
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Incorporation of well-dispersed calcium phosphate nanoparticles into PLGA electrospun nanofibers to enhance the osteogenic induction potential†
Dandan Wang ab,
Liuyang Xuanab,
Huixiang Zhongab,
Yihong Gongab,
Xuetao Shic,
Feng Yed,
Yan Li*ab and
Qing Jiangab
ab,
Liuyang Xuanab,
Huixiang Zhongab,
Yihong Gongab,
Xuetao Shic,
Feng Yed,
Yan Li*ab and
Qing Jiangab
aDepartment of Biomedical Engineering, Guangdong Provincial Key Laboratory of Sensor Technology and Biomedical Instrument, School of Engineering, Sun Yat-sen University, Guangzhou, Guangdong, P. R. China. E-mail: liyan99@mail.sysu.edu.cn; Fax: +86-20-39387890; Tel: +86-20-39387890
bGuangdong Provincial Engineering and Technology Center of Advanced and Portable Medical Devices, Sun Yat-sen University, Guangzhou, Guangdong, P. R. China
cSchool of Materials Science and Engineering, South China University of Technology, Guangzhou, P. R. China
dKey Laboratory of Molecular Biology for Infectious Diseases, Ministry of Education of China, The Second Affiliated Hospital, Chongqing Medical University, Chongqing, P. R. China
First published on 3rd May 2017
Abstract
Poly(lactide-co-glycolide) (PLGA) electrospun composite nanofibers were fabricated to mimic bone extra cellular matrix for applications in bone tissue engineering. Poly(acrylic acid) modified Zn-doped calcium phosphate (PAA-CaP/Zn) nanoparticles were first synthesized. PAA was selected because of the strong bonding ability with calcium and Zn was incorporated to enhance the bioactivity. PAA-CaP/Zn nanoparticles were well dispersed with a hydrodynamic size of 184 nm; the presence of Zn was confirmed and the composition was similar to hydroxyapatite. These nanoparticles were non-cytotoxic against adipose derived stem cells from rats (rADSC) at 0.1 mg ml−1 based on an extraction assay and direct contact method. The nanoparticles were further entrapped into PLGA nanofibers. All electrospun membranes were composed of smooth fibers and for composite membranes, nanoparticles were homogeneously distributed in the fiber matrix. Furthermore, the tensile strength was increased with compromised elasticity, while the degradation was retarded and the biomineralization property was enhanced due to the presence of nanoparticles. rADSC proliferated very well on composite membranes and the one with 5% nanoparticles (P-5) demonstrated the highest osteogenic induction effect as significantly more calcium was deposited after osteogenic induction for 21 days. Sample P-5 was the most suitable for applications in bone tissue engineering.
1. Introduction
Tissue engineering combines the developments in material science and cell biology to repair defects which are congenital or have resulted from accidents/diseases. It is an important technology to minimize patients' pain.1 Bone tissue engineering is one branch, mainly focused on repairing bone defects. For such purposes, scaffold materials are indispensible, which should be biocompatible, bioactive and biodegradable. The degradation by-products of ideal scaffolds need to also be biocompatible so as not to trigger undesired responses such as immune rejection or inflammation, which minimize the influence on the microenvironment of implants and furthermore lead to patients' pain.2 For such applications, there are three kinds of scaffold material: organic, inorganic and organic/inorganic composite materials. Among them, poly(lactide-co-glycolide) (PLGA), which is biocompatible with a controllable degradation rate3,4 and its degradation by-products are also biocompatible, has been widely used. A PLGA scaffold has been applied to repair rat scull defects; on scaffolds, cells proliferated very well and the repair outcome was good.5PLGA scaffolds were prepared with various methods, such as particle leaching, phase separation, lyophilization, self-assembly and electrospinning. Among these techniques, electrospinning has attracted lots of attentions because of its reproducibility, large specific area of the electrospun membrane, maneuverability of fiber diameter (could be in nano-meter or micro-meter range) and flexibility of surface morphology.1,6 The scaffold fabricated with electrospinning not only mimicked the structure of extra cellular matrix (ECM), but also offered sufficient space for cell proliferation and nutrient transportation due to its porous structure, which further improved the biocompatibility of scaffolds.7,8
To further mimic the composition of bone ECM, inorganic materials such as hydroxyapatite (HAp) was embedded into PLGA electrospun fibers to form composite membranes.9–13 The compositions and properties of such membranes were intensively studied. Different amounts of HAp nanoparticles (1, 5, 10 and 20 wt%) were encapsulated into PLGA fibers. With the increase of HAp amount, the average fiber diameter increased from 300 nm (pure PLGA) to 700 nm (with 20% HAp) while the glass transition temperature (Tg) also increased. Regarding the dynamic mechanical behavior, HAp nanoparticles acted as reinforcements at lower concentrations (1% and 5%), but acted as defects at higher concentrations (10% and 20%). HAp is hydrophilic, which influenced the uptake of PBS and the mass loss of composite membranes.11 On PLGA with 5 wt% HAp electrospun composite membrane, MC3T3-E1 cells showed a more spreading morphology and significant higher alkaline phosphatase (ALP) secretion.13 The effect of HAp morphology on cell behaviors was investigated.9 Needle-like or spherical HAp nanoparticles were entrapped in PLGA nanofibers. In vitro tests demonstrated that a higher cell (MC3T3-E1) proliferation and enhanced osteogenesis was observed on composite membranes with needle-like HAp.9 The size of HAp particles also played an important role in cell behaviors.10 Poly(L-lactide) (PLLA) composite scaffolds were prepared by incorporating needle-shaped nano- or micro-sized HAp particles into PLLA nanofibers using electrospinning, which had higher relative cell (rat osteosarcoma ROS17/2.8 cells) viabilities and ALP activities than the pure PLLA scaffold, especially the scaffold with micro-sized HAp particles.10 The presence of HAp nanoparticles in PLGA composite electrospun scaffolds significantly enhanced the formation of bonelike apatites and also the proliferation of primary human osteoblast (HOB). Additionally, such composite scaffold promoted the response of HOB better than MC 3T3 E1.12
Besides incorporating HAp, composite membranes of more complex compositions and structures were fabricated.14,15 The incorporation of gelatin/HAp into PLGA electrospun nanofibers decreased static contact angle, increased swelling ratio, facilitated adhesion, migration and proliferation of bone cells (MC3T3-E1), as well as the osteogenic differentiation of progenitor cells (mesenchymal stem cells, MSC) based on the results of osteogenic genes expression.14 A double membrane consisting of a PLGA layer and an amorphous calcium phosphate/collagen/PLGA composite layer were synthesized using electrospinning, which was used for treatment of bone defects with complex geometries as wound dressing material.15 Biomineralization on such anisotropic membrane was preferably on the composite layer side. Differentiation of human MSC (hMSC) into the osteogenic lineage was better than that in a 2D control.15 All these composite membranes demonstrated better bioactivity. However, none of these reported structures addressed the dispersity of HAp nanoparticles in polymeric matrix, where HAp nanoparticles were not surface modified. Thus when a higher amount of HAp was encapsulated (>5%), agglomeration of HAp was present, the morphology of nanofibers was also affected11,13 and mechanical properties were compromised. Additionally, in these structures, HAp only contained Ca as the cation ion, which was different from the in vivo composition. Hence for better performance in bone tissue engineering, the composite membrane with improved structure needed to be elaborately designed.
Besides calcium, other metal ions such as Mg, Mn and Zn16 are present in bone minerals. Even these elements are in small amount, they are fundamental to bone metabolism. Zn is an essential element for human health and also important for bone metabolism. Zn was reported to increase the proliferation and differentiation of osteoblast cells,17 and also suppressed the activation of osteoclast cells.18 Thus in this paper Zn was selected to fabricate Zn doped HAp-like calcium phosphate (CaP/Zn) nanoparticles for better bioactivity.
To improve the dispersity, organic additives, such as polyethylene glycol (PEG),19 cetyl trimethylammonium bromide (CTAB)20 and citric acid19 were applied to fabricate HAp nanoparticles. However, these additives either did not bind HAp strongly or were surfactants which may not be suitable for scaffolds applications because of the cytotoxicity. Thus poly(acrylic acid) (PAA) with lots carboxyl groups which can bind HAp strongly and flexible long molecular chains which may facilitate the distribution of nanoparticles in polymeric matrix, was selected as the organic additive for the fabrication of HAp-like calcium phosphate nanoparticles.
In this paper, well dispersed Zn doped HAp-like calcium phosphate nanoparticles were fabricated in the presence of PAA (PAA-CaP/Zn), which were further embedded into PLGA nanofibers using electrospinning technique. To our best knowledge, this composite structure was first developed. The structure and properties of PAA-HAp/Zn nanoparticles and PLGA electrospun composite membranes were intensively studied. The electrospun composite membrane was designed for bone tissue engineering applications. As few studies have investigated the degradation behavior and also the osteogenic differentiation potential of adipose-derived stem cells (ADSC) on such membranes, its degradation behavior was monitored, the proliferation and osteogenic differentiation potential of ADSC from rats (rADSC) were also investigated.
2. Materials and methods
2.1 Materials
Acetone (99.5%), calcium chloride (CaCl2), dimethyl formamide (DMF, 99.5%), ethanol (95%), sodium hydrogen phosphate (Na2HPO4·12H2O) and zinc chloride (ZnCl2) were bought from Guangzhou Chemical Reagent Co. (Guangzhou, Guangdong, China). PAA (Mw 5000) was from Shanghai Ryon Biological Technology CO., Ltd (Shanghai, China) and PLGA (lactide/glycolide molar ratio 50/50, Mw = 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–80000) was from Jinan Daigang Biological Technology Co. (Jinan, Shandong, China). Dulbecco's Modified Eagle Media: Nutrient Mixture F-12 (DMEM/F12) and PS (penicillin/streptomycin antibiotics) were purchased from HyClone (Logan, UT, USA). Fetal bovine serum (FBS) and trypsin–EDTA were purchased from Gibco (Carlsbad, CA, USA). Alamar Blue was from Sigma-Aldrich (St, Louis, MO, USA) and a BCIP/NBT ALP Color Development Kit was from Beyotime (Haimen, Jiangsu, China). All chemicals were used without further purification unless otherwise stated. Deionized (DI) ultrapure water was used throughout the experiment.
000–80000) was from Jinan Daigang Biological Technology Co. (Jinan, Shandong, China). Dulbecco's Modified Eagle Media: Nutrient Mixture F-12 (DMEM/F12) and PS (penicillin/streptomycin antibiotics) were purchased from HyClone (Logan, UT, USA). Fetal bovine serum (FBS) and trypsin–EDTA were purchased from Gibco (Carlsbad, CA, USA). Alamar Blue was from Sigma-Aldrich (St, Louis, MO, USA) and a BCIP/NBT ALP Color Development Kit was from Beyotime (Haimen, Jiangsu, China). All chemicals were used without further purification unless otherwise stated. Deionized (DI) ultrapure water was used throughout the experiment.
2.2 PAA modified Zn doped calcium phosphate (PAA-CaP/Zn) nanoparticles
The cytotoxicity of samples was investigated using extraction assay and direct contact assay. rADSC at passage 3 (P3) were seeded into 96-well plates at 5000 cell per cm2. Samples in powder were sterilized by soaking in 75% ethanol for 3–4 h and rinsed with PBS three times. For extraction assay, the sterilized powder was first suspended in DMEM/F12 medium at 200 mg ml−1 for 24 h in an incubator at 37 °C;22,23 the supernatant was then collected by centrifugation, further filtration through 0.22 μm filter and diluted twice with fresh medium. For direct contact assay, the sterilized powder was suspended in medium at 0.1 mg ml−1. When cells reached 80% confluence, the medium was exchanged with extraction medium or sample/medium suspension. Cells cultured in normal medium were negative controls while in medium containing 0.1% Triton X-100 were positive controls. On day 1 and 3, Alamar Blue assay was conducted to quantify cell viabilities.
2.3 PLGA electrospun composite membranes
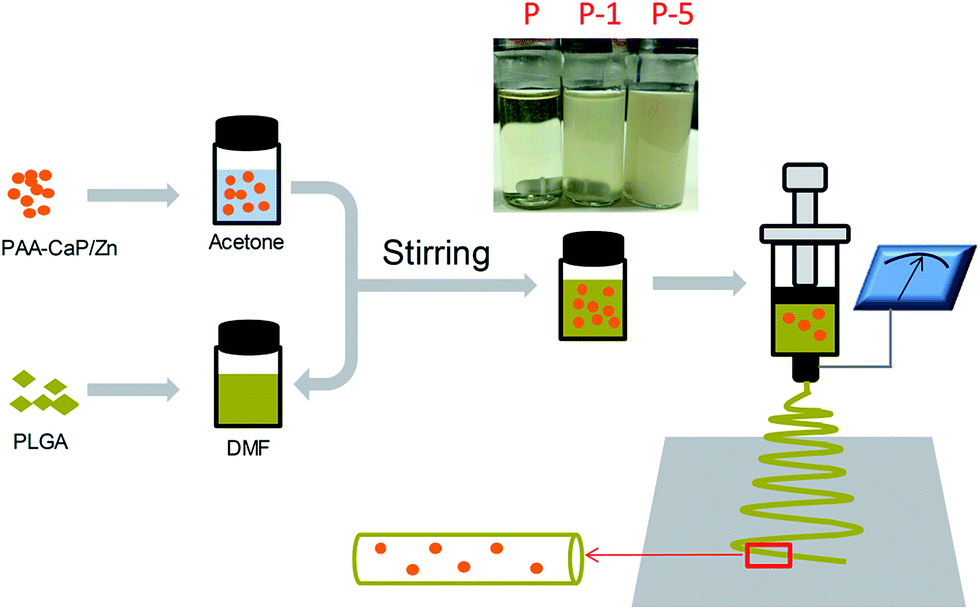 | ||
| Scheme 1 Fabrication of PLGA composite membranes with well-dispersed PAA-CaP/Zn nanoparticles using electrospinning technique. | ||
000 cells per cm2 in proliferation medium. After 24 h, proliferation medium was changed to osteogenic medium which was proliferation medium further supplemented with 10 mM β-glycerophosphate, 0.2 mM L-ascorbic acid and 0.1 μM dexamethasone. Cells were then cultured for 21 days during which osteogenic medium was exchanged every 2 days. Expression of ALP was qualitatively and quantitatively evaluated using a BCIP/NBT ALP Color Development Kit and p-nitrophenylphosphate (pNPP) assay respectively. On day 21, deposition of calcium minerals was investigated using Alizarin red staining. The amount of stains was quantified using a microplate reader based on the absorbance value at 420 nm after dissolved with diluted acidic solution, which represented the amount of deposited minerals.3. Results
3.1 Fabrication and characterization of PAA-CaP/Zn nanoparticles
In this paper, calcium phosphate nanoparticles were fabricated using wet chemical precipitation method. In order to improve the dispersity, PAA was selected because of its high bonding ability with calcium and to be more bioactive, Zn was introduced into the reaction system to obtain PAA modified Zn doped HAp-like calcium phosphate nanoparticles (PAA-CaP/Zn). The crystal phase was investigated using XRD. As shown in Fig. 1a, XRD diffraction peaks were very broad and it was difficult to assign the lattice planes. Even so, the positions of these broad peaks agreed well with that for HAp with PDF #09-0432. The functional groups were investigated using FTIR spectroscopy (Fig. 1b). For PAA-CaP/Zn, the adsorption band at 1576 cm−1 and 1418 cm−1 were due to –C–O–H and –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of PAA, where these bands shifted to lower wavenumbers when compared with that of PAA. It was reasonable that in PAA, –COOH groups were acidic while in PAA-CaP/Zn, there were probably ionic interactions between –COO− groups and Ca.25,26 Additionally, there were adsorption bands at 1065 cm−1 and 572 cm−1, which can be assigned to PO43− groups in HAp.27,28
O of PAA, where these bands shifted to lower wavenumbers when compared with that of PAA. It was reasonable that in PAA, –COOH groups were acidic while in PAA-CaP/Zn, there were probably ionic interactions between –COO− groups and Ca.25,26 Additionally, there were adsorption bands at 1065 cm−1 and 572 cm−1, which can be assigned to PO43− groups in HAp.27,28
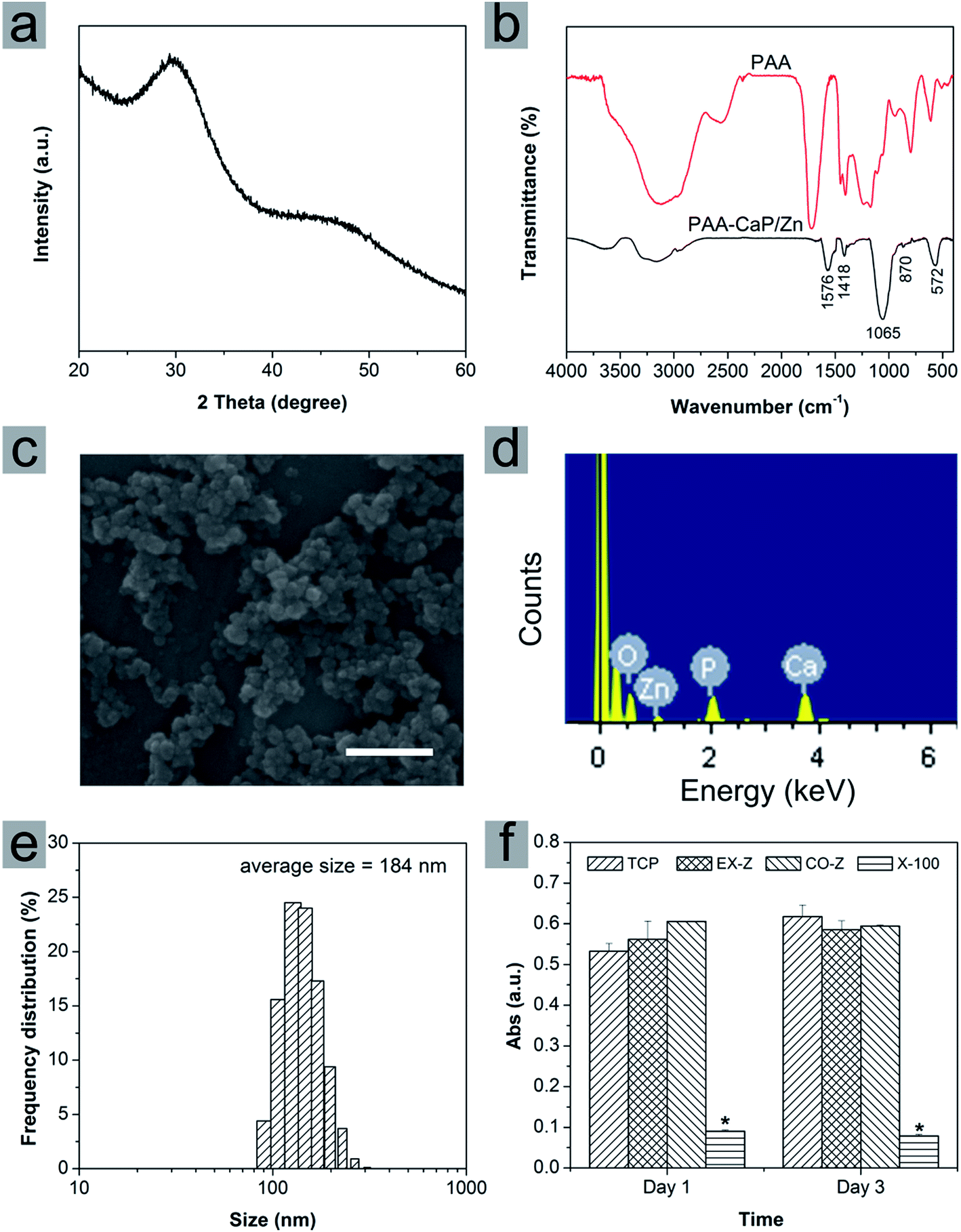 | ||
| Fig. 1 (a) XRD pattern, (b) FTIR spectrum, (c) SEM image, (d) EDS spectrum, (e) size distribution and (f) cytotoxicity results of PAA-CaP/Zn nanoparticles. Cytotoxicity was investigated using extraction assay (EX-Z group, cells were cultured with extract of PAA-CaP/Zn nanoparticles) and direct contact assay (CO-Z group, cells were cultured in nanoparticles/medium suspension). After cell cultured for 1 and 3 days, Alamar Blue assay was conducted. TCP was the group that cells were cultured in normal proliferation medium and as the negative control; X-100 was the positive control group that cells were cultured with medium containing 0.1% Triton X-100. Data = mean ± SD; n = 3, * representing p < 0.05 when compared with TCP group on the same day. Scale bar in (c) represents 500 nm. | ||
As shown in Fig. 1c, the formed nanoparticles were in spheroid morphology of ∼60 nm. Due to the modification of PAA, these PAA-CaP/Zn nanoparticles were well dispersed in aqueous solution. In mother suspension (pH 10) when the reaction was stopped, the average size was determined to be 184 nm based on Zetasizer characterization and the size distribution was narrow. In addition, the zeta potential was measured to be −14.2 mV. The average size measured by Zetasizer was larger than the particle size based on SEM image. There might be two possible reasons. The size determined with Zetasizer was hydrodynamic size. In mother suspension (pH 10), these surface-bonded PAA molecules may form a layer around these nanoparticles and attract some water molecules. Meanwhile, there might be slight aggregation of nanoparticles. Hence the average size from Zetasizer for PAA-CaP/Zn in suspension was larger.
The presence of Zn in PAA-CaP/Zn nanoparticles was confirmed with EDS analysis. Besides Ca and P, a small excitation peak belonging to Zn was observed (Fig. 1d). Furthermore, the atomic ratio was quantified using ICP after nanoparticles were dissolved in nitric acid. For PAA-CaP/Zn, the Zn/Ca atomic ratio was measured to be 5.85% while Ca/P atomic ratio was 1.586. These two values were both close to the theoretical values 5.26%, 1.584, respectively by assuming that all added Zn ions had replaced Ca ions in the structure. All these comparisons showed that PAA-CaP/Zn had the expected composition.
Cytotoxicity of PAA-CaP/Zn nanoparticles was evaluated using extraction assay and direct contact assay. For TCP group (cell cultured in normal proliferation medium), cells proliferated slowly from day 1 to day 3. When compared with TCP group, no matter cells were cultured with nanoparticles extract or suspension, the absorbance values were slightly higher or close to the values for TCP group both on day 1 and 3. Hence, no toxicity effects on rADSC for PAA-CaP/Zn were observed. As for Triton X-100 group, the absorbance values were significantly smaller, showing that Triton X-100 was toxic against rADSC. These nanoparticles were further entrapped into PLGA electrospun nanofibers.
3.2 Fabrication and characterization of PLGA electrospun composite membranes
PLGA electrospun membranes were successfully fabricated following conditions as described in Section 2.3.1. As shown in Fig. 2, all membranes were composed of fibers, where these fibers were smooth, without node-structure and diameters were in the range of 100–1000 nm. The average diameter for P, P-1 and P-5 were 604 ± 220 nm, 415 ± 128 nm and 420 ± 142 nm, respectively. Sample P-8 was also fabricated following the same parameters as described in Section 2.3.1 at weight ratio of (PAA-CaP/Zn)/PLGA as 8%. As shown in Fig. S1 (ESI),† the membrane was still fiber-like. However, the fiber surface was no longer as smooth as that for sample P. Although the average fiber diameter was 410 ± 123 nm (close to that for P-1, P-5), P-8 was not selected for further studies.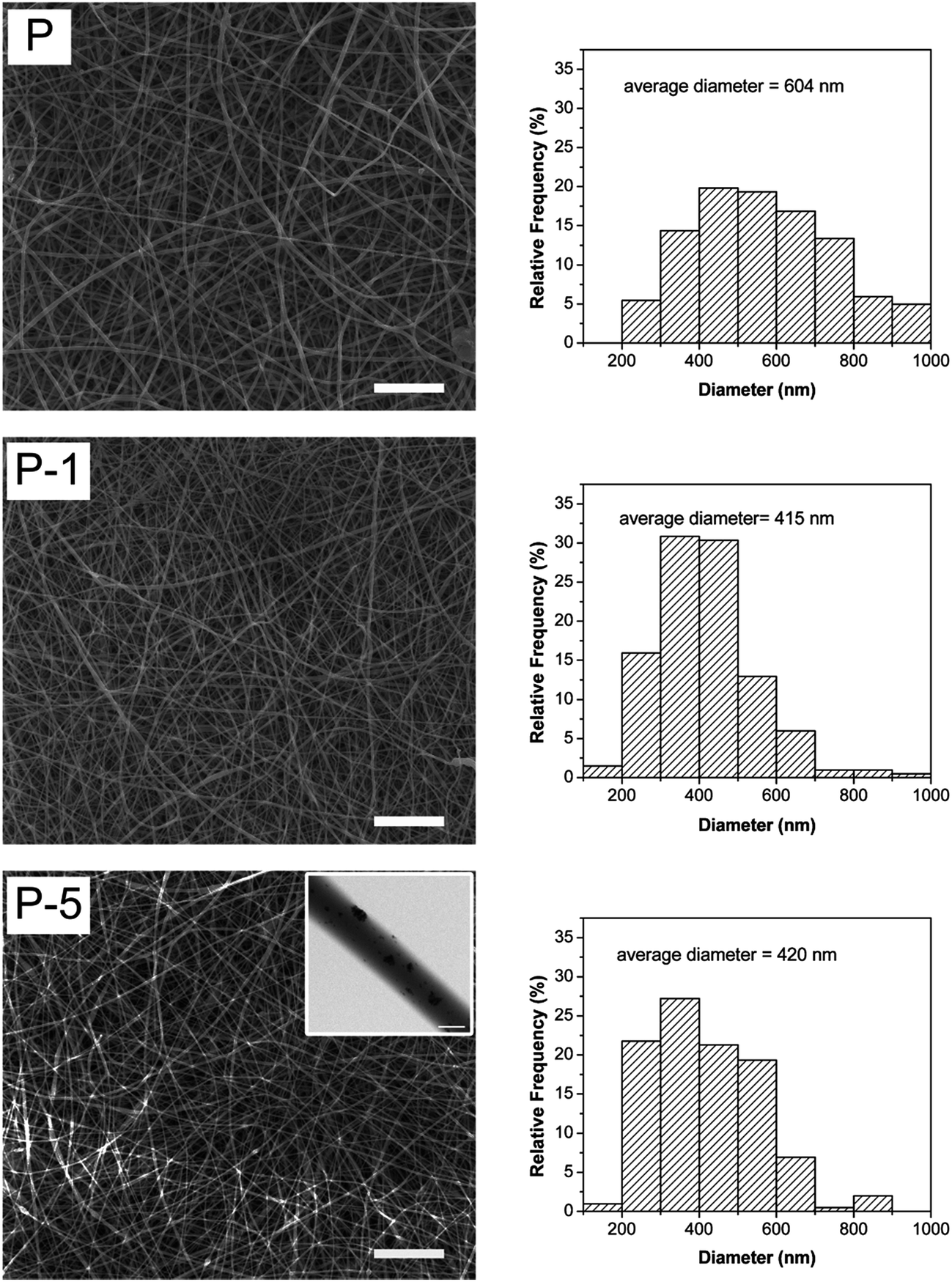 | ||
| Fig. 2 SEM images and fiber diameter distributions of PLGA electrospun membranes. Scale bars are 20 μm (scale bar in the inset is 500 nm). | ||
The entrapment of PAA-CaP/Zn nanoparticles slightly decreased the fiber diameter. The decrement of fiber diameter was probably related to PAA molecules on the nanoparticles since PAA-CaP/Zn nanoparticles were found to be well dispersed in the fiber matrix. To verify the speculation, a sample at weight ratio of PAA/PLGA as 1% was fabricated following the same electrospun parameters. For this sample, the average diameter was 310 ± 114 nm, which was much smaller than that for sample P. Hence when PAA-CaP/Zn nanoparticles were well distributed in polymeric matrix, the fiber diameter was slightly decreased. Using electrospinning method, PLGA composite membranes with similar surface morphology and fiber diameter were obtained.
Mechanical properties of PLGA electrospun membranes (Fig. 3a) were investigated using tensile testing to assess the extensibility of membranes and in turn the interaction between PAA-CaP/Zn nanoparticles and polymeric matrix. For pure PLGA membrane (sample P), the yield stress and strain were 1.9 MPa and 5.83% while the breaking stress and strain were 2.4 MPa and 93%. After encapsulation of PAA-CaP/Zn nanoparticles, the yield stress was increased to 2.23 MPa; the breaking stress increased with the amount of nanoparticles, where for P-1 and P-5, the values were 2.9 MPa and 3.1 MPa respectively. However, the breaking strains of P-1 and P-5 were both less than that for sample P. Elastic modules of these samples were further quantified, which were 2.4 MPa, 3.5 MPa and 3.1 MPa for P, P-1 and P-5, respectively. Based on these comparisons, it was found the presence of nanoparticles increased the tensile strength but compromised elasticity.
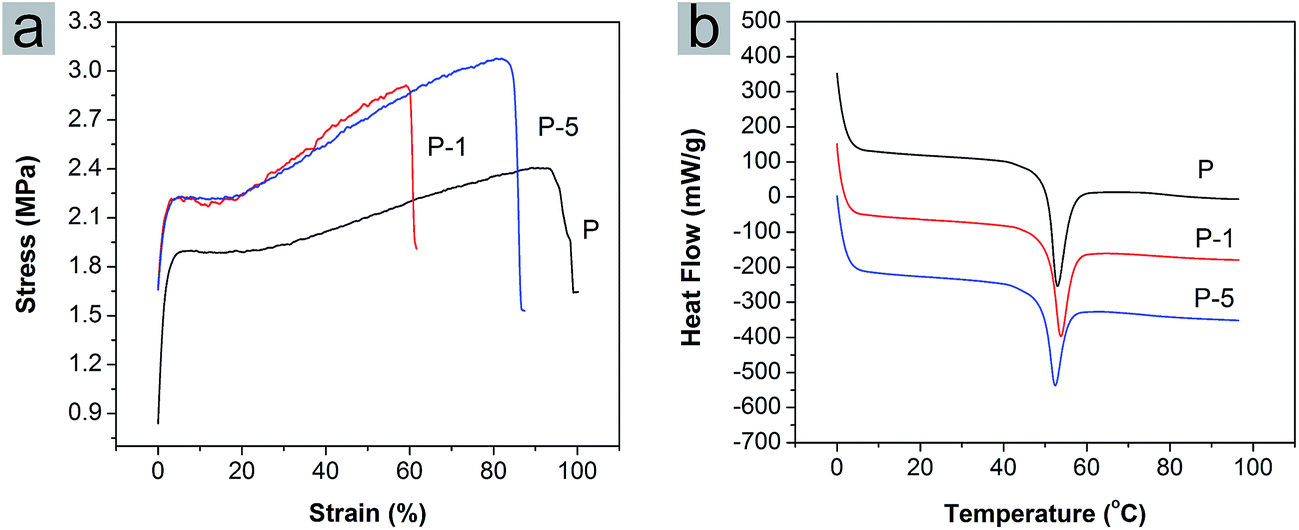 | ||
| Fig. 3 (a) Stress–strain curves and (b) DSC thermographs of PLGA electrospun membranes P, P-1 and P-5. | ||
The glass transition temperature (Tg) of PLGA electrospun membranes were quantified using DSC. The DSC thermographs from the first heating cycle were shown in Fig. 3b. For P, P-1 and P-5, Tg was 53.2 °C, 52.8 °C and 52.4 °C, respectively. The value decreased slightly with the amount of PAA-CaP/Zn increasing. The trend was different from the reported result11 that Tg increased with the amount of HAp in PLGA nanofibers. Here the encapsulated nanoparticles were surface modified with PAA and these nanoparticles were well distributed in the fiber matrix. The presence of only PAA molecules in PLGA electrospun membranes decreased the Tg (for PLGA composite membrane with 5 wt% PAA which was fabricated following the same parameters, the Tg was 52.1 °C). All these factors probably resulted in the difference from the reported data.
PLGA is degradable. Here the degradation behavior of PLGA electrospun membranes was evaluated. Through 28 days of degradation, fiber-like morphology of all membranes was maintained (Fig. 4). From day 7 to 21, there was no significant difference among these three samples. However, on day 28, some fiber adhesions were observed on P and P-1, and these two samples appeared to be more flat and dense than sample P-5. As for P-5, fiber-like surface morphology was preserved throughout 28 days of degradation, which might be beneficial for cell proliferation and differentiation.
 | ||
| Fig. 4 Surface morphology of samples P, P-1 and P-5 after degraded in PBS for 7, 14, 21 and 28 days. Scale bars are 20 μm. | ||
The in vitro biomineralization properties of PLGA electrospun membranes were evaluated by soaking in SBF. As shown in Fig. 5, on day 7, there were some crystals deposited on each sample. On sample P, crystals were mainly formed on surface of nanofibers while on surface of P-1 and P-5, the amount of crystals were much more and fiber-like morphology was covered by crystal deposition. When looking at higher magnification (insets in Fig. 5), deposits on sample P were plate-like and demonstrated the trend to form spherical aggregates; while on surfaces P-1 and P-5, deposits had already formed spherical aggregates, and aggregates on P-5 appeared to be more dense. On day 14, much more deposits were formed on each sample. The morphology of crystals was similar to the reported results.29 The elements of deposits were characterized using EDS. As shown in Fig. 5 (3rd column), both Ca and P were detected on all samples and Ca/P atomic ratio for sample P-5 was the highest. Combining the morphology results, it can be concluded that the deposited minerals were hydroxyapatite. All these observations demonstrated that entrapment of PAA-CaP/Zn nanoparticles in PLGA nanofibers enhanced in vitro biomineralization properties of PLGA electrospun composite membranes.
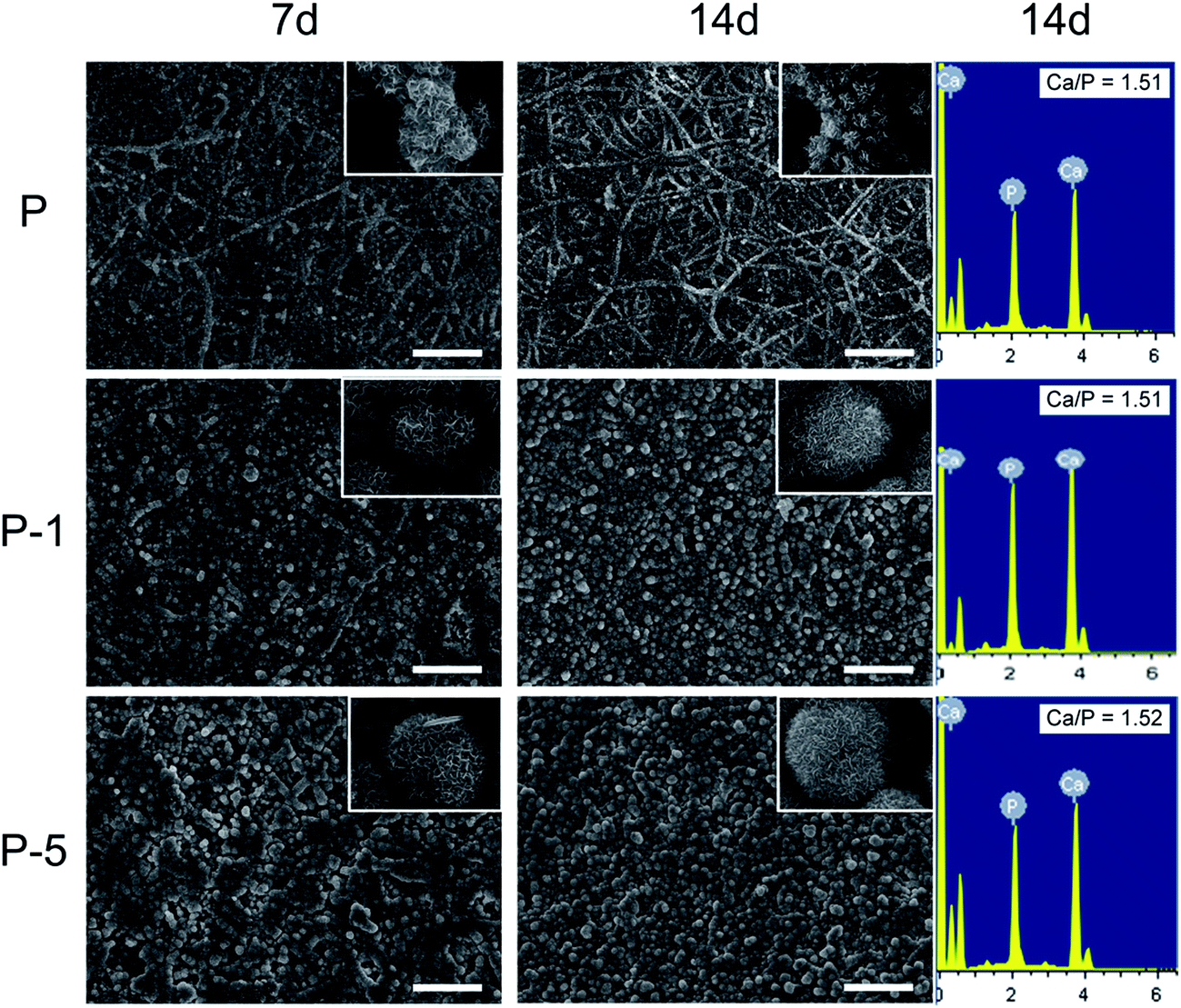 | ||
| Fig. 5 SEM images of PLGA electrospun membranes after soaked in SBF for 7 and 14 days for in vitro biomineralization evaluation. The insets are enlarged images of deposited crystals. Scale bars are 10 μm. | ||
3.3 In vitro proliferation and osteogenic differentiation of rADSC on PLGA electrospun composite membranes
Proliferation of rADSC on PLGA electrospun membranes were monitored using Alamar Blue assay. As shown in Fig. 6a, absorbance values which represented cell numbers gradually increased with culturing time, showing that cells proliferated very well on all three samples through 6 days. At each time point, there was no significant difference among these three samples while composite membranes appeared to have more cells. Cell morphology was further observed under SEM. When compared with cells on sample P, cells on P-1 and P-5 were more flattened with plenty of lamellipodia. All these results demonstrated that PLGA electrospun composite membranes offered a favorable microenvironment for rADSC adhesion and proliferation.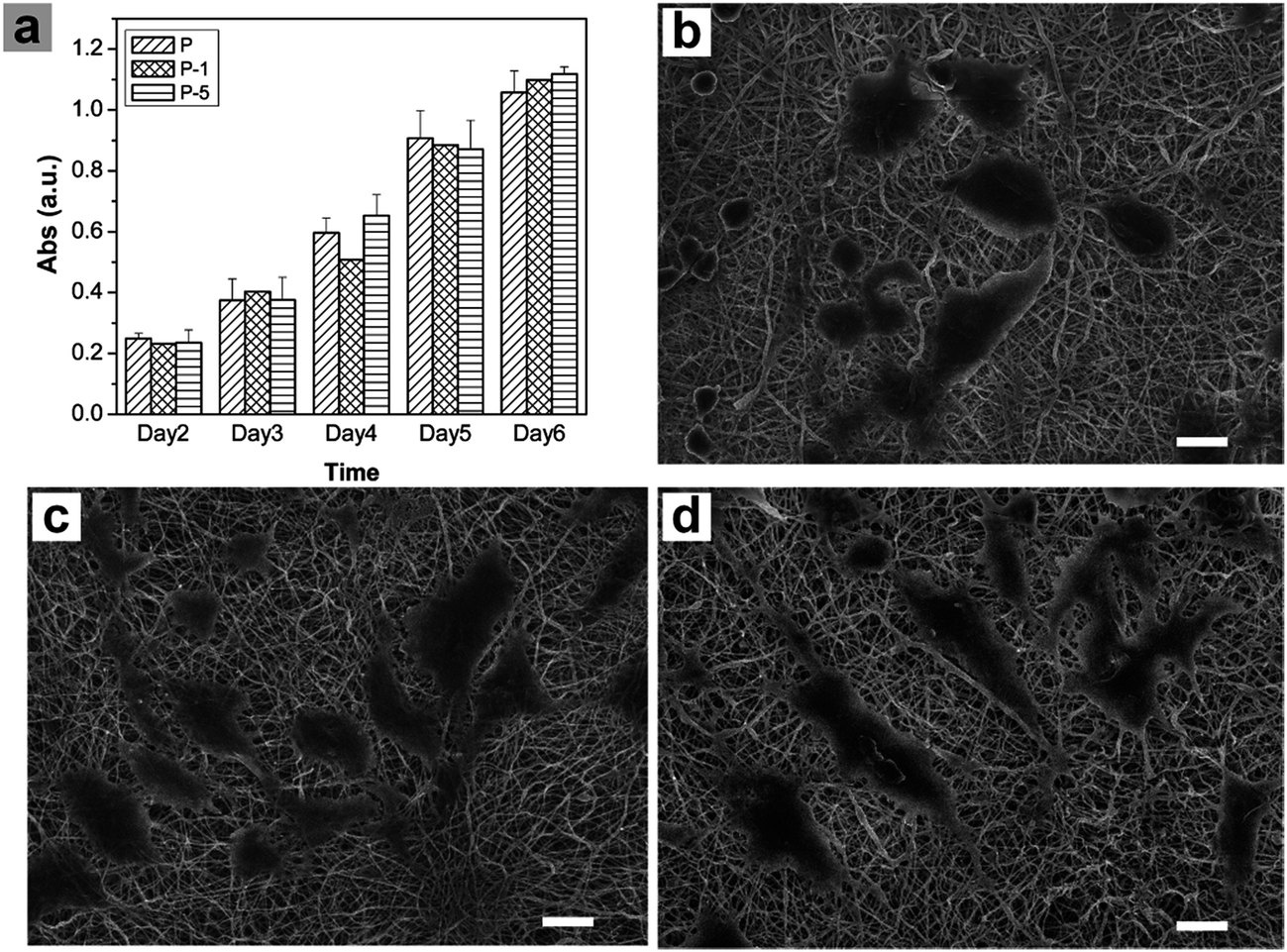 | ||
| Fig. 6 (a) Monitor of rADSC proliferation on PLGA electrospun membranes using Alamar Blue assay, SEM images of rADSC on (b) P, (c) P-1 and (d) P-5 on day 7. Data = mean ± SD; n = 3. Scale bars are 20 μm. | ||
The osteogenic differentiation behavior of rADSC on PLGA electrospun membranes was investigated based on monitoring ALP activity and calcium mineral deposition. During osteogenic induction, ALP activity of all samples gradually increased with culturing time (Fig. 7a). In addition, the expression of ALP was confirmed using BCIP/NBT staining. As shown in Fig. 7c, from day 3 to day 7, the amount of purple stains in microscope images of all samples increased while the color of the digital camera images was darkened. On day 3, there was no significant difference among these three samples (Fig. 7a and c); when culturing time was elongated to 10 days, the ALP activity of sample P-5 was significantly higher than that of P and P-1 (Fig. 7a). After osteogenic induction for 21 days, calcium mineral deposition was quantified with Alizarin red staining. As shown in Fig. 7b, the absorbance value which represented the calcium mineral deposition increased with the amount of PAA-CaP/Zn in membranes. For sample P-5, the value was significantly higher than that for sample P and P-1. Even in the microscope image, the red stains of P-5 were more homogeneous and in digital camera image, the color of P-5 was redder than other samples. Based on these data, it can be concluded that PLGA electrospun composite membranes facilitated rADSC osteogenic differentiation, especially sample P-5 which was fabricated with the appropriate amount of PAA-CaP/Zn nanoparticles.
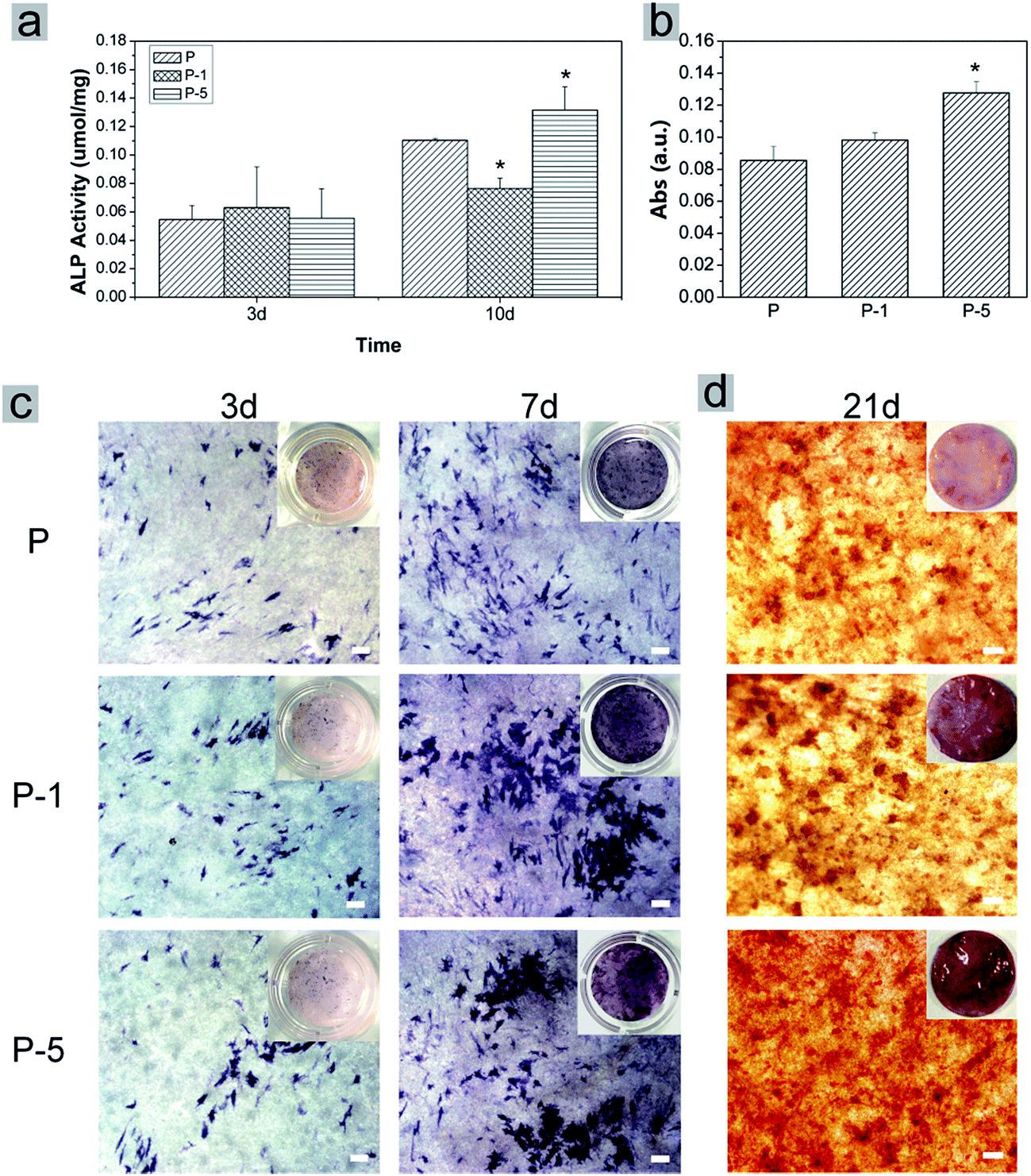 | ||
| Fig. 7 Investigation of rADSC osteogenic differentiation on PLGA electrospun membranes. (a) Measurement of ALP activity on day 3 and 10, (b) quantification of calcium deposition by dissolving alizarin red stain on day 21, (c) representative light microscope images of rADSC after ALP staining on day 3 and 7, and (d) after Alizarin red staining on day 21. The insets were photos taken with a digital camera. Scale bars represent 100 μm. Data was displayed as mean ± SD for n = 3. * representing p < 0.05 when compared with sample P. | ||
4. Discussion
Scaffold is an indispensible component in tissue engineering. To obtain high bioactivity, scaffold was designed to mimic the structure of ECM. Here, electrospinning was selected to fabricate composite membranes as the inter-connecting structure of electrospun fibers is an perfect resemble of nature ECM.6,30 To more closely mimic bone ECM which does not only have collagen nanofibers (organic component) but also contains well-dispersed HAp nanoparticles, PAA modified Zn doped HAp-like calcium phosphate (PAA-CaP/Zn) nanoparticles were first fabricated and then further entrapped in PLGA electrospun nanofibers.Bone HAp is non-stoichiometric at low crystallinity and has other elements in small amounts. Here, the wet chemical reaction was adopted to fabricate HAp nanoparticles31 and Zn was selected for doping to improve the bioactivity. However, the radius of Zn2+ is 0.074 nm, which is slightly smaller than that of Ca2+ (0.099 nm).32 Its presence in the reaction system might affect the crystallization process, leading to the formation of amorphous calcium phosphate nanoparticles, corresponding to the broadened XRD peaks (Fig. 1a). In PAA-CaP/Zn, the presence of Zn was confirmed with EDS. Based on ICP, the atomic ratio of Zn/Ca was quantified as 5.85% and the (Zn + Ca)/P atomic ratio was 1.679. Even though the molar ratio of added (Ca + Zn)/P was set as 1.667, the measured (Zn + Ca)/P atomic ratio was slightly larger. It was highly speculated that there might be some unreacted amorphous Ca(OH)2 molecules adsorbed onto nanoparticles surface since the pH of reaction system was maintained at ∼10. At such a small doping amount, PAA-CaP/Zn nanoparticles were tested as non-cytotoxic (Fig. 1f). Zn was an indispensible trace element for human health, which was also reported to promote osteocalcin secretion of bone cells, enhance the mineralization of bone tissues and facilitate maturation of bone matrix.32 These properties might be beneficial for osteogenic differentiation of rADSC on the fabricated composite membranes.
To improve the dispersity, PAA was used because of its high bonding ability with calcium. The hydrodynamic size of PAA-CaP/Zn nanoparticles was quantified as 184 nm, which was larger than the reported value.31 However, when the same synthetic parameters were followed just without PAA, the hydrodynamic size of the formed particles was quantified as 6817 nm with zeta potential of −4.28 mV in mother suspension (pH 10) when reaction was stopped. Even though there might be slight aggregation of PAA-Ca/P nanoparticles (the hydrodynamic size 184 nm was larger than the size from SEM image), PAA still significantly enhanced the dispersity of calcium phosphate nanoparticles. Based on these characterizations, it can be concluded that well-dispersed PAA-CaP/Zn nanoparticles were successfully fabricated using the simple wet chemical method.
To form composite membranes, PAA-HAp/Zn nanoparticles along with PLGA were dispersed/dissolved in acetone/DMF solvents mixture and then electrospun following the fabrication parameters as described in Section 2.3.1. The diameter of nanofibers was all ∼400 nm, which was similar to the size of collagen fiber in ECM.33,34 The PAA-HAp/Zn nanoparticles were evenly distributed in PLGA matrix and its presence did not significantly affect the surface morphology of membranes. This was very different from the reported result that PLGA with 5 wt% HAp electrospun composite membrane had rough nanofibers with HAp aggregates on surface.13 The difference was probably because here calcium phosphate nanoparticles were modified with PAA and the dispersity of nanoparticles was significantly improved.
PLGA electrospun composite membranes were designed to repair bone defects. Thus the degradation behavior was investigated. Throughout 28 days of immersion in PBS at 37 °C, fiber-like morphology was all maintained. Among all samples, P-5 was the most stable since surface morphology was nearly not changed (Fig. 4). In long term view, the inclusion of PAA-CaP/Zn nanoparticles might further enhance the repairing effect of PLGA membranes as the degradation by-products of PLGA are acidic while calcium phosphate is basic in nature. It would neutralize these acidic by-products, thus improve the biocompatibility of PLGA membranes. The presence of PAA-HAp/Zn nanoparticles also improved the mechanical properties (Fig. 3) and the in vitro biomineralization properties (Fig. 5) of PLGA membranes. All these results demonstrated the benefits of composite materials which combined the advantages of each component together. This observation was similar to published results.35,36
Even though three different ratios of PAA-CaP/Zn nanoparticles were entrapped in PLGA electrospun nanofibers, there was no significant difference regarding surface morphology. Throughout 6 days' proliferation, there was no significant difference among these three samples either. rADSC proliferated well on these membranes for which no surface coating or treatment was applied. It was probably because that the fiber diameter was similar to that of collagen, which resulted in a high specific surface area and hence facilitated cell adhesion.37,38 However, the morphology of rADSC was different on these membranes. As shown in Fig. 6, cell spreading area on sample P-1 and P-5 appeared to be larger than that on sample P. This observation was similar to the reported results that cells responded differently to the membranes with different mechanical properties.39,40
During osteogenic differentiation, rADSC behaved differently on these membranes (Fig. 7). Sample P-5 demonstrated the strongest enhancing effect on osteogenic differentiation of rADSC. Among all samples, P-5 was the most stable through 28 days of degradation. This stable surface morphology might be beneficial for rADSC differentiation. Meanwhile it was reported that within a certain range, the osteogenic enhancing effect of scaffold material was positively related with its mechanical properties.41,42 Here, P-5 had higher elastic modules. The high amount of PAA-CaP/Zn nanoparticles may also contribute to the enhancing effect on osteogenic differentiation of rADSC as Zn2+ has been reported to promote osteocalcin expression of bone cells.32 It was highly speculated that all these factors influenced synergistically on osteogenic differentiation of rADSC. Since more stable microenvironment and higher elastic modules were both due to the high amount of PAA-CaP/Zn nanoparticles in P-5, it can be concluded that the designed composition of P-5 facilitated the osteogenic differentiation of rADSC.
5. Conclusions
In this paper, well dispersed HAp-like PAA-CaP/Zn nanoparticles were successfully synthesized using a simple precipitation method and these nanoparticles were further entrapped into PLGA electrospun nanofibers. PAA-CaP/Zn nanoparticles were spheroids of ∼60 nm. The presence of Zn significantly lowered the crystallinity and these nanoparticles were non-cytotoxic. When included in PLGA nanofibers, these nanoparticles did not compromise the fiber morphology. rADSC proliferated very well on these membranes. The presence of nanoparticles increased mechanical properties and retarded the degradation rate, which together with the appropriate bone ECM mimicking morphology and composition might contribute to the enhancing effect on osteogenic differentiation of rADSC. The fabricated composite membranes demonstrated the potential to be used in bone tissue engineering because of its fiber-like structure and inorganic/organic composition, which needs further in vivo investigations.Acknowledgements
This work was supported in part by National Natural Science Foundation of China [grant numbers 51303216, 51103182 and 31270028], Fundamental Research Funds for the Central Universities [grant number 14lgpy12], Guangdong Natural Science Funds for Distinguished Young Scholar [grant number 2016A030306018], Guangdong Provincial Education Department [grant number 2013KJCX005], and the Australia-China Joint Institute for Health Technology and Innovation.References
- D. W. Hutmacher, Biomaterials, 2000, 21, 2529–2543 CrossRef CAS PubMed.
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS PubMed.
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3, 1377–1397 CrossRef CAS PubMed.
- K. Rezwan, Q. Z. Chen, J. J. Blaker and A. R. Boccaccini, Biomaterials, 2006, 27, 3413–3431 CrossRef CAS PubMed.
- L. I.-R. Susan, M. G.-K. Genevieve and J. Y. Michael, Biomaterials, 1998, 19, 1405–1412 CrossRef.
- W.-J. Li, C. T. Laurencin, E. J. Caterson, R. S. Tuan and F. K. Ko, J. Biomed. Mater. Res., 2001, 12, 613–621 Search PubMed.
- B. M. Baker, A. O. Gee, R. B. Metter, A. S. Nathan, R. A. Marklein, J. A. Burdick and R. L. Mauck, Biomaterials, 2008, 29, 2348–2358 CrossRef CAS PubMed.
- S. Gao, M. Calcagni, M. Welti, S. Hemmi, N. Hild, W. J. Stark, G. M. Buergisser, G. A. Wanner, P. Cinelli and J. Buschmann, Injury, 2014, 45, 974–980 CrossRef PubMed.
- A. Haider, K. C. Gupta and I.-K. Kang, BioMed Res. Int., 2014, 2014, 308306 Search PubMed.
- F. Peng, X. Yu and M. Wei, Acta Biomater., 2011, 7, 2585–2592 CrossRef CAS PubMed.
- M. V. Jose, V. Thomas, K. T. Johnson, D. R. Dean and E. Nyairo, Acta Biomater., 2009, 5, 305–315 CrossRef CAS PubMed.
- M. Li, W. Liu, J. Sun, Y. Xianyu, J. Wang, W. Zhang, W. Zheng, D. Huang, S. Di, Y.-Z. Long and X. Jiang, ACS Appl. Mater. Interfaces, 2013, 5, 5921–5926 CAS.
- L. Lao, Y. Wang, Y. Zhu, Y. Zhang and C. Gao, J. Mater. Sci.: Mater. Med., 2011, 22, 1873–1884 CrossRef CAS PubMed.
- D. Li, H. Sun, L. Jiang, K. Zhang, W. Liu, Y. Zhu, J. Fangteng, C. Shi, L. Zhao, H. Sun and B. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 9402–9410 CAS.
- N. Hild, O. D. Schneider, D. Mohn, N. A. Luechinger, F. M. Koehler, S. Hofmann, J. R. Vetsch, B. W. Thimm, R. Müller and W. J. Stark, Nanoscale, 2011, 3, 401–409 RSC.
- H. B. John and A. Alison, Nutr. Res. Rev., 1992, 5, 167–188 CrossRef PubMed.
- W. Xue, K. Dahlquist, A. Banerjee, A. Bandyopadhyay and S. Bose, J. Mater. Sci.: Mater. Med., 2008, 19, 2669–2677 CrossRef CAS PubMed.
- M. Hie and I. Tsukamoto, Eur. J. Pharmacol., 2011, 668, 140–146 CrossRef CAS PubMed.
- A. Wang, H. Yin, D. Liu, H. Wu, M. Ren, T. Jiang, X. Cheng and Y. Xu, Mater. Lett., 2007, 61, 2084–2088 CrossRef CAS.
- S. Pujari-Palmer, S. Chen, S. Rubino, H. Weng, W. Xia, H. Engqvist, L. Tang and M. K. Ott, Biomaterials, 2016, 90, 1–11 CrossRef CAS PubMed.
- E. Arrigoni, S. Lopa, L. De Girolamo, D. Stanco and A. T. Brini, Cell Tissue Res., 2009, 338, 401–411 CrossRef PubMed.
- Y. Ramaswamy, C. Wu, A. V. Hummel, V. Combes, G. Grau and H. Zreiqat, Biomaterials, 2008, 28, 4392–4402 CrossRef PubMed.
- H. Valiense, M. Barreto, R. F. Resende, A. T. Alves, A. M. Rossi, E. Mavropoulos, J. M. Granjeiro and M. D. Calasans-Maia, J. Biomed. Mater. Res., Part B, 2016, 104, 274–282 CrossRef CAS PubMed.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed.
- K. Kato, Y. Eika and Y. Ikada, J. Mater. Sci., 1997, 32, 5533–5543 CrossRef CAS.
- S.-C. Liou, S.-Y. Chen and D.-M. Liu, Biomaterials, 2003, 24, 3981–3988 CrossRef CAS PubMed.
- R. Gonzalez-McQuire, J.-Y. Chane-Ching, E. Vignaud, A. Lebugle and S. Mann, J. Mater. Chem., 2004, 14, 2277 RSC.
- R. N. Panda, M. F. Hsieh and R. J. Chung, J. Phys. Chem. Solids, 2003, 64, 193–199 CrossRef CAS.
- Z. Cao, D. Wang, L. Lv, Y. Gong and Y. Li, RSC Adv., 2016, 6, 10641–10649 RSC.
- Z.-M. Huang, Y. Z. Zhang, M. Kotaki and S. Ramakrishna, Compos. Sci. Technol., 2003, 63, 2223–2253 CrossRef CAS.
- Q. Wang, Z. Gu, S. Jamal, M. S. Detamore and C. Berkland, Tissue Eng., Part A, 2013, 19, 2586–2593 CrossRef CAS PubMed.
- E. S. Thian, T. Konishi, Y. Kawanobe, P. N. Lim, C. Choong, B. Ho and M. Aizawa, J. Mater. Sci.: Mater. Med., 2013, 24, 437–445 CrossRef CAS PubMed.
- L. A. Smith and P. X. Ma, Colloids Surf., B, 2004, 39, 125–131 CrossRef CAS PubMed.
- E. Tom and B. Jonathan, J. Cell Biol., 1972, 54, 626–637 CrossRef.
- Y. Ito, H. Hasuda, M. Kamitakahara, C. Ohtsuki, M. Tanihara, I. K. Kang and O. H. Kwon, J. Biosci. Bioeng., 2005, 100, 43–49 CrossRef CAS PubMed.
- H. W. Kim, H. H. Lee and J. C. Knowles, J. Biomed. Mater. Res., Part A, 2006, 79, 643–649 CrossRef PubMed.
- M. R. Williamson, R. Black and C. Kielty, Biomaterials, 2006, 27, 3608–3616 CrossRef CAS PubMed.
- J. Stitzel, J. Liu, S. J. Lee, M. Komura, J. Berry, S. Soker, G. Lim, M. Van Dyke, R. Czerw, J. J. Yoo and A. Atala, Biomaterials, 2006, 27, 1088–1094 CrossRef CAS PubMed.
- S. Ouasti, R. Donno, F. Cellesi, M. J. Sherratt, G. Terenghi and N. Tirelli, Biomaterials, 2011, 32, 6456–6470 CrossRef CAS PubMed.
- V. E. G. Diederich, P. Studer and A. Kern, Biotechnol. Bioeng., 2013, 110, 1508–1519 CrossRef CAS PubMed.
- B. Jankovic, J. Pelipenko, M. Skarabot, I. Musevic and J. Kristl, Int. J. Pharm., 2013, 455, 338–347 CrossRef CAS PubMed.
- J. H. Lee, H. K. Park and K. S. Kim, Biochem. Biophys. Res. Commun., 2016, 473, 752–757 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEM image and fiber diameter distribution of P-8 which was fabricated at weight ratio of (PAA-CaP/Zn)/PLGA as 8% and following the same parameters as that for other PLGA electrospun membranes. See DOI: 10.1039/c7ra01865g |
| This journal is © The Royal Society of Chemistry 2017 |