
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntelligent ECM mimetic injectable scaffolds based on functional collagen building blocks for tissue engineering and biomedical applications
R. Ravichandrana,
C. Astrandb,
H. K. Patrac,
Anthony P. F. Turnerc,
V. Chotteaub and
J. Phopase *a
*a
aDivision of Molecular Physics, Department of Physics, Chemistry and Biology (IFM), Linköping University, S-58183, Linköping, Sweden. E-mail: jayph@ifm.liu.se
bSchool of Biotechnology, KTH-Royal Institute of Technology, SE-106 91, Stockholm, Sweden
cBiosensors and Bioelectronics Centre, Department of Physics, Chemistry and Biology (IFM), Linköping University, SE-581 83, Linköping, Sweden
First published on 12th April 2017
Abstract
Hydrogels comprising natural extracellular matrix (ECM) components are very attractive as scaffolds for regenerative medicine applications due to their inherent biointeractive properties. Responsive materials that adapt to their surrounding environments and regulate transport of ions and bioactive molecules manifest significant advantages for biomedical applications. Although there are many exciting challenges, the opportunity to design, fabricate and engineer stimuli-responsive polymeric systems based on ECM components is particularly attractive for regenerative medicine. Here we describe a one-pot approach to fabricate in situ fast gellable intelligent ECM mimetic scaffolds, based on methacrylated collagen building blocks with mechanical properties that can be modulated in the kPa–MPa range and that are suitable for both soft and hard tissues. Physiochemical characterizations demonstrate their temperature and pH responsiveness, together with the structural and enzymatic resistance that make them suitable scaffolds for long-term use in regenerative medicine and biomedical applications. The multifunctionality of these hydrogels has been demonstrated as an in situ depot-forming delivery platform for the adjustable controlled release of proteins and small drug molecules under physiological conditions and as a structural support for adhesion, proliferation and metabolic activities of human cells. The results presented herein should be useful to the design and fabrication of tailor-made scaffolds with tunable properties that retain and exhibit sustained release of growth factors for promoting tissue regeneration.
Introduction
The locally secreted three-dimensional (3D) macromolecular constituent called the extracellular matrix (ECM), composed of protein fibers and filaments (collagen, fibronectin, elastin) surrounded by a hydrated network of polysaccharides, glycosaminoglycans and proteoglycans, regulates the fate of cells from small tissues to large organs.1 The synergistic and antagonistic communication between cells and the dynamic 3D microenvironment confers well-defined matrix properties such as bio (physio-chemical) and biomechanical properties (topography, porosity, stiffness etc.) and also orchestrates numerous cellular events (adhesion, spreading, migration, proliferation, differentiation and signaling) that are interconnected to dictate repair and remodeling of tissues/organs.1,2 Consequently alterations in any ECM component or in its regulation can have a remarkable impact on its properties, leading to a disorganized network and ultimately failure of tissue/organ homeostasis and function leading to need for a scaffolding platform to repair and remodel the damaged site.3The matrix properties of ECM varies between tissues such that the differences in macro–micro and nano level orientation of ECM components are very important to consider in developing and manipulating ECM mimetic scaffolds for tissue specific needs.3,4 Recent advances in tissue engineering and polymer chemistry have led to the development of simple supporting scaffolds into smart dynamic artificial matrices (biomaterials) able to deliver functional biomolecules for ECM remodeling both in vitro and in vivo.4–6 Functional biomaterial that offers the flexibility to design specifically tailored scaffolds with the desired topology, composition and functionality are becoming increasingly attractive for use in tissue engineering and biomedical applications, including drug delivery and diagnostics.7,8 In particular, hydrogels (injectable or implantable) that facilitate predictable manipulation and fine tuning of post-fabrication matrix properties have gained lot of attention in tissue engineering due to their structural similarities to native soft tissues.4,7 Despite their promise, hydrogels suffer from a series of difficulties to administer it in vivo using minimally invasive techniques, including their mechanical properties, limited stability and bio-degradation, all of which are undesirable for long-term applications.9 These limitations have prompted the design of in situ gelling injectable hydrogels, fabricated using physical, chemical or enzymatic crosslinking strategies.
Covalently cross-linked, Intelligent Biomimetic Hydrogels (CIBHs) are an intriguing class of hydrogel scaffold comprising covalently cross-linked interpenetrating polymeric networks made from different polymers, which can switch matrix behavior in response to environmental triggers.10 The superior performance of CIBHs, such as improved bio-compatibility, enhanced elastomechanical properties, tunable degradation kinetics and responsiveness, have recently been a focus of attention for biomedical applications and tissue engineering.9,11,12 Over the last decade, numerous natural and synthetic polymers have been actively investigated for the design of CIBHs, but collagen based CIBHs have remained unexplored to date. Collagen, the main component of mammalian extracellular matrix (ECM), has many distinctive features that play key roles in tissue regeneration. Although use of extracted collagen has been popular in tissue engineering due to its inherent biocompatibility, it suffers from limited functional groups (–NH2 and –COOH), which renders the protein less amenable to manipulation.13 In our previous work, we overcame this limitation by functionalizing type-I collagen with methacrylate groups, without altering the native structure and bioactivity of collagen, which allows collagen to be cross-linked using cell-friendly crosslinking strategies.14
In this study we present a one-pot approach to fabricate dual-stimuli-responsive (thermo and pH), covalently cross-linked double network injectable hydrogels under physiological conditions based on methacrylated type-I collagen (MAC), that can act both as a structural support to cells and a carrier for the tunable controlled release of growth factors and bioactive molecules. The free radical co-polymerization of MAC and N-isopropyl acrylamide (NIPAm) formed a hydrogel comprising covalently linked interpenetrating networks (IPNs) of collagen and thermoresponsive NIPAm polymer (pNIPAm). The resultant hydrogels were characterized in detail to find an optimal hydrogel for long-term biomedical and tissue engineering applications. We have explored the potential of these hydrogels as an intelligent delivery system for the controlled localized delivery of large protein and small drug molecules by encapsulating Bovine Serum Albumin (BSA) and vitamin E succinate (VES) in situ as a model protein and small drug molecules, respectively. Further, the potential of these hybrid hydrogels as a self-supporting injectable scaffolding system for tissue engineering is demonstrated by evaluating in vitro biocompatibility using human skeletal muscle cells (SkMC).
Results and discussion
Design, fabrication and characterization of intelligent ECM mimetic injectable hydrogels
There are few criteria that help user to design an in situ gellable injectable hydrogel, such as: (i.) solubility, viscosity and compatibility of precursor building blocks (polymers); (ii.) therapeutic window (fast gelling time); and (iii.) degradation time and its products.12 Its known that chemical crosslinking offers more advantages than other techniques as it provides unique control over the above mentioned criteria.14 In our design, we have used MAC as an ECM mimicking building block and N-isopropyl acrylamide (NIPAm) as a stimuli-responsive monomer. These were cross-linked under physiological conditions by free-radical copolymerization using the redox initiators ammonium persulfate (APS) and N,N,N,N-tetramethylethylenediamine (TEMED) and N,N′-methylenebis(acrylamide) (MBAA) as the crosslinking agent (Fig. 1), resulting in the formation of dual-stimuli-responsive hydrogels comprising covalently crosslinked double network of thermoresponsive (pNIPAm) and pH responsive biopolymer (collagen). Earlier reports have demonstrated the physical blending of pNIPAm in a collagen sponge and thermal crosslinking of pNIPAm with collagen.15,16 However, to the best of our knowledge, this is the first report to demonstrate the fabrication and characterization of multi-responsive hydrogels comprising covalently crosslinked IPNs of collagen and thermoresponsive polymer. The hydrogels were spontaneously formed in situ after the injection and could be molded into self-supporting hydrogels with the desired shape and size. The design criteria were very much satisfied as our hydrogel precursors are biocompatible, less viscous and soluble in aqueous medium, thus allowing users to encapsulate bioactive molecules inside the hydrogels in situ during fabrication. | ||
| Fig. 1 Fabrication of intelligent ECM mimetic injectable hydrogels using free radical co-polymerization of MAC and NIPAm. | ||
We first investigated the mechanical strength, swelling properties and enzymatic resistance of MAC–NIPAm (CN) hydrogels with varying MAC and NIPAm compositions, in order to find an optimal injectable scaffold having the highest collagen content together with superior thermoresponsive properties. As stated above, the ECM properties of various tissue (for e.g. the mechanics of soft tissues such as brain (0.2–1 kPa) and hard tissues such as cartilage (20–60 kPa)) are dissimilar by several folds.17 The mechanical properties of scaffolds are one of the important design parameters such that the designed scaffold should have specific ECM mimetic properties in order to regulate cellular behavior in the tissue/organ microenvironment.18 The mechanical properties of the CN hydrogels can be easily modulated from Pa to kPa by varying the composition of MAC and NIPAm, demonstrates the versatility of these hydrogels as injectable as well as implantable scaffolds for tissue specific (soft or hard) applications. The mechanical properties of the CN hydrogels were assessed using rheology. The composition details and mechanical properties of the CN hydrogels are given in Table 1. Hydrogels comprising only pNIPAm (PN) and only collagen (MAC) were soft and showed significantly lower storage moduli (6 and 8 kPa, respectively). The formation of IPN after copolymerization of MAC and NIPAm resulted in a significantly higher storage modulus compared to pure collagen or pure pNIPAm hydrogels. Fig. 2A shows a comparison of storage moduli of the CN5 and CN10 hydrogels with respect to PN and MAC hydrogels. The storage modulus of CN5 and CN10 hydrogels increased ≈2–5 times, respectively compared to control PN and MAC hydrogel. The storage modulus further increased as the collagen content in the hydrogels increased, due to the greater crosslinking density between the two polymeric networks.14
| Hydrogel | MAC (mg) | NIPAm (mg) | G′ (kPa) | G′′ (kPa) |
|---|---|---|---|---|
| PN | 0 | 100 | 6 ± 0.13 | 0.2 ± 0.035 |
| CN5 | 5 | 95 | 17.1 ± 2.1 | 0.3 ± 0.1 |
| CN10 | 10 | 90 | 36.6 ± 2.2 | 1 ± 0.4 |
| CN20 | 20 | 80 | 103 ± 17 | 1.3 ± 0.4 |
| CN30 | 30 | 70 | 151 ± 5.8 | 1.5 ± 0.7 |
| CN40 | 40 | 60 | 265 ± 10 | 10 ± 3 |
| CN50 | 50 | 50 | 420 ± 20 | 25 ± 7.2 |
| MAC | 10 | 0 | 8.4 ± 1.7 | 0.3 ± 0.06 |
 | ||
| Fig. 2 Rheological properties (A) storage modulus (G′) of PN, MAC, CN5 and CN10 hydrogels, G′of CN5 and CN10 hydrogels increased ≈2–5 times, respectively compared to control PN and MAC hydrogel. (B) Gelation time of PN, MAC and CN10 hydrogels (inset figure showing flow property of CN10 formulation, droplet coming out of the 21 G needle). | ||
The next parameter that also dictates the scaffold's versatility as an injectable system is the need to have relatively fast gelation kinetics. Very interestingly all our hydrogel formulations had very fast gelation time (>400 s, 37 °C). After injection, fast gelling of CN hydrogels offers potential advantages at the target site such as minimizing the diffusion of precursors to neighboring tissues, that in turn enables site specific delivery of any loaded bioactive moieties.12 Fig. 2B shows the time dependent gelation of CN10, PN and MAC hydrogels. PN and MAC had relatively fast gelation compared to CN10 hydrogel. In most in situ gelling systems, crosslinking density and gelation kinetics are inextricably linked (both governed by the degree of precursor polymer functionalization), representing a challenge in rational hydrogel design of highly cross-linked gels that can still be delivered by injection or weaker gels that need to be gelled quickly. Another important parameter that dictates the efficacy of administration of an injectable formulation is its shear-thinning properties, i.e. that it should have relatively low viscosity to allow for easy injection through a needle.12,19 All the CN hydrogel precursors were highly soluble in aqueous medium and were able to inject easily through the needle. Inset of Fig. 2B illustrates the flow property of CN10 hydrogel precursors through 21 G needle.
The impetus for the development of intelligent hydrogels over common hydrogels is due to their response to external stimulus, hence the evaluation of swelling properties is very important as it plays an important role in the release of drug or bioactives under physiological conditions at specific sites in a controlled and predetermined fashion.20 In order to quantify the responsiveness of CN hydrogels as a function of temperature change, we evaluated their swelling properties at 25 °C and 37 °C (Fig. 3A). The swelling behavior of crosslinked hydrogel matrix depends on many factors such as the: (i) nature of ionizable groups; (ii) polymer composition; (iii) hydrophobicity/hydrophilicity of the polymer backbone; and (iv) crosslinking density. The ratio of MAC with respect to NIPAm monomer had significant influence on the thermoresponsive behavior of the resulting CN hydrogels at 25 °C and 37 °C. In general, the hydrogels with lower collagen content showed a higher swelling ratio (SR) at 25 °C compared to 37 °C, because of the inherent ability of pNIPAm to change hydrophilicity in response to temperature change. The pNIPAm polymer is hydrophobic above its lower critical temperature (LCST) and is hydrophilic below LCST (32 °C). Therefore, the hydrogels showed higher SR at 25 °C compared to the hydrogels at 37 °C. Increasing the concentration of collagen diminished the temperature-stimuli response due to the higher concentration of the hydrophilic component, which increases the crosslinking density of the formulation and also overcomes the hydrophobic interactions of isopropyl groups in pNIPAm, responsible for VPT.21,22 Overall, the CN5 and CN10 hydrogel formulation showed superior thermoresponsive properties compared to other formulations. Fig. 3B illustrates the linear swelling profiles of CN5 and CN10 hydrogels.
 | ||
| Fig. 3 (A) Temperature-dependent swelling profiles of all formulations after 180 h. (B) Temperature-dependent linear swelling profiles of all CN05 and CN10 hydrogels over a time period of 180 h. (C) Collagenase degradation profiles of MAC, CN10, CN20 and CN30 hydrogels. | ||
After administration of hydrogels in vivo it should be stable over a long period in order to achieve sustained therapeutic efficiency.23 Therefore, the stability of CN hydrogels against enzymatic degradation was further investigated. The formation of a covalently linked IPN structure resulted in significantly higher stability when exposed to high concentrations of collagenase (5 U mL−1). Fig. 3C shows a comparison of the collagenase degradation profiles of CN10, CN20, CN30 and MAC hydrogels. 40% and 80% of MAC hydrogel was degraded in 32 and 70 hours, respectively, and was completely degraded after 96 hours, whereas CN hydrogels showed significantly higher resistance against collagenase. 70% of CN10, 75% of CN20, and 89% of CN30 hydrogels remained intact even after a week. The enzymatic resistance of these hydrogels can be attributed to the formation of more compact IPN structure with high crosslinking density.24 The synergetic effect of the covalently linked IPN from the natural and synthetic components enhanced the enzymatic resistance resulting in a stable hydrogel for long-term applications.25 Additionally, the presence of reactive functional groups also provides the flexibility to modulate the rate of degradation of hydrogels by incorporating matrix cleavable sequences inside the bio matrix that could be used for soft tissue applications, and this will be demonstrated in our further studies. Of all the tested compositions (Table 1), the CN10 hydrogel showed the optimal injectable and responsive properties with highest collagen content and was therefore chosen for further studies.
We further investigated the pH-responsive swelling and de-swelling properties and temperature dependent phase-transition properties of the CN10 hydrogel. Fig. 4A shows the temperature dependent response index profiles of CN10 hydrogel at different solution pH and alternating temperatures of 25 °C and 37 °C. The hydrogels in pH 2 and pH 7.4 solution showed no marked difference in swelling at 25 °C, however higher difference (25%) was observed in their shrinking behavior at 37 °C. The hydrogel in pH 7.4 solution showed higher shrinkage compared to the hydrogel at pH 2. This behavior can be attributed to the pH dependence of the carboxylic acid groups present on the collagen side chains. The carboxylic acid groups (–COOH) are converted into carboxylate anions (–COO−) at pH 7.4 leading to the electrostatic repulsion between negatively charged carboxylate anions while under acidic conditions, stabilization of hydrogen bonds between –COOH groups overcomes the repulsive forces, making hydrogels structurally more compact.26 Further the formation of strong hydrogen bonds between –COOH groups of collagen and amide groups of pNIPAm polymer imparted higher structural rigidity at acidic pH leading to lower shrinkage of hydrogels at pH 2 compared to pH 7.4.
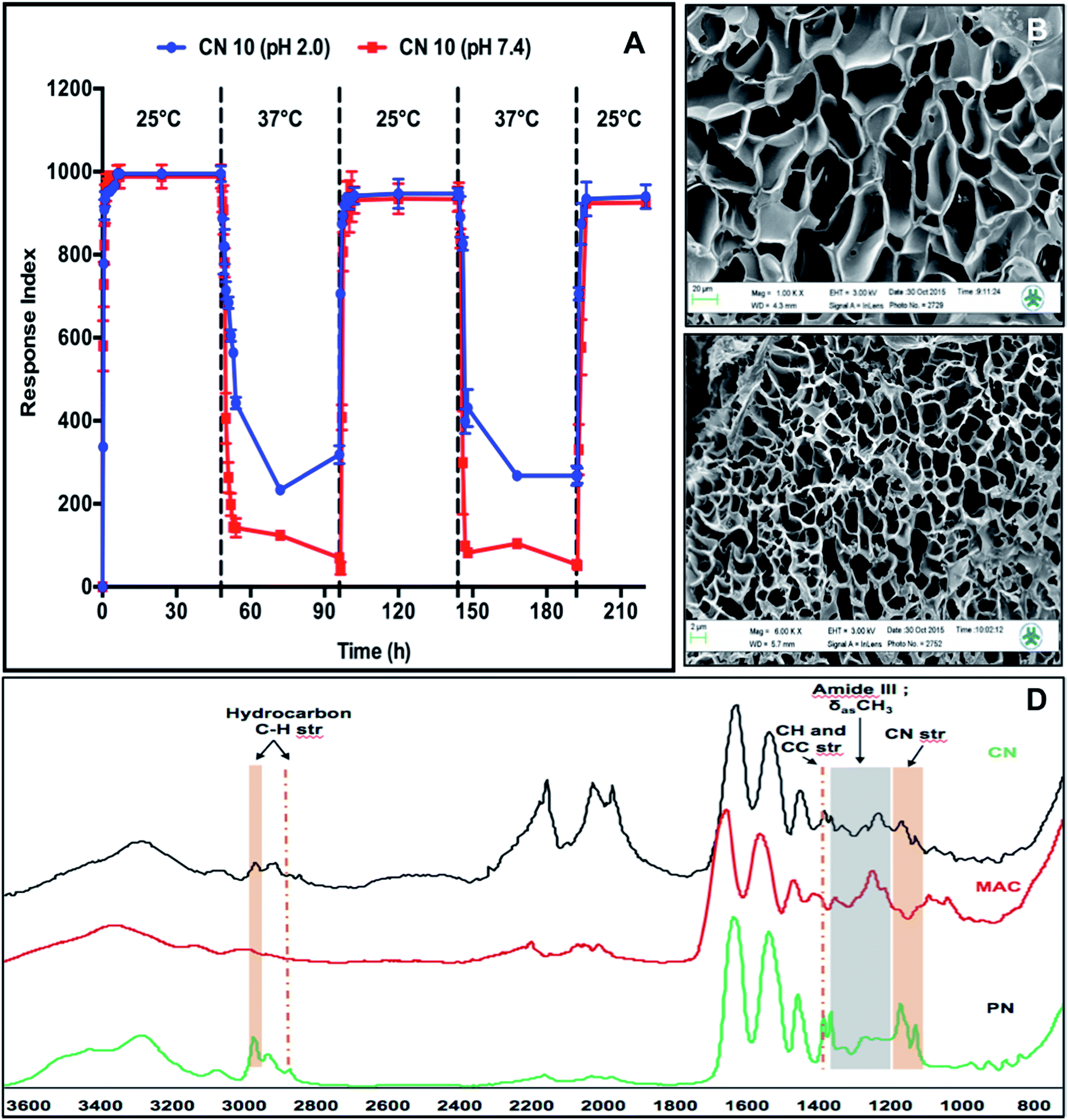 | ||
| Fig. 4 Physio-chemical characterizations of CN10 hydrogels. (A) pH dependent reversible responsiveness of CN10 hydrogels at 25 °C and 37 °C. (B and C) SEM cross-sectional images of CN10 hydrogel at 25 °C and 37 °C respectively (scale bar = 100 μm). (D) FT-IR measurements of CN10, MAC and PN hydrogels. | ||
The phase transition of CN10 hydrogels was further determined using UV-VIS spectrometry over a temperature range of 30–39 °C. The most commonly used temperature-sensitive hydrogel is pNIPAm, which shows a sharp volume phase transition (VPT) at approximately 32–33 °C, but the LCST can be increased towards physiological temperatures by copolymerizing it with hydrophilic polymers.27 Below the LCST, strong hydrogen bonds are formed between hydrophilic amide groups of pNIPAm and water molecules that are responsible for absorption of water. Increasing the temperature above the LCST causes disruption of hydrogen bonds due to hydrophobic aggregation of isopropyl groups, leading to the phase transition of the hydrogel network.28,29 As the relatively hydrophilic MAC proportion increases compared to the pNIPAM proportion, the LCST value also increases due to the strong hydrogen bonding with water.28,29 The LCST of CN10 hydrogels was calculated based on the inflexion point of the curve of hydrogels. MAC hydrogel was used as a control and it showed no signs of volume transition over the observed temperature range (30–39 °C), whereas CN10 hydrogel showed a transition from 33 to 36.95 °C (data not shown), suggesting that the hydrophilic groups of collagen contribute greatly to the increase of the LCST of the hydrogels. This flexibility of CN hydrogels to tune the LCST indicates their potential to design scaffolds with suitable properties for target specific application.
The structural and functional composition in the CN10 hydrogel was further characterized using SEM and IR respectively. The changes in CN10 hydrogel morphology, associated with temperature responsiveness, examined using SEM showed a significant structural difference (Fig. 4B and C). The reticulated porous networks of CN10 hydrogel was more compact at 37 °C compared to 25 °C. The average diameter of pores at 25 °C and 37 °C was around 44 ± 10 μm and 3 ± 0.69 μm respectively. Fig. 4D shows the FT-IR spectra for the CN10, MAC and PN hydrogels, respectively. The CN10 hydrogel spectrum shows the corresponding functional group peaks of both collagen and pNIPAm, indicating the presence of an IPN structure. The absorption peaks in the CN10 hydrogel at 1400 (C–(CH3)2) and 1150 cm−1 (C–N of amide III) resulted from C–H, C–C and C–N stretching from pNIPAm, and the peaks between 2800 and 3000 cm−1 correspond to C–H stretching vibrations of –CH3 and –CH2– groups. Furthermore, the presence of a collagen amide III band at 1250 cm−1 confirmed the presence of the IPN network in the CN10 hydrogel.
Sustained release of BSA and VES from CN10 hydrogels
The dual-stimuli responsiveness (pH and temperature) of CN10 hydrogels with appropriate material properties makes them attractive to be utilized as an injectable delivery vehicle for controlled drug release. The one-pot design of fast-gelling injectable hydrogels allowed large protein and small drug molecules to be encapsulated homogeneously inside the hydrogel matrix during the fabrication process. We first encapsulated Bovine Serum Albumin (BSA) as a model protein to demonstrate the potential of the CN10 hydrogel as an in situ depot-forming system for controlled and sustained localized delivery of large protein molecules. The encapsulation efficiency of BSA was 92% and it exhibited zero-order drug release in response to external stimulus. The release profiles of BSA from the CN10 hydrogels under different pH and temperature conditions are shown in Fig. 5A and B. Higher BSA release from the hydrogel was observed at pH 7.4 compared to pH 2. This behavior can be attributed to the electrostatic repulsions between the negatively charged carboxylate groups in collagen and BSA at pH 7.4, which would accelerate its release.16,30,31 At pH 7.4, 60% and 90% of BSA was released after 120 and 200 hours, respectively, at 37 °C, whereas only 50% and 60% of BSA was released after 120 and 200 hours at 25 °C and this remained static up to 360 hours. Previous studies have shown that, BSA has higher binding affinity on pNIPAm microgels at 25 °C than at 37 °C, which corresponds to our studies.32 Furthermore the higher release at 37 °C can also be attributed to the reduced intermolecular attraction between the protein and polymeric networks due to the shrinkage of pNIPAm, leading to higher diffusion rate.16 On the other hand, the release of BSA was slower under acidic condition due to strong hydrogen bonding within the hydrogel networks as well as electrostatic interactions between BSA and the network.31 In pH 2 solution, 45% and 60% of BSA was released after 120 and 360 hours, respectively, at both 25 °C and 37 °C.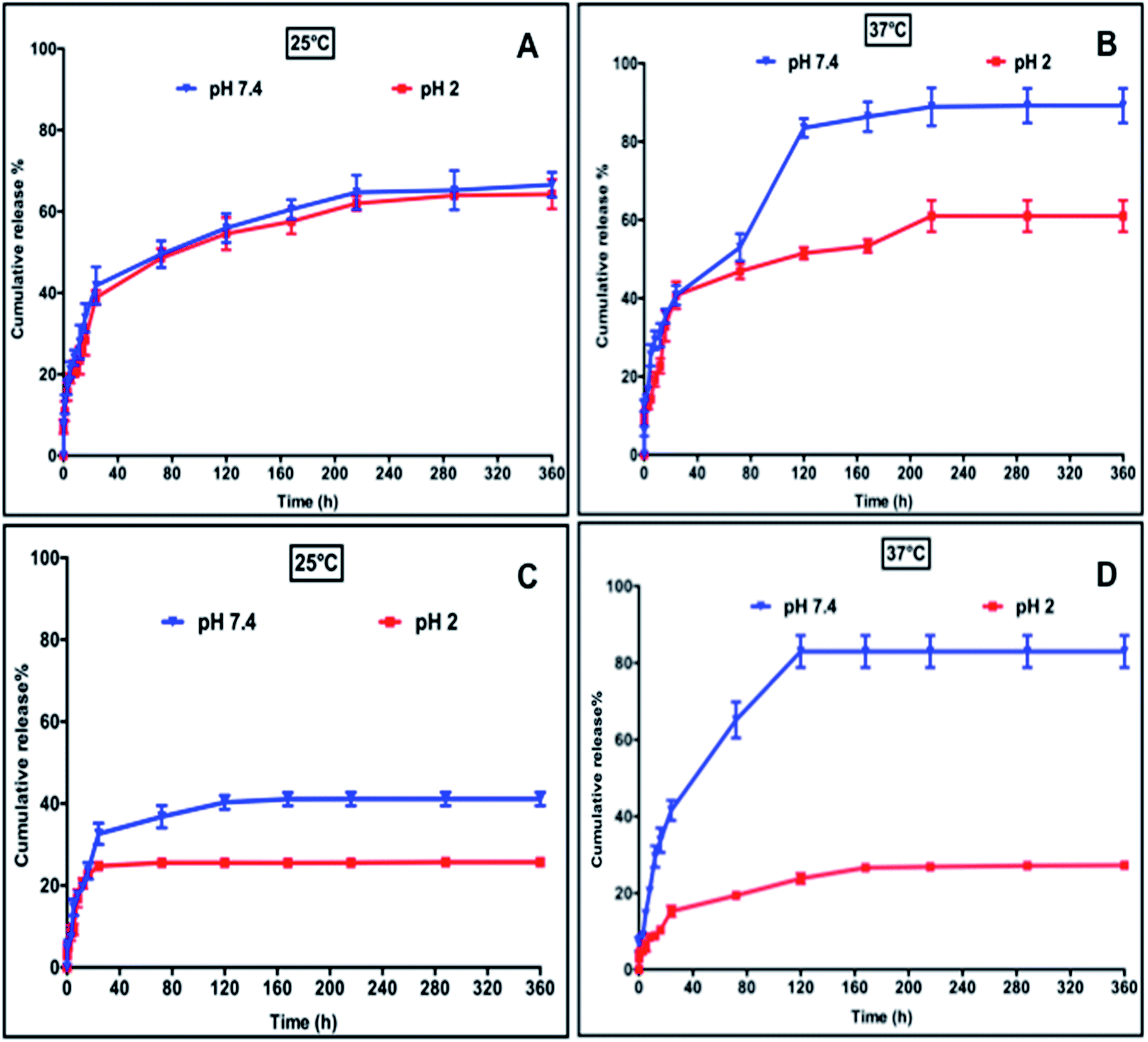 | ||
| Fig. 5 Release profiles of BSA (A and B) and VES (C and D) from CN10 hydrogel matrix at different pH and temperatures. | ||
Vitamin E succinate (VES), a soluble form of vitamin E is one of the most effective anticancer derivatives of the vitamin E family. VES has been shown to be highly selective for malignant cells whereas it is largely non-toxic to normal cells.33 Apart from its potent anti-cancerous properties, VES also possess anti-inflammatory properties33 and was chosen as a model drug to evaluate the release properties of the CN10 hydrogel matrix as a carrier for the site specific delivery of small drug molecules. The release kinetics of VES under different conditions are shown in Fig. 5C and D. In general, the release of VES was significantly slower under acidic conditions. At pH 2, only 25–30% of VES was released at both 25 °C and 37 °C after 120 hours. This is due to the hydrogen bonding between the VES and hydrogel networks. The carboxylic acid groups in collagen and VES become protonated under acidic conditions, resulting in a strong hydrogen bond between the hydrogel and VES, which hinders the release under acidic conditions. On the other hand, the electrostatic repulsion between carboxylate anions of VES and collagen resulted in higher release of VES at pH 7.4. At pH 7.4, 40% and 80% of VES was released at 25 °C and 37 °C, respectively, after 120 hours. The higher release at 37 °C can be attributed to the enhanced diffusion rate resulting from the shrinkage of pNIPAm. In general, any bioactive release from hydrogel matrix depends on (i.) molecular weight of the drug, (ii.) molecular architecture and functionalities of bioactive moieties and (iii.) polymer precursors.27,34 However, in stimuli responsive hydrogels the response of the hydrogel matrix architecture with respect to external stimuli also influences the diffusion rate due to their matrix switching properties.20 In recent years, several types of conventional hydrogels based on natural and synthetic polymeric systems have been widely used for delivering a series of drugs.35 Stimuli responsive hydrogels add further strength to the field as a self-regulating system capable of precisely releasing a drug in response to physiological needs. As stated above, the presence of a hydrophilic component (collagen) tuned the LCST of the CN10 hydrogel close to physiological temperature and renders it more attractive from a self-regulated drug release perspective. The difference in release rate, demonstrated by CN10 hydrogels under different stimuli conditions, can be a result of the interplay between many factors such as interactions between: (i) polymer–polymer and polymer–drug; and (ii) polymer and ionic strength of surrounding release media.35,36
From our results it is evident that all these factors influence the diffusion of drug/protein and demonstrates the use of CN10 hydrogel matrix as a self-regulated tunable release system for bioactive molecules under different environmental conditions.
Biocompatibility
The biocompatibility of CN hydrogels was evaluated by cultivating human skeletal muscle cells (SkMC) on CN10 and CN50 hydrogel surfaces. Post seeding, live SkMC cells were visualized by calcein staining showing viable cells attached on the hydrogel surface at 72 hours of culture on CN10 and CN50 hydrogels where collagen-I pre-coated Tissue Culture Plates (TCP) served as control (Fig. 6A). We observed no immediate signs of toxicity from the CN hydrogels as the cells were attached and successfully cultured for several days. Proliferation of SkMCs growing on CN10 hydrogels was further quantified using MTS assay. As shown in Fig. 6B, no significant differences in metabolic activity of cells grown 6 days on CN10 hydrogel was observed as compared to control TCP. Confluency is an important hallmark for muscle cell progenitors to be able to differentiate by fusion forming multinucleated myotubes in the development of skeletal muscles.37,38 We therefore evaluated the occupancy of cells after 3 and 9 days on CN10 hydrogel surface by phalloidin–DAPI staining (Fig. 6C). After 7–9 days cultivation in proliferation medium on CN10 hydrogel, the SkMCs displayed a densely packed monolayer covering the total surface. Taken together the above results show that, CN10 hydrogels are cyto-compatible, supported proliferation of human SkMCs at the same rate as conventional collagen-coated plates. They also facilitated easy adhesion of cells upon seeding, which remained attached during cultivation at 37 °C.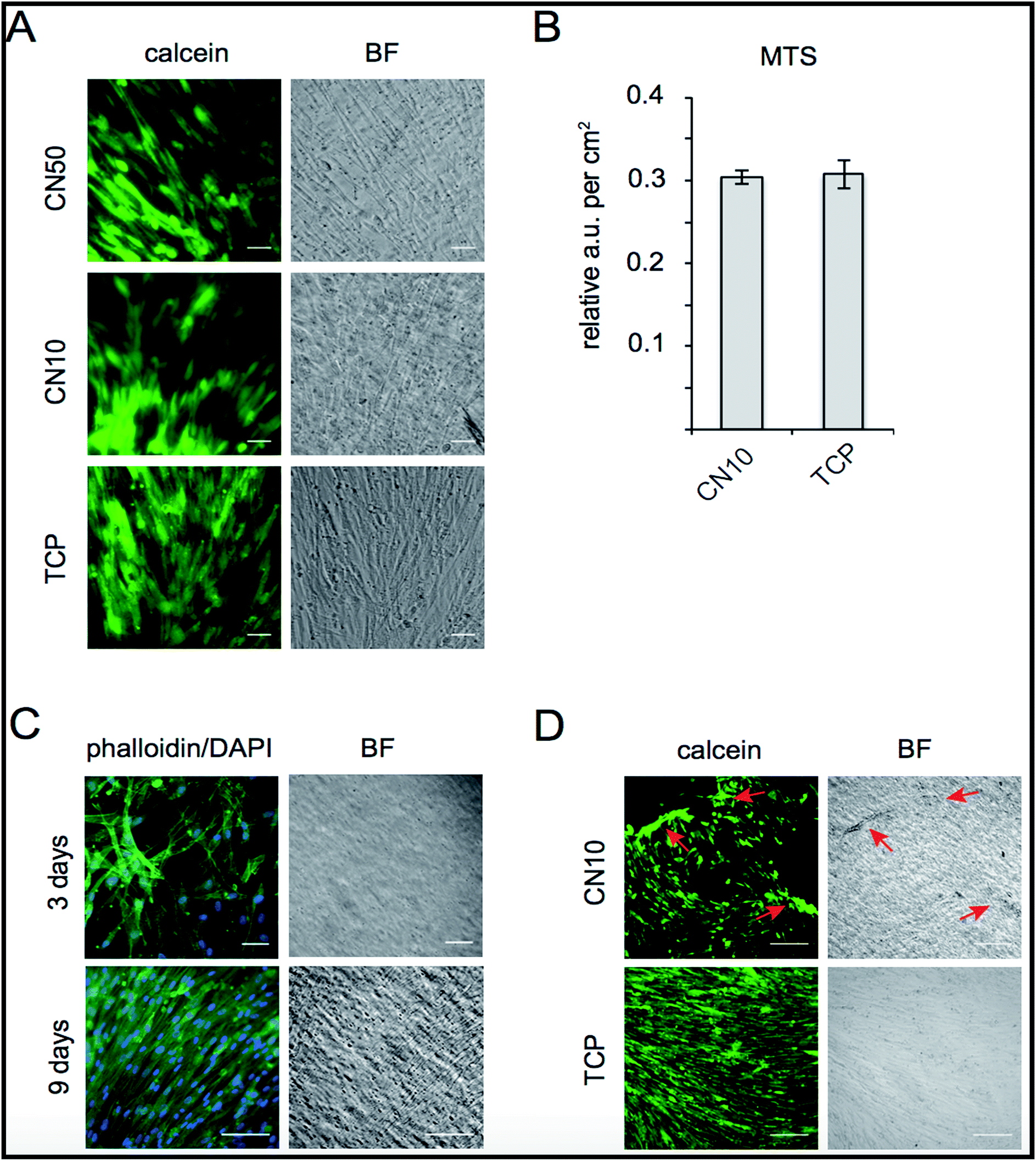 | ||
| Fig. 6 Human SkMC growth characteristics, morphology and detachment. (A) Viable SkMC visualized by calcein staining (green) after 72 h cultivation on CN50, CN10 and TCP (BF-bright field, scale bar = 50 μm). (B) Metabolic activity assessed using MTS assay as a measurement of proliferation at 6 days on CN10 compared to TCP. (C) SkMC morphology on CN10 at 3 days assayed by phalloidin (green) illustrating intra-cellular filaments and nuclear staining with DAPI (blue) SkMC (scale bar = 50 μm) and densely packed SkMC after 9 days (scale bar = 200 μm). (D) Demonstration of cell detachment after 30 min at 25 °C of calcein stained SkMCs (red arrows) from CN10 surface (scale bar = 200 μm). | ||
Interestingly, we also observed that a cellular monolayer could be detached from the CN10 hydrogel surface simply by transferring the hydrogel to room temperature, due to the hydrophobic to hydrophilic transition of pNIPAm (Fig. 6D). This holds great promise for the use of collagen derived hydrogel matrix for cell sheet engineering applications in future.39,40 As mentioned, a sheet of cells could be easily detached by decreasing the temperature, thus preserving cell health and without any enzymatic action (known to be potentially deleterious for the cells). This opens up a myriad of possibilities for applications of cell-based therapy with intelligent properties exploitable both in vivo and/or in vitro as a smart cell detachment method. Cell sheet engineering is considered as one of promising method to treat damaged soft tissue surfaces such as periodontium, ocular epithelium and endothelium, esophageal mucosa, pancreatic islets, hepatic tissue etc,41 This is the first report to demonstrate the compatibility and smart detachment of human skeletal muscle cells from a type-I-collagen-based hydrogel surface and the method could be used to create effective 3D scaffolds for cell sheet engineering applications.
Experimental section
Materials and methods
Synthesis of MAC is reported in our previous work.14 All chemicals were purchased from Sigma, unless otherwise mentioned.Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 SkMCs (ScienCell Research Laboratories, CA, USA) were seeded on every 500 μm CN10, CN50 scaffolds and the control tissue culture plates pre-coated with collagen I (1 mg mL−1, Sigma Aldrich, USA). After 72 hours of cultivation of SkMCs in skeletal muscle cell medium (ScienCell Research Laboratories, CA, USA), live staining was done by adding calcein AM (1 mM, Sigma Aldrich, USA) for 30 minutes to the samples. For morphology analysis cells were fixed after 3 and 9 days' cultivation with 4% paraformaldehyde for 12 minutes at 37 °C and stained with phalloidin-488 (1:1000, CytoPainter Phalloidin-iFluor 488, Abcam). Nuclei were stained with DAPI (Fluorescent Mounting media, Merck, Millipore, MA. USA) All imaging was achieved using an inverted microscope (Leica DMI6000 B). Relative proliferation of SkMCs on the different surfaces was assessed using a MTS assay kit (CellTiter 96® Aqueous assay, Promega Biosciences, CA, USA). 10% MTS mix reagent was added to samples after 6 days' post seeding and the quantity of formazan was measured at 492 nm using CLARIOstar® (BMG LABTECH, Ortenberg, Germany). All cultivations were done in triplicate (n = 3).
000 SkMCs (ScienCell Research Laboratories, CA, USA) were seeded on every 500 μm CN10, CN50 scaffolds and the control tissue culture plates pre-coated with collagen I (1 mg mL−1, Sigma Aldrich, USA). After 72 hours of cultivation of SkMCs in skeletal muscle cell medium (ScienCell Research Laboratories, CA, USA), live staining was done by adding calcein AM (1 mM, Sigma Aldrich, USA) for 30 minutes to the samples. For morphology analysis cells were fixed after 3 and 9 days' cultivation with 4% paraformaldehyde for 12 minutes at 37 °C and stained with phalloidin-488 (1:1000, CytoPainter Phalloidin-iFluor 488, Abcam). Nuclei were stained with DAPI (Fluorescent Mounting media, Merck, Millipore, MA. USA) All imaging was achieved using an inverted microscope (Leica DMI6000 B). Relative proliferation of SkMCs on the different surfaces was assessed using a MTS assay kit (CellTiter 96® Aqueous assay, Promega Biosciences, CA, USA). 10% MTS mix reagent was added to samples after 6 days' post seeding and the quantity of formazan was measured at 492 nm using CLARIOstar® (BMG LABTECH, Ortenberg, Germany). All cultivations were done in triplicate (n = 3).Summary and conclusions
In summary, we have developed a modular platform, based on methacrylated collagen building blocks, to engineer multi-component ECM mimicking scaffolds with pre-defined properties that can be tuned rationally by varying the composition of reacting components. As a proof-of concept, we have demonstrated a one-pot approach to fabricate novel covalently cross-linked thermo- and pH-responsive injectable collagen hydrogels under physiological conditions. The free radical co-polymerization of MAC and NIPAm in presence of MBAA formed a covalently linked double network, a biocompatible hydrophilic network that mimics the natural ECM, and a stimuli-responsive network that undergoes volume-phase transition (VPT) from hydrophilic to hydrophobic in response to temperature. The resultant hydrogels were characterized in detail and their application as a support to cells and a carrier for the controlled release of bioactive molecules was demonstrated. Bovine Serum Albumin (BSA) and vitamin E succinate (VES) were encapsulated easily in the hydrogels as a model protein and small drug molecules, respectively. The hydrogel exhibited a sustained release of BSA and VES over 2 weeks under physiological conditions, indicating the suitability of the hydrogel for the delivery of both, small drug molecules as well as large protein molecules. Additionally, varying the temperature and pH of the release media could also modulate the release of BSA and VES from the hydrogel. Furthermore, the hydrogels were biocompatible as shown by excellent cell viability and proliferation capacity when seeded with human skeletal muscle cells (SkMC). Combinatorial impacts of covalent crosslinking of collagen with other stimuli responsive polymers (light, chemical, biological etc.), to render it as a multi-responsive system, are as yet unexplored and the applications of such advanced polymeric materials are potentially wide spread.44–47 We believe the fundamental design behind our current work and the results of this study illustrated a great potential for methacrylated collagen as a building block for developing ECM mimetic tailor-made scaffolds with tunable properties for a wide range of biomedical and tissue engineering applications.Acknowledgements
This work was funded by the Swedish Research Council Junior Researcher Project Grant dnr (621-2012-4286) to JP, CeNano PhD student salary grant to JP and RR. VC and CA were financially supported by FP7-Health-Innovation 2-2013-601700 program, acronym HESUB. The authors wish to acknowledge Thomas Lingerfelt for helping to record SEM pictures, Dr Daniel Aili, Karl Schenström and Jönas Christofferson for their experimental support.Notes and references
- F. Gattazzo, A. Urciuolo and P. Bonaldo, Biochim. Biophys. Acta, 2014, 1840, 2506–2519 CrossRef CAS PubMed.
- C. C. DuFort, M. J. Paszek and V. M. Weaver, Nat. Rev. Mol. Cell Biol., 2011, 12, 308–319 CrossRef CAS PubMed.
- B. Trappmann and C. S. Chen, Curr. Opin. Biotechnol., 2013, 24, 948–953 CrossRef CAS PubMed.
- H. Geckil, F. Xu, X. Zhang, S. Moon and U. Demirci, Nanomedicine, 2010, 5, 469–484 CrossRef CAS PubMed.
- W. P. Daley, S. B. Peters and M. Larsen, J. Cell Sci., 2008, 121, 255–264 CrossRef CAS PubMed.
- P. Lu, K. Takai, V. M. Weaver and Z. Werb, Cold Spring Harbor Perspect. Biol., 2011, 3, a005058 Search PubMed.
- K. Matyjaszewski, Science, 2011, 333, 1104–1105 CrossRef CAS PubMed.
- F. L. Zhao, D. Yao, R. W. Guo, L. D. Deng, A. J. Dong and J. H. Zhang, Nanomaterials, 2015, 5, 2054–2130 CrossRef CAS PubMed.
- M. Patenaude, N. M. B. Smeets and T. Hoare, Macromol. Rapid Commun., 2014, 35, 598–617 CrossRef CAS PubMed.
- T. Miao, S. L. Fenn, P. N. Charron and R. A. Oldinski, Biomacromolecules, 2015, 16, 3740–3750 CrossRef CAS PubMed.
- Y. Li, J. Rodrigues and H. Tomas, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC.
- E. Bakaic, N. M. B. Smeets and T. Hoare, RSC Adv., 2015, 5, 35469–35486 RSC.
- J. F. Mano, G. A. Silva, H. S. Azevedo, P. B. Malafaya, R. A. Sousa, S. S. Silva, L. F. Boesel, J. M. Oliveira, T. C. Santos, A. P. Marques, N. M. Neves and R. L. Reis, J. R. Soc., Interface, 2007, 4, 999–1030 CrossRef CAS PubMed.
- R. Ravichandran, M. M. Islam, E. I. Alarcon, A. Samanta, S. Wang, P. Lundstrom, J. Hilborn, M. Griffith and J. Phopase, J. Mater. Chem. B, 2016, 4, 318–326 RSC.
- M.-T. Nistor, A. P. Chiriac, C. Vasile, L. Verestiuc and L. E. Nita, Colloids Surf., B, 2011, 87, 382–390 CrossRef CAS PubMed.
- Y. Zheng, B. Wang, M. Liu, K. Jiang, L. Wang and Y. Yu, Colloid Polym. Sci., 2015, 293, 1615–1621 CAS.
- D. E. Discher, D. J. Mooney and P. W. Zandstra, Science, 2009, 324, 1673–1677 CrossRef CAS PubMed.
- J. Zhu and R. E. Marchant, Expert Rev. Med. Devices, 2011, 8, 607–626 CrossRef CAS PubMed.
- M. K. Nguyen and D. S. Lee, Macromol. Biosci., 2010, 10, 563–579 CrossRef CAS PubMed.
- S. Chaterji, I. K. Kwon and K. Park, Prog. Polym. Sci., 2007, 32, 1083–1122 CrossRef CAS PubMed.
- M. Yang and K. Zhao, J. Phys. Chem. B, 2015, 119, 13198–13207 CrossRef CAS PubMed.
- Y. Zhou, H. Tang and P. Wu, Phys. Chem. Chem. Phys., 2015, 17, 25525–25535 RSC.
- R. Vaishya, V. Khurana, S. Patel and A. K. Mitra, Expert Opin. Drug Delivery, 2015, 12, 415–440 CrossRef CAS PubMed.
- A. Borzacchiello, L. Russo, B. M. Malle, K. Schwach-Abdellaoui and L. Ambrosio, BioMed Res. Int., 2015, 2015, 12 Search PubMed.
- K. H. Bae, L. S. Wang and M. Kurisawa, J. Mater. Chem. B, 2013, 1, 5371–5388 RSC.
- K. Hemmati and M. Ghaemy, Int. J. Biol. Macromol., 2016, 87, 415–425 CrossRef CAS PubMed.
- W. B. Liechty, D. R. Kryscio, B. V. Slaughter and N. A. Peppas, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 149–173 CrossRef CAS PubMed.
- S. J. Lue, C.-H. Chen and C.-M. Shih, J. Macromol. Sci., Part B: Phys., 2011, 50, 563–579 CrossRef CAS.
- C. Fucinos, P. Fucinos, M. Miguez, I. Katime, L. M. Pastrana and M. L. Rua, PLoS One, 2014, 9, e87190 Search PubMed.
- X. Cheng, H. E. Canavan, D. J. Graham, D. G. Castner and B. D. Ratner, Biointerphases, 2006, 1, 61 CrossRef CAS PubMed.
- X. L. Zhu, X. L. Gu, L. N. Zhang and X. Z. Kong, Nanoscale Res. Lett., 2012, 7, 519 CrossRef PubMed.
- V. Grabstain and H. Bianco-Peled, Biotechnol. Prog., 2003, 19, 1728–1733 CrossRef CAS PubMed.
- Y. H. Dong, Y. H. Guo and X. B. Gu, Aizheng, 2009, 28, 1114–1118 CAS.
- M. K. Nguyen and E. Alsberg, Prog. Polym. Sci., 2014, 39, 1236–1265 CrossRef PubMed.
- D. Y. Kim, D. Y. Kwon, J. S. Kwon, J. H. Kim, B. H. Min and M. S. Kim, Polym. Rev., 2015, 55, 407–452 CrossRef CAS.
- M. W. Tibbitt, J. E. Dahlman and R. Langer, J. Am. Chem. Soc., 2016, 138, 704–717 CrossRef CAS PubMed.
- R. O. Godinho, J. Morphol. Sci., 2006, 23, 173–186 Search PubMed.
- J. T. Doherty, K. C. Lenhart, M. V. Cameron, C. P. Mack, F. L. Conlon and J. M. Taylor, J. Biol. Chem., 2011, 286, 25903–25921 CrossRef CAS PubMed.
- N. G. Patel and G. Zhang, Organogenesis, 2013, 9, 93–100 CrossRef PubMed.
- C. Somaiah, A. Kumar, D. Mawrie, A. Sharma, S. D. Patil, J. Bhattacharyya, R. Swaminathan and B. G. Jaganathan, PLoS One, 2015, 10, e0145068 Search PubMed.
- M. Egami, Y. Haraguchi, T. Shimizu, M. Yamato and T. Okano, Arch. Pharmacal Res., 2014, 37, 96–106 CrossRef CAS PubMed.
- H. Cicek and A. Tuncel, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 527–541 CrossRef CAS.
- L. Koh, M. Islam, D. Mitra, C. Noel, K. Merrett, S. Odorcic, P. Fagerholm, W. Jackson, B. Liedberg, J. Phopase and M. Griffith, J. Funct. Biomater., 2013, 4, 162 CrossRef PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- P. Schattling, F. D. Jochum and P. Theato, Polym. Chem., 2014, 5, 25–36 RSC.
- O. Parlak, A. P. F. Turner and A. Tiwari, Adv. Mater., 2014, 26, 482–486 CrossRef CAS PubMed.
- O. Parlak, M. Ashaduzzaman, S. B. Kollipara, A. Tiwari and A. P. F. Turner, ACS Appl. Mater. Interfaces, 2015, 7, 23837–23847 CAS.
| This journal is © The Royal Society of Chemistry 2017 |