
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA comprehensive understanding of water photooxidation on Ag3PO4 surfaces†
Zuju Ma a,
Sen Linb,
Rongjian Saa,
Qiaohong Lia and
Kechen Wu*a
a,
Sen Linb,
Rongjian Saa,
Qiaohong Lia and
Kechen Wu*a
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, China. E-mail: wkc@fjirsm.ac.cn
bState Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fuzhou 350002, China
First published on 3rd May 2017
Abstract
The oxygen evolution reaction (OER) is known to be the bottleneck of water-splitting. Ag3PO4 is a highly efficient visible light photocatalyst for dye degradation and water oxidation to O2, with a higher OER rate than BiVO4 and WO3. Despite extensive studies on Ag3PO4, the surface properties including surface electronic states, reaction sites and mechanisms of OER on Ag3PO4 surfaces are not clear at present. Herein, we reported a comparative first-principles density functional theory study of the bulk, surface properties and the mechanism of OER on the three primary low index facets of Ag3PO4: (100), (110) and (111). We revealed for the first time that the rate-limiting step of the OER on Ag3PO4 (100), (110) and (111) surfaces is the dehydrogenation of HO* (HO* → O* + H+ + e−), which is different from most reported metal oxides and nitrides like TiO2 and g-C3N4. The OER process on the (100) surface tends to proceed by following a different mechanism as that on the (110) and (111) surfaces. The illumination of the Ag3PO4 (100), (110), and (111) surfaces with solar light provides enough overpotential for the OER to proceed spontaneously. In particular, the free energy change of removal of the first proton from water on the Ag3PO4 (111) surface is much lower than that on (100) and (110) surfaces, giving an explanation for the experimentally observed higher catalytic activity of the (111) surface. The exposed phosphorus atoms on the Ag3PO4 (111) surface promote the dehydrogenation of H2O and suppress the formation of mid-gap states. Our results are profound for understanding the underlying mechanism of the photocatalytic water oxidation process occurring on Ag3PO4 surfaces, and serve as a foundation for developing new high-performance Ag3PO4 based photocatalysts for water splitting and organic contaminant decomposition.
1 Introduction
Direct splitting of water using an efficient photocatalyst is a clean and sustainable technique to produce renewable fuels and has fascinated researchers for over 40 years.1–3 There has been remarkable progress in developing inorganic and organic systems with high photocatalytic activities for water splitting into H2 and O2 under ultraviolet and visible light, such as n-type TiO2,4 NaTaO3,5 La2Ti2O7 (ref. 6) and Sr2NbO7,7 GaN–ZnO solid solutions,8,9 and Pt–PdS/CdS10 etc. However, these reported photocatalysts for overall water splitting still suffer from very low quantum efficiency in the visible range, with solar-to-hydrogen efficiencies of less than 0.1%.11–14 A main reason is that water oxidation to O2 is difficult. The water splitting reaction can be treated in terms of two coupled half-reactions, the H2 evolution reaction (HER) and O2 evolution reaction (OER). The OER is recognized as a much more complicated process and with a much lower reaction rate since the formation of molecular oxygen involves four-electron transfer and occurs on a timescale of ca. 5 orders of magnitude slower than H2 evolution.15–18 This suggests that a hole would easily recombine with an electron before completion of the OER.In 2010, Yi et al. reported a new use of cubic Ag3PO4 semiconductor, which shows extremely high quantum yield (up to 80–90%) of O2 generation from water oxidation at wavelengths less than 480 nm.17 They demonstrated that Ag3PO4 outperforms BiVO4 and WO3 (two of the most well-studied visible-driven water oxidation photocatalysts) in terms of OER rate and dye degradation.17,19 The origin of its high performance was proposed to be the formation of highly dispersive Ag s–Ag s hybrid bands at the conduction band minimum (CBM), which is advantage for the transfer of carrier to surface.20 Afterwards, Martin and Bi et al. further improved the activity of the Ag3PO4 by controlling the percentage of exposed facets on crystal surfaces.15,21 They found that tetrahedral crystals, composed of (111) facets, show a 10 times higher initial oxygen evolution rate than either (110) or (100) facets.15 Theoretical calculations of the surface energies for these facets have been performed to explain the facet effect on their photocatalytic activity difference.15,21 However, the value of surface energy is a very limited criterion to assess photocatalytic activity. Ag3PO4 nanocrystals with controlled particle size have been found to exhibit high visible light activity due to the increased specific surface area.22 Many Ag3PO4 based composite photocatalysts including TiO2/Ag3PO4,23 graphene/Ag3PO4 (ref. 24 and 25) and Ag/Ag3PO4 (ref. 26–28) have been prepared to improve photocatalytic activity of Ag3PO4 by enhancing separation of photogenerated electron–hole pairs. The efficiency of OER on a semiconductor surface is usually determined by four fundamental processes: (i) light absorption; (ii) separation of photoexcited electron–hole pairs; (iii) migration of electrons and holes towards the surface; and (iv) eventual reduction/oxidation reactions.19 The first three processes take place in the bulk region of the photocatalyst, which have been well studied by the screened hybrid functionals and GW perturbation theory. Nevertheless, the last process, i.e. the oxidation/reduction reaction, on Ag3PO4 surfaces are not at present clear, especially the mechanism of OER. A complete atomic-scale understanding of the OER process on this advancing high efficient visible light photocatalyst is imperative. In this work, particular attentions are focused on the following questions: (i) does the OER take place on Ag3PO4 (100), (110) and (111) surfaces following the same mechanism as that in a few most reported metal oxides such as TiO2, and nitrides like GaN, and g-C3N4?16,18,29–36 If yes, do they have the same rate-limiting step? (ii) What factors lead to the surface-dependent photocatalytic activity of Ag3PO4 (100), (110) and (111) surfaces? (iii) What role does the surface microstructure have in determining the surface electronic structure and the barrier of OER step?
In the work, we reported a systematic study of the bulk, surface properties and the mechanism of OER on three primary low index facets of Ag3PO4: (100), (110) and (111) by combining hybrid DFT calculations and first principles thermodynamics. In Section 2, the computational methods and the surface models are presented. The method to calculate the free energy differences between the different reaction steps is briefly introduced. All possible surface terminations for each surface orientation ([100], [110] and [111]) were optimized. In Section 3, we first investigate the bulk properties of Ag3PO4. They are found to be well reproduced by PBE0 hybrid functional, including accurate band gap and highly dispersive CBM. We then calculate and compare the geometries, surface formation energies, surface electronic structures, and the reaction mechanism steps of the OER of the Ag3PO4: (100), (110) and (111) surfaces. The theoretical overpotential for the OER at an applied potential U and a given pH is determined. Possible explanations for the surface-dependent photocatalytic activity of Ag3PO4 are discussed. Lastly, we investigate another two possible mechanisms of the OER on Ag3PO4 surfaces. We suggested that this study is crucial for the understanding of the photocatalytic process occurring at Ag3PO4 surfaces. It also provides useful insights for designing better anode materials in (photo) electrochemical water splitting cells.
2 Methods and model
2.1 Method
Our calculations on the bulk Ag3PO4 and three primary low index facets of Ag3PO4: (100), (110) and (111) were performed using the plane-wave basis Vienna Ab initio Simulation Package (VASP) code.37–39 The projected augmented wave (PAW)40 potentials with the valence states 2s and 2p for O, 3s and 3p for P, and 4d and 5s for Ag were used to treat the core-valence interaction. The Perdew–Burke–Ernzerhof (PBE) functional41 was used for the structural optimization. It has been found to reproduce the structural properties of bulk Ag3PO4.42 To better describe the exchange–correlation effects of localized electrons, three hybrid functionals (PBE0, HSE03 and HSE06)43 were used for the electronic structure calculations. The PBE functional was also used for comparison. A Γ-centered 5 × 5 × 5 Monkhorst–Pack grid for the Brillouin zone sampling44 and a cutoff energy of 450 eV for the plane wave expansion were found to obtain accurate lattice parameters of the bulk Ag3PO4.2.2 Model
The (100), (110) and (111) surfaces of Ag3PO4 were modeled using a period slab with a vacuum space of 15 Å in the direction of surface normal, which is thick enough for the system to converge to an accurate total energy. The atoms in the slab were allowed to fully relax until the residual forces acting on all the constituent ions dropped below 0.05 eV Å−1. A Γ-centered 5 × 5 × 1 Monkhorst–Pack k-point grid and a cutoff energy of 450 eV were found to get convergent total and adsorption energies. A self-consistent dipole correction was applied in the direction of surface normal in the structural relaxations and total energy calculations.The adsorption energy for adsorbate species A was calculated as the energy difference: ΔEads = E*A − (E* + EA). Where * is the bare surface, *A indicates the adsorption of species A on the surface *, and EA is the energy of A in its gas state.
2.3 Thermodynamics and photocatalysis for OER
The water oxidation reaction can be written as| H2O → ½O2 + 2H+ + 2e− | (1) |
We modeled this reaction using the method developed by Norskov and coworkers,45,46 who have studied the water oxidation on different metal oxide surfaces, such as TiO2, RuO2, and IrO2. The reaction is decomposed into four one-electron steps A–D:
A
| H2O + * → HO* + H+ + e− | (2) |
B
| HO* → O* + H+ + e− | (3) |
C
| H2O + O* → HOO* + H+ + e− | (4) |
D
| HOO* → O2 + * + H+ + e− | (5) |
The above mechanism is the most common one of the water splitting on semiconductor surfaces. We denote this mechanism as mechanism I. It involves the dehydrogenation of H2O, followed by the dissociation of HO* to O*. Subsequently, O* reacts with another H2O to generate HOO* which then dehydrogenates to O2.29,45 Another two possible mechanisms (mechanism II involving the HOOH*, mechanism III involving the interaction between O* and HO*) are discussed in Section 3.3.
The method we used in the work concentrates on the thermochemistry of the reaction. We assume that the reaction proceeds through one-electron transfer steps which depend directly on the applied potential. The overpotential needed for the different elementary steps is restricted to the barriers that come from differences of free energies (ΔG) of the intermediates. Kinetic barriers arising from the diffusion of species is not included in the model. From the results, we can evaluate whether OER may take place thermodynamically at a given pH and an applied potential. The details of the calculation of ΔG are summarized as follows.45
Free energy change ΔG0 = ΔE + ΔZPE − TΔS, with ΔE denoting the total self-consistent energy change (reaction energy) derived from DFT calculations, ΔZPE indicating the differences in zero-point energies, and ΔS indicating the change of entropy. ZPE for the OER intermediates were obtained from the vibrational frequencies. We assume that S = 0 for the intermediates adsorbed on the surface, because of the absence of translational and rotational motions upon adsorption. For free gas-phase molecules, we obtained ZPE and S from thermodynamics database.45
At standard conditions (U = 0, pH = 0, p = 1 bar, T = 298 K), the free energy change of the reaction *AH → H+ + A + e− is equivalent to that of the reaction *AH → ½H2 + A. Thus, the free energy of H+ + e− has been replaced by ½H2. Under the influence of a potential bias U and an infinite pH, the free energy difference was shifted by ΔG(U, pH) = ΔGU + ΔGpH = −eU − kT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln10 × pH.
ln10 × pH.
In comparison with H2O and H2, O2 has a much more complicated electronic structure which cannot be described accurately by DFT. In this work, the free energy change of the total reaction H2O → ½O2 + H2 is fixed at the experimentally found value of 2.46 eV to avoid the direct calculation of O2 molecule. Hence, the free energy of an O2 molecular can be written as: G[O2] = 4.92 − 2E[H2] + 2E[H2O] − (ΔZPE − TΔS)[2H2O → O2 + 2H2]. More details about this method can be found in the reference by Norskov and coworkers.45
Based on above assumptions, the reaction free energy changes (ΔG) for the proposed reaction steps in mechanism I are calculated by the following equations:
|
ΔGA = ΔEA + 1/2EH2 − EH2O + (ΔZPE − TΔS)A − eU − kTln10 × pH
| (6) |
|
ΔGB = ΔEB + 1/2EH2 + (ΔZPE − TΔS)B − eU − kTln10 × pH
| (7) |
|
ΔGC = ΔEC + 1/2EH2 − EH2O + (ΔZPE − TΔS)C − eU − kTln10 × pH
| (8) |
|
ΔGD = 4.92 eV + ΔED − 3/2EH2 + 2EH2O + (ΔZPE − TΔS)D − eU − kTln10 × pH
| (9) |
3 Results and discussion
3.1 Bulk properties
For reference, we first preliminary investigate the geometry and electronic structure of bulk Ag3PO4, which have been extensively studied.20,42,47,48 The optimized equilibrium lattice parameter at PBE level with a Γ-centered 5 × 5 × 5 k-point mesh is 6.087 Å, which is well within the theoretical uncertainties (1.36%) as compared with the experimentally reported values. As illustrated in Fig. 1(a), cubic Ag3PO4 has P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3n symmetry with each atom tetrahedrally coordinated.17 The [PO4]3− tetrahedral unit with strong P–O bonds weakens the covalent nature of Ag–O bonds. Umezawa et al. suggested that this special structural character inhibits the hybridization of 4d orbitals of Ag and 2p orbitals of O, and forming highly dispersive Ag s–Ag s hybrid bands at CBM. The delocalized charge distribution at CBM results in a small mass of the photoexcited electron, which is advantageous for the transfer of carrier to surface.20
3n symmetry with each atom tetrahedrally coordinated.17 The [PO4]3− tetrahedral unit with strong P–O bonds weakens the covalent nature of Ag–O bonds. Umezawa et al. suggested that this special structural character inhibits the hybridization of 4d orbitals of Ag and 2p orbitals of O, and forming highly dispersive Ag s–Ag s hybrid bands at CBM. The delocalized charge distribution at CBM results in a small mass of the photoexcited electron, which is advantageous for the transfer of carrier to surface.20
 | ||
| Fig. 1 (a) Crystal structure representation of bulk Ag3PO4 with indication of PO4 tetrahedral unit in purple. Ag, O, and P atoms are represented by silver, red and purple balls, respectively. (b) Band structure of bulk Ag3PO4 calculated by the PBE0 functional. The Fermi level was set to the zero of energy. (c) Total DOS of bulk Ag3PO4. (d) The enlarged plots of the partial DOS of bulk Ag3PO4 close to the VBM. (e) The enlarged plots of the partial DOS of bulk Ag3PO4 close to the CBM. | ||
It has been proven that the conventional exchange–correlation (XC) functionals such as GGA/PBE and LDA usually underestimate the band gap of semiconductors due to the self-interaction error in the approximate exchange functional.49,50 Previous studies have confirmed that the band gap of bulk Ag3PO4 calculated by the LDA and GGA are far smaller than the experimental value.20,47,48 A hybrid functional that includes a portion of exact exchange can correct much of this error. In this work, three hybrid functionals (PBE0, HSE03 and HSE06) and a PBE functional were adopted for the electronic structure calculation of bulk Ag3PO4. We find the band gap value of 2.46 eV obtained by PBE0 functional is in good agreement with the experimental value of 2.36 eV.17 It is also consistent with our previous study.42 By contrast, the obtained band gap values by using the PBE, HSE03 and HSE06 functionals are 0.18, 1.48, and 1.76 eV, respectively, which all underestimate the band gap. The reason for such a large discrepancy of band gap obtained by different functionals is mainly attributed to the portion of Fock exchange potential included in the calculation of XC energy. The results suggest the importance of using proper XC functional and parameters to obtain an accurate description of electronic structure of Ag3PO4. The obtained band structure of bulk Ag3PO4 at PBE0 level is shown in Fig. 1(b). The corresponding total and partial DOS for CB and VB are shown in Fig. 1(c–e). The most desirable characteristic of the electronic structure of bulk Ag3PO4 is its highly dispersive CBM, which leads to a small effective mass of photoexcited electron. The CBM is mainly composed of the delocalized 5s orbitals of Ag atoms (see Fig. 1(e)). As the formation of the metallic Ag–Ag bond, Ag s –Ag s hybrid orbitals embrace the [PO4]3− tetrahedral units, which is beneficial for the transfer of photoexcited electrons.20 The VBM is predominated by O 2p and Ag 4d orbitals, as shown in Fig. 1(d). The relatively flat energy bands near the VBM result in a large effective mass of photoexcited hole. In our previous work, we have determined the effective mass of the electron 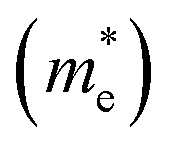 and the hole
and the hole 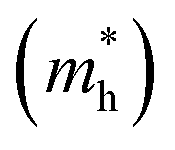 of bulk Ag3PO4 to be 0.47me and 2.13me, respectively.42 The value of
of bulk Ag3PO4 to be 0.47me and 2.13me, respectively.42 The value of 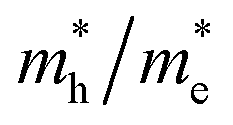 of bulk Ag3PO4 is 4.5, which is much larger than that of some other ternary oxides photocatalysts such as BiVO4
of bulk Ag3PO4 is 4.5, which is much larger than that of some other ternary oxides photocatalysts such as BiVO4 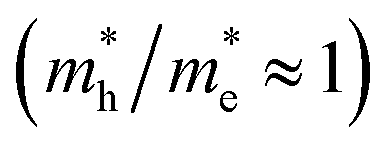 .50 A significantly different mobility between photoexcited electron and hole results in a low recombination rate of electron–hole pairs. The results suggest that Ag3PO4 has an excellent band edge for carrier separation. These special inherent characters of bulk Ag3PO4 actually contributes to a high photocatalytic activity of Ag3PO4. As the water oxidation process takes place on the surface of photocatalyst, the photocatalytic activity is also strongly affected by the surface properties, which is the subject of next section.
.50 A significantly different mobility between photoexcited electron and hole results in a low recombination rate of electron–hole pairs. The results suggest that Ag3PO4 has an excellent band edge for carrier separation. These special inherent characters of bulk Ag3PO4 actually contributes to a high photocatalytic activity of Ag3PO4. As the water oxidation process takes place on the surface of photocatalyst, the photocatalytic activity is also strongly affected by the surface properties, which is the subject of next section.
3.2 Surface properties
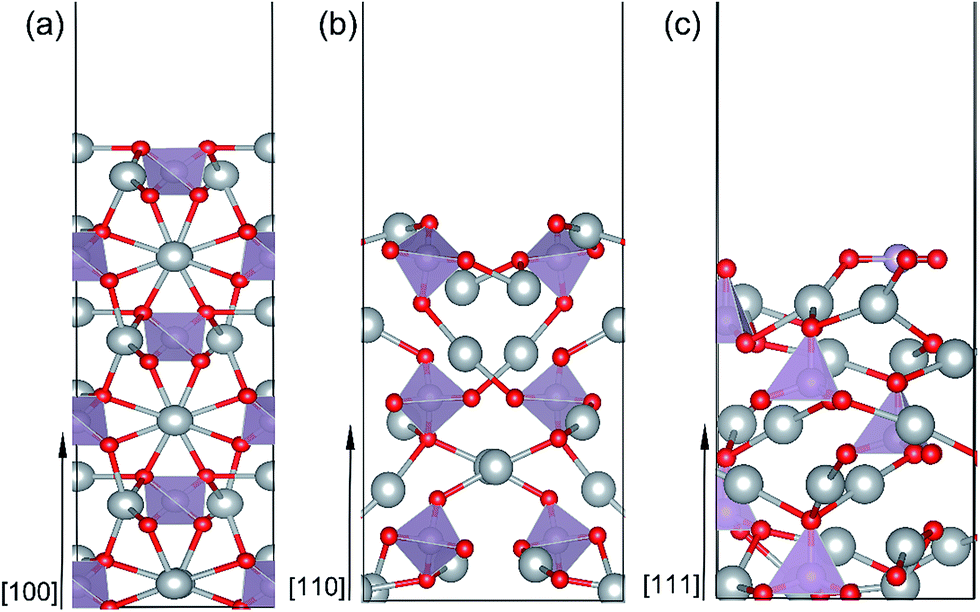 | ||
| Fig. 2 Side view of the optimized Ag3PO4 (100) (a), (110) (b) and (111) (c) surfaces. | ||
All possible termination structures for each surface orientation ([100], [110] and [111]) were built and fully optimized. The termination structures and the corresponding formation energies were listed in Fig. S1 and Table S1 in the ESI.† The surface termination with the lowest formation energy for each surface orientation was identified and selected for further study. Here, we briefly introduce the structural characters of these three surfaces. Fig. 2(a–c) present a side view of the most stable geometries for the Ag3PO4 (100), (110) and (111) surfaces, respectively. It can been seen that the atoms in the top few layers of these surfaces undergo strong rearrangement to minimize the surface energy after fully relaxation. Specifically: (i) (100) surface. The outmost Ag atoms are found to move inward by 0.39 Å after relaxation. The O atoms in the second subsurface tend to move outward. Both the near-surface Ag and O atoms are coordinatively unsaturated. The Ag atoms in the outmost surface are coordinated with two O atoms, while the O atoms are threefold coordinated with two Ag atoms and one P atom. Both Ag and O atoms are fourfold coordinated in the bulk region of Ag3PO4. The tetrahedral structure of the near-surface [PO4]3− unit is not distorted much after surface relaxation, indicating the strong P–O covalent bonds in the (100) surface. (ii) (110) surface. Like (100) surface, the outmost undercoordinated Ag and O atoms in (110) surface tend to lie on a plane. The near-surface O atoms are twofold coordinated, and Ag atoms are threefold coordinated. (iii) (111) surface. In the (111) surface, the P and O atoms are displaced on the outmost layer. This is different from the (100) and (110) surfaces, in which the P atoms are not exposed on the outmost surface. The P atom on the top layer of (111) surface coordinates with three O atoms, forming a triangle with P atom at the center. In addition, one single O–P bond with length of 1.49 Å is found on the second subsurface.
The predicted Ef values for the (100), (110) and (111) surfaces of Ag3PO4 are 0.34 J m−2, 0.38 J m−2 and 0.66 J m−2, respectively. The obtained relative stability of these three surfaces is consistent with the experimental fact that (111) surface is more reactive than (110) and (100) surfaces.15 The results also agree with earlier theoretical data of Martin15 and Bi21 et al. Since the (100) and (110) surfaces have similar atomic arrangement in the outmost surface (containing unsaturated Ag and O atoms), the Ef of (100) surface is thus only a little smaller than that of (110) surface. By contrast, the (111) surface has a significantly larger Ef than that of (110) and (100) surfaces. Such a discrepancy might be ascribed to the presence of more abundant active sites on the (111) surface as compared to the (110) and (100) surfaces.
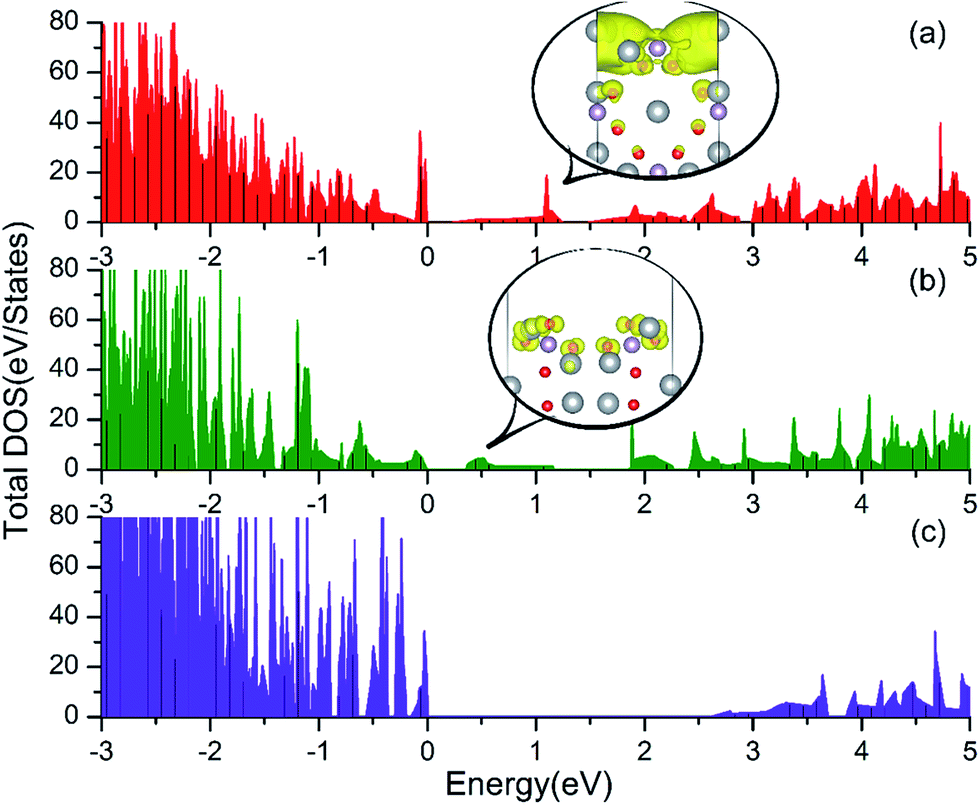 | ||
| Fig. 3 Total DOS of the Ag3PO4 (100) (a), (110) (b) and (111) (c) surfaces. The band decomposed charge density for the mid-gap states in the (100) and (110) surfaces are depicted in the inset of (a) and (b), respectively. Here, the color code for the different atoms is as in Fig. 1. The isosurface (yellow surface) is at 0.002 e bohr−3 and 0.001 e bohr−3 for (100) and (110) surfaces, respectively. | ||
3.3 Mechanisms of OER
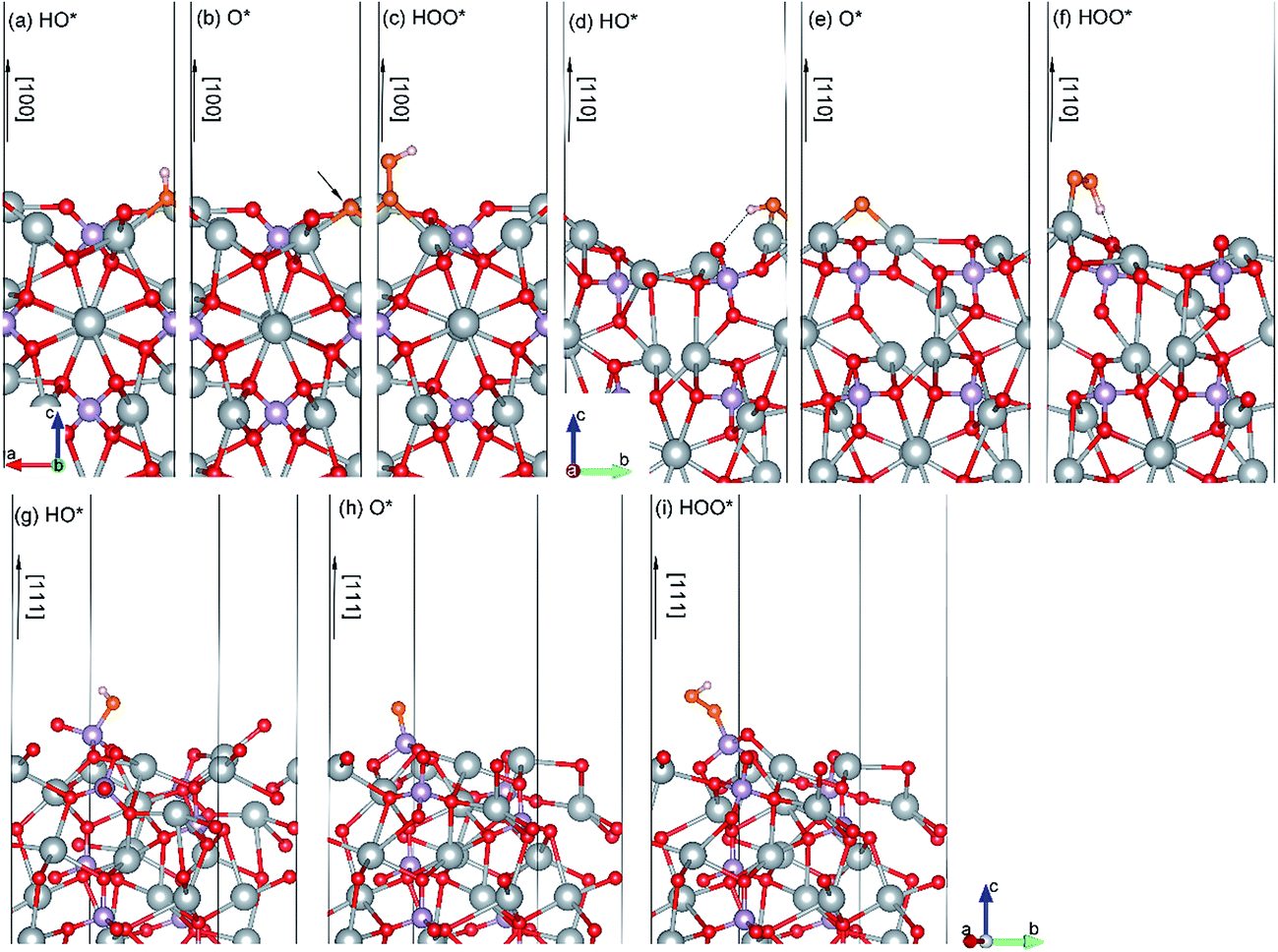 | ||
| Fig. 4 Side views of the optimized geometries of relevant species (HO*, O*, and HOO*) during OER on Ag3PO4 (100) (a–c), (110) (d–f) and (111) (g–i) surfaces. The hydrogen bond is indicated by dashed line. The O atoms in the HO*, O*, and HOO* species are indicated as orange spheres. | ||
| Adsorption energy (eV) | (100) | (110) | (111) |
|---|---|---|---|
| HO | −2.40 | −2.04 | −3.93 |
| O | −2.98 | −2.23 | −4.29 |
| HOO | −1.01 | −1.09 | −2.20 |
HO*. As shown in Fig. 4(a), the HO* species is found to be covalently bond to three Ag atoms with the O–H bond perpendicular to the (100) surface plane. Similarly, the most stable adsorption site of HO* on the (110) surface is bond to two Ag atoms (Fig. 4(d)). Specially, the HO* species on the (110) surface also connects with a neighboring O atom via a hydrogen bond. The calculated length of the hydrogen bond is 2.02 Å. In contrast, the HO* species on the (111) surface is stabilized by the unsaturated P atom with strong P–O covalent bonding (see Fig. 4(g)). The adsorption energies of the HO* species on the (100), (110) and (111) surfaces of Ag3PO4 are calculated to be −2.40, −2.04, and −3.93 eV, respectively. The much lower value of the adsorption energy for the HO* adsorbed (111) surface is attributed to the formation of strong P–O covalent bonds.
O*. The O* species is found to be covalently bond to three (two) unsaturated Ag atoms for (100) ((110)) surface, forming an Ag-oxide group. The average Ag–O* bond length of the Ag-oxide group is 2.14 Å for the (100) surface and 2.02 Å for the (110) surface, respectively. The most stable adsorption site of O* on (111) surface is still the on-top site above the unsaturated P atom (Fig. 4(h)). The P–O* bond length is 1.5 Å, which is smaller than that (1.61 Å) in HO* adsorbed (111) surface. It suggests that the dehydrogenation of hydroxyl makes P–O* covalent bond stronger in the (111) surface. The adsorption energy of O* on the (111) surface is found to be −4.29 eV, also significantly lower than that on the (110) and (100) surfaces (−2.23 eV and −2.98 eV, respectively).
HOO*. The O* species reacts with another H2O molecular and generates an HOO*. The HOO* species on the (100) surface is also found to be covalently bond to three Ag atoms, but with longer Ag–O average bond length of 2.33 Å as compared to the O* adsorbed (100) surface. The adsorption energy (−1.01 eV) is thus significantly larger than that (−2.98 eV) of O* absorbed (100) surface. The HOO* species on the (110) surface is only bound to one Ag atom with Ag–O bond length of 2.18 Å (Fig. 4(f)). Nevertheless, it is also stabilized by a hydrogen bond with H–O (surface) length of 1.34 Å. The adsorption energy for HOO* absorbed (110) surface is calculated to be −1.09 eV, close to that of HOO* absorbed (100) surface. In the (111) surface, a covalent bond between HOO* and P atom with a P–O bond length of 1.65 Å is formed. It still has the largest adsorption energy (−2.20 eV) as compared to that of HOO* absorbed (100) and (110) surfaces.
ln10 × pH), as illustrated in Fig. S7 in ESI.†
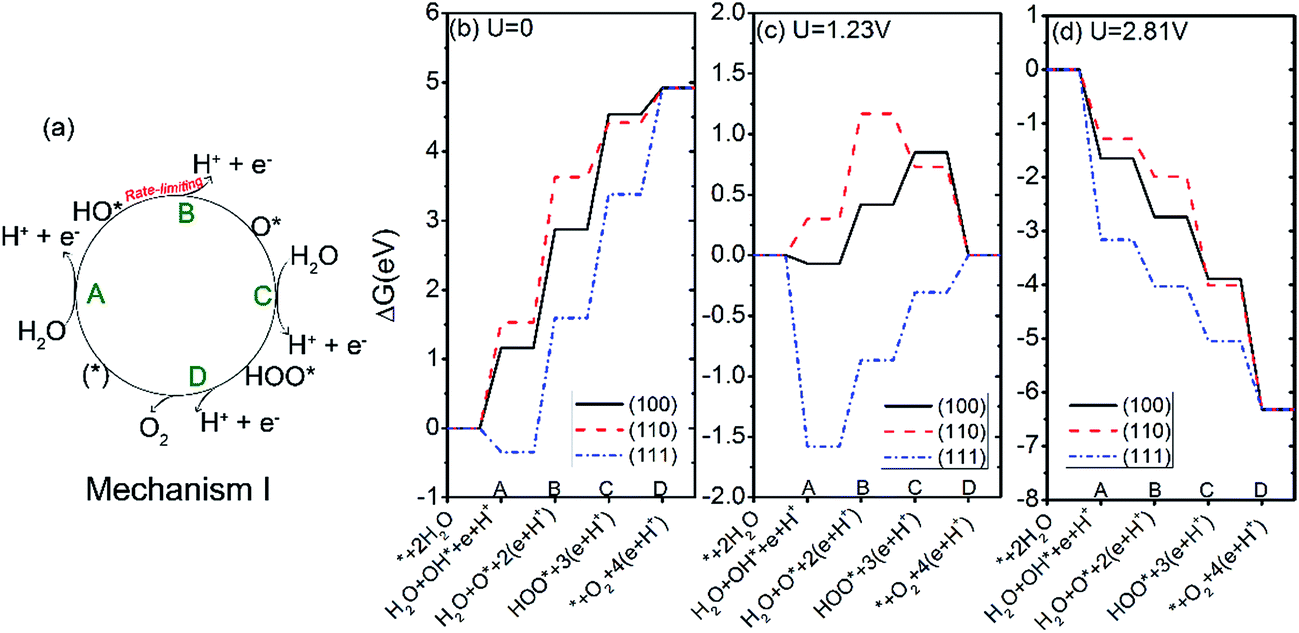 | ||
| Fig. 5 (a) Oxygen evolution reaction mechanism I; (b–d), the free energies of the intermediates on Ag3PO4 (100), (110) and (111) surfaces following mechanism I at pH = 0 and U = 0 (b), 1.23 V (c), and 2.81 V (d). | ||
The free energy changes at the equilibrium potential (U = 1.23 V) and pH = 0 were plotted in Fig. 5(c). We see that the steps of A and D of the OER on (100) surface change from uphill to downhill. For the (110) surface, the steps of C and D change from uphill to downhill. For the (111) surface, the ΔG for the step B, C, and D tend to be less positive compared with U = 0. To have every step downhill, an overpotential of 0.49, 0.87, and 0.71 V is required for (100), (110), and (111) surfaces respectively. These values are in the same magnitude as those of other semiconductor oxides like TiO2 (0.78 V),45 RuO2 (0.37 V)46 and IrO2 (0.56 V).46
Under irradiation, the difference between the O2 evolution redox potential (1.23 V vs. NHE) and the redox potential corresponding to the valence band of Ag3PO4 is assumed to be driving force for the photo-oxidation of water.29,45 The redox level for the valence band edge UVB of Ag3PO4 has been found experimentally to be about 2.81 eV for pH = 0.17 According to Fig. 5(d), the value of UVB provides enough overpotential to compel every step of OER to be downhill for all three surfaces. It suggests that the photoexcited hole in Ag3PO4 can afford sufficient overpotential for OER to proceed spontaneously on (100), (110), and (111) facets of Ag3PO4, in good agreement with the experimental findings by Martin and Yi et al.15,17 In particular, the photo-oxidation of water would be more likely to take place on (111) surface than on (110) and (100) surfaces, due to the much lower overpotential for the first step of OER on the (111) surface (−3.16 V) than that on the (100) and (110) surfaces (−1.65 V and −1.28 V, respectively). The results are consistent with previous experimental findings where the tetrahedral Ag3PO4 crystals, composed of (111) facets, show a higher oxygen evolution rate than either (110) or (100) facets.15
We further studied another two possible mechanisms that have been reported in studies of TiO2 and g-C3N4.29,45 The two new mechanisms are referred to as mechanism II and III, respectively, as illustrated in Fig. 6(a). In mechanism II, the step A is same as that in mechanism I, i.e. the dehydrogenation of H2O. Step B′ is that the absorbed HO* reacts with another water to yield HOOH*. The HOOH* intermediate further dissociates to HOO* in step C′, followed by a dehydrogenation step to O2 in the last step.29 The four steps of mechanism II can be written as:
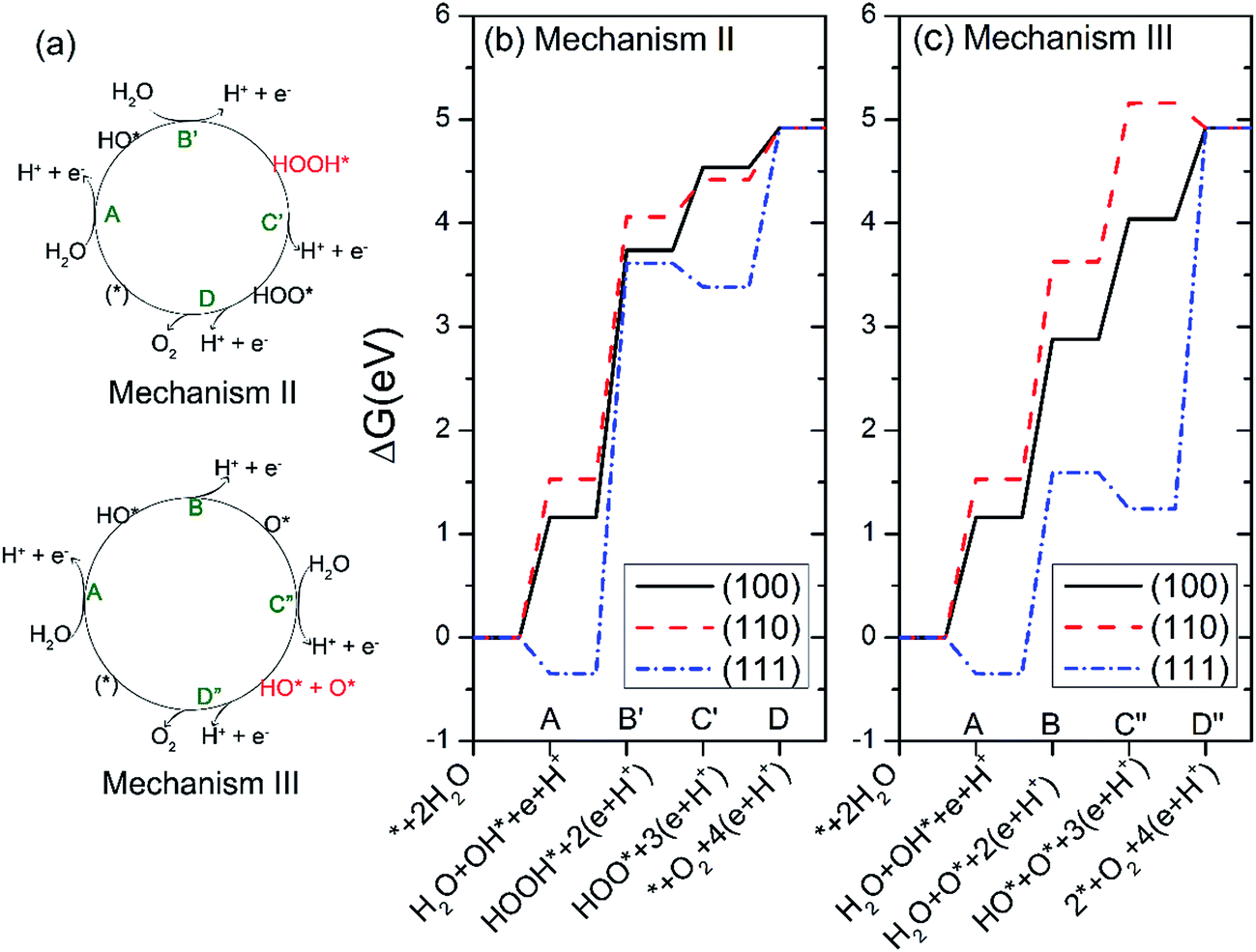 | ||
| Fig. 6 (a) Oxygen evolution reaction (OER) mechanism II and III. (b and c) The free energies of the intermediates on Ag3PO4 (100), (110) and (111) surfaces following mechanism II (b) and III (c) at pH = 0 and U = 0. | ||
A
| 2H2O + * → H2O + HO* + H+ + e− | (10) |
B′
| H2O + HO* → HOOH* + H+ + e− | (11) |
C′
| HOOH* → HOO* + H+ + e− | (12) |
D
| HOO* → O2 + * + H+ + e− | (13) |
The mechanism III involves two steps of dehydrogenation of H2O to produce HO* species (step A and step C″). In step B, one HO* dissociates the hydrogen atom to yield atomic O*. In step D″, the atomic O* eventually reacts with another HO* to generate O2 gas.29 The four steps of mechanism III can be written as:
A
| 2H2O + * → H2O + HO* + H+ + e− | (14) |
B
| H2O + HO* → H2O + O* + H+ + e− | (15) |
C″
| H2O + O* → HO* + O* + H+ + e− | (16) |
D″
| HO* + O* → O2 + 2* + H+ + e− | (17) |
The calculated free energies of the intermediates following mechanism II and III at the potential of U = 0 V and pH = 0 were plotted in Fig. 6(b and c), respectively. If the mechanism II is operated, the rate-limiting step for all three surfaces corresponds to the step B′, i.e. the formation of HOOH*, as seen from Fig. 6(b). Especially, the free-energy difference of step B′ raises as much as 3.96 eV for the Ag3PO4 (111) surface at U = 0 and pH = 0. This value has exceeded the valence band edge of Ag3PO4 (UVB = 2.81 eV), as shown in Fig. 7(a). It indicates that the photoexcited hole in the valence band edge of Ag3PO4 (111) surface cannot provide sufficient overpotential for the step B′ of mechanism II to proceed. For the (100) and (110) surfaces, the thermodynamic barriers of the rate-limiting step (step B′) following mechanism II are 2.58, and 2.53 eV respectively, which are a little smaller than UVB. However, they are much larger than the barriers of rate-limiting step in mechanism I (1.72 eV for (100) surface, 2.1 eV for (110) surface). Therefore, we tend to consider the mechanism II involving a HOOH* intermediate less favorable than mechanism I for the oxidation of water on Ag3PO4 (100), (110), and (111) surfaces.
 | ||
| Fig. 7 The free energies of the intermediates on Ag3PO4 (100), (110) and (111) surfaces following mechanism II (a) and III (b) at pH = 0 and U = 2.81 V. | ||
In mechanism III, the rate-limiting step for the Ag3PO4 (100) and (110) surfaces is still the step B (the dehydrogenation of HO*), similar to the mechanism I, as seen from Fig. 6(c). Interestingly, in comparison with mechanism I, the rate-limiting step for the Ag3PO4 (111) surface changes from step B to step D″, in which O* reacts with an HO* to generate O2 gas. The corresponding barrier of rate-limiting step for (111) surface increases from 1.94 eV to 3.68 eV at U = 0, which also exceeds the maximum potential energy gain from irradiation. Fig. 7(b) shows that step D″ is an endothermic process for Ag3PO4 (111) surface even under the irradiation (U = 2.81 V). Therefore, we conclude that the mechanism II and III are unlikely to take place on the Ag3PO4 (111) surface. For (100) surface, the barrier of step C in mechanism I is larger than that of step C″ in mechanism III. Therefore, the OER process would like to proceed by following mechanism III for (100) surface. For (110) surface, the mechanism I is more likely to take place as ΔGIC < ΔGIIIC″.
4 Conclusions
We report here a comparative study of the bulk, surface properties and the photo-oxidation of water on three low index facets of Ag3PO4: (100), (110) and (111) based on the first-principles DFT and the thermochemistry of the reaction steps. The conclusions are as follows: (1) for all three surfaces, the rate-limiting step for the OER process is the dehydrogenation of HO* (HO* → O* + H+ + e−); (2) oxidation of water on the Ag3PO4 (100), (110), and (111) surfaces requires an overpotential of 0.49, 0.87, and 0.71 V respectively at the equilibrium potential (U = 1.23 V) and pH = 0; (3) the first proton removal of water (step A: H2O + * → HO* + H+ + e−) is sensitive to the crystalline surface structures. At U = 0 and pH = 0, the free energy changes of step A (ΔGA) on (100) and (110) surfaces are endothermic, while ΔGA is calculated to be negative (exothermic) for (111) surface. The much lower adsorption energy of HO* on (111) surface leads to the deprotonation of the first absorbed water molecular becoming to be a favorable step in (111) surface, compared with (100) and (110) surfaces; (4) the illumination of the Ag3PO4 (100), (110), and (111) surfaces with light provides enough overpotential for the photooxidation to proceed spontaneously; (5) the OER process tends to proceed by following mechanism III on the (100) surface. By contrast, the (110) and (111) surfaces would like to oxide water by following mechanism I; (6) the total DOS plots are strongly related to the surface geometry. For the (100) and (110) surfaces, the highly localized states are formed deep in the gap. In contrast, there is no impurity states in the mid-gap of (111) surface. This can largely avoid the formation of charge carrier recombination centers and improve the visible photocatalytic efficiency. Our results are important for understanding the underlying mechanism of the photocatalytic water oxidation process occurring at Ag3PO4 surfaces, and serve as a foundation for developing new high-performance Ag3PO4 based photocatalysts for water splitting.Acknowledgements
This work was supported by the National Science Foundation of China (No. 21501177, 21673240, and 21201165) and the Special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund (the second phase). The authors also gratefully acknowledge the National Supercomputing Center in Shenzhen for providing the computing resources.References
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- S. U. M. Khan, M. Al-Shahry and W. B. Ingler, Science, 2002, 297, 2243–2245 CrossRef CAS PubMed.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS PubMed.
- H. G. Kim, D. W. Hwang, S. W. Bae, J. H. Jung and J. S. Lee, Catal. Lett., 2003, 91, 193–198 CrossRef CAS.
- A. Kudo, H. Kato and S. Nakagawa, J. Phys. Chem. B, 2000, 104, 571–575 CrossRef CAS.
- K. Maeda, K. Teramura, D. L. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- K. Maeda, T. Takata, M. Hara, N. Saito, Y. Inoue, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2005, 127, 8286–8287 CrossRef CAS PubMed.
- H. Yan, J. Yang, G. Ma, G. Wu, X. Zong, Z. Lei, J. Shi and C. Li, J. Catal., 2009, 266, 165–168 CrossRef CAS.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- V. Balzani, A. Credi and M. Venturi, ChemSusChem, 2008, 1, 26–58 CrossRef CAS PubMed.
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed.
- I. Tsuji, H. Kato and A. Kudo, Angew. Chem., Int. Ed., 2005, 44, 3565–3568 CrossRef CAS PubMed.
- D. J. Martin, N. Umezawa, X. Chen, J. Ye and J. Tang, Energy Environ. Sci., 2013, 6, 3380–3386 CAS.
- W. N. Zhao and Z. P. Liu, Chem. Sci., 2014, 5, 2256–2264 RSC.
- Z. G. Yi, J. H. Ye, N. Kikugawa, T. Kako, S. X. Ouyang, H. Stuart-Williams, H. Yang, J. Y. Cao, W. J. Luo, Z. S. Li, Y. Liu and R. L. Withers, Nat. Mater., 2010, 9, 559–564 CrossRef CAS PubMed.
- Y.-F. Li, Z.-P. Liu, L. Liu and W. Gao, J. Am. Chem. Soc., 2010, 132, 13008–13015 CrossRef CAS PubMed.
- D. J. Martin, G. G. Liu, S. J. A. Moniz, Y. P. Bi, A. M. Beale, J. H. Ye and J. W. Tang, Chem. Soc. Rev., 2015, 44, 7808–7828 RSC.
- N. Umezawa, O. Y. Shuxin and J. H. Ye, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 035202 CrossRef.
- Y. Bi, S. Ouyang, N. Umezawa, J. Cao and J. Ye, J. Am. Chem. Soc., 2011, 133, 6490–6492 CrossRef CAS PubMed.
- D. Cao-Thang, N. Thanh-Dinh, F. Kleitz and D. Trong-On, Chem. Commun., 2011, 47, 7797–7799 RSC.
- W. Yao, B. Zhang, C. Huang, C. Ma, X. Song and Q. Xu, J. Mater. Chem., 2012, 22, 4050–4055 RSC.
- Q. Xiang, D. Lang, T. Shen and F. Liu, Appl. Catal., B, 2015, 162, 196–203 CrossRef CAS.
- X. Yang, H. Cui, Y. Li, J. Qin, R. Zhang and H. Tang, ACS Catal., 2013, 3, 363–369 CrossRef CAS.
- Y. Liu, L. Fang, H. Lu, L. Liu, H. Wang and C. Hu, Catal. Commun., 2012, 17, 200–204 CrossRef CAS.
- Y. Liu, L. Fang, H. Lu, Y. Li, C. Hu and H. Yu, Appl. Catal., B, 2012, 115, 245–252 CrossRef.
- Y. Bi, H. Hu, S. Ouyang, Z. Jiao, G. Lu and J. Ye, J. Mater. Chem., 2012, 22, 14847–14850 RSC.
- S. Lin, X. Ye, X. Gao and J. Huang, J. Mol. Catal. A: Chem., 2015, 406, 137–144 CrossRef CAS.
- J. Chen, Y.-F. Li, P. Sit and A. Selloni, J. Am. Chem. Soc., 2013, 135, 18774–18777 CrossRef CAS PubMed.
- Y. Ji, B. Wang and Y. Luo, J. Phys. Chem. C, 2014, 118, 1027–1034 CAS.
- Y. Ji, B. Wang and Y. Luo, J. Phys. Chem. C, 2012, 116, 7863–7866 CAS.
- A. V. Akimov, J. T. Muckerman and O. V. Prezhdo, J. Am. Chem. Soc., 2013, 135, 8682–8691 CrossRef CAS PubMed.
- R. Long and O. V. Prezhdo, J. Am. Chem. Soc., 2014, 136, 4343–4354 CrossRef CAS PubMed.
- W. R. Duncan, W. M. Stier and O. V. Prezhdo, J. Am. Chem. Soc., 2005, 127, 7941–7951 CrossRef CAS PubMed.
- A. V. Akimov, A. J. Neukirch and O. V. Prezhdo, Chem. Rev., 2013, 113, 4496–4565 CrossRef CAS PubMed.
- G. Kresse, Vienna ab initio simulation package (VASP), 5.3.5, 2014, http://cms.mpi.univie.ac.at/vasp/vasp/vasp.html Search PubMed.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter, 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- Z. Ma, Z. Yi, J. Sun and K. Wu, J. Phys. Chem. C, 2012, 116, 25074–25080 CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter, 1976, 13, 5188 CrossRef.
- A. Valdes, Z. W. Qu, G. J. Kroes, J. Rossmeisl and J. K. Norskov, J. Phys. Chem. C, 2008, 112, 9872–9879 CAS.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Norskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- J. J. Liu, X. L. Fu, S. F. Chen and Y. F. Zhu, Appl. Phys. Lett., 2011, 99, 191903 CrossRef.
- X. G. Ma, B. Lu, D. Li, R. Shi, C. S. Pan and Y. F. Zhu, J. Phys. Chem. C, 2011, 115, 4680–4687 CAS.
- S. Lany and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 235104 CrossRef.
- A. Walsh, Y. Yan, M. N. Huda, M. M. Al-Jassim and S.-H. Wei, Chem. Mater., 2009, 21, 547–551 CrossRef CAS.
- B. J. Zheng, X. Wang, C. Liu, K. Tan, Z. X. Xie and L. S. Zheng, J. Mater. Chem. A, 2013, 1, 12635–12640 CAS.
- Z. Ma, K. Wu, R. Sa, Q. Li, C. He and Z. Yi, Int. J. Hydrogen Energy, 2015, 40, 980–989 CrossRef CAS.
- K. Yang, Y. Dai and B. Huang, J. Phys. Chem. C, 2007, 111, 12086–12090 CAS.
- J. Wirth, R. Neumann, M. Antonietti and P. Saalfrank, Phys. Chem. Chem. Phys., 2014, 16, 15917–15926 RSC.
- A. Hussain, J. Gracia, B. E. Nieuwenhuys and J. W. Niemantsverdriet, ChemCatChem, 2013, 5, 2479–2488 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02853a |
| This journal is © The Royal Society of Chemistry 2017 |