
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel macroporous polymer–inorganic nanocomposite as a sorbent for pertechnetate ions
Bojana M. Markovića,
Drina Lj. Jankovićb,
Aleksandar A. Vukadinovićb,
Danijela V. Ranđelovića,
Danijela D. Maksinb,
Vojislav V. Spasojevićb and
Aleksandra B. Nastasović *a
*a
aInstitute of Chemistry Technology and Metallurgy, University of Belgrade, Njegoševa 12, Belgrade, Republic of Serbia. E-mail: anastaso@chem.bg.ac.rs; anastasovic@gmail.com; Fax: +381 11 2636 061; Tel: +381 11 2635 839
bVinča Institute of Nuclear Sciences, University of Belgrade, P. O. Box 522, 11001 Belgrade, Republic of Serbia
First published on 18th April 2017
Abstract
A novel magnetic macroporous poly(glycidyl methacrylate-co-ethylene glycol dimethacrylate) (mPGME) was prepared by suspension copolymerization and functionalized with diethylenetriamine (mPGME-deta). The samples were characterized by Fourier transform infrared spectroscopy (FTIR) analysis, mercury porosimetry, scanning electron microscopy with energy-dispersive X-ray spectroscopy (SEM-EDS), atomic force microscopy (AFM) and SQUID magnetometry. The sorption behavior of mPGME-deta toward pertechnetate ions (TcO4−) from aqueous solution was studied. Experimental results indicated relatively fast TcO4− sorption kinetics that depends on pH and ionic strength, with a high removal efficiency of 99% within 180 min in the pH range 3.0–5.0. The pseudo-second-order model yielded the best fit for the kinetic data.
Introduction
Technetium-99 (99Tc) is a soft β-emitter (Eβmax = 294 keV) and a high yield (6%) fission product of uranium-235 (235U) and plutonium-239 (239Pu) with a long half-life (2.11 × 105 years) and long-term radiological effects.1 In a reducing environment, Tc is present in the +4 state, forming soluble hydroxides TcO2·nH2O, while, under oxidizing conditions, the predominant form of Tc is the pertechnetate oxyanion (TcO4−) which is highly soluble and difficult to eliminate, posing a significant environmental hazard.2,3Several polymeric sorbents have been investigated for their potential use in pertechnetate removal, like commercial anion-exchange resins,4 bifunctional chloromethylstyrene based anion-exchange resins cross-linked with divinylbenzene (DVB) with quaternary ammonium groups (triethylamine and trihexylamine),5 noncovalently immobilized (thia)calix[4]arenes on Amberlite XAD-7,6 4-vinylpyridine-based resins,7 etc.
Magnetic polymers are composed of a magnetic (Fe3O4, Fe2O3, nickel and cobalt) and polymer component that can be easily manipulated with a magnetic field. So far these types of materials have been used in catalysis, biotechnology/biomedicine, magnetic resonance imaging,8 metal chelation,9 etc. Magnetic polymers can be prepared from separately obtained polymers and magnetic cores (phase-separation, solvent evaporation and layer-by-layer process), or in situ by polymerization in a continuous phase in the presence of inorganic particles by suspension, dispersion, emulsion, miniemulsion, and microemulsion polymerization. Various preparation methods provide magnetic polymer microspheres with different morphology, like magnetic core–polymer shell, magnetic multicores homogeneously dispersed within the polymer matrix, polymer core coated magnetic nanoparticles (“strawberry” morphology) and polymer chains attached to the magnetic core (“brush” morphology).10 The most important parameters for application of magnetic copolymers are the size, size distribution, morphology, hydrophobicity/hydrophilicity, density of reactive surface groups and magnetic properties.11
Our previous studies have shown that PGME-deta due to the presence of numerous functional groups (–OH and –NH2) can be used for binding of metal oxo-anion species, like Cr and Mo.12–14 The sorption ability of PGME-deta towards pertechnetate anions was also investigated.15,16 In this study, magnetic macroporous poly(glycidyl methacrylate-co-ethylene glycol dimethacrylate) copolymer (mPGME) was prepared by suspension polymerization, functionalized with diethylenetriamine (mPGME-deta) and tested as a potential technetium-99 sorbent in form of pertechnetate ion (TcO4−) from aqueous solutions. The morphology, chemical and physical properties of mPGME and mPGME-deta were evaluated using Fourier transform infrared spectroscopy (FTIR) analysis, mercury porosimetry, scanning electron microscopy with energy-dispersive X-ray spectroscopy (SEM-EDS), atomic force microscopy (AFM) and SQUID magnetometry.
To the best of our knowledge, the study regarding the TcO4− sorption onto mPGME has not been published. Thus, basic objectives of this study were to study the effect of various experimental parameters such as the contact time, sample pH, competing anions and ionic strength on the sorption of pertechnetate anions by mPGME-deta as well to investigate the kinetics and equilibrium isotherms. Surface-reaction (pseudo-first-order, PFO and pseudo-second-order, PSO) and particle diffusion-based (intraparticle diffusion, IPD and Boyd's) kinetic models were used for analysis of the kinetic data. Adsorption isotherms were studied at varying pertechnetate concentrations and different background solutions.
Experimental section
Materials
Glycidyl methacrylate (GMA), diethylene triamine, 2,2′-azobisiso-butyronitrile, (AIBN), cyclohexanol and 1-tetradecanol were purchased from Merck (Germany). Ethylene glycol dimethacrylate (EGDMA) was supplied from Fluka (USA). Poly(N-vinyl pyrrolidone) (PVP, Kollidone 90) was purchased from BASF (Germany). FeSO4·7H2O and FeCl3·6H2O (Sigma-Aldrich) were used as sources for Fe(II) and Fe(III). All the other chemicals were analytical grade reagents. Technetium (99mTc + 99Tc) was eluted from 99Mo/99mTc generator (Vinča Institute of Nuclear Sciences, Belgrade, Serbia) in the form of sodium pertechnetate using normal saline (0.9% NaCl). The specific activity of 99mTc in sodium pertechnetate solution was determined by a Capintec CRC-15beta dose calibrator. Both 99Tc and 99mTc were contained in the pertechnetate form. A pH meter, Beckman F40 with a combined Ag/AgCl electrode, was employed for adjusting pH values. Adsorption isotherms of pertechnetate (99TcO4− + 99mTcO4−) by the mPGME-deta was carried out in various background solutions including deionized water (Milli-Q Millipore 18 MΩ cm−1 conductivity), 0.01 M KNO3, 0.1 M KNO3, 1 M KNO3 and groundwater from Bukulja mountain near Arandjelovac town (Republic of Serbia). The groundwater chemistry is provided in Table 1. Purified deionized water used in all experiments was prepared by the Milli-Q system (Millipore Co., Billerica, MA, USA).| Ion | mg l−1 |
|---|---|
| Calcium (Ca2+) | 83 |
| Magnesium (Mg2+) | 19 |
| Sodium (Na+) | 9.9 |
| Potassium (K+) | 1.6 |
| Iron (Fe2+/3+) | <0.05 |
| Bicarbonate (HCO3−) | 317 |
| Sulfate (SO42−) | 21.2 |
| Chloride (Cl−) | 11.5 |
| Fluoride (F−) | 0.18 |
Synthesis of magnetite nanoparticles
Superparamagnetic magnetite nanoparticles were prepared via improved chemical co-precipitation method.17 According to this method, 0.016 mol of FeSO4·7H2O and 0.028 mol of FeCl3·6H2O were dissolved in 320 ml of deionized water, stirred under N2 at 80 °C. After 1 h, 40 ml of NH3·H2O solution was injected rapidly into the mixture (at pH ∼ 10), stirred for another 1 h and cooled to room temperature. The precipitated magnetic particles were washed five times with hot water, separated by magnetic decantation and dried under vacuum at 70 °C.mPGME synthesis and amino-functionalization
Macroporous mPGME copolymer was prepared by suspension copolymerization of glycidyl methacrylate (GMA) and ethylene glycol dimethacrylate (EGDMA, crosslinker), in the presence of inert component and suspended magnetite nanoparticles (2 mass%). The aqueous phase consisting of 225.0 g of water and 2.25 g poly(N-vinyl pyrrolidone) was placed in polymerization reactor and heated to 70 °C. The monomer phase containing monomer mixture (19.5 g GMA and 29.1 g EGDMA), AIBN as initiator (0.5 g), 63.8 g of inert component (51.0 g of cyclohexanol and 12.8 g of tetradecanol) and 1.45 g of magnetite (Fe3O4) nanoparticles was sonicated for 30 min at 300/600 W within an ultrasonic water bath (Sonic 12GT). The resulting mixture was dropped into polymerization reactor. The reaction mixture was stirred at 300 rpm during the monomer addition and heating was started. The copolymerization was carried out under nitrogen atmosphere at 75 °C for 2 h and at 80 °C for 2 h with a stirring rate of 250 rpm. After the reaction mPGME was washed with water and ethanol, kept in ethanol for 12 h, and dried in vacuum at 50 °C. Particles with average particle diameter (D) in the range 150–300 μm were used for subsequent functionalization with diethylene triamine as described elsewhere.18 A mixture of 3.6 g of mPGME, 15.7 g of diethylene triamine, and 100 cm3 of toluene was left at room temperature for 24 h and then heated at 80 °C for 6 h. The amino-functionalized sample was filtered, washed with ethanol, dried, and labeled as mPGME-deta.mPGME and mPGME-deta characterization
The pore size distribution was determined by mercury porosimetry (Carlo Erba 2000, software Milestone 200). Fourier transform infrared (FTIR) spectra were recorded using a Nicolet 380 spectrometer equipped with a Smart Orbit™ ATR attachment containing a single-reflection diamond crystal. The scanning electron microscopy (SEM) micrographs were obtained on JEOL JSM-6460LV instrument (Tokyo, Japan), at a working distance of ca. 14 mm and an accelerating voltage of 20 kV. Functionalized samples were coated with gold in a high-vacuum evaporator. The energy dispersive spectroscopic (EDS) analysis was performed on Jeol JSM 5800 instrument operating at 20 kV. AFM characterizations were performed with AutoProbe CP-Research SPM (TM Microscopes-Veeco) using 90 μm large area scanner. Measurements were carried out in air using non-contact AFM mode, at the scan size (2 × 2) μm2 with the resolution of (256 × 256) data points per image. AFM images were created and analysed using two software packages Image Processing and Data Analysis Version 2.1.15 and SPMLab Analysis software, VEECO DI SPMLab NT Ver. 6.0.2. The roughness analysis was performed using Image Analysis 2.1.2 software, while fractal analysis was performed by partitioning method implementing Fractal Dimension tool available within freeware Gwyddion 2.43. Field dependence of isothermal magnetization M(H) at room temperature was measured on a SQUID-based commercial magnetometer Quantum Design MPMS-XL-5, in the applied DC fields up to 5 T.For determination of the amino groups content, 100 mg of mPGME-deta was immersed in 5.2 cm3 0.1 M HCl solution for 24 hours. After this period 2 cm3 of the solution was taken and the excess of acid was titrated with 0.052 M NaOH in the presence of phenolphthalein solution until the solution turned violet.19 The content of amino groups, CAG (mmol g−1) was calculated from:
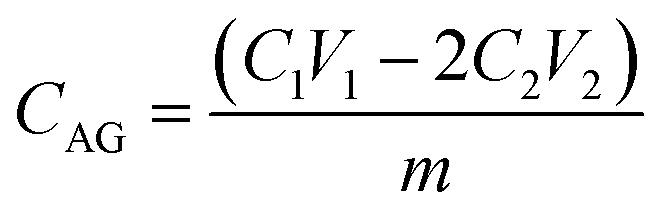 | (1) |
The point of zero charge (pHpzc value) of mPGME-deta was determined by the pH drift method.20 The initial pH (pHi) of test solutions (20 cm3 of 0.01 M NaCl in a series of Erlenmeyer flasks) was adjusted in the pH range between 2 and 12 by adding NaOH or HCl. After that, 50.0 mg of the mPGME-deta was added in the flasks and the final pH values of the solutions (pHf) were measured after 24 h. The pH at which the curve crosses pHi = pHf line was taken as pHpzc.21 pH was measured by Hanna HI 2210 pH meter calibrated before every measure.
Pertechnetate ions sorption onto mPGME-deta
The effect of contact time and pH was studied by shaking (at 120 rpm) 1 mg of mPGME-deta with 4.9 cm3 of the buffer solution and 0.1 ml of TcO4− solution in 10 cm3 borosilicate glass vials. The effect of pH on the TcO4− adsorption on PGME-deta and mPGME-deta was investigated in aqueous buffer solutions in a pH range 1–8. The blanks without copolymer were prepared in the same manner. For sorption isotherms determination, the concentration of TcO4− was in the range 0.2–500 pmol ml−1, while the pH was 5.0 ± 0.5, i.e. in the range of favorable pH. The solutions were equilibrated for 48 h.All uptake experiments were conducted in triplicate. In predetermined time intervals, 100 μl aliquots from the vials containing mPGME-deta as well as from the blanks were taken. The aliquot activity was measured in well-type NaI (Tl) gamma counter (2480 WIZARD Automatic Gamma Counter, Perkin Elmer, USA).
Data treatment
The exact values for the amount of 99mTc and 99Tc in sodium pertechnetate solution, obtained by eluting the 99Mo/99mTc generator, can be calculated using the equations derived for the generator kinetics of the parent–daughter relationship.22,23The amount of 99mTc and 99Tc (pmol) in supernatant and the amount of adsorbed 99mTc and 99Tc were calculated based on the total number of technetium atoms (99Tc and 99mTc) per mCi.
Relative measurements of adsorbed radioactivity were obtained using the following eqn (2):
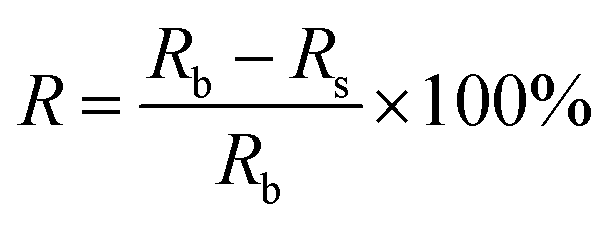 | (2) |
The partition coefficients, Kd (ml g−1), were calculated according to the eqn (3):
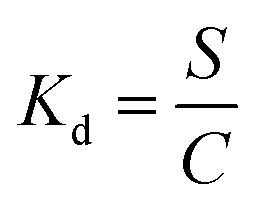 | (3) |
Kinetics analysis
The data for pertechnetate ion sorption by mPGME-deta were analyzed using pseudo-first order (PFO), pseudo-second order (PSO), intraparticle diffusion (IPD) and Boyd's kinetic models.The linearized form of PFO (eqn (4)) and PSO (eqn (5))24 kinetic models used for calculations were as follows:
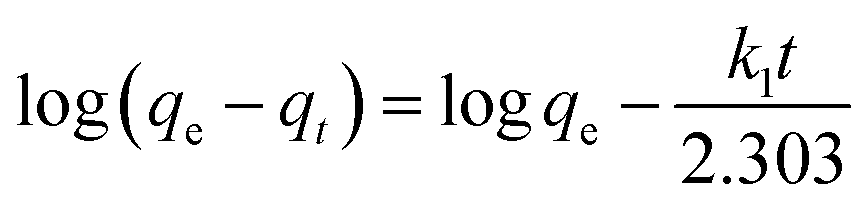 | (4) |
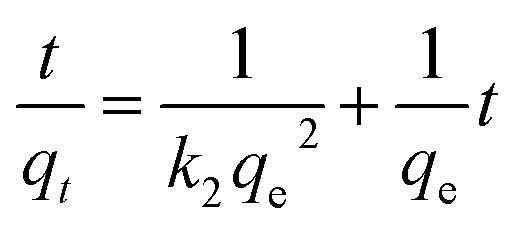 | (5) |
The corresponding equations for intraparticle (IPD) (eqn (6)) and Boyd's (eqn (7))25 kinetic models were as follows:
| qt = kdt1/2 + c | (6) |
| Bt = −0.4997 − ln(1 − G) | (7) |
Results and discussion
Characterization of mPGME and mPGME-deta
The synthesized magnetic mPGME sample was sieved with 0.15, 0.30 and 0.63 mm sieves. The results of sieve analysis were compared with non-magnetic sample PGME and presented in Fig. 1.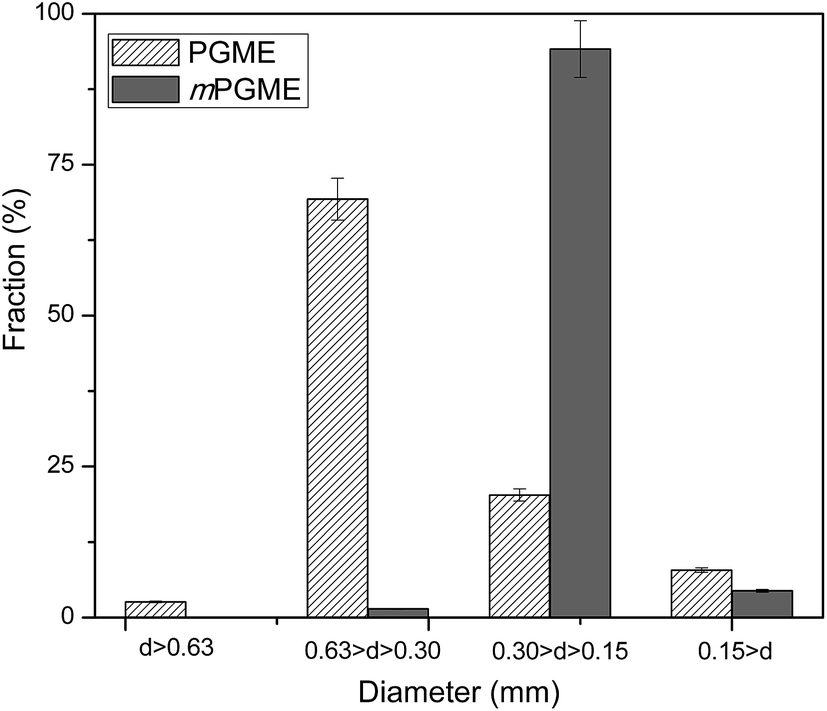 | ||
| Fig. 1 Sieve analysis of non-magnetic and magnetic copolymer samples. | ||
As can be seen, the particle fraction with diameter in 0.15–0.30 mm range is highly dominant for sample mPGME, approximately 95 mass%. It appears that the addition of magnetite in the reaction mixture considerably influences the formation of macroporous structure, narrows the size distribution and shifts it to the smaller particles in comparison with PGME synthesis. We previously observed the similar for PGME/bentonite composites.26
The cumulative pore volume distribution curves for selected samples are presented in Fig. 2. The relevant porosity parameters (specific pore volume, VS, specific surface area, SHg, and pore diameter that corresponds to half of the pore volume, DV/2) for mPGME and mPGME-deta were calculated from the cumulative pore volume distribution curves as described in the literature27 and presented in Table 2.
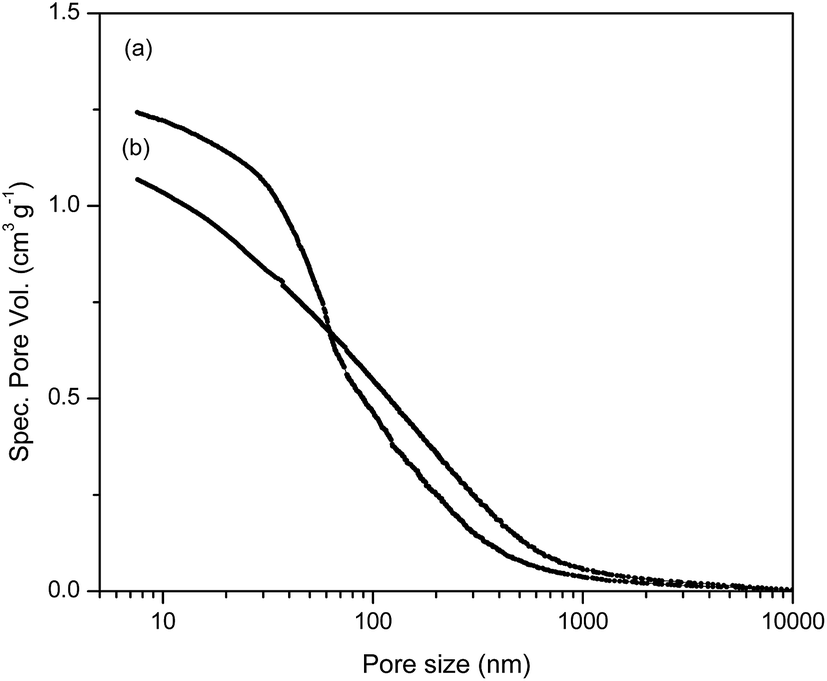 | ||
| Fig. 2 Cumulative pore volume distribution curves for (a) mPGME-deta and (b) mPGME. | ||
| Copolymer sample | Ss,Hg, m2 g−1 | Vs, cm3 g−1 | DV/2, nm |
|---|---|---|---|
| mPGME | 92 | 1.07 | 102 |
| mPGME-deta | 100 | 1.24 | 66 |
It can be seen from porosity data that the amino-functionalization alters the porous structure of magnetic copolymer, causing a shift in the pore size distribution curves of the synthesized samples towards higher specific pore volume and smaller pores, i.e. sample mPGME-deta has almost twice lower DV/2 value. However, the influence of amino-functionalization on SHg value is negligible.
The FTIR spectra of mPGME and mPGME-deta were recorded and presented in Fig. 3.
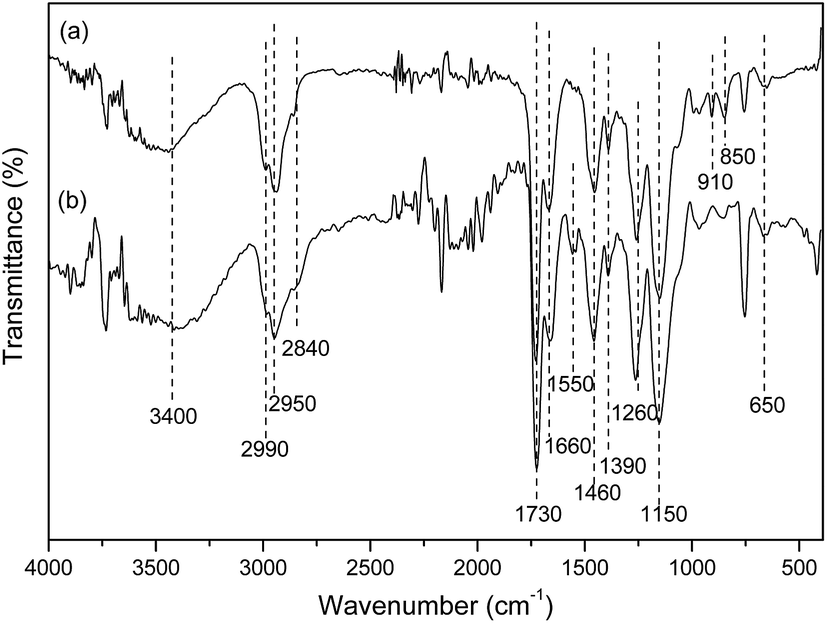 | ||
| Fig. 3 FTIR spectra of (a) mPGME and (b) mPGME-deta. | ||
In the FTIR spectra of magnetic copolymer samples the characteristic absorption bands at ∼2990 cm−1, ∼2950 cm−1 and ∼2840 cm−1 (methyl and methylene stretching vibrations, νC–H), ∼1730 cm−1 (ester carbonyl vibrations, νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), ∼1390 and ∼1460 cm−1 (methyl and methylene bending vibrations, δC–Hasym and δC–Hsym) were observed. Also, the characteristic peaks for epoxy ring vibrations at ∼850 and ∼910 cm−1 and 1260 cm−1 (stretching vibrations, νC–O) and ∼1150 cm−1 (ether stretching vibrations, νC–O–C) were found in the mPGME spectrum.28 The characteristic stretching frequencies of the amino-functionalized sample mPGME-deta appeared at 1550 cm−1 (δNH).
O), ∼1390 and ∼1460 cm−1 (methyl and methylene bending vibrations, δC–Hasym and δC–Hsym) were observed. Also, the characteristic peaks for epoxy ring vibrations at ∼850 and ∼910 cm−1 and 1260 cm−1 (stretching vibrations, νC–O) and ∼1150 cm−1 (ether stretching vibrations, νC–O–C) were found in the mPGME spectrum.28 The characteristic stretching frequencies of the amino-functionalized sample mPGME-deta appeared at 1550 cm−1 (δNH).
The FTIR spectra for both samples confirm the incorporation of magnetite in the macroporous PGME. Namely, the bands in the 650–575 cm−1 range could be attributed to the vibrations of Fe–O bonds in tetrahedral and octahedral sites.29,30 According to the literature, the band at ∼420 cm−1 originates from octahedral Fe and corresponds to the band of Fe–O of bulk magnetite (∼370 cm−1) shifted to a higher wavenumber. The presence of O–H stretching vibration at ∼3400 cm−1 and O–H bending vibrations at 1660 cm−1 could be ascribed to the presence of coordinated OH groups or water molecules with the unsaturated surface Fe atoms.30
For further analysis, the morphology of mPGME and mPGME-deta samples was examined by SEM with energy-dispersive X-ray spectroscopy (SEM-EDS). The results were presented in Fig. 4 and Table 3.
 | ||
| Fig. 4 SEM microphotographs of: mPGME particles (a), particle surface (b), particle cross-section (c) and mPGME-deta particles (d), particle surface (e) particle cross-section (f). | ||
| Element | Particle surface | Particle cross-section | ||||||
|---|---|---|---|---|---|---|---|---|
| mPGME | mPGME-deta | mPGME | mPGME-deta | |||||
| Mass% | Atomic% | Mass% | Atomic% | Mass% | Atomic% | Mass% | Atomic% | |
| C–K | 82.1 | 86.8 | 64.8 | 70.9 | 77.2 | 82.0 | 60.1 | 66.0 |
| O–K | 16.0 | 12.7 | 19.4 | 12.6 | 22.5 | 17.9 | 20.8 | 17.2 |
| N–K | — | — | 13.5 | 15.9 | — | — | 17.4 | 16.4 |
| Fe–K | 1.9 | 0.4 | 2.4 | 0.6 | 0.3 | 0.1 | 1.7 | 0.4 |
The micrographs demonstrate three dimensional porous structures of the samples, composed of a large number of globules and interconnected with channels and pores. Also, from Fig. 4 it is visible that the iron particles are dispersed on the surface of mPGME and mPGME-deta particles. The similar was observed for crosslinked ionic polymers containing strongly basic functional groups adsorb Fe(III)-containing cations31 and magnetic copolymer obtained from suspension polymerization of styrene and divinylbenzene.11 Highly developed internal porous structure is advantageous for the metal ions sorption due to the decrease of the mass transfer resistance, facilitation of the metal ions diffusion which consequently provides high adsorption rate and capacity.32
The SEM-EDS analysis confirmed the presence of all expected elements (C, O, N and Fe). The N percentage was almost the same on the particles surface and in the cross-section indicating that the reaction of epoxy groups with diethylenetriamine occurs on the surface as well as in the interior of the mPGME-deta particles. Although predominantly present at the particle surface, the iron nanoparticles were also embedded in the bulk to a certain extent.
The surface topography of mPGME and mPGME-deta was studied by atomic force microscopy (AFM). The 2D and 3D AFM images of the top view of samples mPGME and mPGME-deta are shown in Fig. 5.
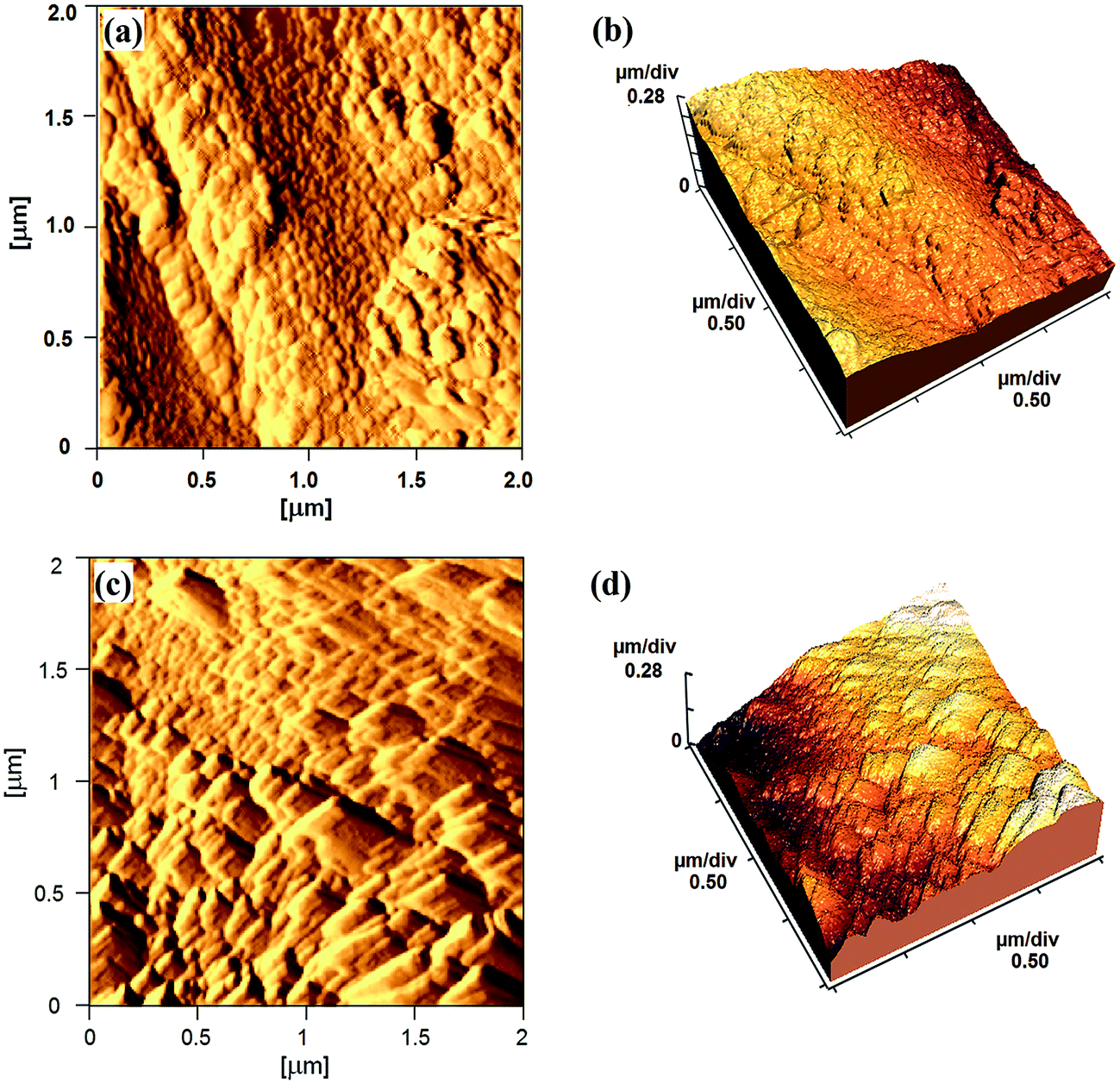 | ||
| Fig. 5 The typical AFM images: (a) 2D and (b) 3D images of the mPGME; (c) 2D and (d) 3D images of the mPGME-deta. | ||
A very important parameter for a potential application of the synthesized copolymers in the adsorption and ion exchange is the surface roughness.33,34 Due to the fact that the roughness of real surfaces is hard to describe theoretically and quantitatively, the surface characterization by fractal dimension (FD) has been proved as useful.35,36 As a measure of surface irregularity, fractal dimension varies between 2 (for a flat sample) and 3 (for an extremely rough sample).37,38 The average roughness (Ra) and root mean square roughness (Rq) as well as fractal dimensions were presented in Table 4.
| Sample | Ra, μm | Rq, nm | Fractal dimensions |
|---|---|---|---|
| mPGME | 265.5 | 314.4 | 2.16 |
| mPGME-deta | 77.33 | 92.99 | 2.27 |
It can be noticed that mPGME surface is significantly rougher than the mPGME-deta surface as indicated by the roughness values presented in Table 4. However, the differences in fractal dimension values for mPGME and mPGME-deta are not so evident.
The magnetization curves measured for magnetite and mPGME at 300 K in a SQUID-based magnetometer in the ±5 T field range are presented in Fig. 6.
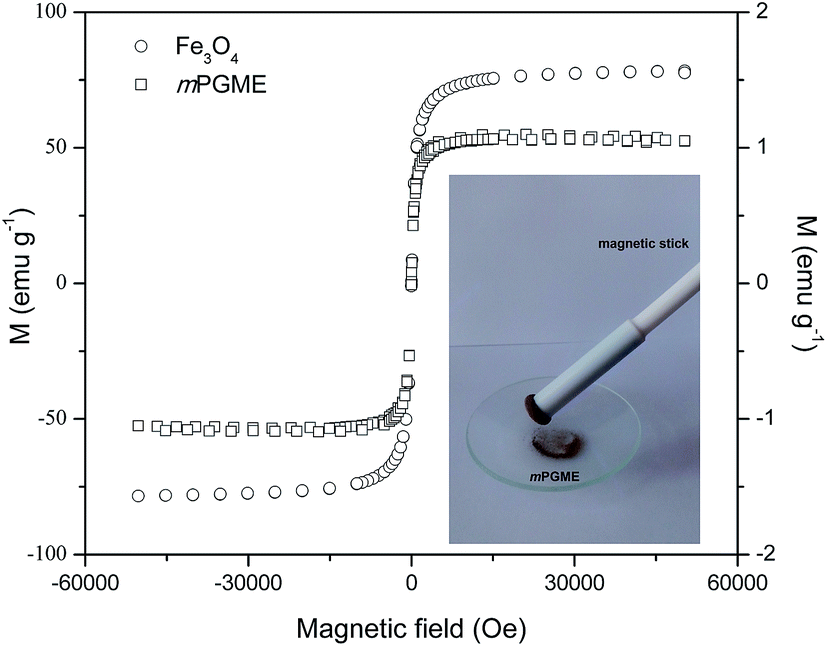 | ||
| Fig. 6 Magnetic hysteresis curves of mPGME and magnetite. | ||
The mPGME sample with the magnetite content of 1.4 mass% shows superparamagnetic behavior with negligible hysteresis loop. Although the saturation magnetization decreased after magnetite incorporation in the magnetic copolymer, complete magnetic separation of mPGME was achieved by placing a magnet near the flask containing the aqueous dispersion of the magnetic copolymer particles (see Fig. 6, inset). Additional amino-functionalization had no effect on PGME magnetization. Similar was observed for silica-coated core–shell magnetite nanoparticles (Fe3O4@SiO2) and nanoparticles with covalently grafted amino groups.39
The effect of contact time and pH on pertechnetate ions sorption onto mPGME-deta
The solution pH and contact time are a crucial parameters that affects the sorbent surface charge, the protonation–deprotonation of surface functional groups, metal speciation in the aqueous solution and consequently, the sorbate/sorbent interactions.40 Thus, the influence of contact time and solution pH on TcO4− removal by mPGME-deta was investigated in the pH range 1.0–8.0 while keeping the other variables constant (shaking speed 120 rpm, sorbent concentration 0.2 g dm−3 and T = 298 K). The effect of the contact time (at pH 5.0) and pH (for contact time 180 min) on the removal efficiency of pertechnetate is presented in Fig. 7.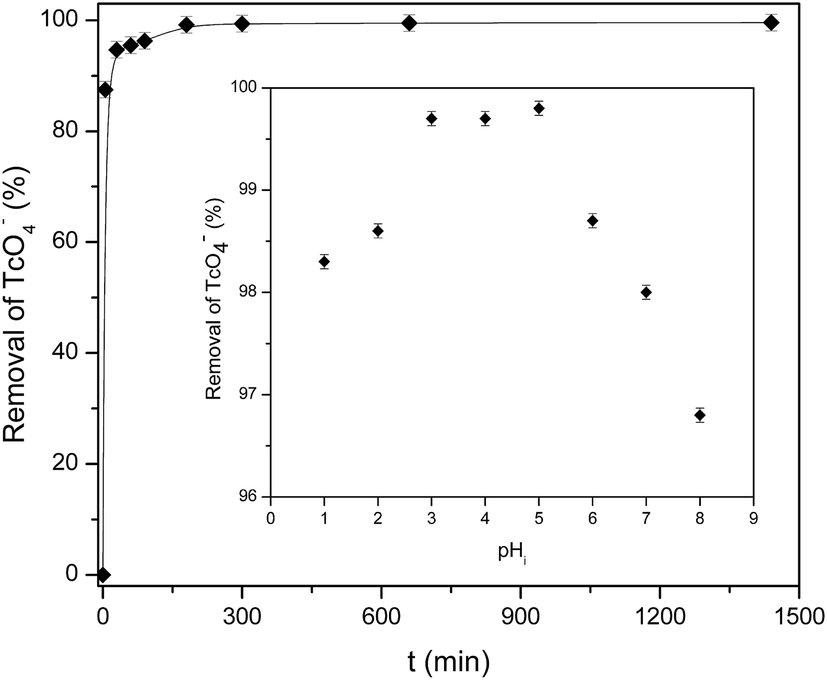 | ||
| Fig. 7 Effect of contact time and pH (inset) on the sorption of pertechnetate from aqueous solution onto mPGME-deta. Vertical error bars represent standard errors. | ||
As can be seen from Fig. 7, pertechnetate ion sorption onto mPGME-deta was rather fast, with 90% and 95% of TcO4− sorbed within the first 5 and 30 min, respectively. In acidic medium (pH 1–3) the removal efficiency increases and then reaches the maximum of >99% in the pH range 3.0–5.0. With further pH increase (pH 6–8), pertechnetate sorption slightly decreased and the removal efficiency of TcO4− fell to 96.5%.
In order to understand the sorption mechanism, the pH values at which the surface has neutral charge (pHpzc) of mPGME-deta were determined by the pH drift method. The results are shown in Fig. 8.
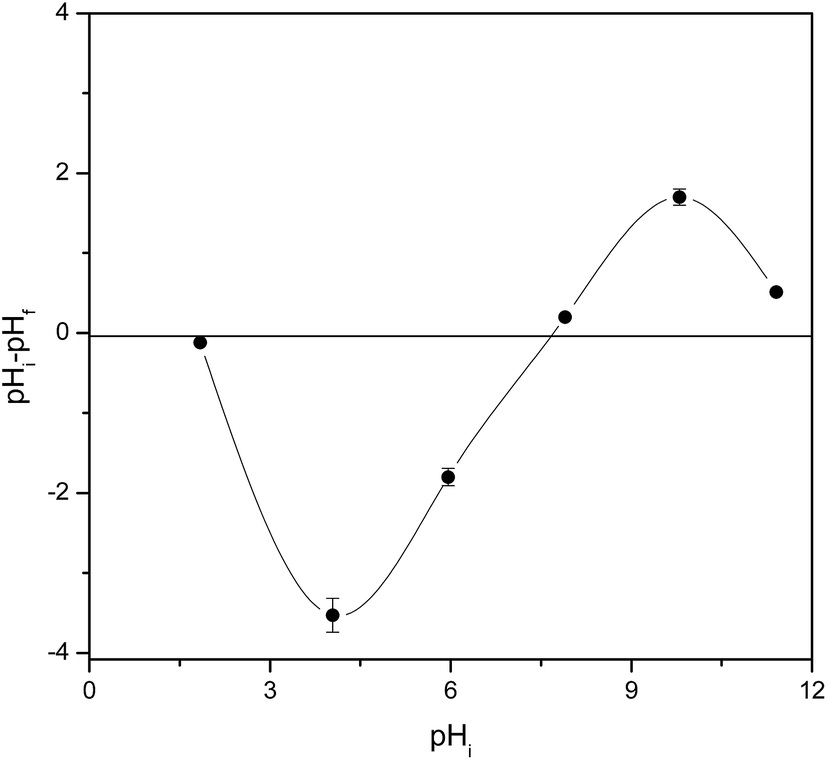 | ||
| Fig. 8 Point of zero charge (pHpzc) of mPGME-deta determined by the pH drift method. Standard errors are shown as vertical error bars. | ||
If the solution pH is higher than pHpzc, the sorbent surface will have a negative charge and sorption of positive ions will be favored due to the electrostatic attraction. On the other hand, the sorption of negatively ions will be favored when pH < pHpzc.41 As can be seen, the pHpzc for the mPGME-deta is 6.8 ± 0.3, high removal efficiency of TcO4− was expected to be achieved at pH below that value. Thus, in acidic medium, the electrostatic interactions between the protonated amino groups of mPGME-deta and TcO4− anions promote high pertechnetate removal. At pH ≥ 6, the TcO4− sorption decreased due to the decrease of the number of protonated amino groups in mPGME-deta.
The influence of solution pH on TcO4− removal by non-magnetic PGME-deta (prepared in the same manner as mPGME, in the absence of magnetite in the reaction mixture) was investigated in the pH range 3.0–7.0 and contact time of 180 min while keeping the other variables constant. The sorption performances of PGME-deta and its magnetic analogue mPGME-deta slightly differ. Namely, the removal efficiency of PGME-deta and mPGME-deta was in the range 95–98%, and 97–99%, respectively. This result was expected if one bears in mind that the TcO4− sorption is predominantly influenced by the surface amino groups attached to the macroporous copolymer and not the magnetization. The role of the magnetization was to enable easy and rapid separation of mPGME-deta particles from the solution after TcO4− sorption by an external magnetic field.
Sorption kinetics
Sorption kinetic data were analyzed using surface-reaction (PFO and PSO) and particle diffusion-based (IPD and Boyd's) kinetic models to determine the nature of sorption kinetics and the rate limiting step for pertechnetate ion were shown in Fig. 9, Tables 5 and 6.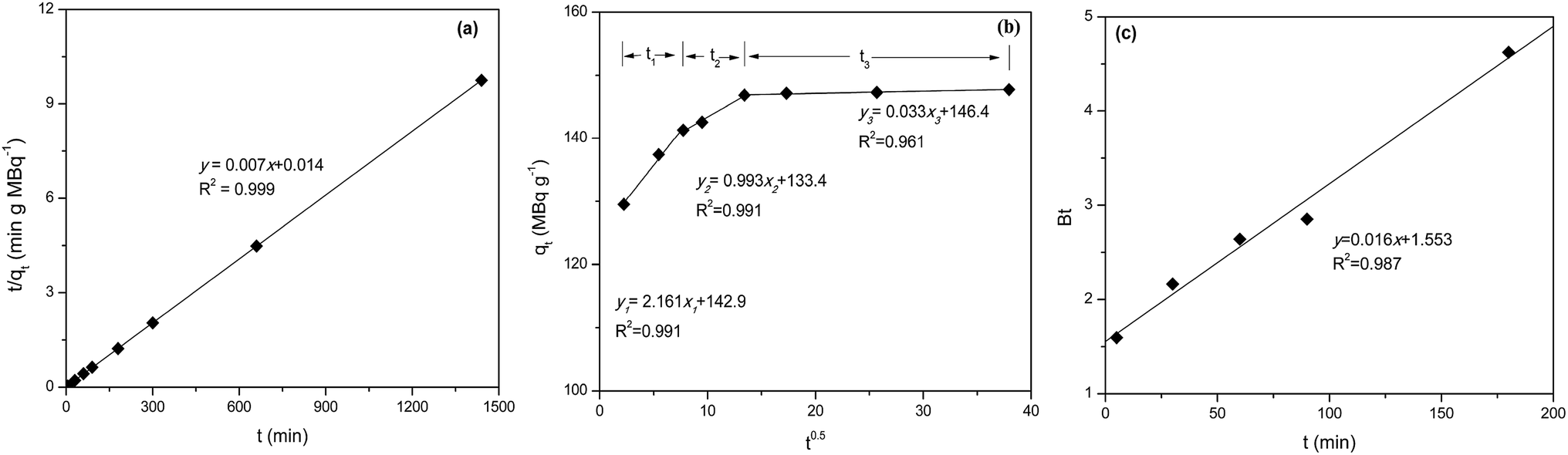 | ||
| Fig. 9 Plots of PSO (a), IPD (b) and Boyd (c) kinetics model for pertechnetate ion sorption on mPGME-deta (pH = 5, t = 24 h, T = 298 K). | ||
| qexpe, MBq g−1 | Pseudo-first-order | Pseudo-second-order | ||||
|---|---|---|---|---|---|---|
| k1 × 102, min−1 | qcalc1, MBq g−1 | R2 | k2 × 103, g MBq−1 min−1 | qcalc2, MBq g−1 | R2 | |
| 148 | 1.612 | 19 | 0.987 | 3.50 | 143 | 0.999 |
| Initial linear portion | Second linear portion | Third linear portion | ||||||
|---|---|---|---|---|---|---|---|---|
| kd1, MBq g−1 min−0.5 | c1, MBq g−1 | R12 | kd2, MBq g−1 min−0.5 | c2, MBq g−1 | R22 | kd3, MBq g−1 min−0.5 | c3, MBq g−1 | R32 |
| 2.161 | 124.9 | 0.991 | 0.993 | 133.4 | 0.991 | 0.033 | 146.4 | 0.961 |
The kinetics of pertechnetate anion sorption onto mPGME-deta is accurately described by PSO model (Fig. 9a) since the calculated qcalc from this model agreed very well with the experimental qexp value, with high value of the correlation coefficients (R2 = 0.999). This implies that chemisorption mechanism plays an important role for the sorption of this anion onto mPGME-deta, i.e. that sorption rate is controlled by both the sorbent capacity and the sorbate concentration.
The qt–t1/2 plot (Fig. 9b) for IPD kinetic model applied on porous sorbents shows multi-linearity, i.e. has three distinct regions.42 The first linear portion included the sorption period of 0–50 min, which represented external mass transfer and binding of pertechnetate ions sorption active sites distributed onto the outer surface of mPGME-deta. The second linear portion included the gradual sorption period of 50–250 min, representing intraparticle diffusion and binding of pertechnetate ions by active sites distributed in the mPGME-deta porous structure. The third linear portion includes the time period from 250 min to 24 h, which denoted establishment of the equilibrium. The value of c provides information about the thickness of the boundary layer, i.e., the larger the intercept, the greater the boundary layer effect. From Fig. 9c it is observed that the Boyd's plot for the pertechnetate ions sorption using mPGME-deta was linear, not passing through the origin, which indicates that external mass transfer mainly governs the rate-limiting process.43
Adsorption isotherms
Pertechnetate ion sorption was studied on mPGME-deta at varying pertechnetate concentrations and different background solutions (at pH = 5, t = 48 h, T = 298 K). The results are presented in Fig. 10.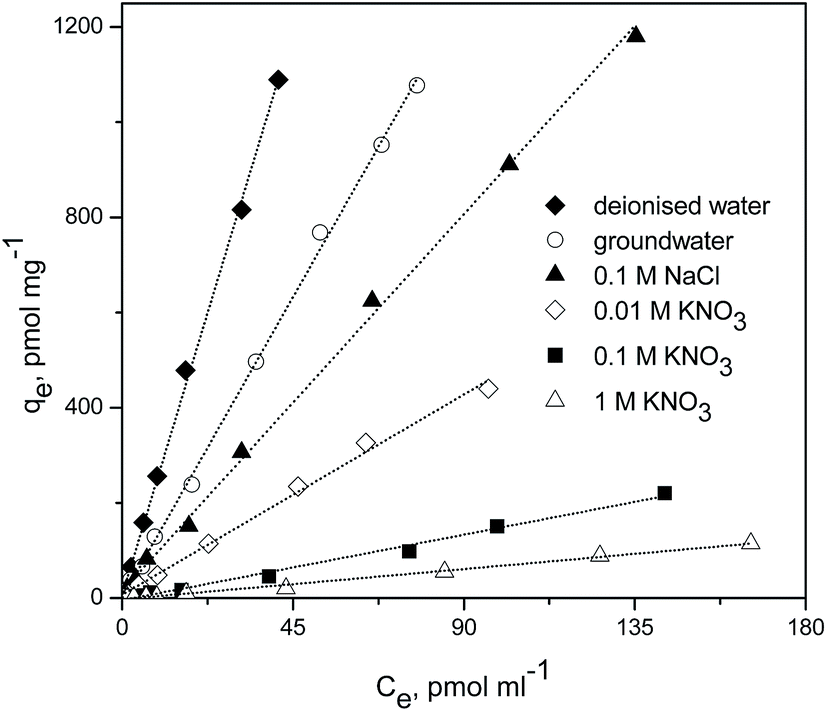 | ||
| Fig. 10 Sorption isotherms of pertechnetate ion sorption on mPGME-deta from different background solutions. | ||
The reaction of TcO4− with mPGME-deta yielded linear adsorption isotherms, with correlation coefficients ranging from R2 = 0.9924 to R2 = 0.9988. The highest correlation coefficients were obtained for deionized water, groundwater and 0.1 M NaCl. The calculated values of partition coefficients, Kd, were presented in Table 7.
| Background solution | Kd, ml g−1 |
|---|---|
| a It should be noted that 99Mo/99mTc generator must be eluted with normal saline. So, when 0.1 ml of pertechnetate solution is added it gives 0.00308 M of NaCl in addition to background solution. | |
| Deionised water | 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 360 360 |
| Ground water | 14056 |
| 0.1 M NaCl | 8801 |
| 0.01 M KNO3 | 4679 |
| 0.1 M KNO3 | 1530 |
| 1 M KNO3 | 707 |
As can be seen from Fig. 10 and Table 7, in 0.1 M NaCl solution and in low ionic strength solution of 0.01 M KNO3, TcO4− can be selectively and effectively removed by mPGME-deta. The observed Kd values ranged from 4679 to 8801 ml g−1. When using the groundwater as a background, the Kd values were even higher, i.e. 14056 ml g−1. Except in a background of 0.003 M NaCl (deionized water), the TcO4− removal efficiency exceeded 99% and the Kd value was the highest, i.e. 26360 ml g−1.
The molar concentration of TcO4− in solution was very low (in the order of 10−8 to 10−9 M) compared to that of Cl− or NO3− (0.01–1 M). The molar ratios of background anions to TcO4− ranged from 3 × 105 to 1 × 109. Under such unfavorable molar concentration ratios, TcO4− was highly competitive to be adsorbed by mPGME-deta among other mentioned anions. The NO3− anions appeared more effective than Cl− in reducing TcO4− adsorption. In high ionic strength background solutions (0.1–1 M NaNO3), the adsorption of TcO4− sharply decreased (Fig. 10 and Table 7).
The obtained results suggest that the main mechanism of TcO4− sorption onto mPGME-deta can be described by ion pair formation between NH3+ surface groups attached to the magnetic polymer (P) and TcO4− anions in solution as presented in eqn (8):
| P-NH3+ + TcO4− ⇔ NH3+TcO4− | (8) |
Eqn (9) describes the sorption of TcO4− in presence of NO3−:
| P-NH3+ + NO3− ⇔ NH3+NO3− | (9) |
Although direct comparison of the reported data on pertechnetate ions sorption by different sorbents is not possible due to the different experimental conditions, some literature data will be mentioned. Previously, we investigated the removal of pertechnetate ions by using PGME-deta samples with different amount of the crosslinking monomer (EGDMA) from aqueous solutions. Linear adsorption isotherms were also found, with partitioning coefficients of 1698 ml g−1 and 2130 ml g−1.16 Popova et al. reported the Kd value of 3 × 105 ml g−1 for sorbent prepared by the noncovalent immobilization of (thia)calix[4]arenes on the Amberlite XAD-7™.6 Bonnesen et al. used a series of monofunctional and bifunctional quaternary ammonium resins of poly(vinylbenzyl chloride)-10% divinylbenzene copolymer with various size of the trialkylammonium group for TcO4− uptake.5 Monofunctional tri-n-methylammonium, tri-n-propylammonium, and tri-n-butylammonium resins showed high 24 h distribution ratios (Kd) for pertechnetate anion from groundwater test solution, i.e. 1680, 9570 and 31800 ml g−1, respectively. On the other hand, bifunctional resins containing exchange sites derived from tri-n-hexylamine in combination with either trimethylamine and triethylamine, showed higher Kd values, i.e. 13200 and 37300 ml g−1, respectively. The Kd values for commercial strong base resins Reillex HPQ and Purolite A-520E were 4540 and 12800 ml g−1, respectively.
Conclusions
A novel magnetic macroporous mPGME sorbent has shown a strong tendency to remove pertechnetate ions (TcO4−) from aqueous solutions in absence and presence of competing anions. Results indicated that sorption kinetics of the pertechnetate ions on mPGME-deta was relatively fast and depends on pH and ionic strength. The amount of sorbed TcO4− slightly decreases with the pH decrease from 2 to 1 and pH increase from 6 to 8, indicating a lowering of the sorption capacity of mPGME-deta. We propose that non-specific sorption of pertechnetate ion via electrostatic interaction takes place at the protonated amine groups NH3+. The removal efficiency higher than 95% and 99% at pH 3–5 within 30 min and 180 min were found, respectively. The pseudo-second order kinetic model fitted very well with the kinetic data of pertechnetate anion sorption onto mPGME-deta, suggesting that the sorption rate is controlled by both sorbent capacity and sorbate concentration. Particle diffusion-based models revealed the strong influence of the intraparticle diffusion and porosity of PGME-deta.The partition coefficient (Kd) of TcO4− exceeded 2.6 × 104 ml g−1 and 1.4 × 104 ml g−1 when deionized water at pH 5 and groundwater were used, respectively. The sample treated with 0.1 M NaCl sorbed TcO4− very well at pH 5 (Kd = 8.8 × 103 ml g−1). The presence of excessive amounts of NO3− ions in the solution reduced the removal of TcO4− for 70 and 95%. The results obtained in this study suggest that mPGME-deta can be effectively used for TcO4− separation from aqueous solutions.
Acknowledgements
This work was supported by the Ministry of Education, Science and Technological Development of the Republic of Serbia (Grants No. III43009, III45015, ON172062, TR32008 and III43001).References
- T. K. Ikäheimonen, V. P. Vartti, E. Ilus and J. Mattila, J. Radioanal. Nucl. Chem., 2002, 252, 309–313 CrossRef.
- K. Shi, X. Hou, P. Roos and W. Wu, Anal. Chim. Acta, 2012, 709, 1–20 CrossRef CAS PubMed.
- Y. Wang, H. Gao, R. Yeredla, H. Xu and M. Abrecht, J. Colloid Interface Sci., 2007, 305, 209–217 CrossRef CAS PubMed.
- B. Gu, G. M. Brown, P. V. Bonnesen, L. Liang, B. A. Moyer, R. Ober and S. D. Alexandratos, Environ. Sci. Technol., 2000, 34, 1075–1080 CrossRef CAS.
- P. V. Bonnesen, G. M. Brown, S. D. Alexandratos, L. B. Bavoux, D. J. Presley, V. Patel, R. Ober and B. A. Moyer, Environ. Sci. Technol., 2000, 34, 3761–3766 CrossRef CAS.
- N. N. Popova, V. P. Morgalyuk, I. G. Tananaev, S. E. Solovieva, E. V. Popova and I. S. Antipin, Russ. Chem. Bull., 2011, 60, 175–178 CrossRef CAS.
- P. Wang, A. Khayambashi, J. Zu, Y. Wei, F. Tang and L. He, J. Radioanal. Nucl. Chem., 2016, 311, 301–306 CrossRef.
- D. Horák, E. Petrovský, A. Kapička and T. Frederichs, J. Magn. Magn. Mater., 2007, 311, 500–506 CrossRef.
- F. Ge, M.-M. Li, H. Ye and B.-X. Zhao, J. Hazard. Mater., 2012, 211–212, 366–372 CrossRef CAS PubMed.
- D. Horák, M. Babič, H. Macková and M. J. Beneš, J. Sep. Sci., 2007, 30, 1751–1772 CrossRef PubMed.
- B. M. Conceição, M. A. S. Costa, L. C. D. S. Maria, M. R. Silva and S. H. Wang, Polimeros, 2011, 21, 409–415 Search PubMed.
- A. Nastasović, Z. Sandić, L. Suručić, D. Maksin, D. Jakovljević and A. Onjia, J. Hazard. Mater., 2009, 171, 153–159 CrossRef PubMed.
- D. D. Maksin, A. B. Nastasović, A. D. Milutinović-Nikolić, L. T. Suručić, Z. P. Sandić, R. V. Hercigonja and A. E. Onjia, J. Hazard. Mater., 2012, 209–210, 99–110 CrossRef CAS PubMed.
- B. M. Ekmeščić, D. D. Maksin, J. P. Marković, Z. M. Vuković, R. V. Hercigonja, A. B. Nastasović and A. E. Onjia, Arabian J. Chem., 2015 DOI:10.1016/j.arabjc.2015.11.010.
- D. D. Maksin, R. V. Hercigonja, M. Ž. Lazarević, M. J. Žunić and A. B. Nastasović, Polym. Bull., 2012, 68, 507–528 CrossRef CAS.
- C. Guo, L. Zhou and J. Lv, Polym. Polym. Compos., 2013, 21, 449–456 CAS.
- K. Can, M. Ozmen and M. Ersoz, Colloids Surf., B, 2009, 71, 154–159 CrossRef CAS PubMed.
- L. Malović, A. Nastasović, Z. Sandić, J. Marković, D. Đorđević and Z. Vuković, J. Mater. Sci., 2007, 42, 3326–3337 CrossRef.
- F. Radovanović, A. Nastasović, T. Tomković, D. Vasiljević-Radović, A. Nešić, S. Veličković and A. Onjia, React. Funct. Polym., 2014, 77, 1–10 CrossRef.
- B. H. Hameed, I. A. W. Tan and A. L. Ahmad, Chem. Eng. J., 2008, 144, 235–244 CrossRef CAS.
- H. Chen, G. Dai, J. Zhao, A. Zhong, J. Wu and H. Yan, J. Hazard. Mater., 2010, 177, 228–236 CrossRef CAS PubMed.
- R. J. Kowalsky and S. Falen, Radiopharmaceuticals in Nuclear Pharmacy and Nuclear Medicine, American Pharmacists Association, Washington DC, USA, 2012 Search PubMed.
- M. Lamson 3rd, C. E. Hotte and R. D. Ice, J. Nucl. Med. Technol., 1976, 4, 21–27 Search PubMed.
- M. I. Khan, T. K. Min, K. Azizli, S. Sufian, H. Ullah and Z. Man, RSC Adv., 2015, 5, 61410–61420 RSC.
- D. K. Singh, V. Kumar, V. K. Singh and S. H. Hasan, RSC Adv., 2016, 6, 56684–56697 RSC.
- M. Žunić, A. Milutinović-Nikolić, A. Nastasović, Z. Vuković, D. Lončarević, I. Vuković, K. Loos, G. Ten Brinke and D. Jovanović, Polym. Bull., 2013, 70, 1805–1818 CrossRef.
- P. A. Webb and C. Orr, Analytical Methods in Fine Particle Technology, Micromeritics Instrument Corp., Norcross, Georgia, United States, 1997 Search PubMed.
- J. Coates, in Encyclopedia of analytical chemistry, ed. R. A. Meyers, John Wiley and Sons Ltd, Chichester, 2006, pp. 10815–10837 Search PubMed.
- A. L. Andrade, D. M. Souza, M. C. Pereira, J. D. Fabris, R. Z. Domingues, U. Federal, D. M. Gerais and B. Horizonte, Nano, 2009, 55, 420–424 CAS.
- S. J. Iyengar, M. Joy, C. K. Ghosh, S. Dey, R. K. Kotnala and S. Ghosh, RSC Adv., 2014, 4, 64919–64929 RSC.
- V. Gutsanu, C. Schitco, G. Lisa and C. Turta, Mater. Chem. Phys., 2011, 130, 853–861 CrossRef CAS.
- G. Bayramoǧlu and M. Yakup Arica, Chem. Eng. J., 2008, 139, 20–28 CrossRef.
- M. Raposo, Q. Ferreira and P. A. Ribeiro, in Modern research and educational topics in microscopy, 2007, pp. 758–769 Search PubMed.
- T. Silk, Q. Hong, J. Tamm and R. G. Compton, Synth. Met., 1998, 93, 65–71 CrossRef CAS.
- B. P. Radoev and B. G. Tenchov, J. Phys. A: Math. Gen., 1987, 20, L159–L162 CrossRef CAS.
- D. Avnir, D. Farin and P. Pfeifer, J. Chem. Phys., 1983, 79, 3566 CrossRef CAS.
- J. D. Miller, S. Veeramasuneni, J. Drelich, M. R. Yalamanchili and G. Yamauchi, Polym. Eng. Sci., 1996, 36, 1849–1855 CAS.
- D. Avnir, D. Farin and P. Pfeifer, J. Chem. Phys., 1983, 79, 3566 CrossRef CAS.
- J. Wang, S. Zheng, Y. Shao, J. Liu, Z. Xu and D. Zhu, J. Colloid Interface Sci., 2010, 349, 293–299 CrossRef CAS PubMed.
- G. M. Rashad, M. R. Mahmoud, A. M. Elewa, E. Metwally and E. A. Saad, J. Radioanal. Nucl. Chem., 2016, 309, 1065–1076 CrossRef CAS.
- M. I. Kandah, R. Shawabkeh and M. A. Al-Zboon, Appl. Surf. Sci., 2006, 253, 821–826 CrossRef CAS.
- A. W. M. Ip, J. P. Barford and G. McKay, Chem. Eng. J., 2010, 157, 434–442 CrossRef CAS.
- Y. Li, Q. Wang, Q. Li, Z. Zhang, L. Zhang and X. Liu, J. Taiwan Inst. Chem. Eng., 2015, 55, 126–132 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |