DOI:
10.1039/C7RA02837G
(Paper)
RSC Adv., 2017,
7, 24282-24290
Bio-based carbonaceous composite materials from epoxidised linseed oil, bio-derived curing agent and starch with controllable functionality†
Received
8th March 2017
, Accepted 27th April 2017
First published on 4th May 2017
Abstract
Development of biomass-derived materials using sustainable practices has been one of the major scientific aims over the last few decades. A new class of bio-derived nanocomposite derived from epoxidised linseed oil, a bio-derived crosslinker and a starch based carbonaceous mesoporous material (Starbon®) has been developed. The use of Starbons® technology enables the incorporation of carbonaceous materials with tuneable surface functionality (from hydrophilic to hydrophobic). The resulting composite demonstrated good thermal stability up to 300 °C, good low temperature modulus, flexibility and uniformity, as demonstrated by TGA, DMA and SEM studies, respectively. Furthermore, the thermoset composites' swelling behaviour in solvents with a high polar index through to non-polar ones was investigated, revealing initially that non polar solvents have a greater impact on swelling than polar solvents and that in all cases the addition of filler reduces the extent of swelling. The inclusion of this carbonaceous material with hierarchical pore structure and high BET surface area may further aid the use of such composites in membrane separation applications.
Introduction
Petroleum resources have been used as the primary source of chemicals and materials from the early 20th century, however, critical issues relating to their finite availability and environmental concerns have resulted in the search for sustainable alternatives.1 Biomass has received significant attention as a feedstock for the production of chemicals and materials due to its inherent renewability and positive environmental credentials.2–4 Plant oils primarily derived of triglycerides and fatty acids are one such potential resource derived from biomass. Epoxidised plants oils have been intensively investigated for the production of novel materials such as alkyd resins, blends and composites.5–8 Thermoset materials are highly-crosslinked polymers that are cured by heat, pressure, light radiation or a combination of these energy sources.9 In many instances epoxidised plant oils are used as precursors for thermoset matertials.10 Bio-derived diacids have been utilised to crosslink epoxidised linseed oil, resulting in highly flexible transparent films, with significant water resistance.11
In addition, fibres and/or particles have been extensively used as reinforcement agents for plant oil based resins, thus enhancing their mechanical properties.12–14 Carbonaceous fillers have also been used to improve thermoset plastic properties e.g. mechanical and thermal behaviours, and thus expand further their range of viable applications.15–17 However, carbonaceous fillers typically have limited surface functionality, are extensively microporous and often their manufacture is energy intensive, and in some cases these materials are costly.18 The polysaccharide-derived carbonaceous materials registered as Starbon®, with a highly developed mesoporous structure and temperature tuneable surface functionality (from hydrophilic to hydrophobic) are one potential alternative.19,20 These materials can be prepared through a simple expansion and carbonisation methodology from the parent polysaccharide.21,22 This flexibility, along with hierarchical pore structure and high BET surface area make Starbon® successful in a wide range of applications including catalysis, chromatography, adsorption of organic compounds (i.e. dyes), precious metals (i.e. gold, and palladium) and CO2 capture.23–28 Furthermore, Starbon® materials have significant aromatic/graphitic character at relatively low temperatures of preparation and therefore less energy is consumed during preparation.
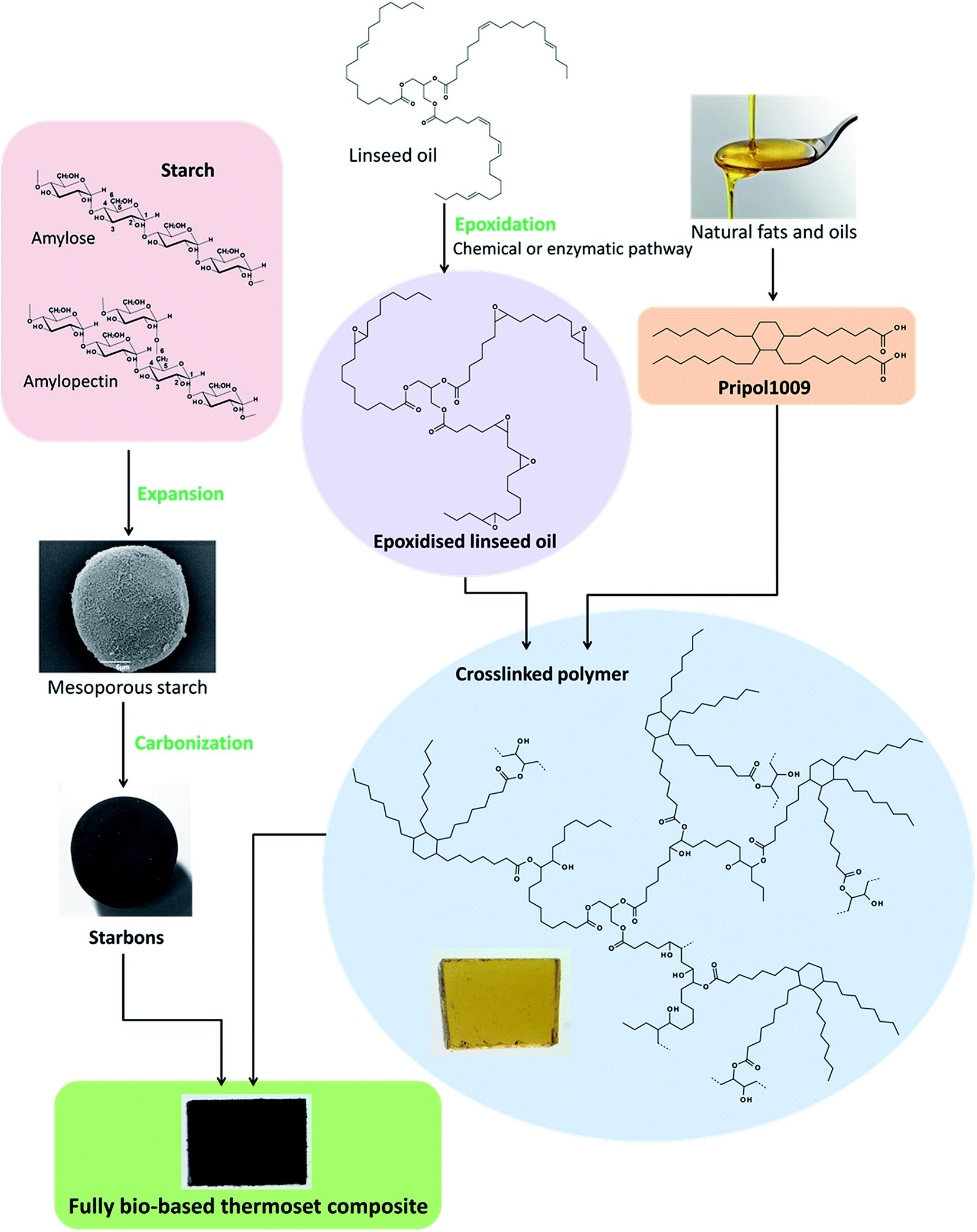
Herein, Starbon® materials carbonised at different temperatures were combined with epoxidised linseed oil and a bio-derived dicarboxylic acid crosslinker to produce thermoset composites. The use of these renewable materials strongly adheres with the principles of Green chemistry, as does the composites synthesis that requires no addition of solvents (Fig. 1). Curing of the resin (control) and composites containing Starbon® with varying functionality (hydrophilic to hydrophobic) was confirmed by both FT-IR and DSC, whilst their morphology, thermal and dynamic mechanical properties were characterised using SEM, TGA and DMA respectively. Furthermore, swelling behaviour of the composites in solvents with different polarities was investigated.
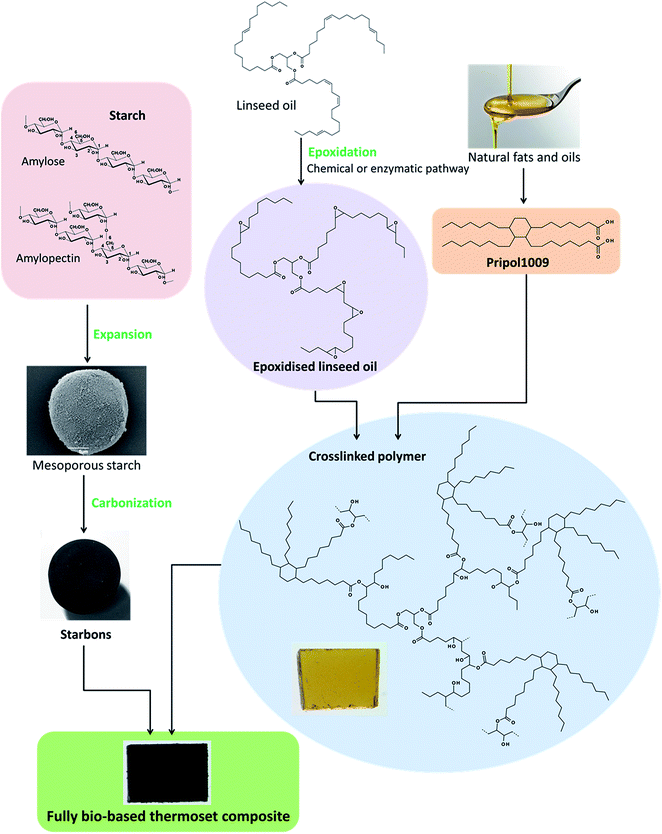 |
| | Fig. 1 Diagram summarising the production of the fully bio-based composites. | |
Experimental
Materials
All materials were used as received without further purification. ELO (Lankroflex L) was obtained from Akcros Chemicals (oxirane oxygen = 9%). Bio-derived dicarboxylic acid (Pripol 1009) was kindly supplied by Croda. 4-N,N-Dimethylaminopyridine (DMAP) was purchased from Fluka. Toluene and ethanol were obtained from Fisher Scientific.
Preparation of Starbon®
Starbon® fillers were synthesised as previously reported in the literature.19 The preparation method comprises three key stages: gelation of starch, retrogradation and water exchange with a lower surface tension solvent, followed by oven-drying to yield a predominantly mesoporous material. In the final stage the mesoporous starch was doped with a catalytic amount of an organic acid (e.g. 0.1 mmol of p-toluenesulfonic acid per 1 g of starch) and heated under vacuum to yield at 300 °C and 800 °C the materials S300 and S800 respectively.
Characterisation of Starbon®
Nitrogen adsorption/desorption isotherms were conducted at 77 K using a Micromeritics ASAP 2010 volumetric adsorption analyser. Prior to analysis, the samples were outgassed at 65 °C for at least 3 h under reduced pressure and mass differences were corrected after the experiment. The surface area of samples was automatically calculated by the Micromeritics ASAP 2010 volumetric adsorption analyser. The isotherm data were used to obtain the BET specific surface area data (SBET) and the Barret–Joyner–Halenda (BJH) pore size distribution was calculated from the adsorption branch of the N2 physisorption isotherms.
Preparation of thermoset composites
A vigorously stirred mixture of epoxidised linseed oil (ELO) (3.5 g), Pripol 1009 (3.5 g) and 4-(dimethylamino) pyridine (DMAP) (0.5% by weight of the total resin weight) was heated to 130 °C for 3 minutes. Subsequently, Starbon® (S300 or S800) was added into the mixture which was maintained at 130 °C with stirring for a further 3 minutes. The mixture was then poured into a circular aluminium tray (inner diameter = 70 mm) and cured in an oven at 140 °C for 20 h.
MDSC of the reaction mixtures
Reaction mixtures were investigated using a TA Q2000 modulated differential scanning calorimeter (MDSC). The mixtures were accurately weighed (10 mg) into high-pressure stainless steel pans, sealed and subjected to a heat/cool cycle from −80 °C to 250 °C at 1 °C min−1 with a modulation cycle of ±0.5 °C every 200 seconds.
Thermal degradation temperature
TG-DTA curves were performed on a Perkin Elmer Pyris Diamond TG-DTA instrument using α-alumina (α-Al2O3) as a standard material. The samples were heated from 25–600 °C at 10 °C min−1 under a nitrogen atmosphere. The initial sample weight was approximately 10–15 mg.
Infrared spectroscopy
In order to study the curing reaction for each formulation, spectra of the samples before and after curing were recorded using a Bruker Vertex 70 spectrophotometer in Attenuated Total Reflectance (ATR) mode with a resolution of 2 cm−1 and 32 scans. All spectra were normalised using OPUS 5.0 software as provided by the instrument manufacturer.
Scanning electron microscopy
Film samples were cut into 2 × 10 mm pieces, and then mounted onto alumina sample holders using double-sided tape. The samples were positioned so that the cross-section of the films can be seen on the top view. Prior to analysis, approximately 5 nm Au/Pd coating was applied to the samples using a high-resolution sputter SC-7640 coating device at a sputtering rate of 1500 V min−1. The samples were viewed using a JEOL JSM-6490LV (JEOL, Japan).
Swelling behaviour7
Composite films of approximate 20 mg weight were immersed in water, ethanol or toluene at room temperature for 24 hours and then removed from the solvent and blotted with a tissue to remove excess liquid before being re-weighed. The average degree of swelling (Gs) was determined from at least three experiments and calculated using the formulation:
where, Wi represents the initial weight of the film and Wf represents the final weight of the film after drying.
Dynamic mechanical analysis
Dynamic mechanical analyser (DMA Q800, TA) in three point bending mode was used for all samples. The dimensions of the samples were 20 × 4 × 1.25 mm3 (length, width, thickness); measurements were carried out at 10 Hz, 20 μm of strain amplitude in bending and a heating rate of 1 °C min−1.
Mechanical test
The composite films were cut into standard dumb-bell shapes with the average thickness in the region of 1–1.5 mm. Tensile studies were conducted using an Instron V.5567A universal testing machine fitted with 1000 N capacity load cell. The initial grip separation was set at 20 mm and the crosshead speed was 20 mm min−1. The mechanical properties reported for each sample were the average of three measurements.
Results and discussion
Characterisation of Starbon®
The textural properties of the Starbon® fillers were quantitatively analysed using N2 adsorption, and are presented in Table 1. It was found that they show type IV behaviour, characteristic of mesoporous materials. Though, with increasing preparation temperature from 300 to 800 °C BET surface areas increased from 115 to 445 m2 g−1, correlating with an increase in micropore formation from 4.5 to 21.6%.21 Furthermore, the formation of an increasingly macroporous structure can also be observed in the presented SEM images of the Starbon® fillers (Fig. 2). Previous findings using FT-IR, 13C-MAS NMR and XPS have also demonstrated that significant differences are observed in the functionality of the Starbon® material dependent on the temperature of preparation, from initially predominately hydroxyl-rich to aliphatic/alkene groups at 300 °C, through to highly aromatic at 800 °C.26,29,30
Table 1 Porosimetry data for Starbon® prepared at 300 °C (S300) and 800 °C (S800)
| Properties |
S300 |
S800 |
| SBET (m2 g−1) |
115 |
445 |
| Total pore volume (cm3 g−1) |
0.67 |
0.74 |
| Average pore diameter (nm) desorption |
29.3 |
23.5 |
| Microporous volume (cm3 g−1) |
0.03 |
0.16 |
| % microporosity |
4.5 |
21.6 |
 |
| | Fig. 2 SEM micrograph of Starbon® prepared at 300 °C (A + B) and 800 °C (C + D). | |
Curing study of the reactive mixtures using FT-IR
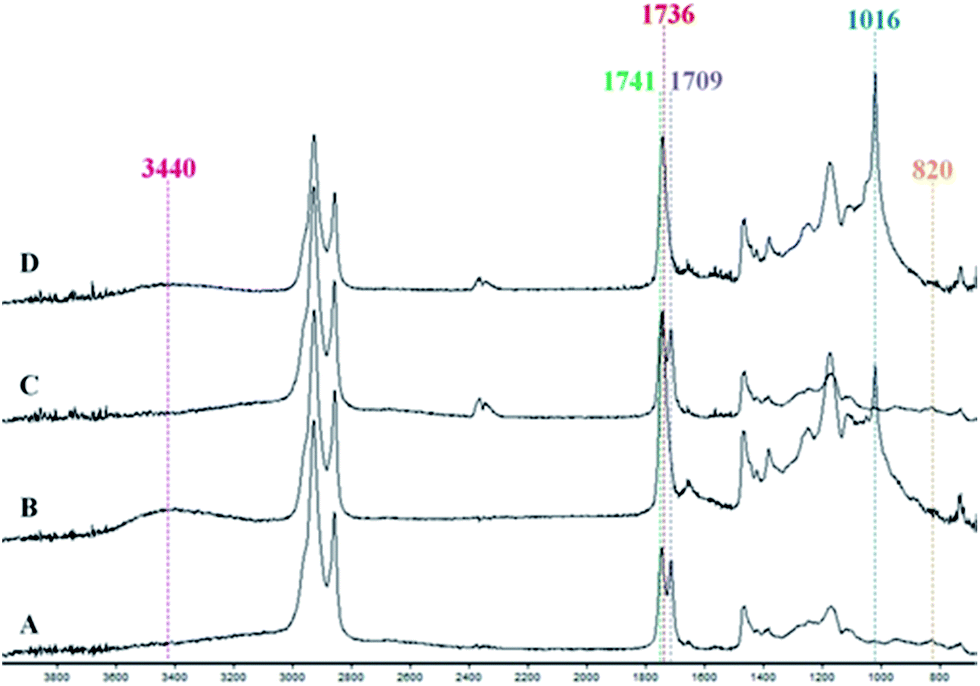
ATR-IR analyses of the reactive mixtures prior to and after heating were carried out in order to determine the potential mechanism of curing (Fig. 3). The disappearance of the peak at 820 cm−1 and the appearance of a band at 3440 cm−1 associated with hydroxyl functionality after curing suggested epoxide ring opening had occurred during the reaction. Furthermore, a new peak at 1016 cm−1 was observed denoting the presence of ether functionality, indicating possible etherification of the alkoxide group, which can then continue to react with other epoxides and form the crosslinked network. During this time reactions involving the diacid crosslinker are also evident from the disappearance of νas(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at 1709 cm−1 corresponding to the carboxylic acid. Furthermore, a band at 1741 cm−1 associated with the CO of the ester group presented in the triglyceride was observed in the spectra of uncured mixture. After curing, a band at 1736 cm−1 could be noted, suggesting esterification. Thus it can be seen that etherification and esterification occurred during the curing process. The addition of Starbon® fillers scarcely altered the spectra of both reactive mixtures and cured films (see ESI†); however, the ester band at 1736 cm−1 of the cured control sample showed a slightly higher intensity than the ones with fillers, suggesting a greater degree of crosslinking as also confirmed by DSC.
O) at 1709 cm−1 corresponding to the carboxylic acid. Furthermore, a band at 1741 cm−1 associated with the CO of the ester group presented in the triglyceride was observed in the spectra of uncured mixture. After curing, a band at 1736 cm−1 could be noted, suggesting esterification. Thus it can be seen that etherification and esterification occurred during the curing process. The addition of Starbon® fillers scarcely altered the spectra of both reactive mixtures and cured films (see ESI†); however, the ester band at 1736 cm−1 of the cured control sample showed a slightly higher intensity than the ones with fillers, suggesting a greater degree of crosslinking as also confirmed by DSC.
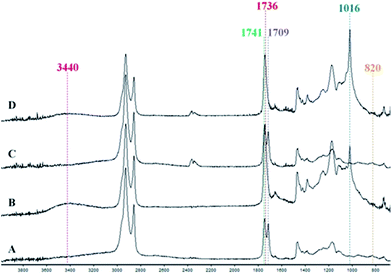 |
| | Fig. 3 FT-IR spectra of (A) control reactive mixture, (B) control thermoset composite, (C) 20% S800 reactive mixture, (D) 20% S800 thermoset composite. | |
Thermal analysis of the reactive mixtures
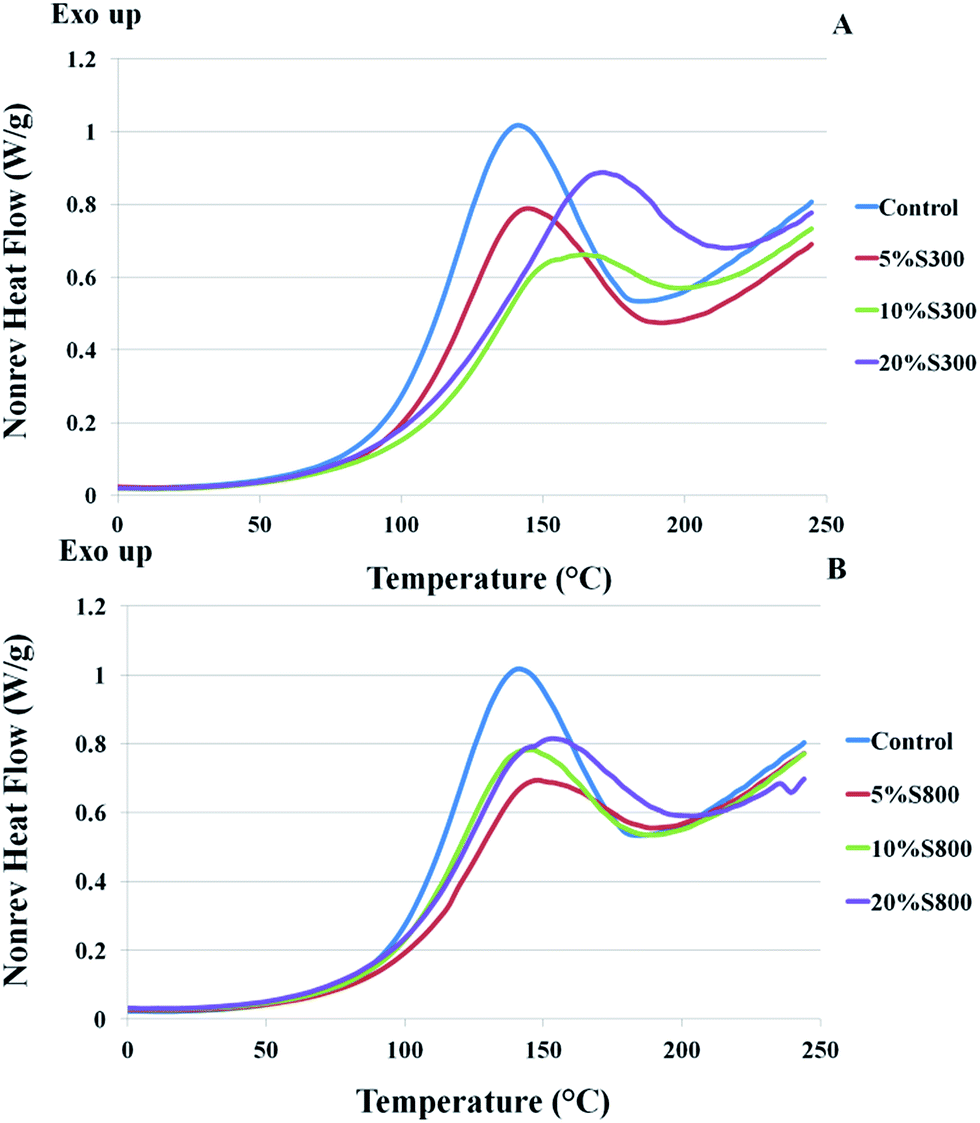
Modulated DSC was used to investigate thermal events that occurred during the curing process of the reactive mixtures. The control formulation was comprised of ELO, Pripol 1009 and DMAP (0.5% of total weight). In other formulations, various amounts of either S300 or S800 (5–20 wt%) was included. Non-reversible heat flow traces of all formulations showed an exothermic transition which corresponded to the curing reaction (Fig. 4). The formulations with Starbon® showed similar or lower total heats of reaction compared to that of the control sample, dependent on the amount of filler added. This was more pronounced for the formulations containing the S300 as the filler. This could be explained by the larger particle size of S300 (Fig. 2) that would inhibit the polymer chains to crosslink, hence the more significant drop in heat of reaction. However, this contradicts the result obtained from a previous study that used starch as filler as the formulation with starch demonstrated higher heat of reaction comparing to ones without.31 In this case it was suggested that the presence of starch might promote additional crosslinking. This postulation was highlighted as being a result of the hydrophilic hydroxyl groups present in the structure of the polysaccharide, which promoted the oxirane ring opening of ELO.31 The carbonisation of the mesoporous starch to obtain Starbon® increases its hydrophobicity and reduces the presence of hydroxyl functionality. At 300 °C, the dominant functionalities present are aliphatic and alkene functionalities. At temperatures of >600 °C aromatic and graphite-type structures are present.21 As such, Starbon® which possesses a lower number of hydroxyl groups (S800) cannot promote the ring-opening as presented in the case of starch, thus resulting in a lower observed heat of reaction.
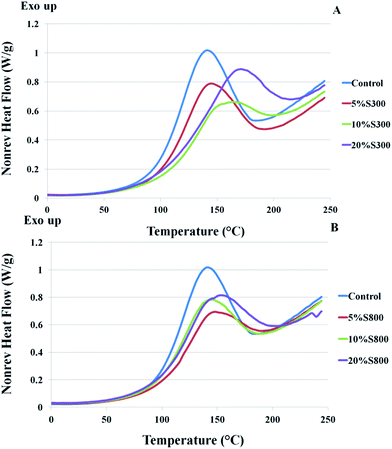 |
| | Fig. 4 DSC thermograms of formulations comprised of ELO, Pripol, DMAP and different amounts of S300 (A) and S800 (B) – 5, 10 and 20%wt. | |
The presence of S300 increases the onset and peak temperatures of the exothermic transition with the amounts of the filler, whilst the S800 formulations demonstrated close values to those of the control. Higher peak temperatures and longer curing time were observed for the Starbon® filled samples, as compared to samples with no filler. This suggests that the addition of Starbon® retarded the curing reaction. Similar observation was reported in carbon nanotubes reinforced polymers.32–34 The Starbon® retardation of cure could relate to the adsorption of polymer chains onto carbonaceous mesoporous surfaces due to a CH–π interaction. Such effects were also observed in a case of carbon nanofiber reinforced bio-based polyester reported by Tang et al.35 The cause of the delay in curing of the polymer was due to the physical hindrance of Starbon® to the mobility of the matrix. Furthermore, a study by Xu et al. revealed that serious retardation indicated better dispersion of a carbonaceous filler in the polymer matrix.32 This results in a better quality of dispersion for the Starbon® materials throughout the matrix and allows more interface area between the filler and polymer, thus increasing the physical hindrance caused by filler particles to the mobility of the polymer chains. In previous studies, starch was also found to cause retardation of the curing process, which may be due to slow diffusion of the molecules whilst starch granules were present in the reactive mixtures.31 It is also noteworthy that the curing curve of the formulation with starch showed two overlapping exothermic transitions between 75 °C to 200 °C, however the formulation with Starbon® gave only one transition in the same region. In a polymeric system, the observation of two transitions is generally associated with the presence of two phases,36 the second transition at higher temperature could correspond to the region that was more influenced by the presence of starch, hence the curing was delayed. The fact that Starbon® fillers are more hydrophobic relative to starch allows better diffusion of the filler particles. The uniform distribution of Starbon® particles results in single curing transition with no evident sign of post-cure in second heating cycle (see ESI†).
The glass transition temperature (Tg) of the S300 filled samples exhibited lower Tg as the loading of the filler increased, whilst the more graphitic S800 filled samples showed a slight increase of Tg compared to that of the control sample. The increase in Tg can be anticipated in composites and this has been reported in numerous studies in the case of carbonaceous filler-polymer composites.37–39 This augmentation of Tg can be associated with a reduction of the mobility of the polymer matrix around Starbon® particles due to the interfacial interactions between the polymer chains and the filler. As such, the segmental motion of the polymer chains is reduced.
However, the decrease in Tg of the more hydrophilic S300 samples may be explained by bundling tendency as observed in composites with single wall carbon nanotubes.40 The tendency of these particles to form close-packed bundles and consequently increase a free volume which provides more mobility of polymer chains, resulting in a drop-off of Tg.40 Also, the heat of reactions for the S300 samples was disrupted to a greater extent compared to the materials containing S800 and as a consequence their subsequent crosslink densities will be lower. It is therefore suggested that a combination of these events led to reduced Tg for the S300 samples compared to the S800 counterparts.
Thermoset composites characterisations
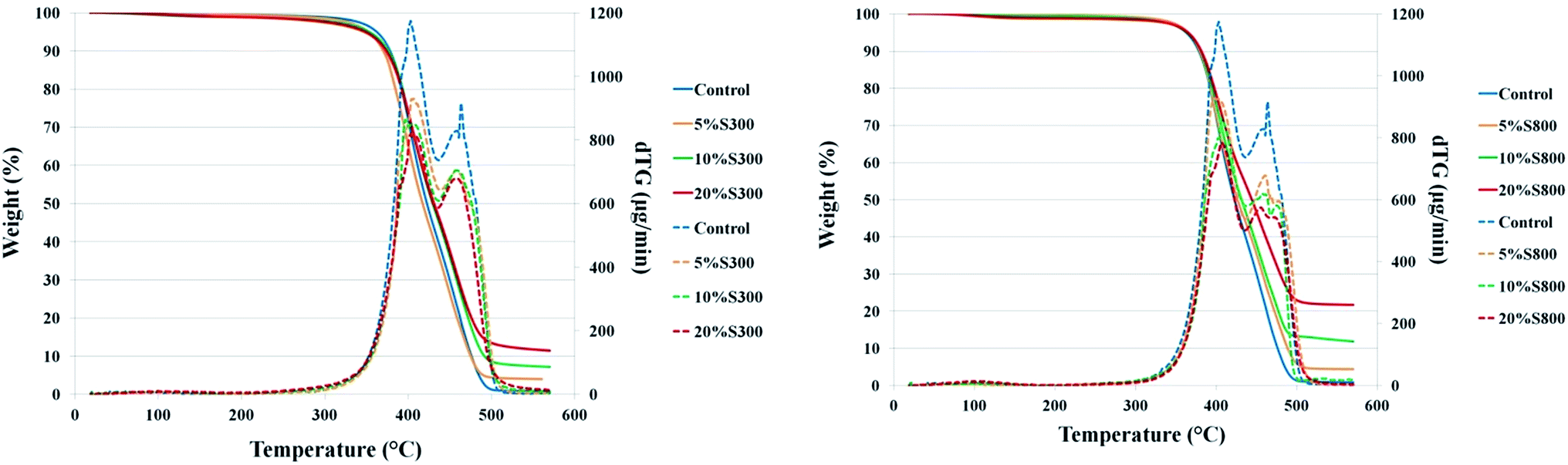
Thermal stability of the composites. TGA thermograms demonstrated that all films have good thermal stability with little to no weight loss up to 300 °C (Fig. 5). The temperature when the original sample weight decreased by 5% (T5%) ranged from 348 °C to 365 °C (see Table S2†), and revealed that addition of the S800 filler did not significantly alter the films initial thermal stability compared to the control (362 °C). However, addition of the S300 filler led to a reduction in initial thermal stability, with 5% S300 filler resulting in a 10 °C decrease, and the 20% S300 a 14 °C diminution. From the TG profile and dTG curves, it is seen that the materials actually experience a two-stage thermal decomposition path; with their respective maximums (TMAX 1 and TMAX 2) occurring at an approximate temperature of 400 °C and 450 °C (Table S2†). Previous work has indicated that the first step of decomposition is associated with the thermolysis of the ether linkages and the second step is likely attributed to the decomposition of the ester moieties.41 The residual mass at 550 °C for samples with fillers increases in both cases. However, it can be seen that the S800 composites had higher residual mass than the S300 films. The cause of this is a combination of the thermally less stable S300† filler, which starts to decompose around 300 °C, whilst the S800 sample is thermally stable up to 800 °C, and also samples with the S300 filler having a T5% some 10 to 14 °C less than the control sample.
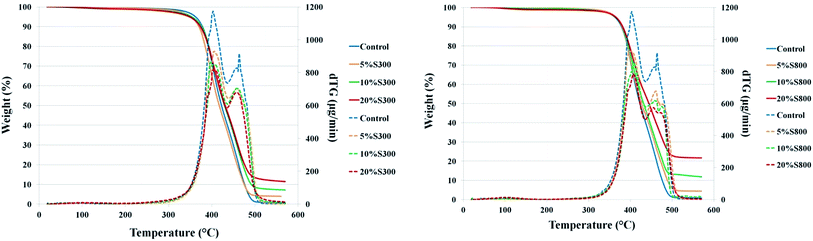 |
| | Fig. 5 TGA and dTG thermograms of films with S300 and S800 at different loadings. | |
Morphological study of the composites. The morphology of the cross-section of composite films were observed using SEM. The cross-section surface of the control film was smooth and homogeneous, whilst those containing both types of Starbon® filler presented some roughness. It can be seen that the S300 and S800 Starbon® particles were thoroughly mixed in the polymer matrix (Fig. 6C and E). At higher magnifications (Fig. 6D and F) however, it is seen that still a few defined spherical granular shapes are observed for the S300 composites and that the S800 filler is more uniformly distributed throughout the composite. This is contrary to previous studies utilising native starch fillers, where it was demonstrated that they retained their spherical granular shape and were poorly dispersed throughout the polymer matrix.31
 |
| | Fig. 6 Scanning electron micrographs of the cross-section of thermoset films – (A) and (B) control, (C) and (D) 20%S300 and (E) and (F) 20%S800. | |
Dynamic mechanical analysis of the composites. In order to obtain information on the effect of the S300 and S800 fillers on the mechanical properties of the cured epoxidised linseed oil, dynamic mechanical analysis (DMA) was carried out. Analysis of the samples was performed over a wide temperature range, with the variation of storage modulus (E′) versus temperature shown in Fig. 7 (and Tables S1 and S2 ESI†). The behaviour of all the samples with temperature is similar, presenting a large decrease in E′ from −120 to −90 °C, due to a secondary relaxation often known as sub-Tg transition (see ESI†), followed by a linear decrease until −20 °C, where the onset of α relaxation (Tg) is observed. Although, the E′ variation does not follow any direct trend with filler concentration, the use of the more hydrophilic S300 filler in general results in a lower E′ than that of the control or that of the hydrophobic graphitic S800 filler. Comparing some key results it can be seen that the 5% S800 composite sample has the highest E′ over the full temperature range. Though, when evaluating the results observed from the MDSC results (Table S1†) it can be seen that this sample has a lower heat of reaction compared to the control and the 5% S300 sample, suggesting that it has a lower resultant crosslink density. This suggests that good filler compatibility and even dispersion, lead to better mechanical properties in these samples. In contrast, 10% S800 composites demonstrate the highest normalised heat of reaction and lowest storage modulus further supporting this conjecture. This suggests that the 10% S800 filler is more agglomerated and as a result not able to impart the fillers mechanical attributes to the composite properties. The crosslink density is an important factor in these materials, where the onset of the α relaxation (associated to Tg) decreases in almost all composites as observed in the tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ results (ESI†). This provides an indication of lower crosslink density due to lower cure grade and worse scattered E′.
δ results (ESI†). This provides an indication of lower crosslink density due to lower cure grade and worse scattered E′.
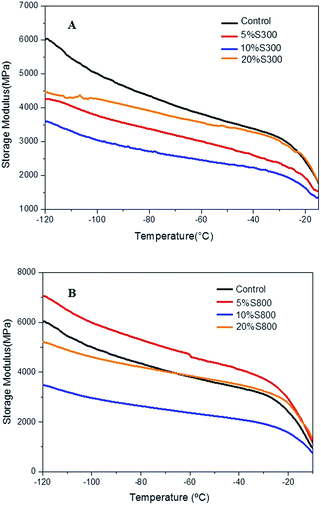 |
| | Fig. 7 Storage modulus of the thermoset films with S300 (A) and S800 (B) at different loadings – 5, 10, 20 wt% as a function of temperature. | |
Though the measurement of the storage modulus is influenced by a number of factors, overall it seems that incorporation of the hydrophobic more graphitic S800 into the resin matrix imparts better mechanical performance for the composites in comparison with the more hydrophilic S300-reinforced materials. Changes in the functionality of starch upon carbonisation from hydroxyl-rich polysaccharides to carboxyl, ether groups at S300 and further into highly aromatic materials with S800 may also explain this different reinforcing effect.25,31 Secondly, higher surface area of the S800 filler would provide a better environment into which the resin could easily adhere, thereby yielding a composite with better dispersion of the filler within the polymer as previously seen on SEM micrographs (Fig. 6D–F).
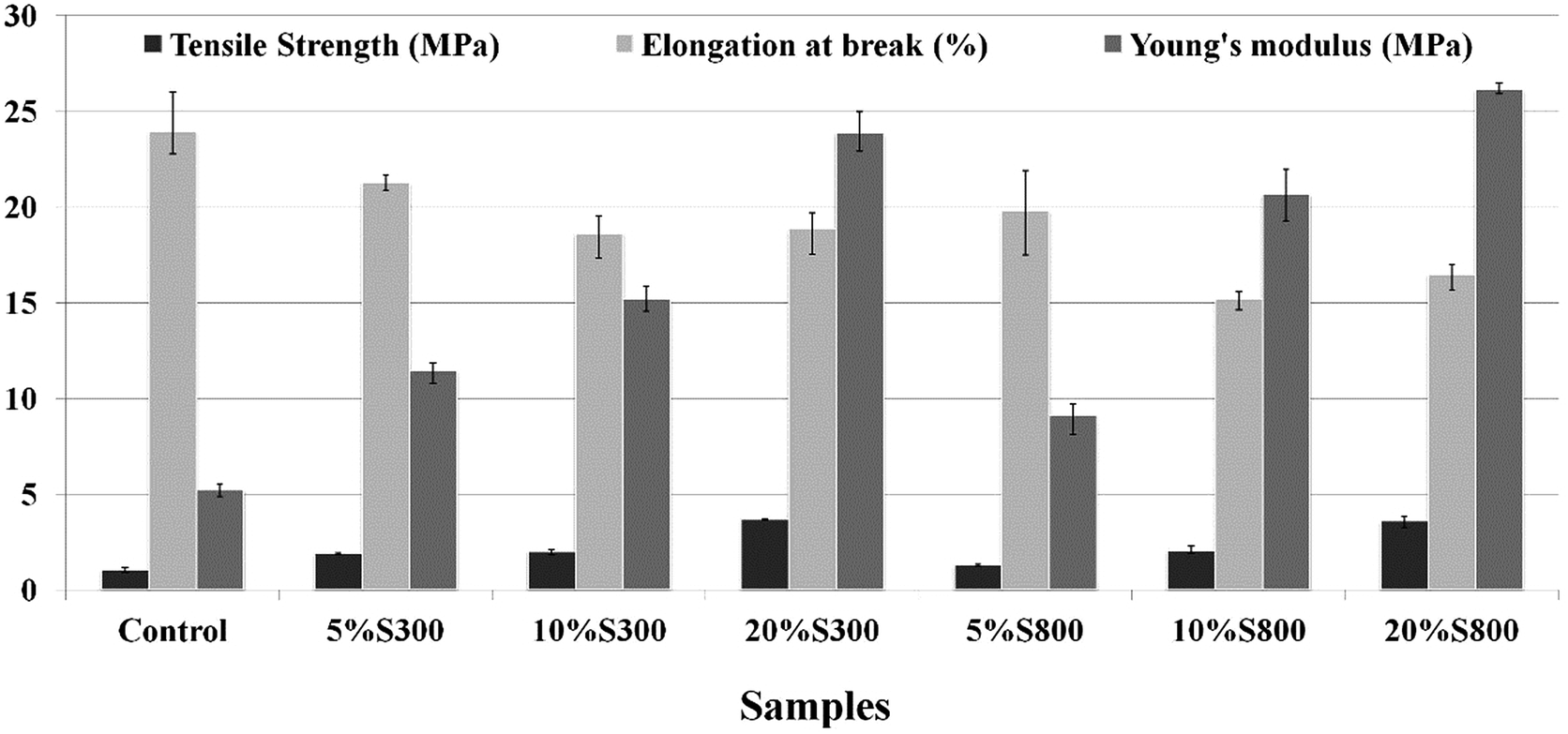
Mechanical properties of the thermoset composites. Tensile testing of the composites clearly demonstrated that Starbon® fillers aided in reinforcing the materials (Fig. 8). The tensile strength and Young's modulus increased with the amount of filler added with the maximum improvement of 260% and 400% respectively. It was noted that the S800 composites had a slightly higher modulus than those of S300 at similar loadings. This could also be attributed to the higher hydrophobicity of S800, resulting in better dispersion of filler in the polymer matrix as previously explained. The elongation at break decreased with the increase in Starbon® loading and the effect was slightly more evident in S800 composites. It is known that the incorporation of fillers can reduce the elongation in composites due to the restriction of the filler on the mobility of the polymer chains.42,43
 |
| | Fig. 8 Mechanical properties of the thermoset composites. | |
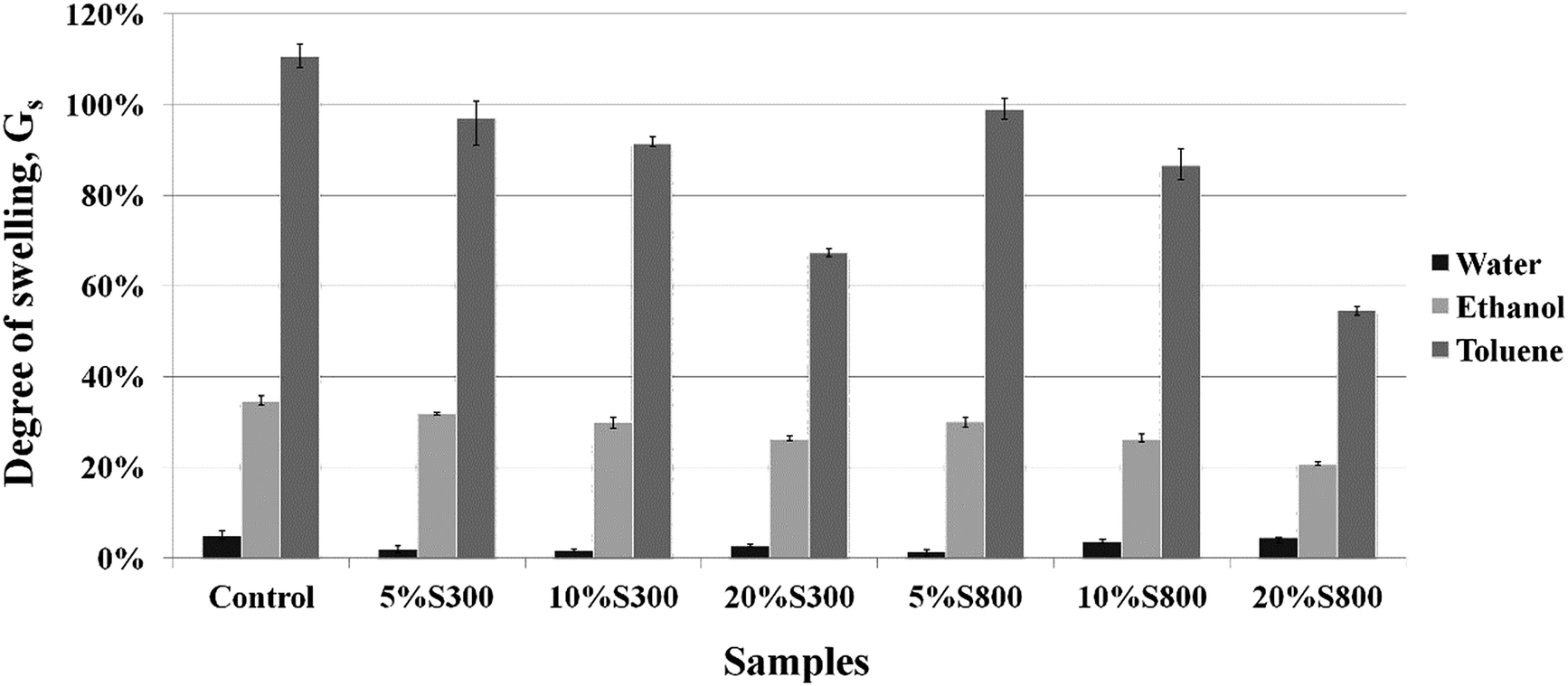
Swelling behaviour of the thermoset composites. Solvents of different polarity were chosen in order to study the swelling behaviour of the composites. The polarity index of water (10.2) is higher than that of ethanol (4.2) and toluene (2.4).44 The thermoset composites demonstrated hydrophobic characteristics with all the samples presenting a degree of swelling less than 5% in water (Fig. 9). The presence of both the S300 and S800 carbonised starch in the composites slightly reduced the water uptake further by 2–3%. When ethanol was used as a media, the degree of swelling increased due to the weaker polarity of the solvent relative to that of water. The sample with no filler showed the highest degree of swelling at 34.5%. The addition of S300 moderately decreased the degree of swelling, which diminished with the amount of filler added. The inclusion of S800 lowered the degree of swelling 2–5% further compared to the formulations with similar amount of the S300. This could be attributed to the aromatic characteristic of the S800 filler. Moreover, the less uniform distribution of the S300 particles throughout the films (as compared to S800) as shown on the SEM images (Fig. 6) might contribute to this higher swelling compared to the more evenly dispersed S800 filler composites. The control sample exhibited the highest degree of swelling with toluene, suggesting a high affinity between the crosslinked polymer and toluene. This was also reported by Zlatanic et al. who studied the swelling properties of six polyurethane networks from 4,4′-diphenylmethane diisocyanate and polyols based on different types of oils. It was found that linseed oil based polyurethane demonstrated the lowest degree of swelling (40%) due to its high crosslinking density, owing to the high unsaturation present in the oil structure.45 However, for the Starbon® composites the degree of swelling was significantly higher. This could be attributed to the saturated hydrocarbon chains present in the structure of ELO and Pripol, which should have a significant affinity with the non-polar solvent. The degree of swelling with toluene decreased with the amount of Starbon® filler added, due to the inability of Starbon® to swell compared to that of the crosslinked polymer. It was also noted that after the immersion in toluene for 48 hours the materials swelled extensively, leading to an eventual deterioration of the specimen, however ones in water and ethanol remained intact.
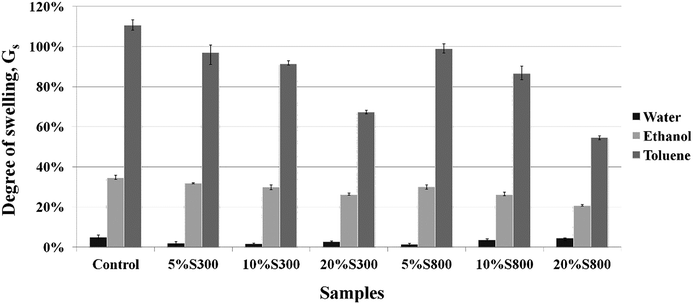 |
| | Fig. 9 Degree of swelling of the thermoset composites in water, ethanol and toluene after 24 h. | |
It is noted that swelling behaviour of the thermoset composites highlights the potential for such materials to be utilised in the development of membranes for water–ethanol separation through pervaporation. This method is considered a promising process in separating azeotropic mixtures. During the process, the penetrants should swell the membrane and as a result its microscopic structure is changed. This phenomenon consequently improves diffusion rates of the swollen membrane as compared with those of non-swollen ones.46 The diffusion of ethanol into a non-swollen membrane is normally difficult due to its large size while the swollen membrane can facilitate its diffusion.47 These thermoset composites should allow good diffusion of ethanol through the membrane and therefore increase the ethanol flux. Also, the production of such materials from bio-based resources which include the incorporation of tuneable mesoporous materials such as Starbon® may open new doors in the separation of higher value products as part of a biorefinery concept and will be the focus of future investigations.
Conclusions
Fully bio-derived thermoset composites were synthesised from epoxidised linseed oil, bio-derived curing agent and carbonaceous starch. The curing was confirmed by ATR-IR and MDSC. Thermal analysis demonstrated that the composites prepared with the more graphitic S800 had a higher Tg than that of the material without filler, implying more interfacial interactions between the polymer chains and the filler. However, for the hydrophilic S300 the bundling tendency seemed to influence the mobility of polymer chains, resulting in the decrease of Tg. Even dispersion of Starbon® filler into the resin imparts good mechanical performance for the composite, with S800 being more pronounced than S300. Starbon® fillers aided in reinforcing the materials, with increased maximum tensile strength and Young's modulus of 260% and 400% respectively. The resulting materials were found to be thermally stable until 300 °C, which enables use in high temperature applications. The use of Starbon® technology enables the incorporation of carbonaceous materials into the composite, with tuneable surface functionality (from hydrophilic to hydrophobic). The inclusion of this carbonaceous material with hierarchical pore structure and high BET surface area may further aid in these composites finding use in a range of applications as a bio-based thermoset material. The swelling tests of the bio-based composites suggested that these materials can potentially be utilised as membranes and will be the focus of future studies.
Acknowledgements
N. S. would like to thank the National Research University Project for their financial support through Biofuels Research Cluster at Khon Kaen University (NRU59006). N. S. and A. J. H. would like to thank the financial support of the Newton Mobility Grant (Royal Society). C. S. gratefully acknowledges funding through The Oil Refinery Contract Contribution Fund and the Ministry of Energy, Thailand. P. S. S. gratefully acknowledges the Spanish Ministry Economy and Competitivity (MINECO) for a Ramón y Cajal senior research fellowship (RYC-2014-16759) and a proyecto de I + D + I para jóvenes investigadores (MAT2014-59674-JIN). H. Y. thanks the financial support from China Scholarship Council. H. Y. is also thankful to the China Postdoctoral Science Foundation. J. P. F.-B. is grateful for the financial support from “Marie Curie” Amarout Europe Programme.
Notes and references
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- V. L. Budarin, P. S. Shuttleworth, J. R. Dodson, A. J. Hunt, B. Lanigan, R. Marriott, K. J. Milkowski, A. J. Wilson, S. W. Breeden, J. Fan, E. H. K. Sin and J. H. Clark, Energy Environ. Sci., 2011, 4, 471–479 CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- J. H. Clark, L. A. Pfaltzgraff, V. L. Budarin, A. J. Hunt, M. Gronnow, A. S. Matharu, D. J. Macquarrie and J. R. Sherwood, Pure Appl. Chem., 2013, 85, 1625–1631 CrossRef CAS.
- P. Gogoi, M. Boruah, S. Sharma and S. K. Dolui, ACS Sustainable Chem. Eng., 2015, 3, 261–268 CrossRef CAS.
- T. Tsujimoto, T. Takayama and H. Uyama, Polymers, 2015, 7, 2165–2174 CrossRef CAS.
- P. Gogoi, H. Horo, M. Khannam and S. K. Dolui, Ind. Crops Prod., 2015, 76, 346–354 CrossRef CAS.
- M. D. Samper, R. Petrucci, L. Sánchez-Nacher, R. Balart and J. M. Kenn, Composites, Part B, 2015, 71, 203–209 CrossRef CAS.
- J. M. Raquez, M. Deléglise, M. F. Lacrampe and P. Krawczak, Prog. Polym. Sci., 2010, 35, 487–509 CrossRef CAS.
- M. Galià, L. Montero de Espinosa, J. C. Ronda, G. Lligadas and V. Cádiz, Eur. J. Lipid Sci. Technol., 2010, 112, 87–96 CrossRef.
- N. Supanchaiyamat, P. S. Shuttleworth, A. J. Hunt, J. H. Clark and A. S. Matharu, Green Chem., 2012, 14, 1759–1765 RSC.
- A. O'Donnell, M. A. Dweib and R. P. Wool, Compos. Sci. Technol., 2004, 64, 1135–1145 CrossRef.
- Z. Liu, S. Z. Erhan, D. E. Akin and F. E. Barton, J. Agric. Food Chem., 2006, 54, 2134–2137 CrossRef CAS PubMed.
- N. Boquillon, J. Appl. Polym. Sci., 2006, 101, 4037–4043 CrossRef CAS.
- M. Yin, J. A. Koutsky, T. L. Bard, N. M. Rodriguez, R. T. K. Baker and L. Klebanov, Chem. Mater., 1993, 5, 1024–1031 CrossRef CAS.
- I. Sedat Gunes, G. A. Jimenez and S. C. Jana, Carbon, 2009, 47, 981–997 CrossRef.
- K. Hilarius, D. Lellinger, I. Alig, T. Villmow, S. Pegel and P. Pötschke, Polymer, 2013, 54, 5865–5874 CrossRef CAS.
- R. H. Baughman, A. A. Zakhidov and W. A. de Heer, Science, 2002, 297, 787–792 CrossRef CAS PubMed.
- V. L. Budarin, J. H. Clark, J. J. E. Hardy, R. Luque, K. Milkowski, S. J. Tavener and A. J. Wilson, Angew. Chem., Int. Ed., 2006, 45, 3782–3786 CrossRef CAS PubMed.
- R. J. White, V. L. Budarin, R. Luque, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 3401–3418 RSC.
- V. L. Budarin, J. H. Clark, F. E. I. Deswarte, J. J. E. Hardy, A. J. Hunt and F. M. Kerton, Chem. Commun., 2005, 23, 2903–2905 RSC.
- A. Muñoz García, A. J. Hunt, V. L. Budarin, H. L. Parker, P. S. Shuttleworth, G. J. Ellis and J. H. Clark, Green Chem., 2015, 17, 2146–2149 RSC.
- J. C. Colmenares, P. Lisowski and D. Łomot, RSC Adv., 2013, 3, 20186–20192 RSC.
- H. L. Parker, J. R. Dodson, V. L. Budarin, J. H. Clark and A. J. Hunt, Green Chem., 2015, 17, 2200–2207 RSC.
- A. S. Marriott, E. Bergström, A. J. Hunt, J. Thomas-Oates and J. H. Clark, RSC Adv., 2014, 4, 222–228 RSC.
- H. L. Parker, A. J. Hunt, V. L. Budarin, P. S. Shuttleworth and J. H. Clark, RSC Adv., 2012, 2, 8992–8997 RSC.
- H. L. Parker, V. L. Budarin, J. H. Clark and A. J. Hunt, ACS Sustainable Chem. Eng., 2013, 1, 1311–1318 CrossRef CAS.
- D. Gema, V. L. Budarin, J. A. Castro-Osma, P. S. Shuttleworth, S. C. Z. Quek, J. H. Clark and M. North, Angew. Chem., Int. Ed., 2016, 55, 9173–9177 CrossRef PubMed.
- A. S. Marriott, A. J. Hunt, E. Bergström, K. Wilson, V. L. Budarin, J. Thomas-Oates, J. H. Clark and R. Brydson, Carbon, 2014, 67, 514–524 CrossRef CAS.
- P. S. Shuttleworth, V. L. Budarin, R. J. White, V. M. Gun'ko, R. Luque and J. H. Clark, Chem.–Eur. J., 2013, 19, 9351–9357 CrossRef CAS PubMed.
- N. Supanchaiyamat, A. J. Hunt, P. S. Shuttleworth, C. Ding, J. H. Clark and A. S. Matharu, RSC Adv., 2014, 4, 23304–23313 RSC.
- J. Xu, K. M. Razeeb and S. Roy, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 1845–1852 CrossRef CAS.
- A. Beigbeder, M. Linares, M. Devalckenaere, P. Degée, M. Claes, D. Beljonne, R. Lazzaroni and P. Dubois, Adv. Mater., 2008, 20, 1003–1007 CrossRef CAS.
- D. Yue, Y. Liu and Z. Shen, J. Mater. Sci., 2006, 41, 2541–2544 CrossRef CAS.
- Z. Tang, D. Sun, D. Yang, B. Guo, L. Zhang and D. Jia, Compos. Sci. Technol., 2013, 75, 15–21 CrossRef CAS.
- F. H. Gojny and K. Schulte, Compos. Sci. Technol., 2004, 64, 2303–2308 CrossRef CAS.
- Z. Jin, K. P. Pramoda, G. Xu and S. H. Goh, Chem. Phys. Lett., 2001, 337, 43–47 CrossRef CAS.
- J. Q. Pham, C. A. Mitchell, J. L. Bahr, J. M. Tour, R. Krishanamoorti and P. F. Green, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 3339–3345 CrossRef CAS.
- A. Allaoui and N. El Bounia, eXPRESS Polym. Lett., 2009, 3, 588–594 CrossRef CAS.
- A. Apicella, C. Migliaresi, L. Nicodemo, L. Nicolais, L. Iaccarino and S. Roccotelli, Composites, 1982, 13, 406–410 CrossRef CAS.
- C. Ding, P. S. Shuttleworth, S. Makin, J. H. Clark and A. S. Matharu, Green Chem., 2015, 17, 4000–4008 RSC.
- W. Liu, K. Xu, C. Wang, B. Qian, Y. Sun, Y. Zhang, H. Xie and R. Cheng, J. Therm. Anal. Calorim., 2016, 123, 2459–2468 CrossRef CAS.
- S. Jeong, S. Yeo and S. Yi, J. Mater. Sci., 2005, 40, 5407–5411 CrossRef CAS.
- P. Hambleton, Support Materials and Solvents, in High Performance Liquid Chromatography: Fundamental Principles and Practice, ed. W. J. Lough and I. W. Wainer, Blackie Academic and Professional, Glasgow, 1996, p. 93 Search PubMed.
- A. Zlatanić, C. Lava, W. Zhang and Z. S. Petrović, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 809–819 CrossRef.
- Y. K. Ong, G. M. Shi, N. L. Le, Y. P. Tang, J. Zuo, S. P. Nunes and T.-S. Chung, Prog. Polym. Sci., 2016, 57, 1–31 CrossRef CAS.
- R. Jiraratananon, A. Chanachai, R. Y. M. Huang and D. Uttapap, J. Membr. Sci., 2002, 195, 143–151 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02837g |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
P. S. Shuttleworth
*a,
P. S. Shuttleworth