DOI:
10.1039/C7RA02760E
(Paper)
RSC Adv., 2017,
7, 27089-27099
Heterostructured Bi2O3/Bi2MoO6 nanocomposites: simple construction and enhanced visible-light photocatalytic performance†
Received
7th March 2017
, Accepted 16th May 2017
First published on 22nd May 2017
Abstract
Heterostructured Bi2O3/Bi2MoO6 nanocomposites with enhanced visible-light photocatalytic activity are successfully constructed by a simple two-step route, employing bismuth nitrate and sodium molybdate as the main raw materials in the presence of polyvinyl pyrrolidone (PVP). The phase and the morphology of the as-prepared composites are characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM), (high-resolution) transmission electron microscopy (TEM/HRTEM), and energy dispersive spectrometry (EDS). It is found that heterostructured Bi2O3/Bi2MoO6 nanocomposites exhibit stronger catalytic activities for the degradation of Rhodamine B (RhB) and 2,4-dinitrophenol (2,4-DNP) under the irradiation of visible light than the single component (Bi2O3 or Bi2MoO6). The above enhanced photocatalytic performance should be attributed to the formation of the p–n junction between the p-type Bi2O3 and n-type Bi2MoO6, which effectively halts the fast recombination of photogenerated electrons and holes. Moreover, the as-obtained Bi2O3/Bi2MoO6 nano-heterostructures also display improved photoelectochemistry performance and high photochemical stability, which is important for conquering the photo-corrosion of photocatalysts. Also, the present work is a useful attempt for the design of new visible-light photocatalytic materials with heterojunction structures.
1. Introduction
Photocatalysis based on semiconductor materials, as a low-cost and sustainable green chemical technology for converting solar energy to chemical energy and completely eliminating organic pollutants, is drawing increasing research interest because of the global energy crisis and environmental pollution.1,2 Over the past decade, various semiconductor-based photocatalysts have been widely investigated, including Bi-based, Ag-based, In-based, TiO2-based and Cu-based ones.3–7 However, some traditional semiconductor photocatalysts such as TiO2 and ZnO can only utilize the UV light below 400 nm due to their large band gaps, which leads to poor photocatalytic efficiency under the irradiation of visible light.8,9 Cu2O is also unsatisfied due to its wide size distribution and the high recombination rate of electron–hole pairs.10 As typical visible-light photocatalysts, Bi-based semiconductor materials, such as Bi2WO6, BiVO4, Bi2MoO6, and BiOX (X = Br, Cl, I),11–14 have been paid much attention in recent years because of their layer structures and high catalytic properties. Among them, Bi2MoO6 is an n-type semiconductor with the layered Aurivillius structure, which consists of [Bi2O2]2+ layers sandwiched between MoO42− slabs.15 Its band gap locates at 2.5–2.8 eV, which largely depends on its structure, shape and components.16 Undoubtedly, Bi2MoO6 is a promising photocatalyst in organic pollutant degradation and water splitting. However, the photocatalytic efficiency of individual Bi2MoO6 is still low due to the rapid recombination of photogenerated electrons and holes. Therefore, it is still necessary to further optimize its photocatalytic performance.17
In order to overcome the intrinsic defects of single component and to improve the photocatalytic performance, different types of Bi2MoO6-based heterostructures with appropriate band positions have been attempted by several groups. For example, heterostructured Bi2MoO6/TiO2 nanocomposites, in which Bi2MoO6 nanosheets grew on the surface of TiO2 nanobelts, were synthesized by Tian and coworkers.18 Against single Bi2MoO6 and TiO2, the improved photocatalytic activity of heterostructures was attributed to the larger specific surface areas and the formation of heterojunction. In particular, the formation of the p–n heterojunction can generate the internal electric field in a semiconductor photocatalyst, which greatly promotes the migration rates of the photogenerated electrons and holes.19 At present, forming a p–n junction in semiconductor photocatalysts has been considered to be the most effective strategy to significantly optimize the photocatalytic activity of photocatalysts. As a p-type semiconductor, Bi2O3 is a fascinating material for the photooxidation of pollutants and the water splitting due to its high refractive index, dielectric permittivity, thermal stability and direct band gap of 2.8 eV.20,21 Obviously, it is probable that the formation of a p–n junction between Bi2MoO6 and Bi2O3 can largely promote the photocatalytic efficiency of the photocatalyst.
To date, few reports are found in the literature on heterostructured Bi2O3/Bi2MoO6 nanocomposites.22,23 For instance, Xu et al. successfully prepared hierarchical β-Bi2O3/Bi2MoO6 heterostructured flower-like microspheres assembled from nanoplates by a facile one-step template-free solvothermal route.22 Hao and coworkers designed an alkali-etching route to synthesize α-Bi2O3/Bi2MoO6 composite nanoflakes using α-Bi2O3 nanoflakes as the original materials.23 The as-obtained heterostructures both exhibited better photocatalytic activities for the degradation of RhB than pure Bi2O3 or Bi2MoO6 samples. In the current work, we successfully construct a novel heterostructured Bi2O3/Bi2MoO6 nanocomposite through a two-step solution route. α-Bi2O3 nanorods were first obtained via the hydrothermal treatment of Bi(NO3)3 in diluted HNO3 solution with PVP. Then, heterostructured Bi2O3/Bi2MoO6 nanocomposites were solvothermally prepared through treating a glycol–water mixed system containing α-Bi2O3 nanorods, Bi(NO3)3 and Na2MoO4. Interestingly, Bi2O3/Bi2MoO6 nanocomposites exhibited obviously different morphology from pure Bi2O3 and Bi2MoO6, and increasing BET surface area. TEM observations uncovered that spherical Bi2MoO6 was distributed on the surface of the thin Bi2O3 nanoflakes to form Bi2O3/Bi2MoO6 heterostructures, which differed from the previous reports.22,23 Furthermore, the photocatalytic experiments showed the as-obtained Bi2O3/Bi2MoO6 nanocomposites presented significantly enhanced visible-light-driven photocatalytic activity for the degradation of RhB and 2,4-dinitrophenol. Compared with some reported works, the as-obtained Bi2O3/Bi2MoO6 nanocomposites also presented better photocatalytic activity.
2. Experimental
2.1 Materials
Bismuth nitrate pentahydrate (Bi(NO3)3·5H2O), sodium molybdate dihydrate (Na2MoO4·2H2O), polyvinylpyrrolidone (PVP; MW 29000), nitric acid (HNO3), sodium hydroxide (NaOH), absolute ethanol, ethylene glycol (EG), rhodamine B (RhB), 2,4-dinitrophenol (2,4-DNP) were used in this work. All chemicals were analytically pure and used without further purification. Deionized water was used throughout experiments.
2.2 Synthesis of Bi2O3 nanorod bundles
In a typical experiment, 1.45 g of Bi(NO3)3·5H2O was firstly dissolved in 30 mL 1.5 mol L−1 of HNO3 under magnetic stirring. Then, 0.1 g of PVP was added under continuously stirring. After the mixed system was continuously stirred for 15 min, the pH of the system was adjusted to 11 with a NaOH solution of 5 mol L−1. Finally, the above mixture was transferred into a 50 mL Teflon-lined stainless steel autoclave and kept at 100 °C for 4 h. The white product was centrifuged, washed with deionized water and ethanol for several times, and dried in a vacuum oven at 60 °C.
2.3 Preparation of heterostructured Bi2O3/Bi2MoO6 nanocomposites
To obtain Bi2O3/Bi2MoO6 nanocomposites, 0.1 g of Bi2O3 nanorods were added into 15 mL EG with 0.485 g Bi(NO3)3·5H2O under magnetic stirring. Subsequently, a 15 mL solution containing 0.121 g of Na2MoO4·2H2O was introduced. After continuously stirring for another 30 min, the system was sealed in a Teflon-lined stainless steel autoclave and heated at 160 °C for 20 h. Subsequently, the system was cooled down to room temperature naturally. The final product was collected and washed several times with deionized water and absolute ethanol, and dried at 60 °C for 4 h.
As a control, pure Bi2MoO6 was also prepared through the above process in the absence of Bi2O3. Simultaneously, a mixture of Bi2O3 and Bi2MoO6, labeled as PM-Bi2O3/Bi2MoO6, was prepared by the simple physical mixing method.
2.4 Visible-light-driven photocatalytic performance
To investigate the photocatalytic activities of as-prepared samples, RhB and 2,4-DNP were used as the pollutants and a 500 W xenon lamp with a cutoff filter at 420 nm as the visible light source. In a typical experiment, 30 mg of catalyst was added to 50 mL of RhB (or 2,4-DNP) solution with a concentration of 10 mg L−1. Then, the system was continuously stirred in the dark for 60 min to establish an adsorption–desorption equilibrium between the catalyst and the pollutant. Finally, the system was irradiated under a high-pressure xenon lamp equipped with a UV filter for desired durations (during irradiation, the system was ceaselessly stirred). The concentration change of the pollutant was monitored with a Metash 6100 UV-vis absorption spectrophotometer (Shanghai).
2.5 Photoelectrochemical measurements
Photocurrent density was measured in a standard three-electrode cell. The product deposited on the ITO glass was used as the working electrode, a platinum coil (0.5 mm × 4 cm) and an Ag/AgCl electrode as the counter and the reference electrodes, respectively. A 0.5 M of Na2SO4 solution that was purged with nitrogen for 10 min prior to the measurement was selected as the electrolyte. The working electrode was prepared as follows: 2 mg of the as-prepared samples were firstly dispersed into 1 mL of twice distilled water under ultrasonication. Next, 100 μL of the sample suspension was uniformly dropped onto the surface of the ITO glass. Afterwards, the ITO glasses were heated at 60 °C for 2 h to evaporate the solvent. The photoelectrochemical properties were monitored with a computer-controlled CHI660D electrochemical workstation under ambient conditions through illumination of a 500 W Xe lamp with a cut-off filter (λ = 420 nm).
2.6 Characterization
The X-ray powder diffraction patterns of the products were carried out on a Bruker D8 Advance X-ray diffractometer equipped with Cu Kα radiation (λ = 0.154060 nm), employing a scanning rate of 0.2 °s−1 and 2θ ranges from 10° to 80°. Transmission electron microscopy (TEM) images were carried out on a Hitachi HT7700 transmission electron microscope, employing an accelerating voltage of 100 kV. High resolution transmission electron microscopy (HRTEM) images were obtained on an FEI Tacnai G2 transmission electron microscope, employing an accelerating voltage of 200 kV. Field emission scanning electron microscopy (FESEM) images and energy dispersive spectrometry (EDS) analysis were obtained on a Hitachi S-4800 field emission scanning electron microscope, employing an accelerating voltage of 5 kV and 15 kV, respectively. UV-vis absorption spectra were recorded on a Metash 6100 UV-vis absorption spectrophotometer (Shanghai). The solid UV-vis diffuse reflection was performed on Shimadzu UV-2450 spectrophotometer. X-ray photoelectron spectroscopy (XPS) of the product was obtained on a Thermo ESCALAB 250 instrument, employing monochromic Al Kα (hν = 1486.6 eV) at a power of 150 W. Photoluminescence (PL) spectra were recorded on a FLSP 920 with a Xe lamp at room temperature, employing the excitation wavelength of 335 nm from a He–Cd laser. The Brunauer–Emmett–Teller (BET) surface areas of the product were measured on a Micromeritics ASAP 2460 analyzer.
3. Results and discussion
3.1 Structure and morphology characterization
Fig. 1a shows the XRD patterns of as-prepared Bi2O3, Bi2MoO6, and Bi2O3/Bi2MoO6. According to the diffraction patterns, pure Bi2O3 and Bi2MoO6 prepared in the present work can be separately determined as the monoclinic α-Bi2O3 phase (PDF no. 76-1730) and the orthorhombic Bi2MoO6 one (PDF no. 72-1524). The diffraction peaks of the as-prepared Bi2O3/Bi2MoO6 nanocomposites are very similar to those of pure Bi2MoO6, but they markedly widen against those of pure Bi2MoO6. This implies that the final product possibly owns smaller particle size. Furthermore, the main diffraction peak situations of Bi2O3 and Bi2MoO6 are very close. Their overlapping each other is maybe another reason causing the wideness of diffraction peaks. Fig. 1b gives the EDX analysis the final nanocomposites. Only O, Bi and Mo elements are detected, and the atomic ratio of Bi/Mo is calculated to be ∼3.25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which is bigger than 2:1 in Bi2MoO6. This fact implies the presence of Bi2O3 in the final product.
:1, which is bigger than 2:1 in Bi2MoO6. This fact implies the presence of Bi2O3 in the final product.
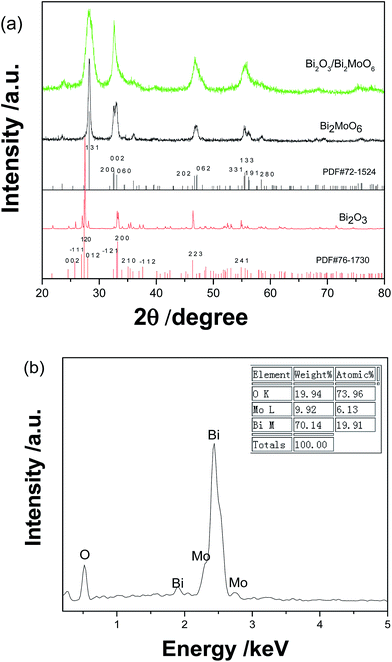 |
| | Fig. 1 (a) XRD patterns of Bi2O3, Bi2MoO6 and Bi2O3/Bi2MoO6 nanocomposites and (b) the EDS analysis of Bi2O3/Bi2MoO6 nanocomposites. | |
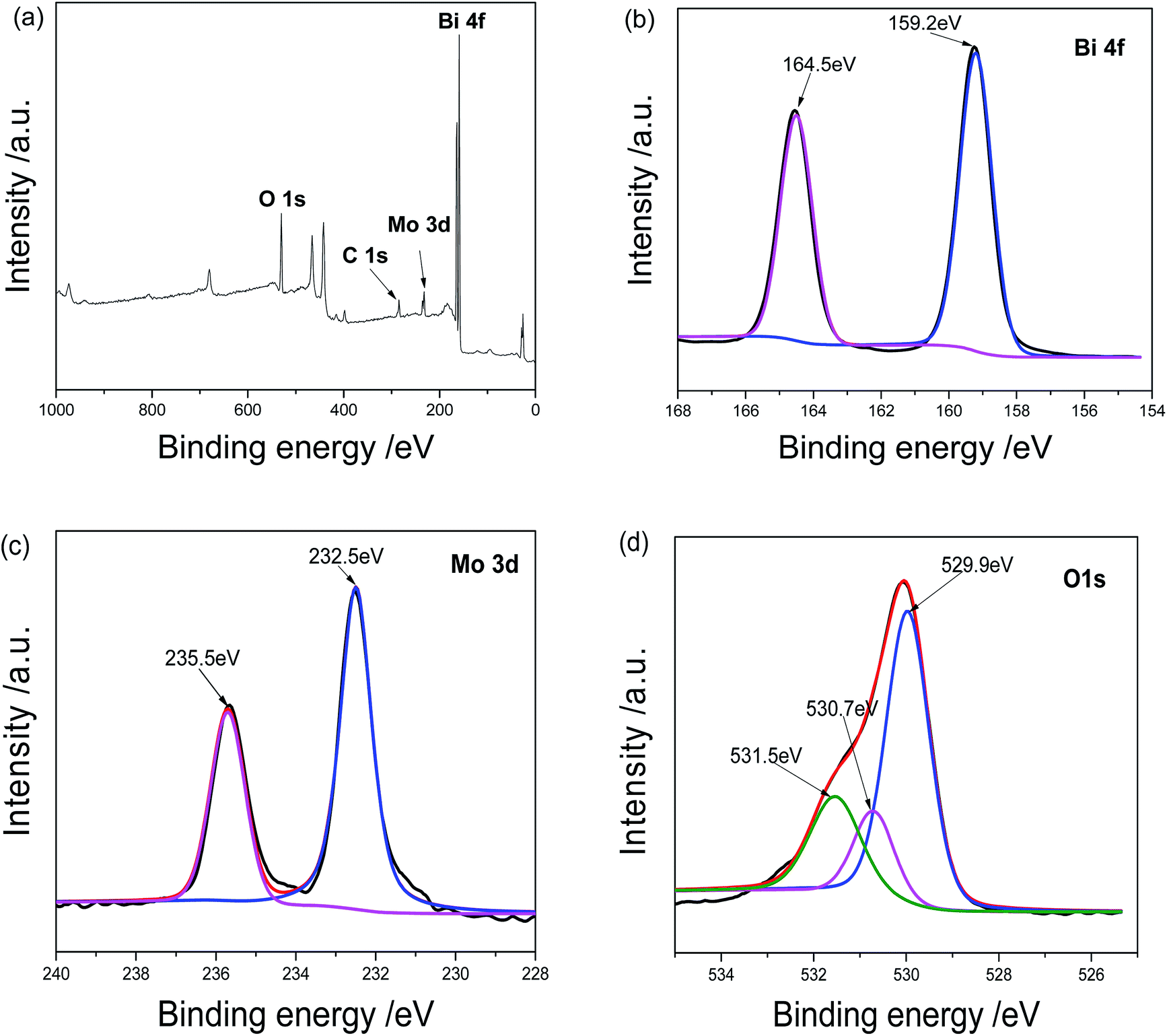
Further evidences came from the XPS analysis of the product. As shown in Fig. 2a, the sample contains O, C, Mo and Bi elements. The C peak should be ascribed to the XPS instrument itself. The Bi 4f spectrum is displayed in Fig. 2b. The peaks centered at 164.5 and 159.2 eV are assigned to Bi 4f5/2 and Bi 4f7/2 of Bi3+, respectively.16 However, two Bi 4f peaks in pure Bi2O3 locate at 164.2 and 158.6 eV and the values in pure Bi2MoO4 164.6 and 159.3 eV.23 The above facts confirm the formation of Bi2O3/Bi2MoO6 composites. In Mo 3d spectrum shown in Fig. 2c, the peaks at 235.5 and 232.5 eV are ascribed to Mo 3d3/2 and Mo 3d5/2 of Bi(VI), respectively.16 The O 1s spectrum contains three peaks (see Fig. 2d). The peaks at 529.9 and 530.7 eV are assigned to the crystal lattice oxygen of Bi2MoO4 and Bi2O3; and the one at 531.5 eV should be ascribed to the adsorbed oxygen.24
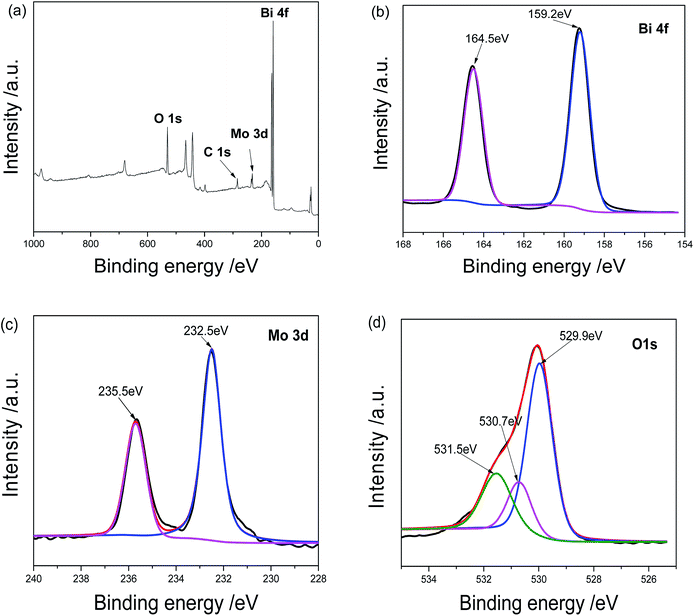 |
| | Fig. 2 XPS analyses of Bi2O3/Bi2MoO6 nanocomposites: (a) survey spectrum, (b) Bi 4f, (c) Mo 3d and (d) O 1s. | |
Fig. 3 depicts the typical FESEM images of as-prepared Bi2O3, Bi2MoO6 and Bi2O3/Bi2MoO6 composite. Pure Bi2O3 samples prepared by the hydrothermal route are composed of abundant nanorod bundles (see Fig. 3a and its inset). Pure Bi2MoO6 samples obtained by the solvothermal approach comprise of plentiful irregular nanoplates with the length of ∼50 nm (Fig. 3b). However, the final Bi2O3/Bi2MoO6 composites present flake-like nanostructures and the arrangement of the nanoflakes are unordered (see Fig. 3c). Obviously, the morphology of the final composite is different from the nanorod bundles of Bi2O3 and the nanoplates of Bi2MoO6. Furthermore, FESEM observations showed that only micro-rods were obtained when no extra Bi(NO3)3 was added into the system with Bi2O3 and Na2MoO4 (see Fig. 3d). TEM observations further proved the above difference of the morphology. As shown in Fig. 4a, the nanorod bundles of Bi2O3 can be readily distinguished. The lengths of nanorods are polydispersed and the diameter of single nanorod is ∼20 nm. Similarly, the plate-like nanostructures of Bi2MoO6 are also clearly visible (see Fig. 4b), which is in good agreement with the result of FESEM. Fig. 4c displays a representative TEM image of the final composite. Abundant nanoparticles with the mean size of ∼10 nm are distributed on the surfaces of the nanoflakes. Comparing with the morphologies of Bi2O3 and Bi2MoO6, one can find that the final composite has obviously different morphology. Combining the results of Fig. 3d and b, one can find that Bi2O3 nanorods and Bi(NO3)3 are necessary in the formation of the final composites. The shape change of Bi2O3 nanorod bundles implies than nanorods take part in the reaction of forming Bi2MoO6 and change the nucleation environment of Bi2MoO6. Thus, the final composite owns markedly different morphology from single component. Fig. 4d shows a HRTEM image of the composite. Owing to different contrasts, one can find that several near-spherical nanoparticles strew the surface of a thin nanoflake. The d-spacing of the thin nanoflake is measured to 0.32 nm, which is very close to 0.32434 nm of the (120) plane of α-Bi2O3. The distance between planes of a spherical nanoparticle is ∼0.316 nm, which is close to 0.31562 nm of the (131) plane of orthorhombic Bi2MoO6. The above facts confirm the formation of Bi2O3/Bi2MoO6 composites. Furthermore, the selected area electron diffraction (SAED) pattern shown in the inset of Fig. 4d presents concentric rings, implying the polycrystalline nature of the composite.
 |
| | Fig. 3 SEM images of as-prepared Bi2O3 nanorod bundles (a), Bi2MoO6 nanoplates (b), Bi2O3/Bi2MoO6 nanocomposites (c) and the product from the system with Bi2O3 nanorods and Na2MoO4 (d). | |
 |
| | Fig. 4 TEM images of (a) Bi2O3 nanorod bundles, (b) Bi2MoO6 nanoplates, and (c) Bi2O3/Bi2MoO6 nanocomposites; (d) HRTEM image and SEAD pattern of Bi2O3/Bi2MoO6 nanocomposites. | |
The above shape change strongly affected the surface areas and the pore volumes of the products. Fig. 5 depicts the N2 adsorption–desorption isotherms and the pore size distributions of the as-obtained products. The BET surface areas of Bi2O3, Bi2MoO6 and Bi2O3/Bi2MoO6 composites are 0.4288 m2 g−1, 18.97 m2 g−1 and 54.32 m2 g−1 in turn. The pore volumes of Bi2MoO6 and Bi2O3/Bi2MoO6 composites are calculated to be 0.22 cm3 g−1 and 0.49 cm3 g−1, respectively. However, no pore volume of Bi2O3 was obtained in BET measurement since its surface area was too small to meet BJH criteria. Obviously, the Bi2O3/Bi2MoO6 composites bear the larger BET surface area and pore volume than Bi2MoO6 nanoplates. Furthermore, the Bi2O3/Bi2MoO6 composites also presented different optical properties from pure Bi2O3 and Bi2MoO6. Fig. 6a exhibits UV-vis diffuse-reflectance spectra (DRS) of Bi2O3, Bi2MoO6 and Bi2O3/Bi2MoO6 composites. The absorption edges of three samples are in turn at 443, 477 and 461 nm. Namely, the absorption edge of the composite locates between those of two components. The PL spectra of three samples are displayed in Fig. 6b, from which one can easily find that the Bi2O3/Bi2MoO6 composite bears the weakest luminescent emission under the excitation of 310 nm UV light. The above facts show that the fast recombination of photo-induced electron–hole pairs in pure Bi2O3 and Bi2MoO6 has been efficiently restrained after forming the Bi2O3/Bi2MoO6 composite. Obviously, this is favorable for the enhancement of the photocatalytic performance.
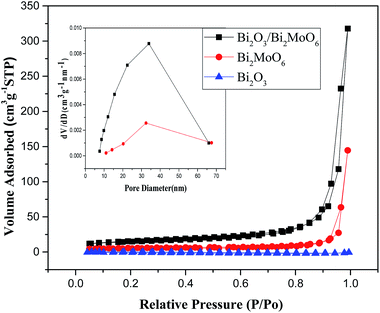 |
| | Fig. 5 N2 adsorption–desorption isotherms and corresponding pore size distributions of the as-obtained products. | |
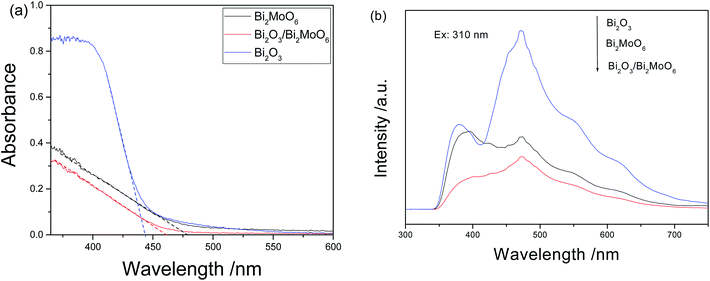 |
| | Fig. 6 (a) UV-vis diffuse reflectance and (b) PL spectra of Bi2O3, Bi2MoO6 and Bi2O3/Bi2MoO6 nanocomposites. | |
3.2 Photocatalytic performance
To investigate the photocatalytic activity of the as-prepared Bi2O3/Bi2MoO6 composites, toxic organic compounds such as 2,4-dinitrophenol (2,4-DNP) and RhB were employed as the pollutants. For comparison, Bi2O3 nanorod bundles, Bi2MoO6 nanoplates and their physical mixture (PM-Bi2O3/Bi2MoO6) were used as photocatalysts under the same experimental conditions, too. Fig. 7a depicts the UV-vis absorption spectra of 2,4-DNP solution irradiated by the visible light for various durations in the presence of Bi2O3/Bi2MoO6 composites. Markedly, the peak intensity of 2,4-DNP gradually decreases with the prolonging of the irradiation time, indicating good photodegradation activity of the as-obtained composites. Fig. 7b depicts the correlation curves between the concentration changes of 2,4-DNP and the irradiation durations in the presences of various photocatalysts. After irradiating for 80 min, the degradation efficiencies of four catalysts for 2,4-DNP are in turn 27.6% for Bi2O3, 4.3% for Bi2MoO6, 63.9% for PM-Bi2O3/Bi2MoO6 and 88.9% for Bi2O3/Bi2MoO6 composites. Although the PM-Bi2O3/Bi2MoO6 presents the appreciably higher photocatalytic activity than pure Bi2O3 and Bi2MoO6, its catalytic ability is still much lower than that of Bi2O3/Bi2MoO6 composites. Obviously, the highly efficient separation of photogenerated carriers in the composite is main reason to promote the photocatalytic activity of the catalyst. Additionally, the high surface area and the big pore volume of the composite are also favorable for the adsorption of organic molecules and the acceptance of the incident light. Hence, the Bi2O3/Bi2MoO6 composites present the enhanced photocatalytic performance.
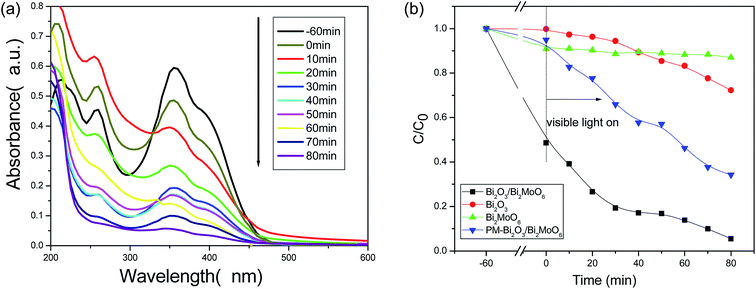 |
| | Fig. 7 (a) The absorption spectra of 2,4-DNP solution solution after irradiated by the visible light for various durations under the presence of 30 mg Bi2O3/Bi2MoO6 nanocomposites; (b) the concentration–time curves of 2,4-DNP irradiated by the visible light in the presence of 30 mg various catalysts. | |
Similar experimental phenomena were also found on the degradation of RhB dye. As seen from Fig. 8a, under the presence of 30 mg Bi2O3/Bi2MoO6 nanocomposites, the maximal absorbance (λmax) gradually shifts from the initial 554 nm towards shorter wavelength and finally to 496 nm with the expansion of the irradiation time. This is in good agreement with the degradation of RhB catalyzed by BiVO4.25 The blue-shift of absorption band should be mainly attributed to the stepwise removal of the N-ethyl group during the degradation of RhB. After the de-ethylated process is fully completed, the produced rhodamine is further decomposed due to the gradual destruction of the conjugated structure.25 Finally, the color of the solution is faded (see the inset in 8a). Fig. 8b shows the concentration–time curves of RhB dye in the presences of various catalysts. When Bi2O3/Bi2MoO6 composites were used as the photocatalyst, RhB was fully degraded after irradiation for 60 min under the visible light. The degradation efficiency is far higher than 37.7% of Bi2O3, 45.9% of Bi2MoO6 and 70.8% of PM-Bi2O3/Bi2MoO6. Moreover, compared with some previous reports (Table 1), the present Bi2O3/Bi2MoO6 composites also presented better photocatalytic activity for the photodegradation of RhB.
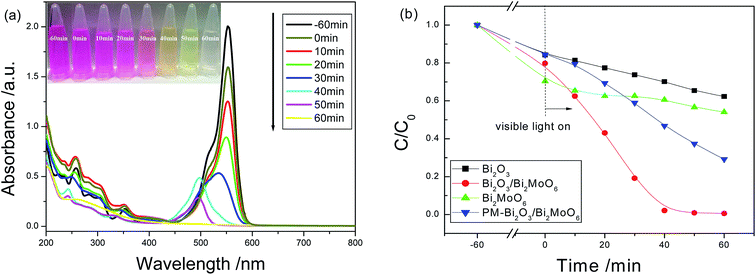 |
| | Fig. 8 (a) The absorption spectra of RhB solution irradiated by the visible light for various durations under the presence of Bi2O3/Bi2MoO6 composites; (b) the concentration–time curves of RhB solution irradiated by the visible light in the presences of various catalysts. The inset in (a) is the photographs of RhB solution irradiated by the visible light for various durations under the presence of Bi2O3/Bi2MoO6 composites. | |
Table 1 Photocatalytic efficiency of RhB over various Bi-based ternary oxides photocatalysts under visible light irradiation
| Photocatalyst |
CRhB |
Light source |
Photocatalytic efficiency |
Ref. |
| Bi2O3/Bi2MoO6 (30 mg) |
10 mg L−1 |
500 W Xe lamp |
100% 60 min |
This work |
| Bi2MoO6/ZnTiO3 (50 mg) |
10 mg L−1 |
150 W Xe lamp |
82% 240 min |
26 |
| Fe3O4/SiO2/Bi2MoO6 (100 mg) |
10 mg L−1 |
300 W Xe lamp |
100% 120 min |
27 |
| BiIO4/Bi2MoO6 (50 mg) |
0.01 mM |
1000 W Xe lamp |
55% 300 min |
28 |
| Bi2MoO6/BiOCl (50 mg) |
10 mg L−1 |
500 W Xe lamp |
100% 300 min |
29 |
| Bi2WO6/ZnWO4 (20 mg) |
10 mg L−1 |
500 W mercury lamp |
80%, 80 min |
30 |
| Ag–graphene–Bi2WO6 (50 mg) |
0.01 mM |
350 W Xe lamp |
100% 90 min |
31 |
| C3N4@Ag–Bi2WO6 (50 mg) |
10 mg L−1 |
350 W Xe lamp |
100% 90 min |
32 |
| CuO/BiVO4 (200 mg) |
10 mg L−1 |
300 W Xe lamp |
100% 150 min |
33 |
| BiVO4/BiOI (30 mg) |
0.02 mM |
500 W Xe lamp |
100% 300 min |
19 |
To ascertain the mineralization degree of organic species in the photodegradation of RhB by Bi2O3/Bi2MoO6 composites, the total organic carbon (TOC) value was measured. As shown in Fig. S1,† the TOC value slowly decreases with the increase of irradiation time from 0–100 min; and then rapidly decreases, indicating that RhB is continuously mineralized. After irradiating for 160 min, the TOC value changes from 14.951 to 6.157 mg L−1. The mineralization ratio of ∼59% is reached.
Furthermore, the stability and reusability of the present photocatalyst were also investigated. As shown in Fig. 9, the photocatalytic efficiency of Bi2O3/Bi2MoO6 composites after 5 cycles under the same conditions still reaches ∼91%, implying the good stability of the present photocatalyst. The above facts clearly indicate that the as-obtained Bi2O3/Bi2MoO6 composites are outstanding photocatalyst and have potential applications in the field of environmental treatment and protection.
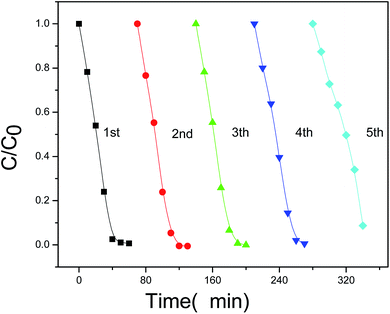 |
| | Fig. 9 The photocatalytic efficiency of Bi2O3/Bi2MoO6 composites to RhB dye after five cycles. | |
To faithfully demonstrate the stability of the present catalyst, we suppose that the photodegradation process of RhB by Bi2O3/Bi2MoO6 composites in each cycle is the pseudo-first-order model. Thus, the rate constant, k, could be obtained according to the below equation:
where,
C0 and
C are the concentrations of the dye solution at time 0 and
t, respectively. Fig. S2a
† exhibits the plots of the rate constant
vs. the irradiation time for five cycles. The
k value is calculated from the slope of the plots of ln(
C/
C0)
vs. t. The
k value has marked decrease after 4 cycles. Fig. S2b
† gives the histogram of the kinetic constant
vs. the cycle number, which faithfully shows the above change of the
k value.
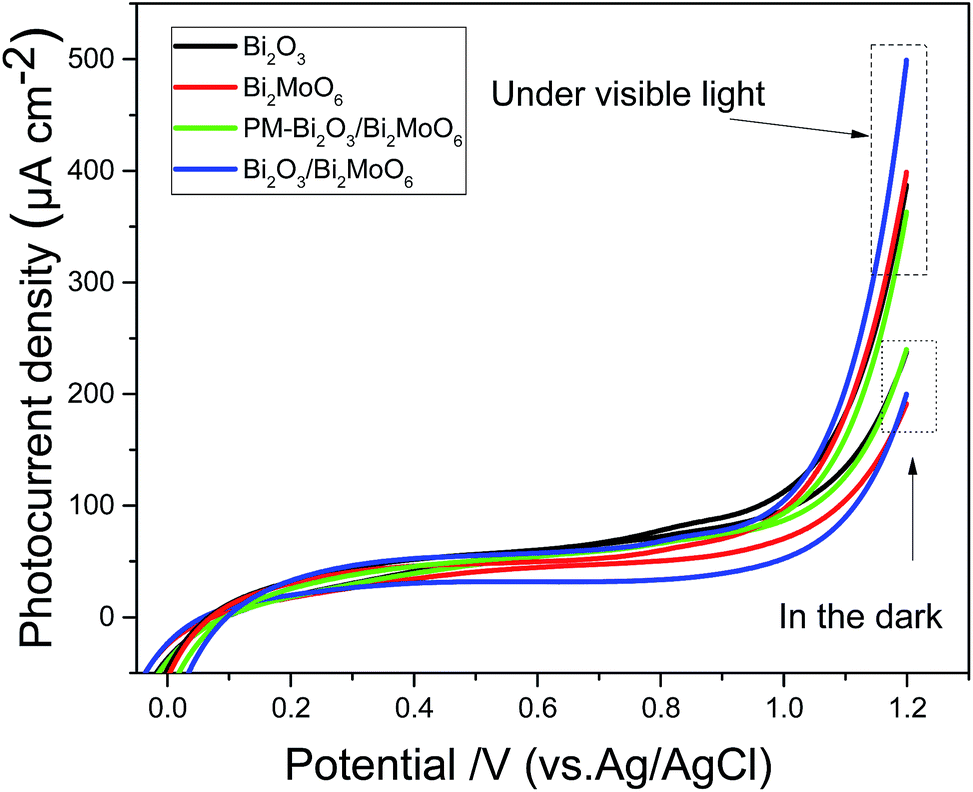
In the PL spectra of the samples shown in Fig. 6b, Bi2O3/Bi2MoO6 composites own the weakest emission intensity, implying the efficient charge separation of the photocatalyst. To further evaluate the charge separation efficiencies of different photocatalysts, the photo-current densities of various catalysts were measured before and after visible light illumination. As shown in Fig. 10, all of the catalysts exhibit small current densities in the dark. However, under visible light irradiation the photocurrent densities of all catalysts markedly increase. At 1.2 V, for example, the photocurrent density changes from 237 to 388 μA cm−2 for Bi2O3, from 194 to 400 μA cm−2 for Bi2MoO6, from 243 to 366 μA cm−2 for PM-Bi2O3/Bi2MoO6 and from 200 to 500 μA cm−2 for Bi2O3/Bi2MoO6. Among them, the Bi2O3/Bi2MoO6 composites present the highest photocurrent density and the biggest increase. Mentioned by the previous text, Bi2O3/Bi2MoO6 composites have the biggest surface area, which is beneficial for light harvesting.34 Thus, more electron–hole pairs are generated in Bi2O3/Bi2MoO6 composites. Simultaneously, the recombination of the produced electron–hole pairs can be effectively suppressed, resulting in the marked increase of the photocurrent density of Bi2O3/Bi2MoO6 composites. Moreover, it is worthy pointing out that PM-Bi2O3/Bi2MoO6 has smaller photocurrent density than Bi2O3 and Bi2MoO6, but it presents stronger photocatalytic activity. To ascertain the reason causing the above contradiction, the BET surface area of PM-Bi2O3/Bi2MoO6 was investigated. Fig. S3† depicts N2 adsorption–desorption isotherm and corresponding pore size distributions of PM-Bi2O3/Bi2MoO6. The BET surface area of PM-Bi2O3/Bi2MoO6 was 10.45 m2 g−1, lower than 18.97 m2 g−1 of Bi2MoO6. Thus, PM-Bi2O3/Bi2MoO6 should absorb fewer incident photons than Bi2MoO6, which resulted in the lower photocurrent density. In the photocatalytic degradation, however, PM-Bi2O3/Bi2MoO6 owned stronger ability to restrain the fast recombination between the photoelectrons and poles than single component. As a result, PM-Bi2O3/Bi2MoO6 exhibited better photocatalytic activity.35
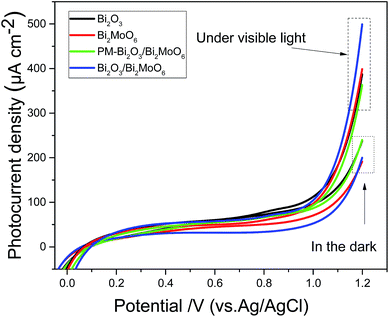 |
| | Fig. 10 Photocurrent densities of Bi2O3, Bi2MoO6, PM-Bi2O3/Bi2MoO6 and Bi2O3/Bi2MoO6 composites measured under a 500 W Xe lamp with a cut-off filter (λ > 420 nm) in a 0.5 mol L−1 Na2SO4 solution. | |
3.3 Photocatalytic mechanism
In the present Bi2O3/Bi2MoO6–RhB system, to ascertain the definite oxidative species during the photodegradation process, some scavengers including iso-propanol (IPA), ethylene diamine tetraacetic acid disodium salt (EDTA-2Na), and benzoquinone (BQ) were employed, respectively. As seen from Fig. 11, when IPA was used as the scavenger, the photocatalytic degradation of RhB dye was only slightly affected, implying that ˙OH group hardly had contribution to the RhB oxidation. When BQ was selected as the scavenger, the photodegradation of RhB dye was markedly inhibited, indicating that ˙O2− played the important role in the photodegradation of RhB. While EDTA-2Na was employed as the scavenger, the photocatalytic degradation of RhB dye was strongly restrained, showing that h+ played the crucial role in the photodegradation of RhB.
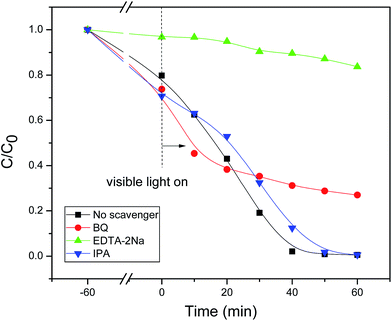 |
| | Fig. 11 Photocatalytic degradation efficiency of RhB solutions with various scavengers in the presence of 30 mg Bi2O3/Bi2MoO6 composites. | |
Usually, when p-type and n-type semiconductors contact each other, carriers will diffuse due to the concentration gradient of carries between p–n junctions. Holes transfer from the p-type semiconductor to the n-type semiconductor side, and electrons enter the p-type side from the n-type one. Hence, the region in the n-type semiconductor that loses electrons will have a net positive charge; and the region in the p-type semiconductor that accepts the electrons will have a net negative charge. At the interface of the semiconductors, the charge redistribution creates an internal electric field in the space charge region from the n-type side to the p-type side.17 The direction of the internal electric field is opposite to the direction of the diffusion current, so when the redistribution of mobile electrons or holes will reach a balance, which is called as the Fermi energy level (EF) equilibrium state between p–n junctions. In the present work, the optical band gap of pure Bi2O3 and Bi2MoO6 could be calculated from the absorption spectra shown in Fig. 6a through the equation of αhν = A(hν − Eg) n/2. Where α, h, ν, A and Eg are in turn the absorption coefficient, Planck constant, light frequency, constant and band gap. And n depends on the characteristics of transition in a semiconductor (n = 4 for indirect band gap and = 1 for direct band gap). Here, n = 4 for Bi2O3 and n = 1 for Bi2MoO6.36 Fig. 12 depicts the plots of (αhν)2/n ∼ hν of Bi2O3 and Bi2MoO6. The band gaps of Bi2O3 and Bi2MoO6 are 2.67 and 2.80 eV, respectively.
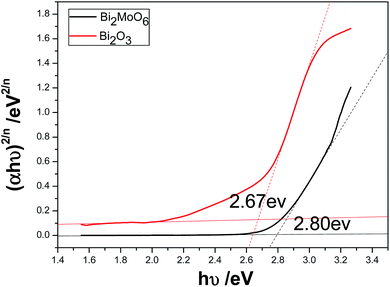 |
| | Fig. 12 The plots of (αhν)2/n vs. hν of Bi2O3 and Bi2MoO6. | |
Thus, the positions of conduction and valence band can be determined by the following equation:
where,
ECB is the CB edge potential.
X is the electronegativity of the semiconductor, which is the geometric mean of the electronegativity of the constituent atoms. The
X values for Bi
2O
3 and Bi
2MoO
6 are ∼5.99 and 5.55 eV,
1,24 respectively. The
Ee value is the energy of free electrons on the hydrogen scale (∼4.5 eV). According to the
eqn (2) and
(3),
ECB and
EVB values of Bi
2MoO
6 are determined to be −0.35 eV and 2.45 eV; and those of pure Bi
2O
3 are 0.16 eV and 2.83 eV. The energy band structures of Bi
2O
3 and Bi
2MoO
6 are shown in the left of
Fig. 13. The difference of CB and VB positions between Bi
2O
3 and Bi
2MoO
6 will facilitate the move of the photogenerated electrons and holes in opposite directions, which can effectively inhibit their recombination. Moreover, as a p-type semiconductor, the
EF of Bi
2O
3 is on the verge of the valence band, whereas the
EF of n-type Bi
2MoO
6 is close to the conduction band. Before the hybrid of p-type Bi
2O
3 with n-type Bi
2MoO
6, the Fermi level of Bi
2O
3 lower than that of the Bi
2MoO
6 (see the left in
Fig. 13). After the hybrid of Bi
2MoO
6 and Bi
2O
3, the
EF of Bi
2O
3 shifts upward, whereas the
EF of Bi
2MoO
6 shifts downward until an equilibrium state is formed. Eventually, the conduction band bottom of Bi
2MoO
6 becomes lower in energy than that of Bi
2O
3, resulting in the formation of a p–n junction (see the right in
Fig. 13). Under the visible light irradiation, Bi
2O
3 with a narrow band gap is excited easily, and generates photoelectrons and holes. The electrons in the CB of Bi
2O
3 can easily migrate to that of Bi
2MoO
6 and meanwhile, the produced holes remain in the VB of Bi
2O
3. Since the CB potential (−0.35 eV) of Bi
2MoO
6 is more negative than that of E
0 (O
2/˙O
2−) (−0.046 eV
vs. NHE),
37 the active species ˙O
2− can be produced due to the reduction of adsorbed O
2 by photogenerated electrons. Subsequently, the h
+ and ˙O
2− take part in photoredox reactions to degrade organic pollutants directly or indirectly. Furthermore, the internal electric field in the Bi
2O
3/Bi
2MoO
6 p–n junction increases the migration of photogenerated electrons and holes,
38 which also promotes the photocatalysis of organic pollutants.
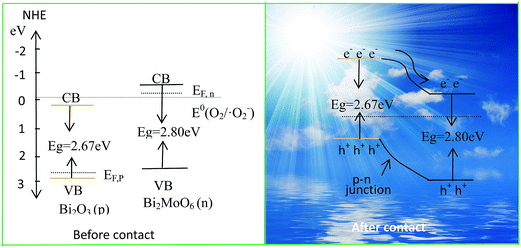 |
| | Fig. 13 Schematic diagrams for the energy band of Bi2O3 and Bi2MoO6 (the left) and the possible charge separation in Bi2O3/Bi2MoO6 composites (the right). | |
4. Conclusion
In summary, the Bi2O3/Bi2MoO6 composites have been successfully prepared via a two-step solvothermal route. Electron micrographs showed that the as-synthesized Bi2O3/Bi2MoO6 composites owned the obviously different morphology from pure Bi2O3 and Bi2MoO6. N2 sorption–desorption experiments and photoelectrochemical measurements showed that Bi2O3/Bi2MoO6 composites had the biggest surface area and the highest photocurrent density. It was found that as-obtained Bi2O3/Bi2MoO6 composites presented stronger catalytic activities to the degradation of RhB and 2,4-DNP under the irradiation of the visible light than single component (Bi2O3 or Bi2MoO6) and their physical mixture. The above enhanced photocatalytic performance originated from the p–n junction between p-type Bi2O3 and n-type Bi2MoO6, in which the fast recombination of photogenerated electrons and holes was efficiently restrained. Simultaneously, experiments uncovered that the hole (h+) was the main oxidative species and ˙O2− radicals also acted as the important role in the photodegradation of organic pollutants. The present photocatalyst also presented good cycling stability. After 5 cycles, the catalytic efficiency still retained ∼91%, indicating that the as-obtained photocatalyst has potential application in environmental protection and wastewater treatment.
Acknowledgements
The authors thank the National Natural Science Foundation of China (21571005), High School Leading talent incubation programme of Anhui province (gxbjZD2016010) and The Recruitment Program for Leading Talent Team of Anhui Province for the fund support.
References
- H. Fan, Y. Li, B. Liu, Y. Lu, T. Xie and D. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 4853–4857 CAS.
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed.
- X. Wang, J. Ran, M. Tao, Y. He, Y. Zhang, X. Li and H. Huang, Mater. Sci. Semicond. Process., 2016, 41, 317–322 CrossRef CAS.
- M. Vinod, V. Biju and K. Gopchandran, Superlattices Microstruct., 2016, 89, 369–377 CrossRef CAS.
- C. Yang, X. You, J. Cheng, H. Zheng and Y. Chen, Appl. Catal., B, 2017, 200, 673–680 CrossRef CAS.
- S. Sakthivel and H. Kisch, Angew. Chem., Int. Ed., 2003, 42, 4908–4911 CrossRef CAS PubMed.
- J. Di, J. Xie, S. Yin, H. Xu, L. Xu, Y. Xu, M. He and H. Li, RSC Adv., 2014, 4, 14281–14290 RSC.
- J. Ma, K. Wang, L. Li, T. Zhang, Y. Kong and S. Komarneni, Ceram. Int., 2015, 41, 2050–2056 CrossRef CAS.
- B. Liu, L. Liu, X. Lang, H. Wang, X. Lou and E. Aydil, Energy Environ. Sci., 2014, 7, 2592–2597 CAS.
- Y. Wang, M. Zheng, S. Liu and Z. Wang, Nanoscale Res. Lett., 2016, 11, 390 CrossRef PubMed.
- S. Obregón, A. Caballero and G. Colón, Appl. Catal., B, 2012, 117–118, 59–66 CrossRef.
- J. Sheng, X. Li and Y. Xu, ACS Catal., 2014, 4, 732–737 CrossRef CAS.
- J. Li, X. Liu, Z. Sun, Y. Sun and L. Pan, J. Colloid Interface Sci., 2015, 452, 109–115 CrossRef CAS PubMed.
- Y. Wang, L. Lin, F. Li, L. Chen, D. Chen, C. Yang and M. Huang, Photochem. Photobiol. Sci., 2016, 15, 666–672 CAS.
- C. Guo, J. Xu, S. Wang, L. Li, Y. Zhang and X. Li, CrystEngComm, 2012, 14, 3602–3608 RSC.
- J. Yang, X. Niu, S. An, W. Chen, J. Wang and W. Liu, RSC Adv., 2017, 7, 2943–2952 RSC.
- L. Li, P. Salvador and G. Rohrer, Nanoscale, 2014, 6, 24–42 RSC.
- J. Tian, P. Hao, N. Wei, H. Cui and H. Liu, ACS Catal., 2015, 5, 4530–4536 CrossRef CAS.
- H. Huang, Y. He, X. Du, P. Chu and Y. Zhang, ACS Sustainable Chem. Eng., 2015, 3, 3262–3273 CrossRef CAS.
- J. Yang, X. Wang, J. Dai and J. Li, Ind. Eng. Chem. Res., 2014, 53, 12575–12586 CrossRef CAS.
- M. Gui, W. Zhang, Q. Su and C. Chen, J. Solid State Chem., 2011, 184, 1977–1982 CrossRef CAS.
- Y. Xu, Z. Zhang and W. Zhang, Mater. Res. Bull., 2013, 48, 1420–1427 CrossRef CAS.
- Z. Hao, L. Xu, B. Wei, L. Fan, Y. Liu, M. Zhang and H. Guo, RSC Adv., 2015, 5, 12346–12353 RSC.
- H. Jiang, J. Liu, K. Cheng, W. Sun and J. Lin, J. Phys. Chem. C, 2013, 117, 20029–20036 CAS.
- S. Liu, K. Yin, W. Ren, B. Cheng and J. Yu, J. Mater. Chem., 2012, 22, 17759–17767 RSC.
- P. Zhang, C. Shao, M. Zhang, Z. Guo, J. Mu, Z. Zhang, X. Zhang and Y. Liu, J. Hazard. Mater., 2012, 217–218, 422–428 CrossRef CAS PubMed.
- X. Hou, Y. Tian, X. Zhang, S. Dou, L. Pan, W. Wang, Y. Li and J. Zhao, J. Alloys Compd., 2015, 38, 214–220 CrossRef.
- H. Huang, L. Liu, Y. Zhang and N. Tian, J. Alloys Compd., 2015, 619, 807–811 CrossRef CAS.
- D. Yue, D. Chen, Z. Wang, H. Ding, R. Zong and Y. Zhu, Phys. Chem. Chem. Phys., 2014, 16, 26314–26321 RSC.
- D. He, L. Wang, D. Xu, J. Zhai, D. Wang and T. Xie, ACS Appl. Mater. Interfaces, 2011, 3, 3167–3171 CAS.
- J. Low, J. Yu, Q. Li and B. Cheng, Phys. Chem. Chem. Phys., 2014, 16, 1111–1120 RSC.
- X. Xiao, J. Wei, Y. Yang, R. Xiong, C. Pan and J. Shi, ACS Sustainable Chem. Eng., 2016, 4, 3017–3023 CrossRef CAS.
- W. Wang, J. Wang, Z. Wang, X. Wei, L. Liu, Q. Ren, W. Gao, Y. Liang and H. Shi, Dalton Trans., 2014, 43, 6735–6743 RSC.
-
(a) Y. Hao, X. Dong, S. Zhai, X. Wang, H. Ma and X. Zhang, Chem. Commun., 2016, 52, 6525–6528 RSC;
(b) Y. C. Hao, X. L. Dong, S. R. Zhai, X. Y. Wang, H. C. Ma and X. F. Zhang, RSC Adv., 2016, 6, 35709–35718 RSC.
- D. D. Qin, C. L. Tao, S. In, Z. Y. Yang, T. E. Mallouk, N. Bao and C. A. Grimes, Energy Fuels, 2011, 25, 5257–5263 CrossRef.
- H. Li, J. Liu, W. Hou, N. Du, R. Zhang and X. Tao, Appl. Catal., B, 2014, 160–161, 89–97 CrossRef CAS.
- H. Li, Q. Deng, J. Liu, W. Hou, N. Du, R. Zhang and X. Tao, Catal. Sci. Technol., 2014, 4, 1028–1037 CAS.
- W. Wang, J. Wang, Z. Wang, X. Wei, L. Liu, Q. Ren, W. Gao, Y. Liang and H. Shi, Dalton Trans., 2014, 43, 6735–6743 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02760e |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Shiyong Ye
* and
Shiyong Ye