
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Natural gas regeneration of carbonate melts following SO2 capture from non-ferrous smelter emissions
Nurlan Dosmukhamedovb,
Valery Kaplan *a,
Yerzhan Zholdasbayb,
Ellen Wachtela and
Igor Lubomirskya
*a,
Yerzhan Zholdasbayb,
Ellen Wachtela and
Igor Lubomirskya
aWeizmann Institute of Science, Israel. E-mail: Valery.Kaplan@weizmann.ac.il; Igor.Lubomirsky@weizmann.ac.il
bKazakh National Research Technical University after K. I. Satpayev, Kazakhstan
First published on 18th April 2017
Abstract
Sulfur emission in the form of SO2 in flue gases is the one of the most serious atmospheric pollutants associated with coal combustion and non-ferrous metal production. The carbonate eutectic method for removing SO2 from flue gases at 723–923 K was initially proposed in the 1970's but despite its great efficiency (SO2 concentration in the flue gas after purification reached 0.003 vol%) it could not be implemented by industry due to the complexity of the carbonate melt regeneration stage. Earlier we proposed a method suited to coal-firing power stations where the melt was regenerated using CO as a reducing agent. However, most metallurgical plants do not use coal and therefore lack a large source of CO. Here we propose a method for removing sulfur from the carbonate eutectic melt by purging it with natural gas or a natural gas/air mixture, which are available in the vast majority of metallurgical plants. This reaction leads to the reduction of sulfate to H2S gas that leaves the melt. The experiments we conducted show that nearly complete sulfur removal from the melt is possible at 823 K and that the reaction rate is sufficiently high for a large scale process. The proposed modifications provide solutions to two major problems previously encountered: (i) high temperature corrosion of the reaction cell can be avoided, since a stainless steel cell with high chromium content is stable with respect to the carbonate eutectic melt at 823 K, and (ii) removal of sulfur in the form of H2S provides considerable freedom in choosing the final industrially useful product: either sulfuric acid, using H2S dry combustion, or elemental sulfur via the Claus process. One can foresee that this carbonate melt-based SO2 removal technique may become a practical and economically attractive method for limiting sulfur emission to the atmosphere from non-ferrous metallurgical processing plants.
1 Introduction
Mines and metallurgical plants produce large amounts of waste because the ore constitutes only a small fraction of the total volume of the mined material. In the metallurgical industry, production of Cu, Pb, Ni, and Zn causes the greatest damage to the environment.1–3 Gases emitted with modern smelting technology (10–30 vol% SO2) can be processed to elemental sulfur at a ratio of one ton of sulfur for each ton of metal.4 Production of one ton of non-ferrous metal by traditional, older processes produces flue gas with a relatively low SO2 content (<10 vol%): these gases are directed to the production of sulfuric acid. In fact, existing purification methods can process gases containing as little as 2 vol% SO2. However, flue gas with less than 2 vol% SO2 is often emitted into the atmosphere2 because SO2 scrubbing with an aqueous suspension of powdered calcium carbonate or hydroxide as the neutralizing agent is often considered to be too costly.5Carbonate eutectic-based melt scrubbing was proposed more than four decades ago.6–8 However, despite its ability to scrub SO2 from sulfur-poor flue gases (<2 vol% SO2) down to 0.003 vol%,6 it was deemed impractical for industrial use due to the complexity of the carbonate melt regeneration process. In its original version, the regeneration was multi-stage and required heating to 1173 K, a temperature at which the carbonate melt is too corrosive to contain without damage to reaction crucible. In our previous study, we proposed a process for regeneration of carbonate/sulfate melts after sulfur scrubbing at 753–823 K, specifically suited for coal-fired power plants.9–11 At these temperatures, a stainless steel reaction chamber is stable with respect to the carbonate/sulfate melt, thereby avoiding the problem of chamber corrosion at high temperatures.6–8,12 In this method, the sulfates, formed during SO2 scrubbing, are removed from the melt at 753–823 K by carbon monoxide which is readily produced by incomplete coal combustion in a separate boiler. For non-ferrous metallurgical plants emitting dilute sulfurous flue gas, carbonate regeneration by CO purging is not applicable; these plants do not use coal in their production process. However, natural gas is often used.13–15 Here, we propose, and experimentally verify, a process for regeneration of carbonate melts via purging with natural gas or a mixture of natural gas and air. We find that the final product is hydrogen sulfide gas, which may be then readily converted into industrially useful products.16
2 Interaction of sulfates in the carbonate melt with natural gas or a mixture of natural gas and air: thermodynamics
The absorption reaction of SO2 in the carbonate eutectic melt can be viewed as ion exchange to produce sulfites, followed by temperature dependent disproportionation to sulfides and sulfates, and finally followed by oxidation of the sulfites and sulfides to give sulfates as the final product.9 For preliminary evaluation of the interaction between alkali metal carbonates and sulfates in the mixed ionic (Na, K, Li)2CO3/(Na, K, Li)2SO4 melt, calculations of the Gibbs free energy were performed for reactions (1)–(3), based on standard values for the pure substances17 (Fig. 1a). As quantitative information concerning the high temperature ionization of the melt constituents and resulting electrostatic interactions is not available (but may involve energies of tens of kJ mol−1 (ref. 18)), calculations of the Gibbs free energy of individual chemical reactions of molecules provide estimates which aid in determining which reactions are most likely to take place under given conditions of temperature and pressure.| Li2CO3 (melt) + Na2SO4 (melt) ↔ Li2SO4 (melt) + Na2CO3 (melt) | (1) |
| Li2SO4 (melt) + K2CO3 (melt) ↔ Li2CO3 (melt) + K2SO4 (melt) | (2) |
| K2CO3 (melt) + Na2SO4 (melt) ↔ Na2CO3 (melt) + K2SO4 (melt) | (3) |
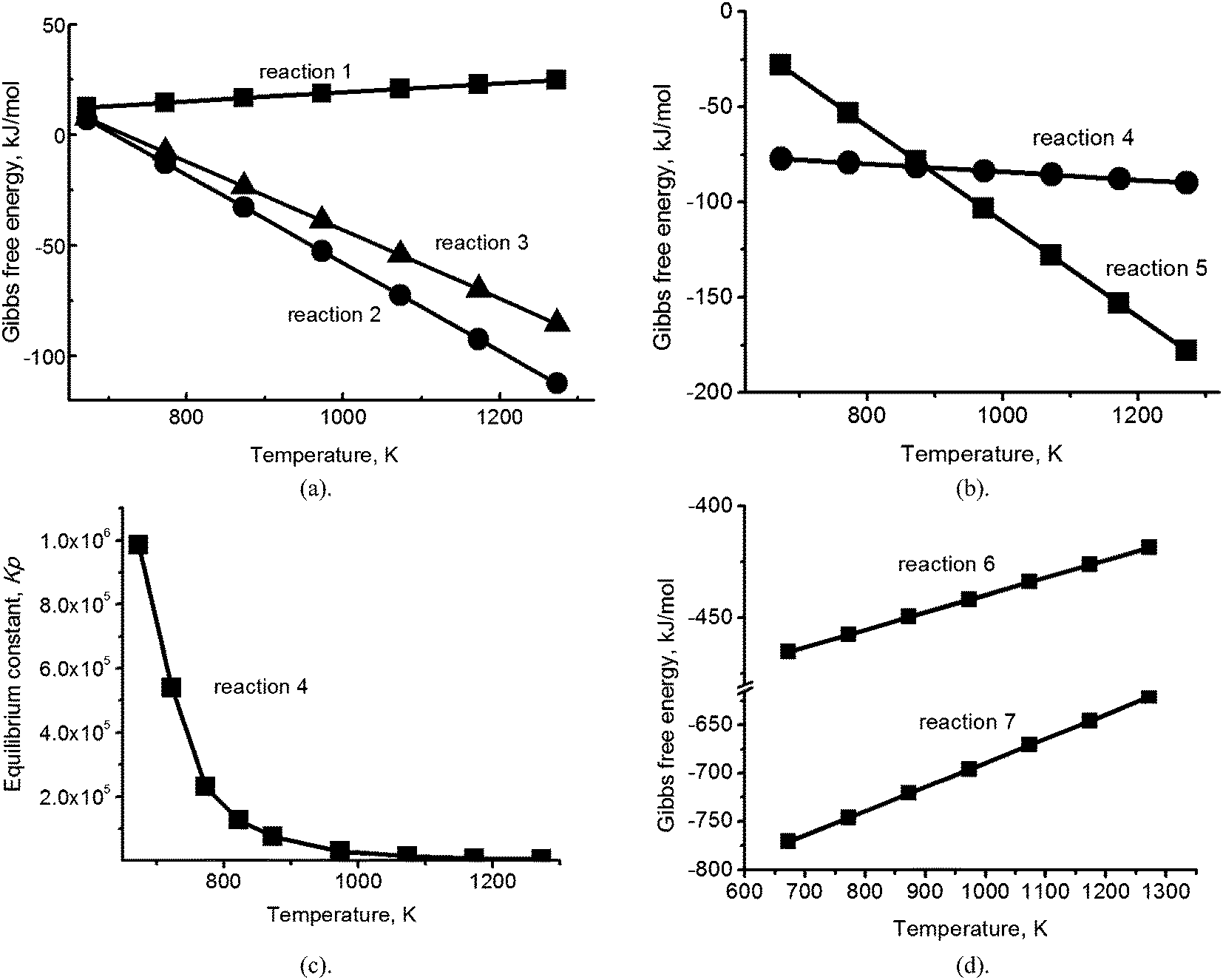 | ||
| Fig. 1 (a) Temperature dependence of the Gibbs free energy for reactions (1)–(3); (b) temperature dependence of the Gibbs free energy for reactions (4) and (5); (c) the dependence on temperature of the equilibrium constant Kp for reaction (4); (d) temperature dependence of the Gibbs free energy for the reaction between hydrogen sulfide and oxygen (reaction (6)) and between potassium sulfide and oxygen (reaction (7)). | ||
The reactions yielding K2SO4 (eqn (2) and (3)) have significant negative Gibbs free energy (∼−25 kJ mol−1; Fig. 1a) in the temperature range of interest, indicating that, of the three sulfates in the melt, K2SO4 is the most stable. Therefore, the thermodynamic calculations of the reduction reactions with natural gas, or a mixture of natural gas and air, were performed only for K2SO4 dissolved in the eutectic (Fig. 1b).
Calculation of the Gibbs free energy for the reduction of sulfates with natural gas (CH4) (Fig. 1b) showed that there are two thermodynamically favorable reaction pathways between 673 K and 1273 K (Fig. 1b): reduction of potassium sulfate to hydrogen sulfide gas (4) and reduction of potassium sulfate to potassium sulfide (eqn (5)), with the former obviously being the more industrially attractive of the two.
| K2SO4 (melt) + CH4 = K2CO3 (melt) +H2O↑ + H2S↑ | (4) |
| K2SO4 (melt) + CH4 = K2S + CO2↑ + 2H2O↑ | (5) |
Fig. 1b shows that below 923 K, the Gibbs free energy values for reaction (4) are a factor of 2–3 more negative than those for reaction (5) and in the temperature range of melt regeneration (753–823 K), the equilibrium constant Kp for reaction (4) is very large: 1–2 × 105 (Fig. 1c). We therefore might expect that the reaction would shift strongly towards the production of hydrogen sulfide. However, we note that a methane molecule is non-polar and therefore is not expected to dissolve in the ionic carbonate melt to any significant extent (cf. 10−7 mol (cm−3 bar−1) for CH4 in the (Na, K) nitrate eutectic19), while the acidic hydrogen sulfide gas molecule is polar (0.97 D) and therefore, has significant solubility.20 Adding air to the natural gas flow could oxidize residual H2S and remove it from the melt by reaction (6), although with SO2 as a product:
| H2S + 1.5O2 = SO2 + H2O↑ | (6) |
The Gibbs free energy of this reaction is very large and negative (Fig. 1d) and it is well known that it proceeds to completion in the gas phase. However, the solubility of H2S and the facile ion exchange of SO2 in the carbonate melt could significantly modify both the rate and extent of the reaction. Considering the effect of air flow on the products of reaction (5), potassium sulfide in the melt (if formed) may, in principle, be oxidized back to sulfate (reaction (7)).
| K2S + 2O2 = K2SO4 (melt) | (7) |
The Gibbs free energy for this reaction is also very large and negative (Fig. 1d), but as for reaction (6), oxidation does not contribute to the removal of sulfur from the melt. Thus, based solely on thermodynamic analysis and solubility considerations, we cannot predict which reactions will take place during the regeneration process or how successful the regeneration will be. In view of this ambiguity, we proceeded to verify the reaction pathways which are actually followed in the mixed alkali carbonate/sulfate melt by performing the experiments described below.
3 Experimental
A ternary carbonate eutectic mixture composed of Li2CO3 (43.5 mol%), Na2CO3 (31.5 mol%), and K2CO3 (25.0 mol%) was prepared by melting together the individual carbonates (98.5% purity) at 823 K in the appropriate quantities to obtain a homogeneous molten carbonate. Potassium sulfate, K2SO4, was introduced into the melt in an amount corresponding to 5.6 wt% sulfur to simulate the melt after SO2 scrubbing of flue gas. Preparation of the alkali metal carbonate/sulfate melt and all subsequent experiments were carried out at a temperature of 823 K in a crucible welded from stainless steel AISI 201 (austenitic chromium–nickel–manganese stainless steel), which is stable with respect to the molten carbonate mixture. Preliminary experiments showed that the stainless steel crucible is stable in the carbonate-sulfate melt up to 1273 K. At that temperature, no corrosion of the crucible was detected during 5 hours. Dimensions of the crucible were: height – 100 mm, diameter – 60 mm, wall thickness – 6 mm. The schematic of the laboratory setup is presented in Fig. 2. Initial weight of the carbonate–sulfate melt in all experiments was 351.4 g. Natural gas (volume%: CH4 – 92.6; C2H6 – 4.07; C3H8 – 1.07; C4H10 – 0.44; C5N12 – 0.42; N2 – 0.9) was used. Natural gas and air were supplied to the melt through separate stainless steel tubes (12H15G9ND chromium–manganese stainless steel) with a diameter of 10 mm. In all experiments, the natural gas was supplied at a rate of 0.38 l min−1. When natural gas was mixed with air, the flow rate of the air was 3.8 l min−1, i.e. ∼0.8 liters of oxygen per min. The duration of each test was 60 minutes. For chemical analysis of the melt, 15–16 g samples were taken with a stainless steel rod. The cooled samples were ground and their composition determined using X-ray fluorescence spectrometry (XRF; PLP-21, AspapGeO, Almaty, Kazakhstan).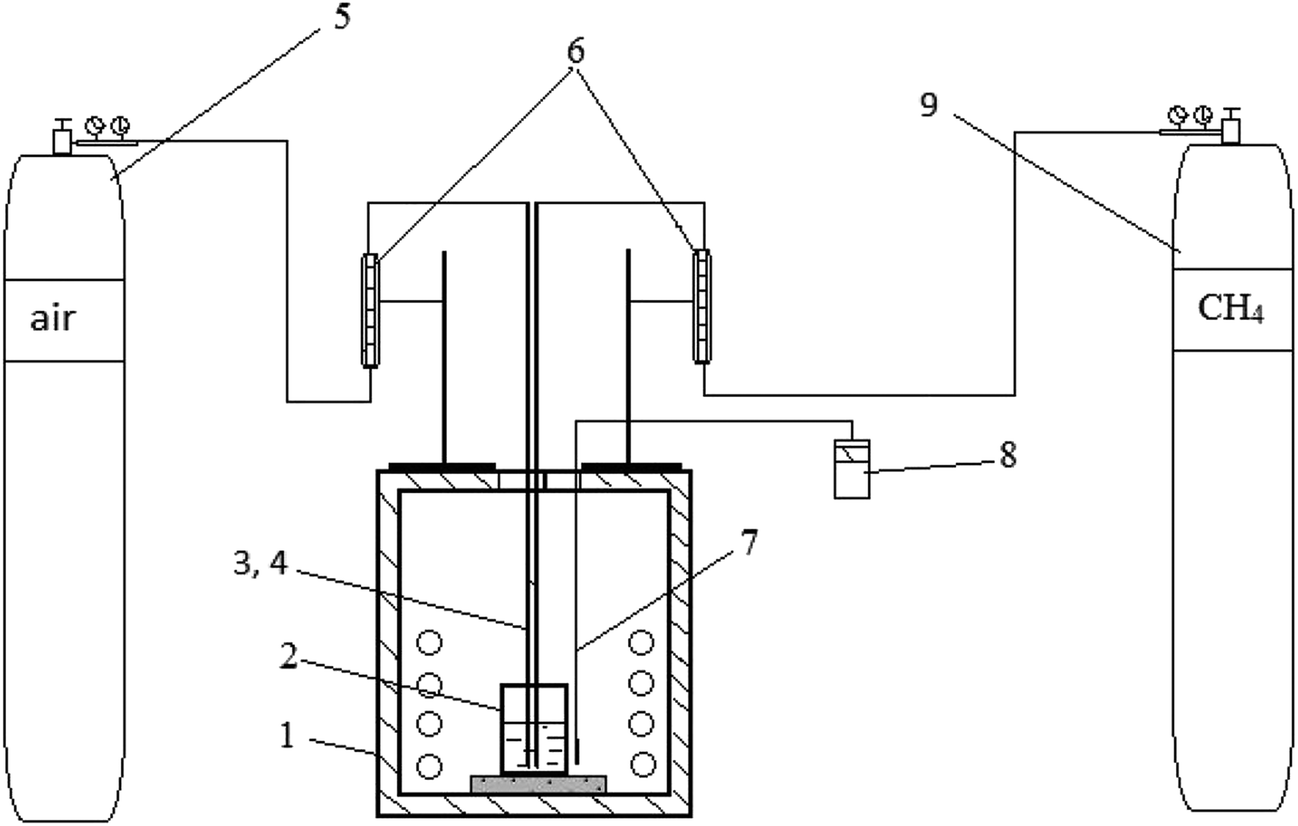 | ||
| Fig. 2 Scheme of the laboratory setup. 1 – furnace; 2 – crucible; 3 and 4 – tubes for separately supplying air and natural gas; 5 – air cylinder; 6 – flowmeters for air and natural gas; 7 – thermocouple type K; 8 – temperature controller; 9 – natural gas cylinder. | ||
In the laboratory, safety considerations do not permit working with a closed gas–melt interface with in situ analysis of the exiting gas (cf.9). The ignition temperature of methane in air is ∼1073 K, whereas ignition of H2S occurs at 573 K.21,22 Therefore, the experiments were carried out in an open crucible, thus preventing quantitative analysis of the exhaust gas.
4 Results and discussion
Bubbling of CH4 into the carbonate eutectic melt produced a strong odor of hydrogen sulfide and the gases exiting the crucible formed a clearly visible flame above the melt surface. XRF measurements were used to monitor the time dependence of the quantity of sulfur remaining in the carbonate melt during the purging process. Short time analyses of cooled melt probes (15–16 g; 3 or 5 min after the initiation of purging) are shown in Table 1 for both CH4 and CH4/air mixtures. One hour of bubbling natural gas alone through the melt was successful in completely removing the sulfur (Table 2). Apparently, metal sulfides do not form in the melt (reaction (5)) since, once formed, they cannot be removed by natural gas. No solid phase was detected. However, under the experimental conditions used, a ≈20% excess of natural gas relative to stoichiometry in reaction (4) was required for sulfur removal (Fig. 3a). Since, as discussed above, natural gas is not expected to readily dissolve in the melt, we suggest that reaction (4) is occurring at the surface of gas bubbles in the melt. The necessity of supplying excess natural gas may derive from the fact that (i) either the rate of reaction (4) is slow; or (ii) reaction (4) is reversible and accumulation of H2S in the melt produces a shift back towards the initial reagents. Since the excess of natural gas required for complete removal of sulfur is small, the accumulation of H2S in the melt (ii), rather than reaction kinetics (i), is the more likely cause. In that case, shifting reaction (4) to the right requires removal of H2S from the reaction zone.| Gas for carbonate melt regenerationa | Time (min) | Elementb,c | Amount (wt%) |
|---|---|---|---|
| a Flow rate of methane 0.38 l min−1; flow rate of air 3.8 l min−1.b Only elements present in amounts approx. ≥0.5 wt% are included in the table.c Li, Na, C and O are not detected by XRF. The most probable sources of Fe, Se and the minute quantities of other metals which are not listed, are the steel crucible, steel gas inlet tubes or the probe rod. | |||
| Methane | 5 | K | 3.73 |
| S | 3.94 | ||
| Se | 1.10 | ||
| Fe | 0.46 | ||
| Methane/air mixture | 3 | K | 3.94 |
| S | 2.81 | ||
| Se | 1.24 | ||
| Fe | 0.47 | ||
| Gas for carbonate melt regeneration | Test duration (min) | Natural gas (liters) | Oxygen (liters) | Sulfur in the melt (wt%) | Sulfur in the melt (grams) | Sulfur extraction yield (%) |
|---|---|---|---|---|---|---|
| CH4 | 0 | 0.0 | — | 5.68 | 20.0 | 0.0 |
| 5 | 1.9 | — | 3.94 | 13.3 | 33.6 | |
| 10 | 3.8 | — | 3.20 | 10.3 | 48.5 | |
| 20 | 7.6 | — | 2.35 | 7.2 | 63.9 | |
| 35 | 13.3 | — | 1.44 | 4.2 | 79.0 | |
| 45 | 17.1 | — | 0.20 | 0.6 | 97.2 | |
| 60 | 22.8 | — | 0.002 | 0.004 | 100.0 | |
| CH4/air | 0 | 0.0 | 0.0 | 5.68 | 20.0 | 0.0 |
| 3 | 1.14 | 2.4 | 2.85 | 9.6 | 52.0 | |
| 6 | 2.28 | 4.8 | 2.31 | 7.4 | 62.8 | |
| 11 | 4.18 | 8.8 | 1.53 | 4.7 | 76.5 | |
| 21 | 7.98 | 16.8 | 1.25 | 3.6 | 81.8 | |
| 36 | 13.68 | 28.8 | 0.04 | 0.1 | 99.4 | |
| 56 | 21.28 | 44.8 | 0.002 | 0.004 | 100.0 |
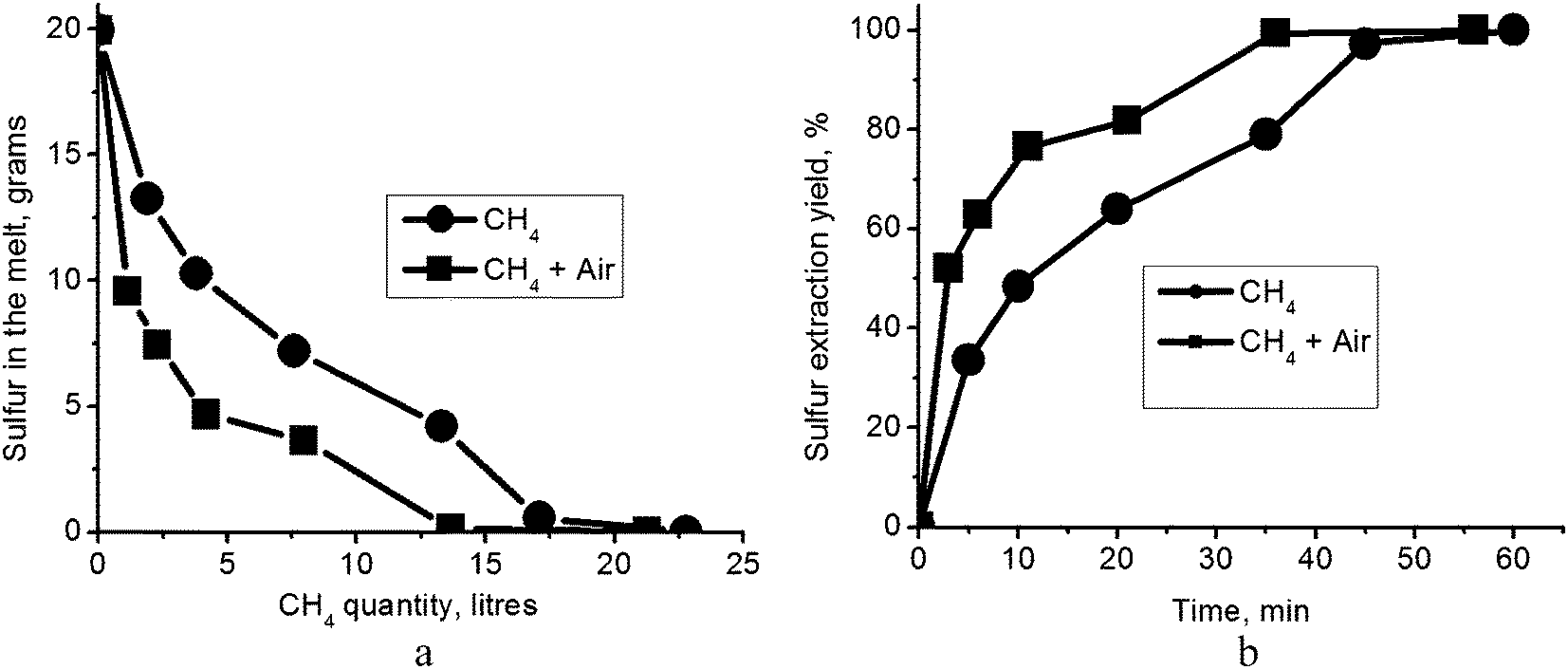 | ||
| Fig. 3 (a) Sulfur content in the melt as a function of the volume of supplied natural gas; (b) sulfur extraction yield as a function of test duration. | ||
Indeed, we found that bubbling air through the melt along with the CH4, and at ten times the flow rate of the CH4, does accelerate removal of sulfur. The quantity of natural gas required for complete regeneration according to reaction (4) then approximates the stoichiometric amount (Fig. 3a). Supported by our earlier observation that oxygen reacting with H2S would only retard sulfur removal (reaction (6)), this finding affirms the contention that the carbonate melt regeneration depends on the rate of removal of H2S by diffusion and convection rather than on reaction kinetics at the bubble/melt interface. It also leads to the suggestion that the proper design of a regeneration chamber, for instance as a shower–tower producing small drops of melt, may even eliminate the necessity for the addition of air; in the case of sufficiently small drops, the total area of the gas bubble/melt interface may be large enough to allow efficient diffusion of H2S out of the melt. We used a linear approximation to calculate the initial (short time) sulfur extraction rate (Fig. 3b). For tests with natural gas flow of 0.38 l min−1 at 823 K, sulfur extraction yield was 6.7% min−1. With the addition of air flow at 3.8 l min−1, the short time extraction yield was sharply accelerated to 17.3% min−1. Given these figures, we find that the rate of carbonate melt regeneration is quite suitable for a large, industrial-scale process.
5 Conclusions
(1) Removal of sulfate from the carbonate melt via bubbling with natural gas can be achieved within the operating temperature range of the flue gas scrubbing tower, i.e. ∼823 K. At these temperatures, high temperature corrosion of the sulfate reduction chamber does not pose a problem for industrial implementation of carbonate melt scrubbing.(2) Removal of sulfur from the carbonate melt by natural gas is a relatively simple, one-stage process, which occurs at a rate that is sufficiently high that the small volume of the melt regeneration chamber can be integrated within the scrubbing tower.
(3) The removal of sulfur in the form of H2S provides considerable freedom in choosing the final product: either sulfuric acid (by H2S dry combustion) or elemental sulfur (by the Claus process), both of which have considerable commercial value (see Table 3 in the Appendix).
(4) One can foresee that carbonate melt-based SO2 removal may become a practical and economical scrubbing method for sulfur-poor flue gases emitted by non-ferrous metals production plants, thereby contributing to the limiting of sulfur emission into the atmosphere.
Appendix
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m3 per day natural gas is needed. In addition, in order to maintain a temperature of 823 K in the regeneration unit, approx. 11000 m3 per hour of natural gas is also required; a similar amount is necessary for maintaining 723 K in the scrubber. For initial melting of the carbonates, 4300–5200 m3 of natural gas are needed (305–370 US $), but this is a one-time expense
000 m3 per day natural gas is needed. In addition, in order to maintain a temperature of 823 K in the regeneration unit, approx. 11000 m3 per hour of natural gas is also required; a similar amount is necessary for maintaining 723 K in the scrubber. For initial melting of the carbonates, 4300–5200 m3 of natural gas are needed (305–370 US $), but this is a one-time expense
| Scrubbing method | Initial investments (US $) | Daily renewal of chemicals (ton) | Daily cost of chemical renewal (US $) | Daily cost of additional natural gas for maintaining the melt temperature in the scrubber and in the regeneration unit (US $ per day) | Output products (ton per day) | Value of output products (US $ per day) |
|---|---|---|---|---|---|---|
| a Prices are obtained from the Alibaba online market.b The price of the natural gas is taken from the US Energy. | ||||||
| Limestone | — | 350 | 43000a |
— | 490 (contaminated gypsum CaSO4·2H2O) | None |
| Eutectic melt (500 ton) and regeneration by natural gas | 350000 (carbonate melt)a, 50000a (molten salt pump for melt recirculation between the scrubber and the regeneration unit) |
112000 m3 natural gas |
8000b | 37488b (528000 m3 of natural gas) |
250 (H2SO4) | 75000a |
Acknowledgements
This work was supported by the Weizmann Institute of Science, via Yeda Ltd. This research is made possible in part by the historic generosity of the Harold Perlman Family.References
- S. Dudka and D. Adriano, Environmental Impacts of Metal Ore Mining and Processing: A Review, J. Environ. Qual., 1995, 26(3), 590–602 CrossRef.
- W. G. Davenport, et al., Extractive Metallurgy of Copper, Pergamon, Oxford, 2002, p. 417 Search PubMed.
- F. K. Crundwell, et al., Extractive Metallurgy of Nickel, Cobalt and Platinum-Group Metals, Elsevier, Oxford, 2011, p. 583 Search PubMed.
- F. Habashi, Copper metallurgy at the crossroads, J. Min. Metall., Sect. B, 2007, 43(1), 1–19 CrossRef CAS.
- P. Nolan. Flue Gas Desulfurization Technologies for Coal-Fired Power Plants, in Coal-Tech 2000 International Conference, Indonesia, Jakarta, 2000 Search PubMed.
- R. A. Mcillroy, G. A. Atwood and C. J. Major, Absorption of Sulfur-Dioxide by Molten Carbonates, Environ. Sci. Technol., 1973, 7(11), 1022–1028 CrossRef PubMed.
- K. A. Moore, Recovery of Sulfur Values from Molten Salt, US Pat., 3867514, 1973.
- S. J. Yosim, et al., Chemistry of Molten Carbonate Process for Sulfur Oxides Removal from Stack Gases, Adv. Chem. Ser., 1973,(127), 174–182 CAS.
- V. Kaplan, E. Wachtel and I. Lubomirsky, Carbonate melt regeneration for efficient capture of SO2 from coal combustion, RSC Adv., 2013, 3(36), 15842–15849 RSC.
- I. Lubomirsky and V. Kaplan, Apparatus and method for removing sulfur dioxide from flue gases, US Pat., 8852540, 2014.
- I. Lubomirsky and V. Kaplan, Apparatus and method for removing sulfur dioxide from flue gases, EP Patent 2723473, 2016.
- T. Krebs and G. M. Nathanson, Reactive collisions of sulfur dioxide with molten carbonates, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(15), 6622–6627 CrossRef CAS PubMed.
- M. E. Schlesinger, et al., Extractive Metallurgy of Copper, Elsevier, Amsterdam, 2011, p. 455 Search PubMed.
- R. J. Sinclair, The Extractive Metallurgy of Zinc, The Australasian Institute of Mining and Metallurgy, Carlton Victoria, Australia, 2005, p. 303 Search PubMed.
- R. J. Sinclair, The Extractive Metallurgy of Lead, The Australasian Institute of Mining and Metallurgy, Carlton Victoria, Australia, 2009, p. 311 Search PubMed.
- N. G. Ashar and K. R. Golwalkar, A Practical Guide to the Manufacture of Sulfuric Acid, Oleums, and Sulfonating Agents, London Springer, New York, 2013, p. 152 Search PubMed.
- E. T. Turkdogan, Physical Chemistry of High Temperature Technology, Academic Press Inc., New York, 1980, p. 462 Search PubMed.
- V. Kaplan, E. Wachtel and I. Lubomirsky, Conditions of stability for (Li2CO3 + Li2O) melts in air, J. Chem. Thermodyn., 2011, 43(11), 1623–1627 CrossRef CAS.
- F. Paniccia and P. G. Zambonin, Interaction of Inert-Gases with Ionic Melts – Solubility of He, Ar, N2, O2 and CH4 in (Na,K)No3 Eutectic Solvent, J. Chem. Soc., Faraday Trans., 1972, 68(11), 2083–2089 RSC.
- M. O. Kawase and M. Otaka, Removal of H2S using molten carbonate at high temperature, Waste Manag., 2013,(33), 2706–2712 CrossRef CAS PubMed.
- A. V. Arseev, Combustion of industrial gases, Metallurgizdat, Moscow, 1952 Search PubMed.
- R. H. Perry, Chemical Engineers Handbook, McGraw-Hill Book Company, New York, Fifth Edition, 1969 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |