole blood. Furthermore, the cells are concentrated in a volume suitable for downstream processing. For cancer cell separation, we used the fact that cells are differentially focused based on size to direct the leukocytes to outlet two. It was previously shown that leukocytes can withstand osmotic shock for up to 30 to 60 seconds before lysing63 and we show here that cancer cell lines (A549) can withstand more than 2 minutes of osmotic shock (Video S1†). However, it will be important to test osmotic shock on CTCs from real patient samples, which will be a subject of a future study.
While, we obtained high yield of cancer cells, the depletion of leukocytes need to be further improved for future clinical application. To separate cancer cells we used the same device that was used for leukocyte separation by simply adding resistance to outlet 1. Future work will involve improving the separation efficiency between cancer cells and leukocyte by modifying the design of the integrated device with improved spiral geometry. For instance, Kim et al.35 recently reported the effective separation of cancer cells from leukocytes after off-chip removal of RBCs. Similar results have been achieved in spiral devices with off-chip dilution. Hence, we anticipate high yield and purity of cancer cells in future designs of the integrated device.
Experimentally, we found that cell viability is maintained during the separation. However, it is important that pre-processing techniques do not perturb the cell populations. This is because cell activation can alter gene and protein expression patterns, so the cells can be non-representative of those in the starting blood, which limits the usefulness of the information gained by analysis of the cell population. Because the blood cells travel at high speed in our system, we tested cell damage/stress via ex vivo LPS stimulation study of the leukocyte subpopulation. LPS stimulation mimics cellular changes presented by individuals with bacterial infections, and is frequently used as a model for both in vivo and ex vivo clinical studies. The differential profiles of surface markers for the leukocyte subpopulations reveal that whole blood processed through the spiral device and control generate similar results, both for the unstimulated and LPS stimulated samples (Fig. 7). This indicates that the device does not activate the cells and does not introduce artifacts affecting stimulated blood sample.
4. Conclusion
Here we present a simple method that combined inertial microfluidics with selective RBC lysis to continuously focus, order and sort live nucleated cells from unprocessed whole blood. The method exploits the osmotic property of cells and inertial migration forces to separate nucleated cells from whole blood. We have developed an integrated device that continuously removes RBCs via osmotic lysis and at the same time concentrates nucleated cells by size-based inertial focusing. Leukocytes were isolated from whole blood with a separation efficiency of 99%. By simply changing the resistance at the outlet, we were able to differentially separate spiked cancer cells with a yield of 88% while 80% of the leukocytes were depleted from the same outlet. Using an integrated device with three outlets, leukocyte was fractionated into subpopulation with purity of 86% for granulocytes and 91% for lymphocytes. Furthermore, the isolated cells remain viable and un-activated, providing the opportunity for further downstream studies.The main text of the article should appear here with headings as appropriate.
Conflict of interest
The authors declare no competing financial interest.Acknowledgements
This work has been sponsored in part by the Swedish Childhood Cancer Foundation, European Commission through the FP7 project CanDo and Swedish Research Council.References
- D. P. Braun and J. E. Harris, J. Natl. Cancer Inst., 1981, 67, 809–814 CAS.
- J. Nairn, G. Hodge and P. Henning, Pediatr. Nephrol. Berl. Ger., 2005, 20, 190–196 CrossRef PubMed.
- L. Vettore, M. C. de Matteis and P. Zampini, Am. J. Hematol., 1980, 8, 291–297 CrossRef CAS PubMed.
- P. Madyastha, K. R. Madyastha, T. Wade and D. Levine, J. Immunol. Methods, 1982, 48, 281–286 CrossRef CAS PubMed.
- H. N. Chang, W. G. Lee and B. S. Kim, Biotechnol. Bioeng., 1993, 41, 677–681 CrossRef CAS PubMed.
- S. Baratchi, K. Khoshmanesh, C. Sacristán, D. Depoil, D. Wlodkowic, P. McIntyre and A. Mitchell, Biotechnol. Adv., 2014, 32, 333–346 CrossRef CAS PubMed.
- J. Yang, Y. Huang, X.-B. Wang, F. F. Becker and P. R. C. Gascoyne, Biophys. J., 2000, 78, 2680–2689 CrossRef CAS PubMed.
- K. E. McCloskey, J. J. Chalmers and M. Zborowski, Anal. Chem., 2003, 75, 6868–6874 CrossRef CAS PubMed.
- O. Lara, X. Tong, M. Zborowski and J. J. Chalmers, Exp. Hematol., 2004, 32, 891–904 CrossRef PubMed.
- S. Wang, K. Liu, J. Liu, Z. T.-F. Yu, X. Xu, L. Zhao, T. Lee, E. K. Lee, J. Reiss, Y.-K. Lee, L. W. K. Chung, J. Huang, M. Rettig, D. Seligson, K. N. Duraiswamy, C. K.-F. Shen and H.-R. Tseng, Angew. Chem., Int. Ed., 2011, 50, 3084–3088 CrossRef CAS PubMed.
- X. Cheng, D. Irimia, M. Dixon, K. Sekine, U. Demirci, L. Zamir, R. G. Tompkins, W. Rodriguez and M. Toner, Lab Chip, 2007, 7, 170 RSC.
- X. Cheng, D. Irimia, M. Dixon, J. C. Ziperstein, U. Demirci, L. Zamir, R. G. Tompkins, M. Toner and W. R. Rodriguez, JAIDS, J. Acquired Immune Defic. Syndr., 2007, 45, 257–261 Search PubMed.
- Y. Xu, J. A. Phillips, J. Yan, Q. Li, Z. H. Fan and W. Tan, Anal. Chem., 2009, 81, 7436–7442 CrossRef CAS PubMed.
- A. A. Adams, P. I. Okagbare, J. Feng, M. L. Hupert, D. Patterson, J. Göttert, R. L. McCarley, D. Nikitopoulos, M. C. Murphy and S. A. Soper, J. Am. Chem. Soc., 2008, 130, 8633–8641 CrossRef CAS PubMed.
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS PubMed.
- S. Matsusaka, M. Kozuka, H. Takagi, H. Ito, S. Minowa, M. Hirai and K. Hatake, Cancer Lett., 2014, 355, 113–120 CrossRef CAS PubMed.
- H. K. Lin, S. Zheng, A. J. Williams, M. Balic, S. Groshen, H. I. Scher, M. Fleisher, W. Stadler, R. H. Datar, Y.-C. Tai and R. J. Cote, Clin. Cancer Res., 2010, 16, 5011–5018 CrossRef CAS PubMed.
- K. Kolostova, Y. Zhang, R. M. Hoffman and V. Bobek, J. Fluoresc., 2014, 24, 1531–1536 CrossRef CAS PubMed.
- P. Sethu, M. Anahtar, L. L. Moldawer, R. G. Tompkins and M. Toner, Anal. Chem., 2004, 76, 6247–6253 CrossRef CAS PubMed.
- A. Russom, A. K. Gupta, S. Nagrath, D. D. Carlo, J. F. Edd and M. Toner, New J. Phys., 2009, 11, 075025 CrossRef PubMed.
- A. Russom, A. K. Gupta, S. Nagrath, D. Di Carlo, J. F. Edd and M. Toner, New J. Phys., 2009, 11, 75025 CrossRef PubMed.
- D. Di Carlo, J. F. Edd, D. Irimia, R. G. Tompkins and M. Toner, Anal. Chem., 2008, 80, 2204–2211 CrossRef CAS PubMed.
- D. A. Drew, Chem. Eng. Sci., 1988, 43, 769–773 CrossRef CAS.
- E. S. Asmolov, J. Fluid Mech., 1999, 381, 63–87 CrossRef CAS.
- J.-P. Matas, J. F. Morris and É. Guazzelli, J. Fluid Mech., 2009, 621, 59 CrossRef.
- J. Matas, J. Morris and E. Guazzelli, Oil Gas Sci. Technol., 2004, 59, 59–70 Search PubMed.
- B. Chun and A. J. C. Ladd, Phys. Fluids, 2006, 18, 031704 CrossRef.
- S. A. Berger, L. Talbot and L. S. Yao, Annu. Rev. Fluid Mech., 1983, 15, 461–512 CrossRef.
- D. Di Carlo, D. Irimia, R. G. Tompkins and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18892–18897 CrossRef CAS PubMed.
- J. Hansson, J. M. Karlsson, T. Haraldsson, H. Brismar, W. van der Wijngaart and A. Russom, Lab Chip, 2012, 12, 4644 RSC.
- H. Ramachandraiah, S. Ardabili, A. M. Faridi, J. Gantelius, J. M. Kowalewski, G. Mårtensson and A. Russom, Biomicrofluidics, 2014, 8, 034117 CrossRef PubMed.
- V. VanDelinder and A. Groisman, Anal. Chem., 2007, 79, 2023–2030 CrossRef CAS PubMed.
- S. Bose, R. Singh, M. Hanewich-Hollatz, C. Shen, C.-H. Lee, D. M. Dorfman, J. M. Karp and R. Karnik, Sci. Rep., 2013, 3, 2329 CrossRef PubMed.
- H.-C. Tseng, R.-G. Wu, H.-Y. Chang and F.-G. Tseng, High-throughput particle separation and concentration using spiral inertial filtration, IEEE, 2012, pp. 835–838 Search PubMed.
- M. Kim, S. Mo Jung, K.-H. Lee, Y. Jun Kang and S. Yang, Artif. Organs, 2010, 34, 996–1002 CrossRef PubMed.
- J. Chen, J. Li and Y. Sun, Lab Chip, 2012, 12, 1753–1767 RSC.
- N. Nivedita and I. Papautsky, Biomicrofluidics, 2013, 7, 054101 CrossRef PubMed.
- B. K. Lin, S. M. McFaul, C. Jin, P. C. Black and H. Ma, Biomicrofluidics, 2013, 7, 034114 CrossRef PubMed.
- N. M. Karabacak, P. S. Spuhler, F. Fachin, E. J. Lim, V. Pai, E. Ozkumur, J. M. Martel, N. Kojic, K. Smith, P. Chen, J. Yang, H. Hwang, B. Morgan, J. Trautwein, T. A. Barber, S. L. Stott, S. Maheswaran, R. Kapur, D. A. Haber and M. Toner, Nat. Protoc., 2014, 9, 694–710 CrossRef CAS PubMed.
- J. Sun, C. Liu, M. Li, J. Wang, Y. Xianyu, G. Hu and X. Jiang, Biomicrofluidics, 2013, 7, 011802 CrossRef PubMed.
- S. Song and S. Choi, Appl. Phys. Lett., 2014, 104, 074106 CrossRef.
- G. Guan, L. Wu, A. A. Bhagat, Z. Li, P. C. Y. Chen, S. Chao, C. J. Ong and J. Han, Sci. Rep., 2013, 3, 1475 CrossRef PubMed.
- I. Cima, C. Wen Yee, F. S. Iliescu, W. Min Phyo, K. Hon Lim, C. Iliescu and M. Han Tan, Biomicrofluidics, 2013, 7, 011810 CrossRef PubMed.
- T. M. Geislinger and T. Franke, Biomicrofluidics, 2013, 7, 044120 CrossRef PubMed.
- H.-S. Moon, K. Kwon, K.-A. Hyun, T. Seok Sim, J. Chan Park, J.-G. Lee and H.-I. Jung, Biomicrofluidics, 2013, 7, 014105 CrossRef PubMed.
- Z. Liu, F. Huang, J. Du, W. Shu, H. Feng, X. Xu and Y. Chen, Biomicrofluidics, 2013, 7, 011801 CrossRef PubMed.
- T. H. Kim, H. J. Yoon, P. Stella and S. Nagrath, Biomicrofluidics, 2014, 8, 064117 CrossRef PubMed.
- J. Sun, C. Liu, M. Li, J. Wang, Y. Xianyu, G. Hu and X. Jiang, Biomicrofluidics, 2013, 7, 011802 CrossRef PubMed.
- J. M. Burke, R. E. Zubajlo, E. Smela and I. M. White, Biomicrofluidics, 2014, 8, 024105 CrossRef PubMed.
- N. Nivedita and I. Papautsky, Biomicrofluidics, 2013, 7, 054101 CrossRef PubMed.
- F. A. W. Coumans, G. van Dalum, M. Beck and L. W. M. M. Terstappen, PLoS One, 2013, 8, e61770 CAS.
- A. A. S. Bhagat, S. S. Kuntaegowdanahalli and I. Papautsky, Lab Chip, 2008, 8, 1906–1914 RSC.
- S. S. Kuntaegowdanahalli, A. A. S. Bhagat, G. Kumar and I. Papautsky, Lab Chip, 2009, 9, 2973–2980 RSC.
- H. W. Hou, M. E. Warkiani, B. L. Khoo, Z. R. Li, R. A. Soo, D. S.-W. Tan, W.-T. Lim, J. Han, A. A. S. Bhagat and C. T. Lim, Sci. Rep., 2013, 3, 1259 CrossRef PubMed.
- Y. Xia and G. M. Whitesides, Annu. Rev. Mater. Sci., 1998, 28, 153–184 CrossRef CAS.
- A. A. S. Bhagat, H. W. Hou, L. D. Li, C. T. Lim and J. Han, Lab Chip, 2011, 11, 1870–1878 RSC.
- L. Wu, G. Guan, H. W. Hou, A. A. S. Bhagat and J. Han, Anal. Chem., 2012, 84, 9324–9331 CrossRef CAS PubMed.
- E. W. Majid, G. Guan, B. L. Khoo, W. C. Lee, A. A. S. Bhagat, D. S.-W. Tan, W. T. Lim, S. C. Lee, P. C. Y. Chen, C. T. Lim and J. Han, Lab Chip, 2014, 14, 128–137 RSC.
- G. Guan, L. Wu, A. A. Bhagat, Z. Li, P. C. Y. Chen, S. Chao, C. J. Ong and J. Han, Sci. Rep., 2013, 3, 1475 CrossRef PubMed.
- P. Sethu, L. L. Moldawer, M. N. Mindrinos, P. O. Scumpia, C. L. Tannahill, J. Wilhelmy, P. A. Efron, B. H. Brownstein, R. G. Tompkins and M. Toner, Anal. Chem., 2006, 78, 5453–5461 CrossRef CAS PubMed.
- A. Russom, P. Sethu, D. Irimia, M. N. Mindrinos, S. E. Calvano, I. Garcia, C. Finnerty, C. Tannahill, A. Abouhamze, J. Wilhelmy, M. C. Lopez, H. V. Baker, D. N. Herndon, S. F. Lowry, R. V. Maier, R. W. Davis, L. L. Moldawer, R. G. Tompkins and M. Toner, Inflammation and Host Response to Injury Large Scale Collaborative Research Program, Clin. Chem., 2008, 54, 891–900 CAS.
- V. Parichehreh, R. Estrada, S. S. Kumar, K. K. Bhavanam, V. Raj, A. Raj and P. Sethu, Biomed. Microdevices, 2011, 13, 453–462 CrossRef PubMed.
- V. Parichehreh, R. Estrada, S. S. Kumar, K. K. Bhavanam, V. Raj, A. Raj and P. Sethu, Biomed. Microdevices, 2011, 13, 453–462 CrossRef PubMed.
- M. E. Warkiani, B. L. Khoo, L. Wu, A. K. P. Tay, A. A. S. Bhagat, J. Han and C. T. Lim, Nat. Protoc., 2016, 11, 134–148 CrossRef CAS PubMed.
- S. Zelenin, J. Hansson, S. Ardabili, H. Ramachandraiah, H. Brismar and A. Russom, Biotechnol. Lett., 2015, 37, 825–830 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of inertial microfluidics experimental, cancer cell separation results (PDF) and a video of cell swelling. See DOI: 10.1039/c7ra02992f |
| This journal is © The Royal Society of Chemistry 2017 |