
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvates and polymorphs of rebamipide: preparation, characterization, and physicochemical analysis†
Xinnuo Xiong,
Qiaohong Du,
Xia Zeng,
Jiawei He,
Hongqin Yang and
Hui Li *
*
College of Chemical Engineering, Sichuan University, Chengdu, Sichuan, China. E-mail: lihuilab@sina.com; Fax: +86 028 85401207; Tel: +86 028 85405149
First published on 27th April 2017
Abstract
This study aimed to investigate the solid forms of the peptic ulcer agent rebamipide (RBM). Four RBM solvates, including dimethyl sulfoxide solvate (RBM/DMSO), dichloromethane solvate (RBM/CH2Cl2), hydrate (RBM/H2O), and hemiethanol hemihydrate, were prepared via solvent crystallization. Single crystals of RBM/DMSO and RBM/CH2Cl2 were successfully obtained at 4 °C. These solid forms were both monoclinic with the space group P21/c. Hirshfeld surface and 2D fingerprint plot analyses indicated that RBM/DMSO and RBM/CH2Cl2 showed similarities in hydrogen bonds but exhibited differences in subtle intermolecular interactions. RBM/CH2Cl2 transformed into a novel solid form (form 3) after solvent removal and transformed into another solid form (form 4) when heated at 220 °C. RBM/H2O was converted to form 5 after dehydration. Forms 3–5 were RBM polymorphs. The stability results indicated that RBM/H2O, form 3, and form 4 showed good stability at 40 °C and 75% relative humidity. Furthermore, the three solid forms displayed higher solubility than commercial RBM (form 1) in both water and phosphate buffer solution (PBS). Compared with form 1, RBM/H2O and form 4 showed a comparable and faster dissolution rate in water and PBS, respectively.
1. Introduction
Polymorphism is the ability of a compound to crystallize in different forms, such as polymorphs, solvates, hydrates, and co-crystals, and is a common phenomenon with important applications in academic and industrial research.1–3 Various solid forms of active pharmaceutical ingredients (APIs) possess significantly different physicochemical properties, including solubility, dissolution rate, stability, and bioavailability.4–7 Therefore, medical researchers must select the optimal solid form of APIs to maximize therapeutic benefits. In addition, different polymorphs may be granted individual patent protection, and the commercial interests of a company could be threatened by the discovery of novel polymorphs.8 Thus, investigating the polymorphism of APIs is crucial and has practical utility in pharmaceutical research and development.9,10Solvates, the crystal forms of a compound that differ in the species or stoichiometry of the included solvent molecules, are widely applied in the pharmaceutical industry.10 Accidental solvate formation is undesirable during drug processing because of potentially uncontrollable changes in the physicochemical properties of the drug product.11 Conversely, solvates with pharmaceutically relevant solvents may improve the physicochemical properties of drugs.12 The developmental capacity of APIs have been improved with solvates, such as darunavir ethanolate,13 indinavir sulfate ethanolate,14 acetone solvate of cabazitaxel,15 canagliflozin hydrate,16 and dimethyl sulfoxide (DMSO) solvate of trametinib.17 Moreover, novel modifications that may be unattainable via ordinary crystallization techniques can be formed by the desolvation (dehydration) of solvates (hydrates).18,19 Thus, investigating the solvates of APIs and corresponding solid forms after desolvation is of great importance.
Rebamipide (RBM) is used to treat gastric ulcers, acute gastritis, exacerbated chronic gastritis, and dry eye.20–22 Two polymorphs (forms 1 and 2),23 one monohydrate,24 two co-crystals,25 one solvate (methanol solvate), and several salts26 of RBM have been reported. In our previous study, we compared the amorphous forms of RBM that were obtained via milling and spray drying.27 The existence of RBM polymorphs and solvates, however, has not been systematically investigated.
Thus, this study aimed to conduct a detailed investigation on the solid forms of RBM, including solvates and polymorphs. Four RBM solvates, namely, dimethyl sulfoxide solvate (RBM/DMSO), dichloromethane solvate (RBM/CH2Cl2), hydrate (RBM/H2O), and hemiethanol hemihydrate (RBM/EtOH/H2O), were prepared through solvent crystallization. After the single crystals of RBM/DMSO and RBM/CH2Cl2 were successfully obtained, their structures were determined via single-crystal X-ray diffraction (SXRD). Hirshfeld surface (HS) and 2D fingerprint plot (FP) were applied to analyze the intermolecular interactions of the two solvates. Although RBM/H2O has been previously reported in a Chinese patent,24 more detailed investigations were performed in the present study. In addition, the phase transitions of the solvates upon heating were investigated, and three novel polymorphs of RBM (forms 3–5) were obtained in this process. The stability of the solvates and polymorphs was explored. Based on the results, we further investigated the solubility and dissolution rate of RBM/H2O, form 3, and form 4, the stable solid forms of RBM.
2. Experimental
2.1. Materials
RBM (99% pure) was purchased from YiTai Technology Co., Ltd. (Shanghai, China) and was consistent with form 1 as reported by Jeon and Sohn (Fig. S1†).23 Ultrapure water (18 MΩ resistivity from a Millipore system) was used throughout the experiment. DMSO, CH2Cl2, methanol (MeOH), ethanol (EtOH), and other solvents were of analytical grade and were purchased from Kelong Co., Ltd. (Chengdu, China).2.2. Preparation of RBM solvates and polymorphs
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) solvent mixture via ultrasonication. The resulting solution was then slowly evaporated at 4 °C. Finally, colorless single crystals suitable for SXRD were obtained.:1 v/v) with heating and stirring at 55 °C. Then, the solution was cooled to room temperature and slowly evaporated under ambient conditions. White globular solids were obtained after approximately 10 days. However, we failed to obtain single crystals suitable for SXRD.
:1 v/v) solvent mixture via ultrasonication. The resulting solution was then slowly evaporated at 4 °C. Finally, colorless single crystals suitable for SXRD were obtained.:1 v/v) with heating and stirring at 55 °C. Then, the solution was cooled to room temperature and slowly evaporated under ambient conditions. White globular solids were obtained after approximately 10 days. However, we failed to obtain single crystals suitable for SXRD.2.3. Analytical methods
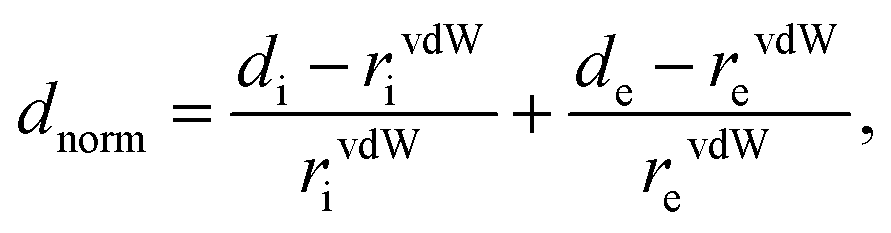 | (1) |
TGA was performed using an analyzer (TG209F1 Iris, NETZSCH, Germany). Samples (5–8 mg) were determined in aluminum crucibles by heating from 30 °C to 500 °C at a rate of 10 °C min−1 under N2 purging (60 mL min−1).
2.4. Accelerated stability tests
The stability of RBM solid forms was investigated at 40 °C and 75% relative humidity (RH) International Conference on Harmonization conditions for three months.36 In this study, 75% RH was achieved with saturated sodium chloride solution in a desiccator.372.5. Solubility and dissolution studies
Solubility studies were conducted by adding excess amounts of RBM solid forms to 10 mL of triple distilled water and phosphate buffer solution (PBS, pH 6.8) in Erlenmeyer flasks.The suspensions were stirred for 72 h at constant temperature (37 °C) and then were centrifuged at 10000 rpm for 20 min using a high-speed refrigerated centrifuge (TGL-16M, Xiangyi, China). The supernatant was filtered through a 0.45 μm Millipore filter, appropriately diluted, and analyzed using a UV-vis spectrophotometer (TU-1901, Beijing, China) at 327 nm. Solid residues obtained by filtration were analyzed through XRPD. All solubility measurements were performed in triplicate.
Dissolution studies were conducted using the paddle method on a ZRC-8D dissolution tester (Chuangxing, Tianjin, China) at 100 rpm and 37 ± 0.5 °C. Samples (30 mg) were added to 900 mL of dissolution media (triple distilled water and pH 6.8 PBS). Aliquots (5 mL) of the sample solutions were withdrawn at 2, 5, 7, 10, 15, 20, 30, 45, 60, 90, and 120 min, and then an equivalent amount of fresh media were added. The sample solutions were filtered through a 0.45 μm filter and analyzed with a UV-vis spectrophotometer (327 nm). Each sample was performed in triplicate.
3. Results and discussion
3.1. Characterization of RBM solvates
| Parameters | RBM/DMSO | RBM/CH2Cl2 |
| Empirical formula | C23H27ClN2O6S2 | C20H17Cl3N2O4 |
| Formula weight | 527.03 | 455.71 |
| Temperature (K) | 148.00(10) | 142.95(10) |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c |
| a (Å) | 8.9468(9) | 14.3285(16) |
| b (Å) | 28.971(3) | 9.7542(4) |
| c (Å) | 9.8022(11) | 21.475(2) |
| α (°) | 90.00 | 90.00 |
| β (°) | 97.029(10) | 136.08(2) |
| γ (°) | 90.00 | 90.00 |
| V (Å3) | 2521.6(5) | 2081.9(3) |
| Z | 4 | 4 |
| ρcalc (g cm−3) | 1.388 | 1.454 |
| μ (mm−1) | 0.358 | 0.470 |
| Crystal size (mm3) | 1104.0 | 936.0 |
| F (000) | 0.4 × 0.3 × 0.25 | 0.4 × 0.35 × 0.3 |
| 2θ for data collection (°) | 5.94 to 52.74 | 5.86 to 52.74 |
| Index ranges | −11 ≤ h ≤ 11, −36 ≤ k ≤ 36, −12 ≤ l ≤ 12 | −17 ≤ h ≤ 12, −12 ≤ k ≤ 11, −18 ≤ l ≤ 26 |
| Reflns collected | 9808 | 8491 |
| Unique reflns | 9808 | 8491 |
| Goodness-of-fit on F2 | 1.049 | 0.912 |
| R1 (I ≥ 2(I)) | 0.0772 | 0.0590 |
| wR2 (all) | 0.2210 | 0.1755 |
| Crystal form | Interactions | H⋯A (Å) | D⋯A (Å) | <DHA (°) | Symmetry code |
|---|---|---|---|---|---|
| RBM/DMSO | O3–H3⋯O1 | 1.685 | 2.505 | 164.92 | x, y, z + 1 |
| N1–H1⋯O2 | 2.005 | 2.869 | 166.83 | x, y, z − 1 | |
| N2–H2⋯O6 | 2.097 | 2.890 | 149.64 | ||
| RBM/CH2Cl2 | O4–H4⋯O1 | 1.602 | 2.505 | 172.14 | x, y − 1, z |
| N1–H1⋯O3 | 1.940 | 2.821 | 173.33 | x, y + 1, z | |
| N2–H2⋯O1 | 2.352 | 3.073 | 139.20 | −x + 1, y − 1/2, −z + 3/2 |
RBM/DMSO was solved in space group P21/c. The stoichiometric ratio of the RBM and DMSO molecules was 1:2. RBM molecules were connected to each other through R22(8) dimer motifs (O3–H3⋯O1 and N1–H1⋯O2) and weak hydrogen bonds of C–H⋯Cl (3.734 Å, 145°) [Fig. 1(a)].38 One DMSO molecule was connected to RBM molecules through N2–H2⋯O6 hydrogen bonds and three weak auxiliary C–H⋯O6 interactions, while another DMSO molecule contacted with RBM molecules through C20–H20B⋯Cl1 weak interactions [Fig. 1(b)]. RBM molecules were arranged in layers that stretched in a “W” shape along the ab plane. DMSO molecules were located between the layers of RBM molecules [Fig. 1(c)]. In addition, π⋯π stacking was observed in the double-layer part of the “W”.
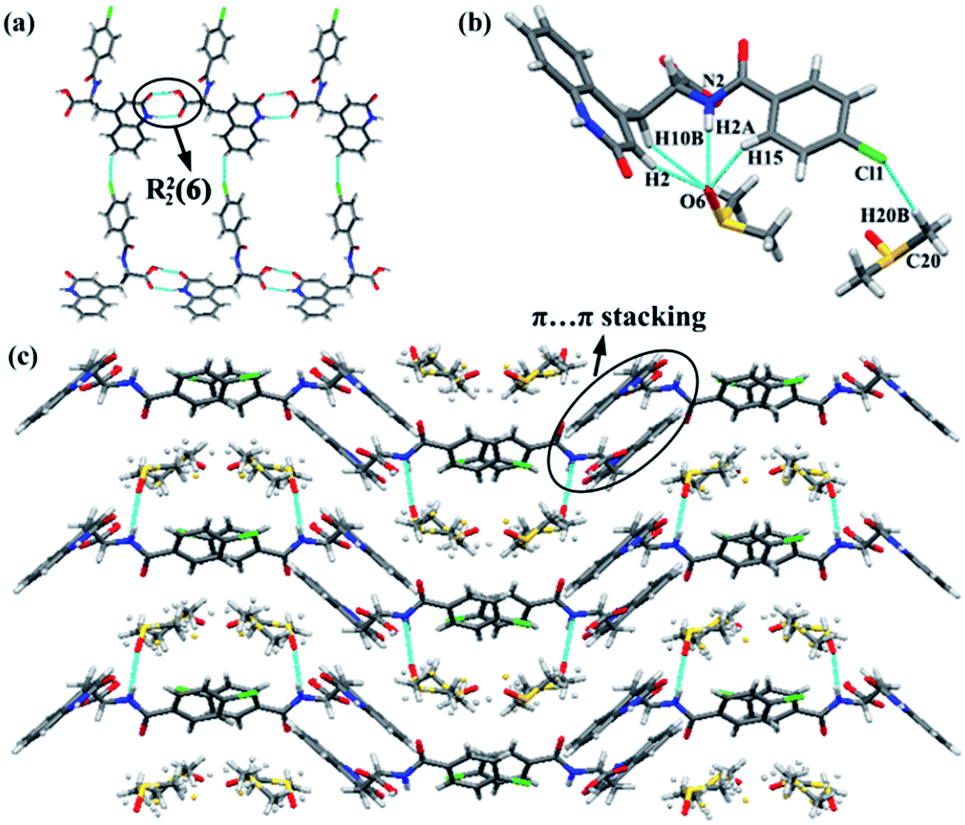 | ||
| Fig. 1 (a) Hydrogen bond network of RBM/DMSO sustained by R22(8) dimer motifs and weak C–H⋯Cl hydrogen bonds. (b) Interactions between DMSO and RBM molecules. (c) View of the 2D packing of RBM/DMSO down the c-axis. DMSO molecules were located between the W-shaped layers. | ||
RBM/CH2Cl2 was solved in space group P21/c. The stoichiometric ratio of the RBM and CH2Cl2 molecules was 1:1. RBM molecules were also connected to each other through R22(8) dimer motifs (O4–H4⋯O1 and N1–H1⋯O3) [Fig. 2(a)].38 Each dimer was connected to neighboring molecules through N2–H2⋯O1 hydrogen bonds and weak C2–H2A⋯O4 hydrogen bonds (3.355 Å, 159°), thus resulting in a sheet-like structure. The sheets were connected to each other through weak C–H⋯Cl (3.629 Å, 138°) hydrogen bonds, which were stronger than those in RBM/DMSO (Fig. S2†). CH2Cl2 molecules interacted with RBM molecules through the weak hydrogen bonds of C20–H20B⋯O2 (3.146 Å, 176°) [Fig. 2(b)]. In 2D packing, the molecules were arranged in a corrugated sheet when viewed down the c-axis [Fig. 2(c)]. π⋯π stacking was observed in the circular part of RBM/CH2Cl2 [Fig. 2(d)].
 | ||
| Fig. 2 (a) Hydrogen bond network of RBM/CH2Cl2 sustained by R22(8) dimer motifs, N2–H2⋯O1 hydrogen bonds, and the weak hydrogen bonds of C2–H2A⋯O4. (b) Interactions between CH2Cl2 and RBM molecules. (c) Corrugated sheet viewed down the c-axis. (d) π⋯π stacking in RBM/CH2Cl2. | ||
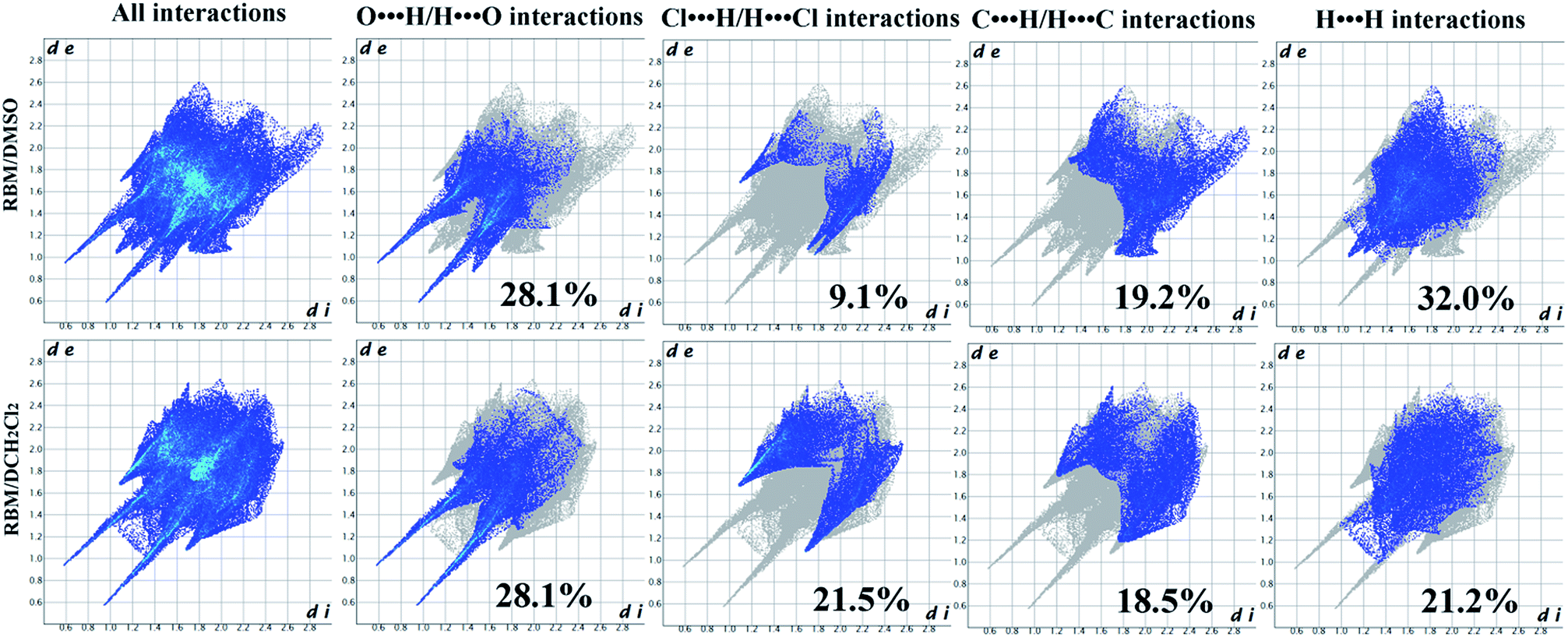 | ||
| Fig. 3 2D FPs of RBM/DMSO and RBM/CH2Cl2. | ||
 | ||
| Fig. 4 Morphological images of RBM solvates: (a) RBM/DMSO, (b) RBM/CH2Cl2, (c) RBM/H2O, and (d) RBM/EtOH/H2O. | ||
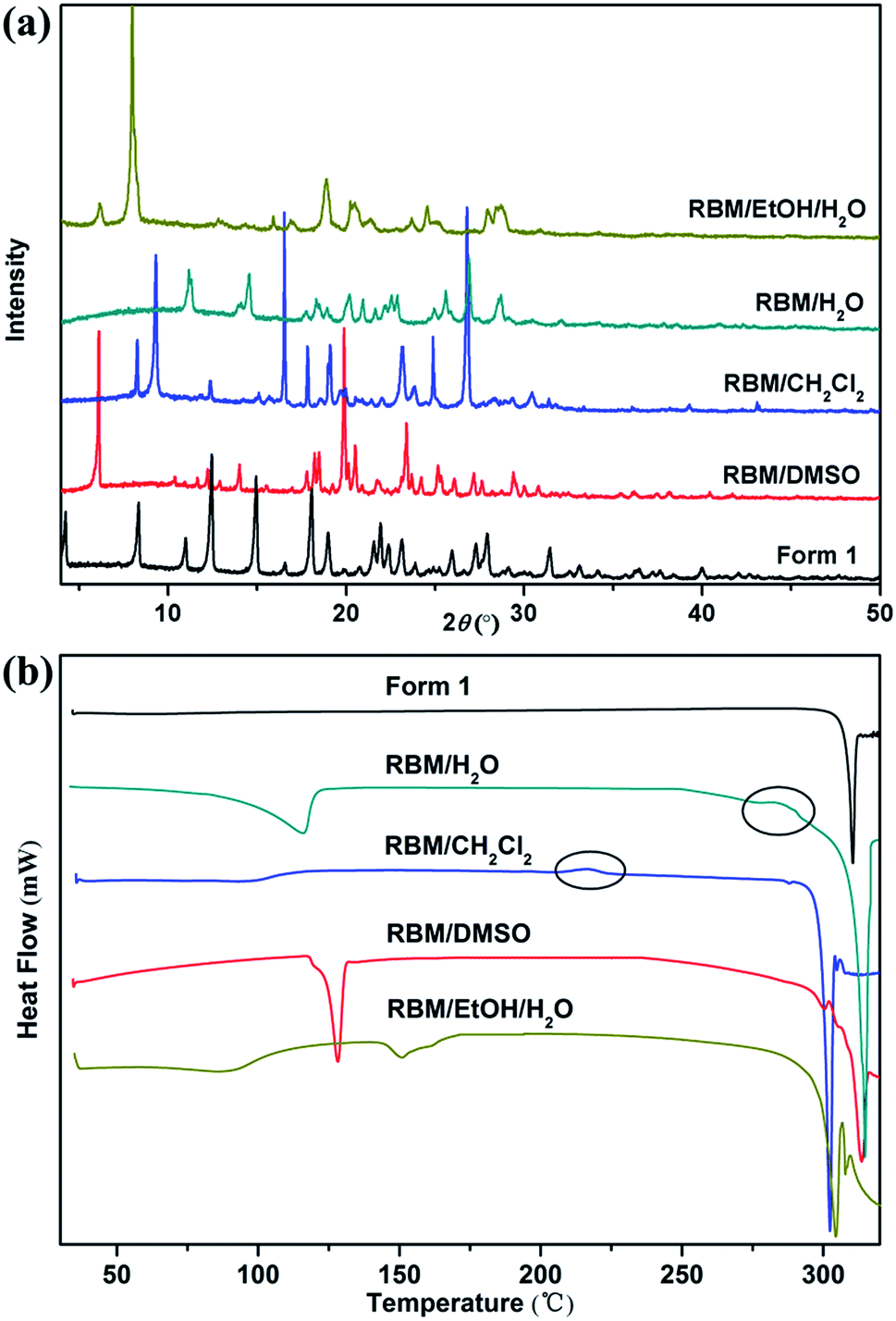 | ||
| Fig. 5 (a) XRPD patterns and (b) DSC curves of the four solvates and form 1 of RBM. | ||
:2 stoichiometry. RBM/CH2Cl2 achieved a mass reduction of 12.55% during the first period, which was attributed to the removal of CH2Cl2 molecules. The experimental weight loss was lower than the theoretical weight loss (18.64%), thus indicating that molecules were partially lost before the TGA test because of the very low boiling point of CH2Cl2. In the DSC curve, RBM/CH2Cl2 exhibited a broad endothermic peak before 120 °C (removal of CH2Cl2) and further showed an exothermic phase transformation at 210–220 °C. The DSC curve of RBM/H2O showed a broad endothermic peak before 120 °C and a small exothermic peak at 280–290 °C, which were ascribed to dehydration and transition, respectively. The weight loss of 4.28% during the first stage indicated that the stoichiometric ratio of RBM to water molecules was 1:1 [Δmcal. (–H2O) = 4.63%]. RBM/EtOH/H2O showed two mass steps of 5.83% and 2.80% at 50–100 °C and 140–160 °C, respectively. This phenomenon suggested the possible presence of 0.5EtOH [Δmcal. (−0.5EtOH) = 5.85%] and 0.5H2O [Δmcal. (−0.5H2O) = 2.37%] molecules in the crystal structure of RBM/EtOH/H2O. The two broad endothermic peaks before 200 °C in the DSC curve further confirmed the hemiethanol hemihydrate structure of RBM/EtOH/H2O. Furthermore, these two peaks were attributed to the removal of EtOH and H2O molecules. The peak at approximately 300 °C for each DSC curve was broad or split, indicating that the solid forms obtained through desolvation or transformation melted with decomposition. This phenomenon was consistent with that indicated by the TGA results.
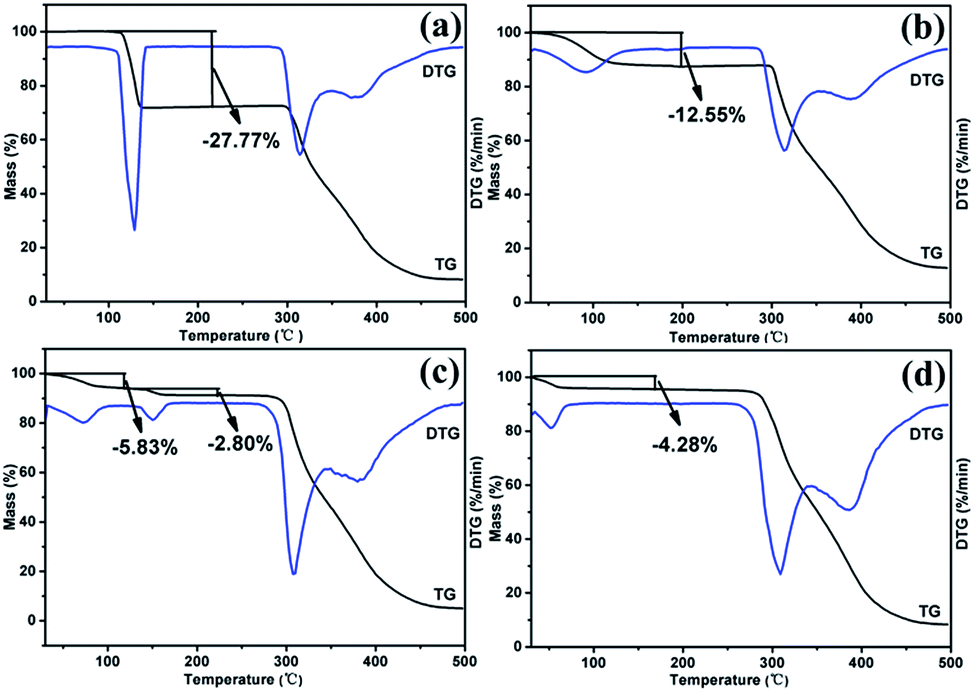 | ||
| Fig. 6 TGA curves of the RBM solvates: (a) RBM/DMSO, (b) RBM/CH2Cl2, (c) RBM/H2O, and (d) RBM/EtOH/H2O. | ||
3.2. Phase transitions upon heating
The modifications formed in solvates by solvent removal must be determined for screening novel solid forms. The phase transitions upon heating were investigated based on the thermal events displayed in the DSC curves.RBM/DMSO and RBM/EtOH/H2O transformed to form 1 after desolvation (Fig. S8†). The XRPD patterns detected at 150 °C and 220 °C for RBM/CH2Cl2 were considerably different from those for RBM/CH2Cl2 [Fig. 7(a)]. No weight loss was observed in the TGA curves before melting and decomposition, and no desolvation peak was detected in the DSC curves [Fig. 7(a) and (c)]. Moreover, the two XRPD patterns were different from that of forms 1 and 2 (Fig. S9†).23 Thus, two novel polymorphs (forms 3 and 4) were generated while heating RBM/CH2Cl2. Together with the DSC result, form 3 was generated by the removal of CH2Cl2 (150 °C) and was transformed to form 4 when further heated at 220 °C [Fig. 7(d)]. A novel polymorph (form 5) was obtained after dehydration of RBM/H2O, as indicated by the XRPD, TGA, and DSC results at 150 °C. The exothermic peak at 280–290 °C demonstrated that form 5 transformed into form 4 when heated at high temperature. In conclusion, three novel RBM polymorphs were obtained by the desolvation and transformation of the solvates. This phenomenon is significant for RBM development because the novel polymorphs may possess desirable physical and chemical properties.
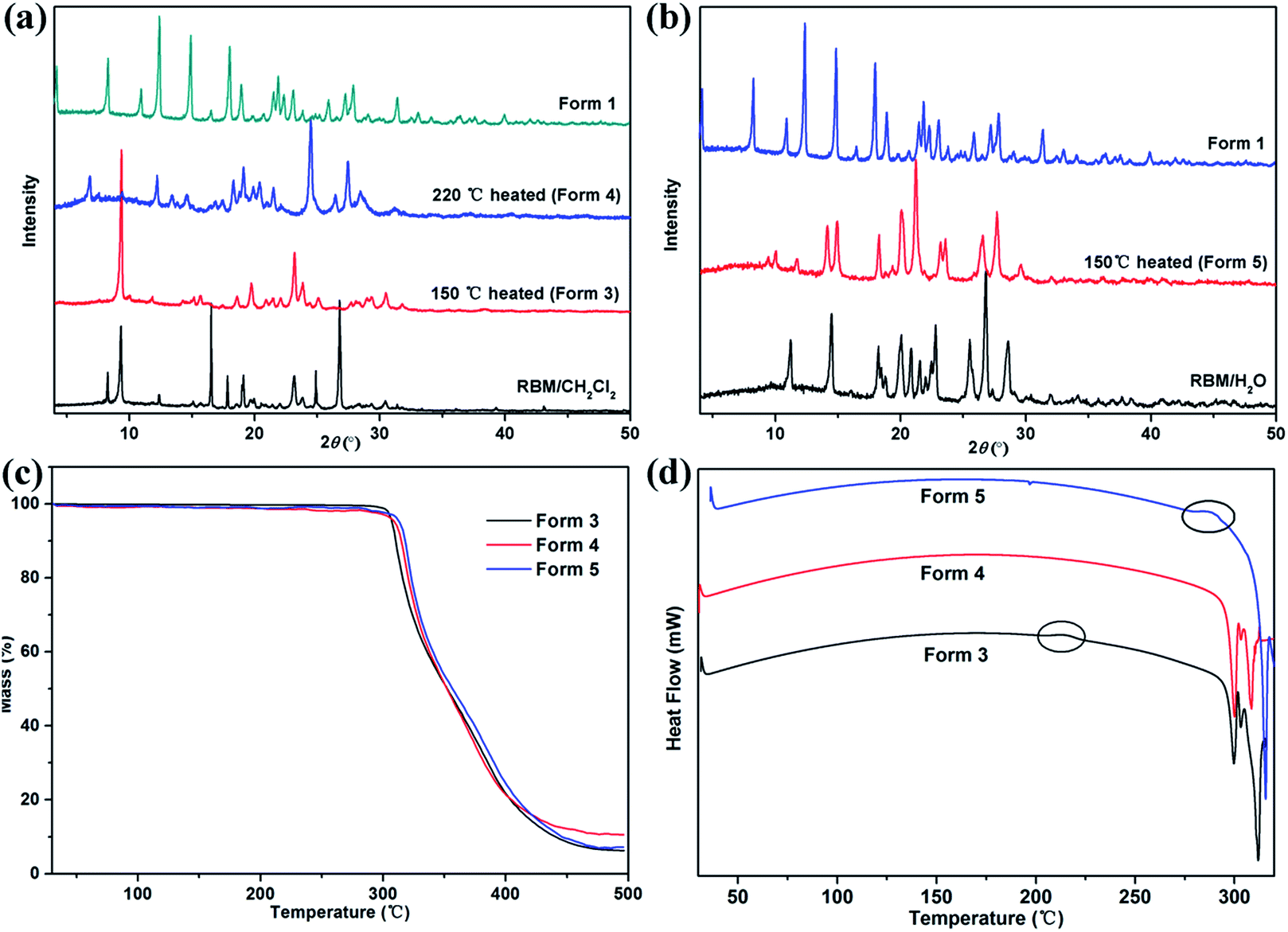 | ||
| Fig. 7 XRPD patterns of (a) RBM/CH2Cl2 heated at 150 °C and 220 °C, (b) RBM/H2O heated at 150 °C, (c) TGA, and (d) DSC curves of forms 3–5. | ||
3.3. Accelerated stability tests
Fig. 8 displays the XRPD patterns of the RBM solid forms, which were monitored at 40 °C and 75% RH. RBM/DMSO and RBM/EtOH/H2O were both converted to form 1 within three days [Fig. 8(a) and (d)]. RBM/CH2Cl2 transformed to form 3, which could remain stable for three months at 40 °C and 75% RH [Fig. 8(b)]. Both RBM/H2O and form 4 showed relatively good stability because their XRPD patterns were nearly unchanged for three months [Fig. 8(c) and (e)]. Form 5 was unstable and could easily absorb water for reconversion to RBM/H2O [Fig. 8(f)].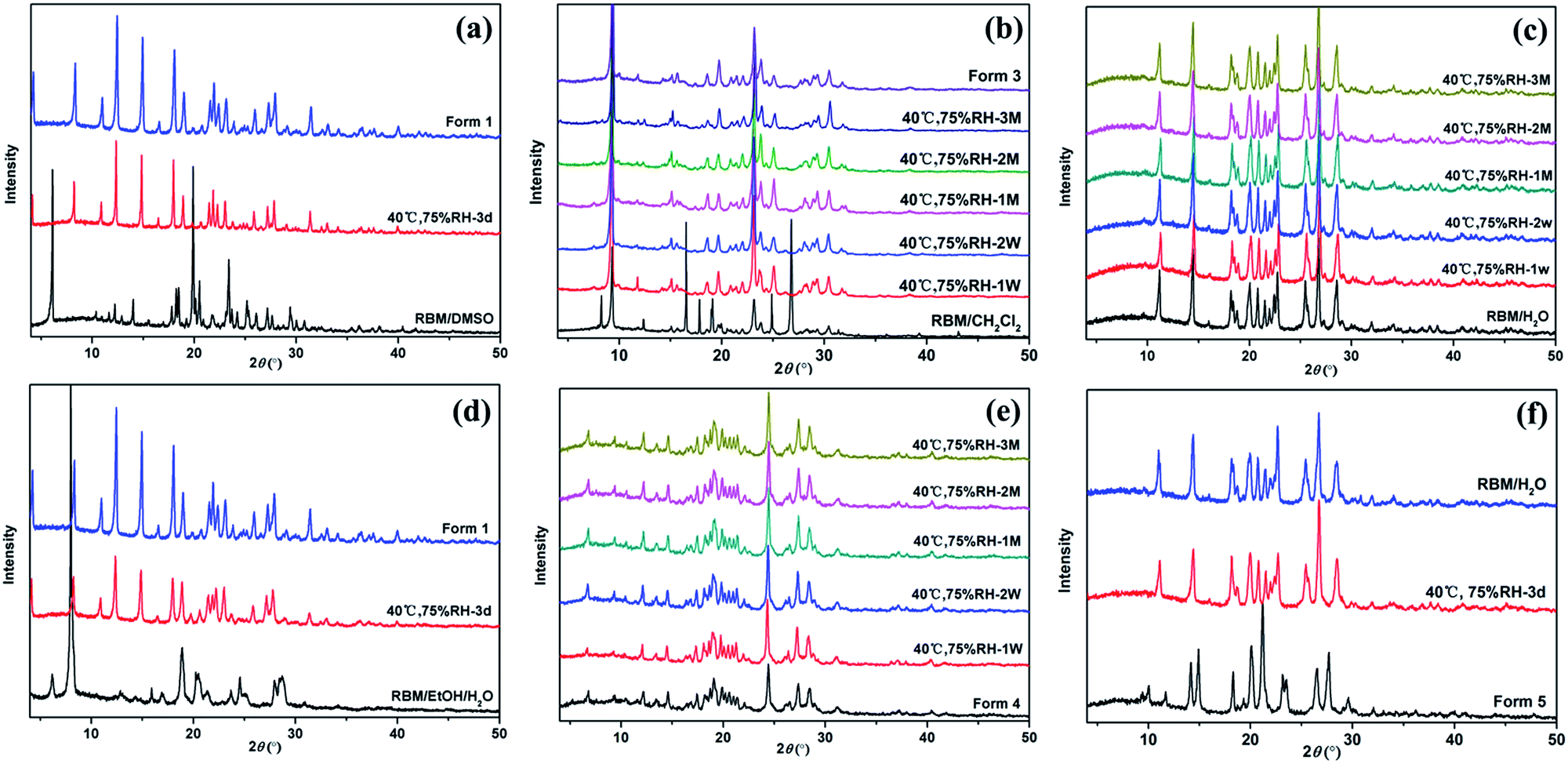 | ||
| Fig. 8 Stability of (a) RBM/DMSO, (b) RBM/CH2Cl2 and form 3, (c) RBM/H2O, (d) RBM/EtOH/H2O, (e) form 4, and (f) form 5 monitored with XRPD at 40 °C and 75% RH. (d = day, W = week, M = month). | ||
3.4. Solubility and dissolution studies
Considering stability and security, the apparent solubility and dissolution rate of RBM/H2O, form 3, and form 4 were investigated and compared with those of form 1. RBM/H2O, form 3, and form 4 demonstrated higher solubility than form 1 in water and PBS [Fig. 9(a)]. Especially RBM/H2O, the solubility of which was 1.62 and 2.23 times higher than form 1 in water and PBS, respectively. The solid residues obtained by centrifugation and filtration after solubility determination were analyzed by XRPD (Fig. S10†). Although forms 3 and 4 were converted to form 1, the apparent solubility of the two forms remained higher than that of the original form 1 in water and PBS. No transformation occurred for RBM/H2O. Moreover, its solid residues showed increased crystallinity after solubility examination. Thus, RBM/H2O is a potential substitute for form 1 in clinical treatment given its dominant solubility and good stability.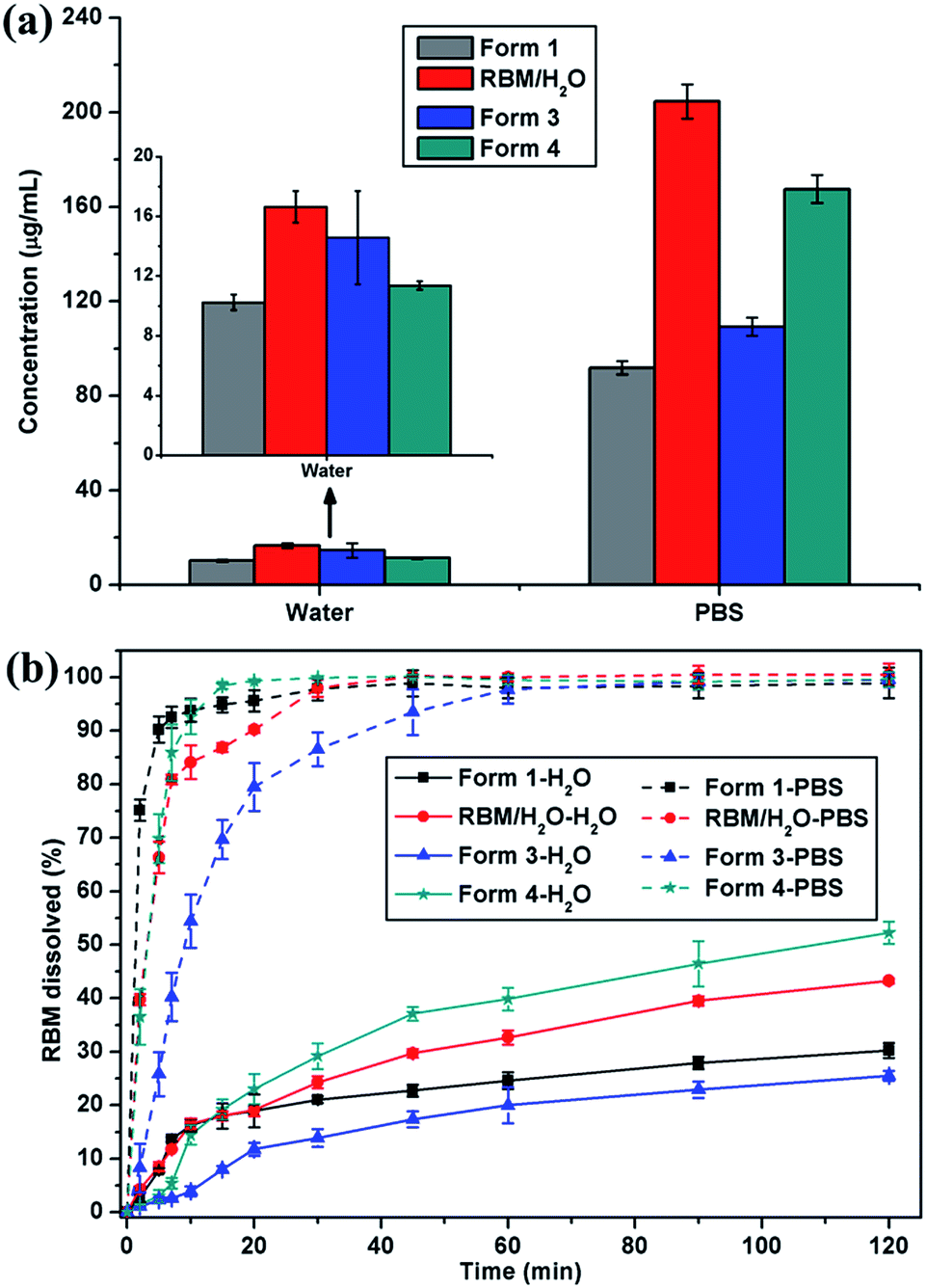 | ||
| Fig. 9 (a) Solubility histograms and (b) dissolution profiles of form 1, RBM/H2O, form 3, and form 4 in water and PBS (mean ± standard deviation, n = 3). | ||
RBM/H2O and form 4 showed a faster dissolution rate than form 1 after 20 min of dissolution in water; moreover, the cumulative drug release of the forms was considerably higher than that of form 1 after 120 min [Fig. 9(b)]. When dissolved in PBS, the three RBM solid forms exhibited a slower dissolution than form 1. However, their cumulative dissolution reached approximately 100% within 60 min. RBM/H2O and form 4, in particular, almost completely dissolved after 30 min. These results indicated that RBM/H2O and form 4 had a comparative or even better solubility and dissolution characteristics than form 1. Thus, RBM/H2O and form 4 may be developed as novel solid forms of RBM for clinical applications.
4. Conclusions
Four solvates and three novel polymorphs of RBM were obtained. The crystal structures of RBM/DMSO and RBM/CH2Cl2 were determined via SXRD. Both structures were sustained by R22(8) dimer motifs which formed through O–H⋯O and N–H⋯O hydrogen bonds. The main difference between the two forms was that additional hydrogen bonds (N2–H2⋯O1) which were absent from RBM/DMSO, were present in RBM/CH2Cl2. The 2D FP result indicated that the subtle intermolecular interactions of RBM/DMSO and RBM/CH2Cl2 were significantly different. The stability results at 40 °C and 75% RH indicated that RBM/DMSO and RBM/EtOH/H2O transformed to form 1 and that form 5 was converted to RBM/H2O. RBM/H2O, form 3, and form 4 showed good stability. In addition, RBM/H2O, form 3, and form 4 displayed a higher solubility than form 1 in water and PBS. Furthermore, the dissolution rates of RBM/H2O and form 4 in water were faster than that of form 1. Thus, RBM/H2O and form 4 exhibited potential to be developed as novel solid forms of RBM for clinical applications, which is meaningful for the drug development of RBM.Acknowledgements
This work was supported by the Applied Basic Research Project of Sichuan Province (Grant No. 2014JY0042), the National Development and Reform Commission and Education of China (Grant No. 2014BW011), and the Large-scale Science Instrument Shareable Platform Construction of Sichuan Province (Grant No. 2015JCPT0005-15010102).References
- C. Näther, I. Jess, P. G. Jones, C. Taouss and N. Teschmit, Cryst. Growth Des., 2013, 13, 1676–1684 Search PubMed.
- R. Bobrovs, L. Seton and N. Dempster, CrystEngComm, 2015, 17, 5237–5251 RSC.
- J. P. Brog, C. L. Chanez, A. Crochet and K. M. Fromm, RSC Adv., 2013, 3, 16905 RSC.
- N. Gong, K. Hu, G. Jin, G. Du and Y. Lu, RSC Adv., 2016, 6, 38709–38715 RSC.
- S. H. Thorat, M. V. Patwadkar, R. G. Gonnade and R. Vaidhyanathan, CrystEngComm, 2014, 16, 8638–8641 RSC.
- K. Xu, S. Zheng, Y. Zhai, L. Guo, P. Tang, J. Yan, D. Wu and H. Li, Int. J. Pharm., 2015, 486, 185–194 CrossRef CAS PubMed.
- D. E. Braun, P. G. Karamertzanis, J. B. Arlin, A. J. Florence, V. Kahlenberg, D. A. Tocher, U. J. Griesser and S. L. Price, Cryst. Growth Des., 2011, 11, 210–220 CAS.
- S. Datta and D. J. Grant, Nat. Rev. Drug Discovery, 2004, 3, 42–57 CrossRef CAS PubMed.
- D. Maddileti, B. Swapna and A. Nangia, Cryst. Growth Des., 2015, 15, 1745–1756 CAS.
- C. Näther, I. Jess, L. Seyfarth, K. Bärwinkel and J. Senker, Cryst. Growth Des., 2015, 15, 366–373 Search PubMed.
- A. O. Surov, K. A. Solanko, A. D. Bond, A. Bauer-Brandl and G. L. Perlovich, CrystEngComm, 2015, 17, 4089–4097 RSC.
- U. J. Griesser, in The importance of solvates, Polymorphism in the Pharmaceutical Industry, 2006, pp. 211–233 Search PubMed.
- B. N. Patel, N. Bhanubhai and C. N. Suhagia, Int. J. Pharm. Pharm. Sci., 2012, 4, 270–273 CAS.
- N. Variankaval, A. S. Cote and M. F. Doherty, AIChE J., 2008, 54, 1682–1688 CrossRef CAS.
- W. Xu, N. Gong, S. Yang, N. Zhang, L. He, G. Du and Y. Lu, J. Pharm. Sci., 2015, 104, 1256–1262 CrossRef CAS PubMed.
- H. X. Ding, C. A. Leverett, R. E. Kyne, K. K. C. Liu, S. J. Fink, A. C. Flick and C. J. O'Donnell, Bioorg. Med. Chem., 2015, 23, 1895–1922 CrossRef CAS PubMed.
- X. Wang, N. Gong, S. Yang, G. Du and Y. Lu, J. Pharm. Sci., 2014, 103, 2696–2703 CrossRef CAS PubMed.
- Z. Yin, Z. Li, W. Z. Yang, J. He and J. P. Cheng, Struct. Chem., 2005, 16, 641–647 CrossRef CAS.
- P. W. Cains, in Polymorphism in pharmaceutical solids: classical methods of preparation of polymorphs and alternative solid forms, 2009, pp. 76–138 Search PubMed.
- R. Pradhan, T. H. Tran, J. Y. Choi, I. S. Choi, H. G. Choi, C. S. Yong and J. O. Kim, Arch. Pharmacal Res., 2014, 38, 522–533 CrossRef PubMed.
- N. T. Tung, C. W. Park, T. O. Oh, J. Y. Kim, J. M. Ha, Y. S. Rhee and E. S. Park, J. Pharm. Pharmacol., 2011, 63, 1539–1547 CrossRef CAS PubMed.
- S. Koh, Y. Inoue, T. Sugmimoto, N. Maeda and K. Nishda, Cornea, 2013, 32, 1219–1223 CrossRef PubMed.
- S. H. Jeon and Y. T. Sohn, Arch. Pharmacal Res., 2016, 39, 508–515 CrossRef CAS PubMed.
- X. Zeng and C. Jia, Preparation of a rebamipide monohydrate crystal form for use in drugs for treating diseases related to gastric mucosa injury, China Patent, CN 104418802, March 18, 2015.
- J. Holland, D. Gooding and A. Chorlton, Novel rebamipide complexes and cocrystals, U.S. Patent, 20140039005, February 6, 2014.
- Y. Chi, C. Liu, T. Ren, X. Wang, Q. Yang, Z. Yang, Y. Yang, S. Yang, J. Gu and C. Hu, Cryst. Growth Des., 2016, 16, 3180–3189 CAS.
- X. Xiong, K. Xu, S. Li, P. Tang, Y. Xiao and H. Li, Drug Dev. Ind. Pharm., 2017, 43, 283–292 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis and R. J. Gildea, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- M. A. Spackman and P. G. Byrom, Chem. Phys. Lett., 1997, 267, 215–220 CrossRef CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- J. Bojarska, A. Fruziński and W. Maniukiewicz, J. Mol. Struct., 2016, 1116, 22–29 CrossRef CAS.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, Crystal explorer, The University of Western Australia, 2012 Search PubMed.
- W. H. Organization, in World Health Organization Technical Report Series: Annex 2: Stability testing of active pharmaceutical ingredients and finished pharmaceutical products, 2009, pp. 87–130 Search PubMed.
- T. Lu and C. Chen, Measurement, 2007, 40, 591–599 CrossRef.
- J. Bernstein, R. E. Davis, L. Shimoni and N. L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1500324 and 1500325. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra02895d |
| This journal is © The Royal Society of Chemistry 2017 |