
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnetic, structural and surface properties of functionalized maghemite nanoparticles for copper and lead adsorption†
Juan A. Ramos Guivar *a,
Elahe Sadrollahib,
Dirk Menzelb,
Edson G. Ramos Fernandesc,
Elvis O. Lópezd,
Marco Morales Torrese,
Jesús M. Arsuagaf,
Amaya Arencibiaf and
F. Jochen Litterstbd
*a,
Elahe Sadrollahib,
Dirk Menzelb,
Edson G. Ramos Fernandesc,
Elvis O. Lópezd,
Marco Morales Torrese,
Jesús M. Arsuagaf,
Amaya Arencibiaf and
F. Jochen Litterstbd
aFaculty of Physical Sciences, National University of San Marcos, P. O. Box 14-0149, Lima 14, Peru. E-mail: aguivar@gmail.com; juan.ramos5@unmsm
bInstitut für Physik der Kondensierten Materie, Technische Universität Braunschweig, 38110 Braunschweig, Germany
cInstituto de Ciência e Tecnologia, Universidade Federal de São Paulo, Av. Cesare Mansueto Giulio Lattes, 1201, 12247-014, São José dos Campos, SP, Brazil
dBrazilian Center for Research in Physics (CBPF), Rio de Janeiro, RJ 22290-180, Brazil
eDepartamento de Física Teórica e Experimental, UFRN, Natal, RN 59078-970, Brazil
fEnvironmental and Chemical Engineering Group, Universidad Rey Juan Carlos, C/Tulipán s/n, Móstoles, Madrid, Spain
First published on 1st June 2017
Abstract
In this study, magnetic nanocomposites were developed and used as adsorbents for lead and copper from aqueous media. Structural, surface, magnetic and textural properties of functionalized maghemite nanoparticles synthesized by alkaline co-precipitation were studied. The surfaces of the iron oxide nanoparticles (Nps) were modified with different chemical agents such as fatty and amino acids, silica (SiO2), mesoporous silica (SBA-15), hydroxyapatite, multiwall carbon nanotubes (MWCNTs) and ethylenediaminetetraacetic acid (EDTA), obtaining NPs with mean particle sizes ranging from 7 to 16 nm according to Rietveld refinement and TEM images analysis. The physicochemical surface properties of the functionalized materials were studied via zeta potential (ζ) and Fourier transform infrared (FTIR) spectroscopy. Mössbauer spectroscopy (MS) as a function of temperature and DC magnetometry were used to study the magnetic properties. The superparamagnetic relaxation was studied by MS. The resolved spectra at 20 K confirm the presence of nanomaghemite phase. Besides, the saturation magnetization varies from 12 to 62 emu g−1. A nitrogen adsorption–desorption technique was used to determine the specific surface area and to study the porous structure. The functionalized γ-Fe2O3 Nps exhibited a Brunauer–Emmett–Teller (BET) specific surface area ranging from 74 to 214 m2 g−1 and revealed remarkable uptake capacities to remove Cu(II) and Pb(II) species from aqueous solutions.
Introduction
Copper and lead are relevant contaminants in the environment, which can affect significantly human health.1–3 Some mental illnesses caused by constant uptake via ingestion, inhalation and dermal contact of these metals at high concentrations are neurodegenerative disorders, Alzheimer type II astrocytosis, Parkinsonism and ataxia.1,2 Therefore, the presence in water of these metals, due to natural processes and human activities, should be avoided. Many efforts have been developed to find an adequate process for the removal of Cu(II) and Pb(II), such as chemical precipitation, ion-exchange processes, membrane filtration, electrodialysis, electrochemical treatments and adsorption.4,5 The last one is recommended as an effective and reliable method due to its reproducibility, low-cost and simplicity. Thus, in this worrying context some metal-sorbent prototypes based on magnetic nanoparticles (Nps) are being developed for environmental purposes, for instance in the removal of organic and inorganic metals pollutants from contaminated water.2,3 Ali summarized some of these current adsorbents proposed for water treatment, some of them are FeOOH-coated maghemite, gum arabic modified iron oxide magnetic Nps (13–67 nm), zero-valent iron (10–30 nm) among others.2 These modified magnetic nanoadsorbents have high capability for metal traces adsorption, low toxic effects and are easy to separate magnetically.2,3 Among a wide variety of nanoparticles, pure iron oxide Nps exhibit adsorption affinity to metallic traces.2 Besides, their adsorption capabilities can be improved by functionalization with other inorganic and organic materials containing functional groups that help for the uptake of these metals.2 In addition, the coating prevents the Nps from flocculation and agglomeration caused by dipole–dipole and van der Waals force interactions, thus being presented as a very important group of adsorbents for metal removal. However, some conditions like colloidal stability, critical particle size and pore diameter, saturation magnetization, specific surface area, etc., should be thoroughly studied before their use in environmental applications.2,6Several techniques are reported in the abundant literature to synthesize magnetic Nps.7 For example, thermal decomposition is used to produce mono-disperse Nps with very high narrow particle size distribution and variety of sizes (5–20 nm).7 The synthesis is carried out in organic environment where solvents with high melting point and expensive surfactants are used as dispersive medium to produce hydrophobic Nps with attractive and defined morphologies.7,8 But, very cautious laboratory conditions and higher temperatures are also employed and subsequent surface modifications should be followed to make the Nps hydrophilic.9,10 In addition, the resultant mass is less than 1 g, making this method inadequate and expensive to be used for environmental applications.10 However, the co-precipitation method seems to be the most appropriate method to give scalability and future industrial applications to the iron oxide Nps, this method was firstly proposed by Kang et al. without the presence of surfactants.11 It consists in the homogenous nucleation and growth of Nps using iron precursors that precipitate in alkaline medium. The method yields particles with a wide particle size distribution where the mean diameters vary between 6–15 nm, and the subsequent functionalization with other agents can be achieved.7
In this paper, the synthesis and characterization of functionalized maghemite (γ-Fe2O3) Nps with different inorganic and organic agents were studied, achieving a detailed properties description including structural, surface and magnetic characterization. The characterization of the textural properties (specific surface area, pore diameter) of our maghemite nanoparticles functionalized with different agents was also carried out. Their application as magnetic nanoadsorbents were tested for Cu(II) and Pb(II) heavy metals removal from aqueous solution.
Experimental
Materials
Iron(II) sulphate heptahydrate (FeSO4·7H2O), iron(III) chloride anhydrous (FeCl3), iron(III) chloride hexahydrate (FeCl3·6H2O), iron(II) chloride tetrahydrate (FeCl2·4H2O), ammonium hydroxide (NH4OH, 28–30%), tetraethyl orthosilicate (TEOS), lauric acid (LA), oleic acid (OA), copper(II) nitrate trihydrate extrapure (Cu(NO3)2·3H2O) and lead(II) nitrate (Pb(NO3)2); were of analytical grade and obtained from Sigma Aldrich and used without further purification. L-Arginine (L-arg) monohydrochloride (98%) was purchased from Alfa Aesar. Multi Walled Carbon Nanotubes (MWCNTs) were obtained from Cheaptubes with outer diameter: 20–30 nm, inner diameter: 5–10 nm, ash: <1.5 wt%, purity: >95 wt%, length: 10–30 μm and BET specific surface area: 110 m2 g−1. Ultrapure water (resistivity of 18.3 MΩ cm) was obtained from a Millipore Milli-Q Water System (Millipore Inc.), and was used for rinsing and to prepare all aqueous solutions.Adsorbents synthesis
To deposit γ-Fe2O3 Nps onto the SBA-15 pores, mesoporous SBA-15 synthesized by the previously reported method was used.13 Briefly, an amount of 1.5 g of pure SBA-15 was dispersed in water by sonication for 30 min, then an aqueous solution containing iron precursors at molar ratio Fe2+/Fe3+ ≈ 0.5 mol mol−1 was added.12 Then, the same procedure described above to obtain γ-Fe2O3 Nps was followed. The obtained sample was labeled as γ-Fe2O3-SBA15.
The syntheses of γ-Fe2O3-2 Nps functionalized with oleic acid, OA, and lauric acid, LA, (labeled as γ-Fe2O3@OA and γ-Fe2O3@LA, respectively) were carried out by dispersing a certain amount of the γ-Fe2O3-2 Nps into OA and LA solutions. This mixture was stirred for 30 min at 80 °C. The final dispersions were filtered and washed several times up to neutral pH. Then, it was dried at 60 °C for 12 h.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:0.5, respectively. This solution was stirred and heated up to reach 80 °C in a N2 reflux system. Then, ammonium hydroxide (NH4OH) was added to the solution and the mixture was maintained under stirring for 60 min. The sample was washed several times to remove the excess of free amino acid molecules and to reduce pH to 7. The final solution was filtered and washed several times till pH = 7. Then, it was dried at 60 °C for 12 h.
:2:0.5, respectively. This solution was stirred and heated up to reach 80 °C in a N2 reflux system. Then, ammonium hydroxide (NH4OH) was added to the solution and the mixture was maintained under stirring for 60 min. The sample was washed several times to remove the excess of free amino acid molecules and to reduce pH to 7. The final solution was filtered and washed several times till pH = 7. Then, it was dried at 60 °C for 12 h.The Nps were deposited onto the o-MWCNTs by the deposition–precipitation method using iron precursors solution with molar ratio Fe2+/Fe3+ ≈ 0.5 in alkaline conditions.12 Briefly, 200 mg of o-MWCNTs were dispersed in ultrapure water for 15 min and then kept in air at 80 °C under vigorous stirring. After that, FeCl3 (1.24 mmol) and FeSO4·7H2O (0.62 mmol) were added to the solution containing o-MWCNTs. Immediately, a solution of NaOH (1.5 M) was added dropwise till the pH reached 12. The mixture was left to react for a period of 2 h through the expected following chemical reaction:15
| Fe2+ + 2Fe3+ + 8OH− + o-MWCNTs Fe3O4/o-MWCNTs + 4H2O |
The obtained dispersion was cooled to RT, magnetically separated and then filtered using a 2 μm membrane filter to remove free magnetic Nps, the obtained solid was washed several times with ultrapure water till the pH was 7. The sample was dried at 80 °C for 12 h and the material was labeled as γ-Fe2O3@MWCNTs.
where qt indicates the amount in mg of adsorbate per gram of adsorbent for a certain time t. C0 and Ct are the initial and final concentrations (mg L−1), respectively, m is the adsorbent mass and V is the volume used for the adsorption test.
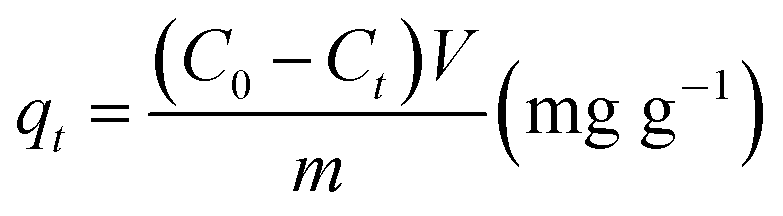
Characterization
The X-ray diffraction (XRD) data were obtained by using a diffractometer Philips X-PERT MPD, operating with CuKα radiation (1.5406 Å). Powder diffraction patterns were obtained in step scanning mode, 2θ = 10–80° with a step of 0.01° and 4 seconds per step. Rietveld refinement was performed using the software package FullProf. All parameters were refined by the least-squares method. The pseudo-Voigt function modified by Thompson-Cox-Hastings (TCH) was used as peak profile function. For estimating the mean crystallite sizes we took care of corrections due to instrumental broadening that was obtained from corundum (Al2O3) as standard material. The transmission electron micrographs (TEM) were acquired in a JEOL JEM-2000 FX instrument, working at 200 kV. Samples were analyzed using a carbon-coated copper grid as support of the acetone dispersions of samples prepared by sonication in an ultrasonic bath solution. The ζ-potential values of the dispersion were obtained using a Zeta sizer (Malvern Zs 90, U.K.) equipment. Fourier Transform Infrared spectroscopy (FTIR) (Varian EXCALIBUR SERIES 3100 – UMA 600) measurements were performed in transmission mode with a resolution of 4 cm−1. N2 adsorption–desorption isotherms at 77 K were measured by using a Micromeritics Tristar 3000 sorptometer to determine textural properties. Surface area was calculated by using the B.E.T. equation and the pore size distribution was obtained from the adsorption branch by means of the B.J.H. model with cylindrical geometry of the pores; pore volume was taken at P/P0 = 0.97. Measurements by X-ray photoelectron spectroscopy (XPS) at high-energy resolution were carried out in this study. The equipment used was a PHOIBOS 100/150 of the SPECS Company. A polychromatic X-ray from Al Kα at excitation energy of 1486.6 eV with binding-energy resolution of 0.84 eV was used in these experiments. To calibrate the spectra the adventitious carbon (C 1s = 284.6 eV) was used as reference. 57Fe Mössbauer absorption spectra have been collected in transmission geometry using a standard spectrometer with sinusoidal velocity sweep. The powder absorbers were enclosed into nylon containers. Absorber thicknesses were chosen equivalent to ca. 0.1 mg 57Fe per cm2. Absorber temperatures were varied between 20 K and 300 K using a variable temperature He-flow cryostat (Cryovac). As 14.4 k eV γ-radiation source we used about 40 mCi of 57Co in a Rh matrix kept at RT. Magnetic measurements were performed as a function of temperature (M–T) and magnetic field (M–H) by using a commercial VSM – Physical Properties Measurement System (PPMS) Dynacool from Quantum Design. The zero field cooling (ZFC) and field cooling (FC) magnetization measurements were recorded in the temperature range from 5 to 300 K and under a small magnetic field of 80 Oe.Results and discussion
The XRD diffractograms for all the synthesized samples are shown in Fig. 1. The comparison of patterns obtained for fatty acids functionalized γ-Fe2O3-2 Nps with the original Nps can be observed in Fig. 1a–c. The XRD pattern of γ-Fe2O3@L-arg Nps is shown in Fig. 1d. In general, all diffractograms showed line broadenings related to the nanoscale size of the synthesized compound. The crystallographic identification was done using the PDF card # 39–1346 for γ-Fe2O3.12 The main diffraction peak in all patterns associated to γ-Fe2O3 phase is at 2θ = 35.5°. Other peaks related to pure maghemite phase are observed at 2θ = 30.2°, 43.2°, 53.1°, 57.1°, and 62.9° from the (220), (400), (422), (511) and (440) crystallographic planes.12 The crystal parameters for all samples obtained from Rietveld refinement are summarized in Table 1. The Rietveld refinement confirmed the presence of inverse spinel cubic structure in the whole set of samples with spatial group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m and cell parameter ranging from a = 8.25 to 8.47 Å (Table 1). The diffractogram for sample γ-Fe2O3@SiO2 (Fig. 1e) shows a broad peak in the range of 2θ = 20–30° due to amorphous silica. Fig. 1f and g show the XRD pattern for un-treated and treated MWCNTs. It can be seen that the acidic treatment has not affected the crystal structure. In that case, the main (hkl) diffractions planes (002, 100, 101) are observed.19 After functionalization of MWCNTs with γ-Fe2O3 Nps the diffractogram given in Fig. 1h revealed the presence of both phases. The γ-Fe2O3-SBA15 sample only exhibits the peaks of γ-Fe2O3 Nps, in the 2θ region between 20–25°, the amorphous phase of silica is not observed (see Fig. 1i). The presence of the SBA15 was proven by means of FTIR and TEM measurements.
m and cell parameter ranging from a = 8.25 to 8.47 Å (Table 1). The diffractogram for sample γ-Fe2O3@SiO2 (Fig. 1e) shows a broad peak in the range of 2θ = 20–30° due to amorphous silica. Fig. 1f and g show the XRD pattern for un-treated and treated MWCNTs. It can be seen that the acidic treatment has not affected the crystal structure. In that case, the main (hkl) diffractions planes (002, 100, 101) are observed.19 After functionalization of MWCNTs with γ-Fe2O3 Nps the diffractogram given in Fig. 1h revealed the presence of both phases. The γ-Fe2O3-SBA15 sample only exhibits the peaks of γ-Fe2O3 Nps, in the 2θ region between 20–25°, the amorphous phase of silica is not observed (see Fig. 1i). The presence of the SBA15 was proven by means of FTIR and TEM measurements.
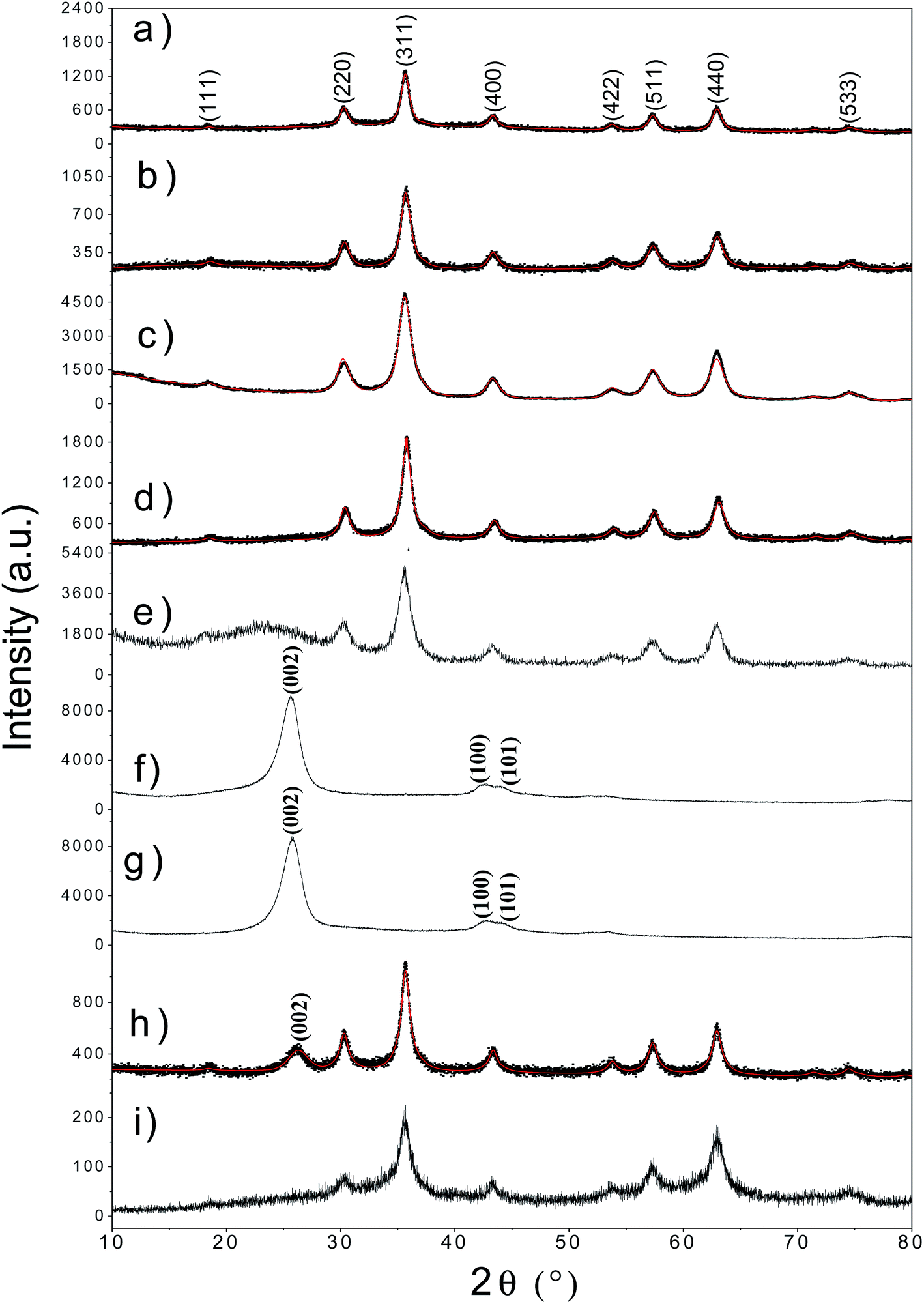 | ||
| Fig. 1 Rietveld refinement for uncoated maghemite nanoparticles (a) and functionalized with OA (b), LA (c) and L-arg (d). Maghemite nanoparticles coated with SiO2 (e). XRD pattern for untreated MWCNTs (f) and activated with HNO3 (g), γ-Fe2O3@MWCNTs hybrid Nps (h) and γ-Fe2O3-SBA15 (i). The principal Miller indices for each phase are also indicated. | ||
| Sample | a (Å) | dXRD (nm) | dTEM (nm) |
|---|---|---|---|
| γ-Fe2O3-1 (ref. 12) | 8.357 | 6.4 | 6.8 |
| γ-Fe2O3-2 | 8.302 | 7.1 | 9 |
| γ-Fe2O3@SiO2 | 8.465 | 10 | 10.4 |
| γ-Fe2O3-SBA15 | 8.345 | 6.2 | 5–9 |
| γ-Fe2O3@OA | 8.253 | 5.9 | 6.3 |
| γ-Fe2O3@LA | 8.317 | 4.5 | 5.5 |
| γ-Fe2O3@L-arg | 8.330 | 5.9 | 7.7 |
| γ-Fe2O3@HAp (ref. 16) | * | 8 | 16 |
| γ-Fe2O3-EDTA1 (ref. 17) | 8.364 | 3 | 4 |
| γ-Fe2O3@MWCNTs | 8.359 | 7.3 | 7.5 |
TEM micrographs for all systems are shown in Fig. 2a–h in which a similar morphology of Nps can be observed. The coating of Nps with silica induced an increment in diameter size (∼10.4 nm for γ-Fe2O3@SiO2 Nps) with respect to the γ-Fe2O3 Nps.12 This is related to the drying and following re-dispersion of Nps in water for functionalization promoting the agglomeration among the Nps. From the particle size histogram, that was fitted to a Gaussian distribution function (see Fig. S1a–f†), a mean diameter of ∼9 nm was estimated for the γ-Fe2O3-2 Nps which is greater than for the previously synthesized Nps (∼6.8 nm)12 using the same method but a different base such as NH4OH (weak) and NaOH (strong base), indicating that the used base can slightly influence the final morphology of the obtained γ-Fe2O3 Nps.17 These results show that the coprecipitation method is a convenient procedure to obtain Nps with diameters less than 10 nm. After coating with OA and LA, the mean particle diameter slightly decreased, what is expected since carboxylic acids act as dispersant or surfactants and size control agents during or after synthesis.20,21 The L-arg aminoacid seems to have the same effect on the γ-Fe2O3 Nps. This size-decreasing trend is also noticed from the Rietveld's refinement where a smaller mean crystallite size was also detected after functionalization (see Table 1). However, some diameters differ a little from the TEM values meaning that some Nps are composed at least of one or two crystallites. A similar result has been reported by López et al. using chitosan-coated magnetic Nps and also in our previous work for γ-Fe2O3@HAp Nps.16,22 In addition, if OA and LA acids would have been added during the Nps synthesis the size distribution would be narrower, since these acids act as size control agents.17,20 It is also observed in TEM images (Fig. 2) that functionalized Nps show well-defined morphology in comparison to the uncoated one (Fig. 2b).
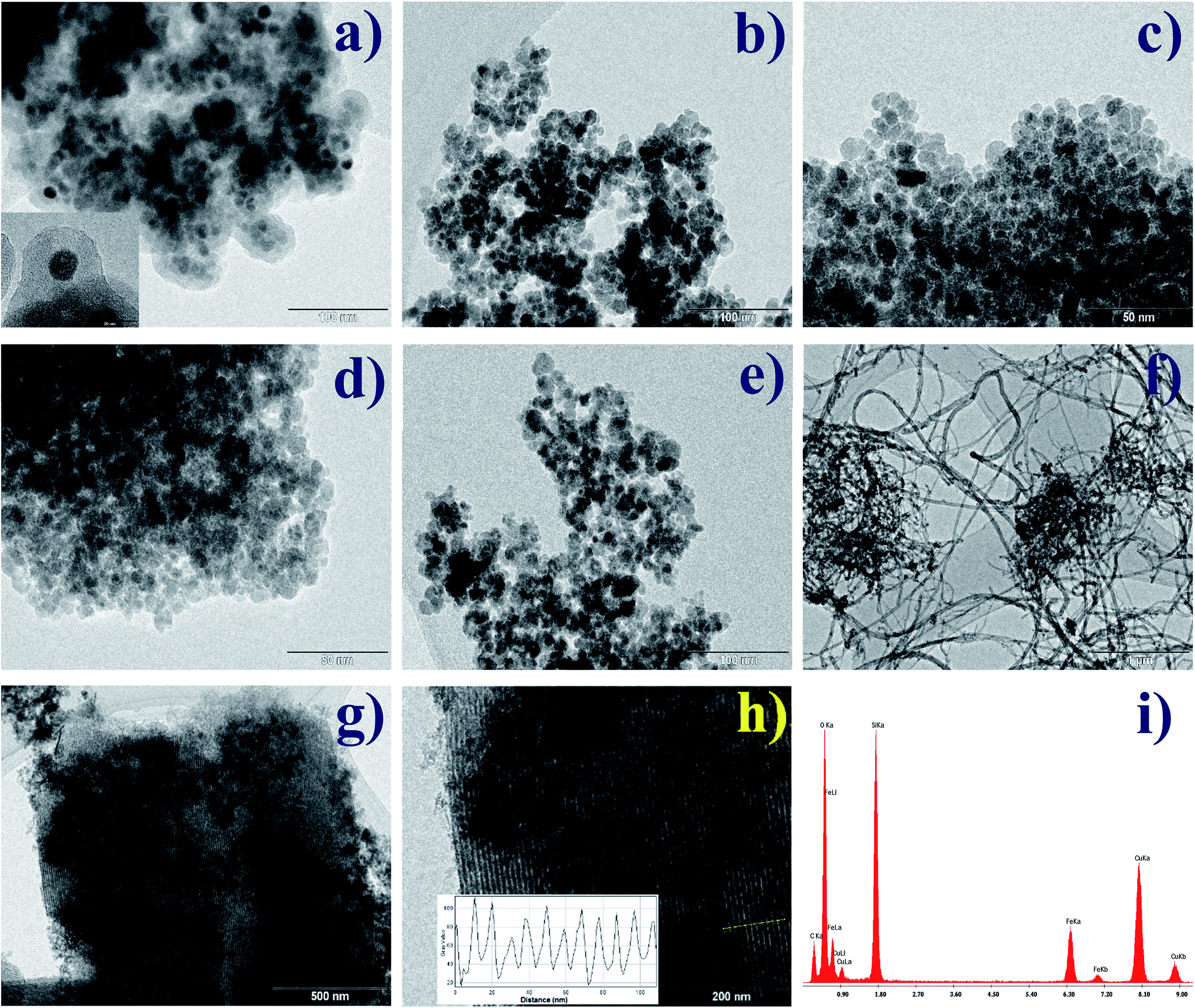 | ||
| Fig. 2 TEM images for γ-Fe2O3@SiO2 (bar length of 100 nm) (a) γ-Fe2O3-2 (bar length of 100 nm) (b), γ-Fe2O3@OA (bar length of 50 nm) (c), γ-Fe2O3@LA (bar length of 50 nm) (d), γ-Fe2O3@L-arg (bar length of 100 nm) (e), γ-Fe2O3@MWCNTs Nps (bar length of 1 μm) (f) and γ-Fe2O3-SBA15 (bar length of 500 and 200 nm) (g and h). EDX analysis for γ-Fe2O3-SBA15 (i). | ||
On the other side, the γ-Fe2O3@MWCNTs hybrid Nps clearly show the coexistence between o-MWCNTs and γ-Fe2O3 Nps with quasi-spherical morphology. The Fig. 2g and h show the Nps embedded in the pores of SBA-15 which mean pore size was estimated by using the B.J.H. model and gave an average of 9 nm.13 Besides, as one can notice the SBA15 retained its 2D hexagonal structure after functionalization with γ-Fe2O3 Nps as also shown by Yiu et al.23 The γ-Fe2O3-SBA15 contains γ-Fe2O3 Nps with sizes ranging from 5–9 nm. In addition, the Energy-dispersive X-ray spectroscopy (EDX) spectrum (Fig. 2i) also confirms the presence of Si, Fe and O atoms in the sample.
The ζ-potential provides information about the charges located on the surface of the functionalized Nps, which is an important parameter that favors the colloidal stability in water. It is known that magnetic Nps have chemical affinity for amino and carboxyl groups.17,20 Thus, the ζ-potential can give information on the nature of functional groups bound to the surface of the γ-Fe2O3@L-arg Nps. The ζ-potential values measured at pH = 7 are summarized in Table 2. The bare γ-Fe2O3 Nps showed values of −5 and −10 mV, indicating a negative surface at this pH, probably due to the slight predominance of negative O− sites. The Nps coated with fatty acids showed negative ζ-potential (−40 mV) indicating a negative charge on the outermost particle surface. Capping agents such as fatty acids form a protective monolayer, where carboxylate (R–COO−) groups are strongly bonded to the particles surface.24 Since zeta-potential was measured at pH = 7, the negative charge is more likely to be from a second layer of fatty acid where their carboxylate (R–COO) groups are facing the water, thus, providing hydrophilic properties to the particle. Recently, Chen et al. showed the presence of a bilayer oleic acid coated iron oxide Nps, where the outer layer is physically adsorbed on the first one through hydrophobic interaction of the surfactants tails.24
| Sample | ζ (mV) | SSABET (m2 g−1) | DP (nm) | VP (cm3 g−1) | Ms (emu g−1) |
|---|---|---|---|---|---|
| γ-Fe2O3-1 (ref. 12) | −5 | 129.7 | 8.8 | 0.29 | 62 (ref. 12) |
| γ-Fe2O3-2 | −10 | 88.3 | 17.1 | 0.33 | 59 |
| γ-Fe2O3@SiO2 | −28.5 | 42.3 | 9.4 | 0.09 | 51 |
| γ-Fe2O3-SBA15 | 214 | 14.6 | 0.55 | 20 | |
| γ-Fe2O3@OA | −40 | 74.8 | 4.7 | 0.09 | 46 |
| γ-Fe2O3@LA | −42 | 62.3 | 10 | 0.19 | 44 |
| γ-Fe2O3@L-arg | −28.6 | 77.5 | 16.9 | 0.26 | 54 |
| γ-Fe2O3@HAp (ref. 16) | −21 | 95.51 | 16.5 | 0.29 | 12 |
| γ-Fe2O3-EDTA1 (ref. 17) | 108.6 | 7.8 | 0.25 | 22 | |
| γ-Fe2O3@MWCNTs | −52 | 129.6 | 13.1 | 0.34 | 37 |
The mechanism of bonding of the first monolayer of fatty acids coating Fe3O4 Nps have been studied previously by FTIR and it was found that the chemisorption occurred through a covalent bonding between iron and carboxyl's oxygens.25 The wavenumber difference, Δ, between the antisymmetric νas(COO−) and symmetric νs(COO−) vibrations was used to determine the mode in which carboxylate binds to the metal oxide surface, a value of 194 cm−1 was calculated for both acids coating the Nps. Similar behaviors were observed by Bloemen et al. and Baccile et al. for carboxylate interacting with iron oxide surfaces.26,27 Besides, the pH stability of this hydrophilic –COO− group was studied by Bloemen et al.,26 the COOH group coating the Nps showed to retain its negative charge even at low values of pH.
On the other hand, the ζ-potential for the γ-Fe2O3@SiO2 and γ-Fe2O3@L-arg Nps have values of −28 mV. These values are near to the threshold of the colloidal stability in aqueous medium. Rehana et al. showed a positive zeta potential value of +3.8 mV for Nps coated with L-arg.20 This last result suggest that our method facilitate the bonding with the Nps through the amine functional groups because carboxylic groups are externally exposed in the surface, explaining the negative value. On the other hand, the γ-Fe2O3@MWCNTs hybrid Nps exhibited a value of −52 mV, a result that is in close agreement with previous work for acid-treated MWCNTs indicating also the presence of carboxyl and hydroxyl groups.28
The mechanism of bonding between L-arg aminoacid and iron oxide Nps is rather more complex to explain; it is due to the presence of carboxylic, amine and guanidinium functional groups in L-arg. Park et al. have studied the interaction between Fe cations on the surface of magnetite Nps and L-glutamic and L-lysine aminoacids and showed that the aminoacid–metal oxide interaction can be studied by ζ-potential, proposing several bonding configurations.29
In order to get additional information and to investigate the plausible interaction between aminoacids and Nps in the present work, the FTIR spectrum for γ-Fe2O3@L-arg Nps and pure arginine are displayed in Fig. 3a and b. For pure L-arg, the peaks at 2943 and 2862 cm−1 correspond to CH2 methylene antisymmetric and symmetric modes of stretching vibration, respectively. Fig. 3a depicts a wide band appearing centered around 3100 cm−1 assigned to ν-NH stretching. The peaks in the region from 1000 to 1800 cm−1 are attributed to different alkyl, carboxyl and to amino vibration modes.30 Those bands below 1700 cm−1 are assigned to νasCOO−, δ-NH2 and NH3+ groups.
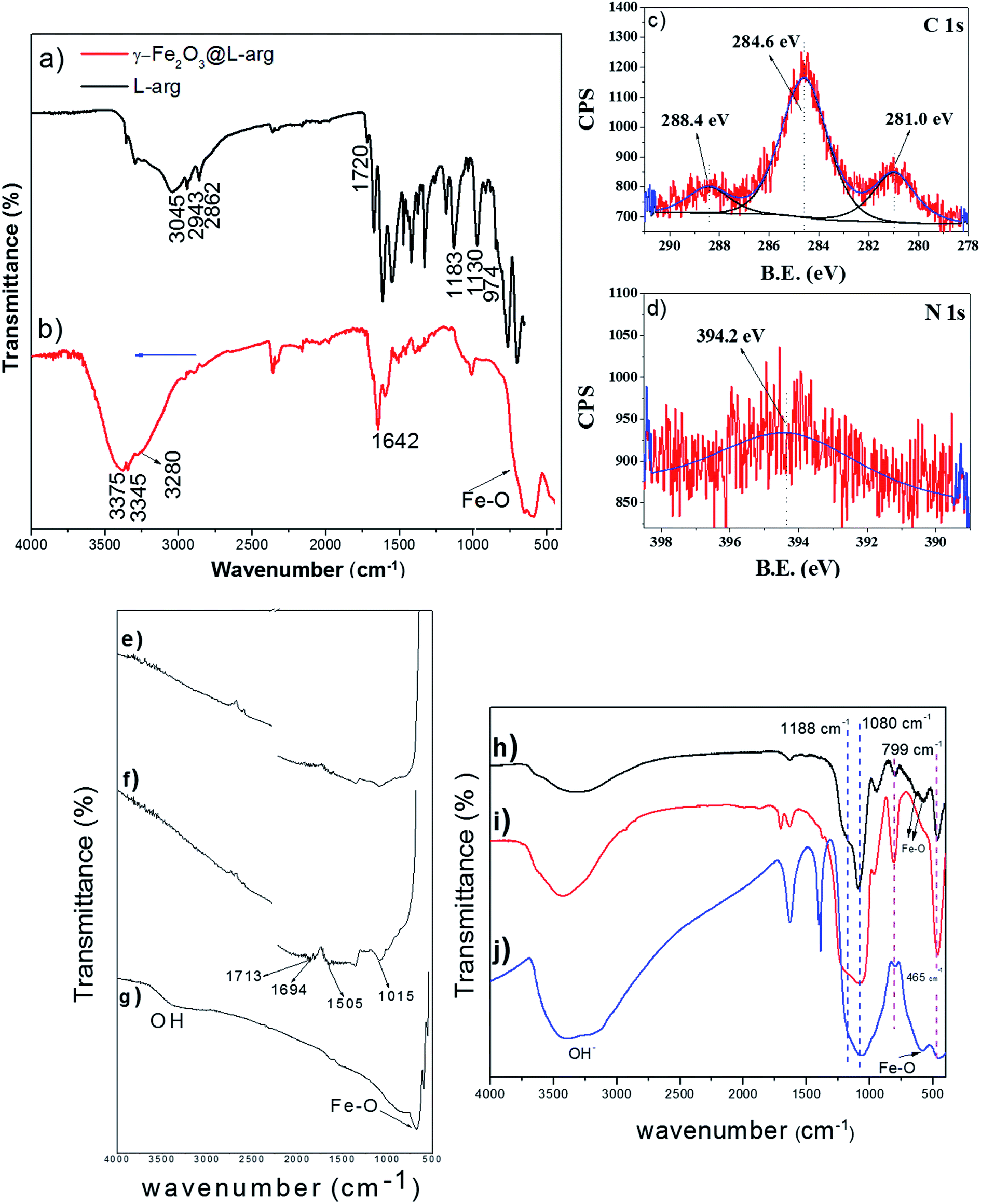 | ||
| Fig. 3 FTIR of pure L-arg (a), γ-Fe2O3@L-arg (b), C 1s and N 1s XPS regions for γ-Fe2O3@L-arg Nps (c and d), MWCNTs, FTIR signal in the range 2700–400 cm−1 is 5× enhanced (e), o-MWCNTs, FTIR signal in the range 2700–400 cm−1 is 5× enhanced (f), γ-Fe2O3@MWCNTs (g), γ-Fe2O3@SiO2 Nps (h), pure SBA15 (i) and γ-Fe2O3-SBA15 (j). | ||
On the other side, in the FTIR spectrum for γ-Fe2O3@L-arg Nps (Fig. 3b) the low IR region shows the characteristic Fe–O bands of γ-Fe2O3 Nps located at 650–700 cm−1. Besides, the strong peaks related to carboxyl vibrations signals, for pure L-arg, in the region 1700 to 1000 cm−1 are significantly reduced. The decrease in the spectrum intensity is probably due to the partial dilution process of L-arg in the synthesis of the Nps; thus, although the spectrum confirms the presence of L-arg, it is not possible to establish a bonding coordination due to the complexity of the spectrum.
Besides, the C 1s XPS shown in Fig. 3c exhibits a peak at ∼288.4 eV related to bidentate carboxylate carbon of the L-arg molecule onto Nps surface.31 Moreover, the presence of a broad N 1s peak at ∼394.2 eV in the XPS spectra (Fig. 3d) indicates that the nitrogen (NH) from the L-arg is also coordinating with the Nps since according to Wang et al. the binding energy of N 1s should show a shift from 398.9 eV to lower values by 1–3 eV when nitrogen is bound to metals, because of a transfer of electron density from nitrogen to metals.32,33 This is in agreement with the zeta potential results. Wang et al. mentioned that during Nps nucleation the Fe3O4–amino/guanidine complexes exist transitorily.32 However, at the end of the synthesis the carboxyl groups of L-arg should replace the amine/guanidine groups to form carboxyl-capped Fe3O4 Nps. The molar ratio used in our work for Fe2+/Fe3+/L-arg was 1:2:0.5 and the reaction time was 1 h. Wang et al.32 used a molar ratio of 1:2 for Fe2+/L-arg and a reaction time of 3 h for their synthesis as an optimum time parameter to get colloidal stability between carboxyl and amine/guanidine groups. In our case, this lack of stability is observed in the zeta potential value of −28 mV where some carboxyl groups are found in a free configuration mode.
The FTIR spectrum displayed in (Fig. 3g) indicates the sorption of Nps onto MWCNTs backbone. The IR spectrum for pristine MWCNTs (Fig. 3e) shows a broader peak at around 1500 cm−1 related to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching of carbon skeleton.34 In the spectrum of Fig. 3f, the vibrations modes located within 1500–1000 cm−1 belong to C–O bonds in the o-MWCNTs. The carboxyl groups (COOH) formed after acidic treatment of MWCNTs with HNO3 produced IR peaks at 1694 and 1713 cm−1 due to stretching vibrations of δ(OH) and ν(CO). In Fig. 3g, a strong band of γ-Fe2O3 at 683 cm−1 was noted. However, the bands related to MWCNTs treated with acid practically disappeared. This decrease could be attributed to an ester linkage configuration between the o-MWCNTs and γ-Fe2O3 NPs, as suggested by Shan et al.34 However, the reduction of intensity for some bands of the o-MWCNTs because of the amount of iron on the sample, resulting from the synthesis, and reflected in the strong broad band between 600–700 cm−1 that cannot be ignored.
C stretching of carbon skeleton.34 In the spectrum of Fig. 3f, the vibrations modes located within 1500–1000 cm−1 belong to C–O bonds in the o-MWCNTs. The carboxyl groups (COOH) formed after acidic treatment of MWCNTs with HNO3 produced IR peaks at 1694 and 1713 cm−1 due to stretching vibrations of δ(OH) and ν(CO). In Fig. 3g, a strong band of γ-Fe2O3 at 683 cm−1 was noted. However, the bands related to MWCNTs treated with acid practically disappeared. This decrease could be attributed to an ester linkage configuration between the o-MWCNTs and γ-Fe2O3 NPs, as suggested by Shan et al.34 However, the reduction of intensity for some bands of the o-MWCNTs because of the amount of iron on the sample, resulting from the synthesis, and reflected in the strong broad band between 600–700 cm−1 that cannot be ignored.
The FTIR spectra of SiO2 coated γ-Fe2O3 Nps are presented in Fig. 3h. The IR position of SiO2 vibration bands are similar to those observed for pure SBA15 and γ-Fe2O3-SBA15 (Fig. 3i and j). The characteristic peaks of the γ-Fe2O3 Nps at 635, 573 and 464 cm−1 are due to the stretching vibrations of Fe–O in octahedral and tetrahedral sites of γ-Fe2O3.35 The peak at 799 cm−1 is related to the bending vibration of Si–O–Si and the overlap shoulder at 465 cm−1 to O–Si–O bond, the strong band and the shoulder at 1080 cm−1 and 1188 cm−1 reflect the presence of asymmetrical and symmetrical Si–O–Si stretching modes.36 It can be seen that the characteristic peaks of γ-Fe2O3 Nps are shifted to higher frequencies from 634 to 636 cm−1 and 567 to 576 cm−1, respectively. However, the band at 464 cm−1 for Fe–O vibrations remained unchanged after coating and deposition of the Nps with SiO2 and SBA15.
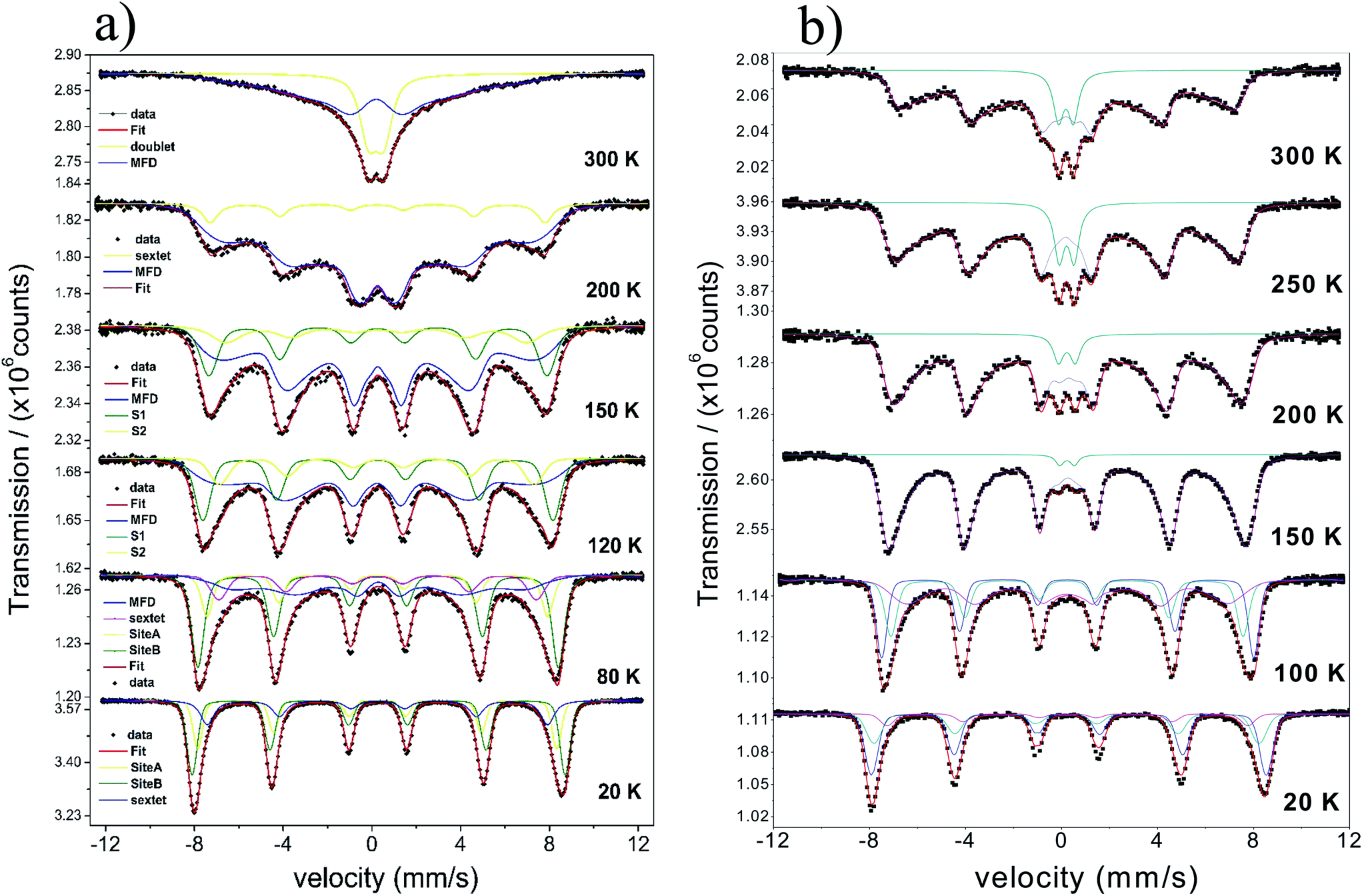
The temperature dependence of the Mössbauer spectra of pure γ-Fe2O3-1, γ-Fe2O3@OA and γ-Fe2O3-LA are typical for Nps with diameters smaller than 10 nm (see Fig. 4b, 5a and 6a). At 20 K the spectra can be fitted with two sextets related to A (Fe(III) in a tetrahedral oxygen coordination) and B sites (Fe(III) in a octahedral coordination) (see hyperfine parameters in Table 3) and the third sextet is related to surface Fe atoms with canted spin structure. At temperatures above 200 K the spectral peaks become broadened indicating the onset of superparamagnetic fluctuations overcoming the magnetic anisotropy barrier of magnetic particles and at 300 K the relaxation-broadened magnetic pattern has similar area as the superparamagnetic contribution with collapsed hyperfine splitting. The rather continuous spectral development with temperature is typical for Nps that are magnetically interacting. Therefore, a distinct separation of magnetically frozen and superparamagnetic fractions cannot be made.
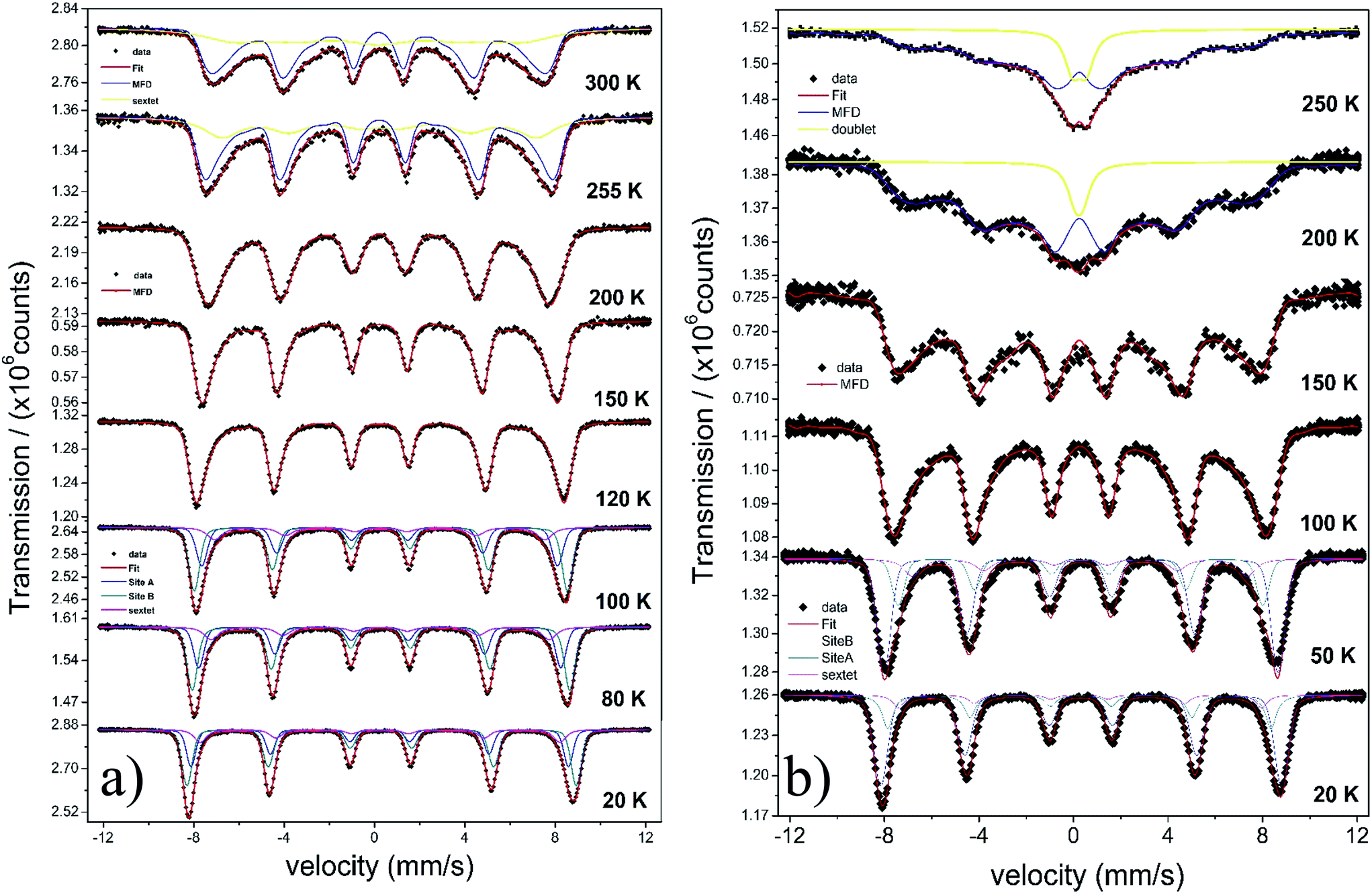 | ||
| Fig. 4 Temperature dependence of Mössbauer spectra for γ-Fe2O3-2 (a) and γ-Fe2O3@OA (b). | ||
 | ||
| Fig. 5 Temperature dependence of Mössbauer spectra for (a) γ-Fe2O3@LA and (b) γ-Fe2O3@MWCNTs Nps. | ||
| sample | Component | RAA (%) | δ (mm s−1) | Bhf (T) | QS (mm s−1) |
|---|---|---|---|---|---|
| γ-Fe2O3-1 | A | 26 | 0.40 | 50.4 | −0.021 |
| B | 59 | 0.41 | 52.4 | 0.02 | |
| Sextet | 15 | 0.36 | 47.7 | 0.004 | |
| γ-Fe2O3@SiO2 | MFD | 100 | 0.42 | 52.9 | 0.002 |
| γ-Fe2O3-2 | A | 31 | 0.38 | 51.9 | 0 |
| B | 52 | 0.45 | 53.6 | 0.03 | |
| Sextet | 17 | 0.40 | 49.5 | 0 | |
| γ-Fe2O3@OA | A | 25 | 0.39 | 50.4 | −0.072 |
| B | 65 | 0.43 | 52.7 | 0.013 | |
| Sextet | 10 | 0.42 | 47.8 | 0.028 | |
| γ-Fe2O3@LA | A | 29 | 0.40 | 51.7 | −0.01 |
| B | 48 | 0.45 | 53.7 | 0.03 | |
| Sextet | 23 | 0.41 | 49.7 | −0.01 | |
| γ-Fe2O3@L-arg | A | 28 | 0.33 | 49.4 | −0.018 |
| B | 64 | 0.50 | 51.4 | 0.02 | |
| Sextet | 8 | 0.35 | 47 | −0.005 | |
| γ-Fe2O3@MWCNTs | A | 31 | 0.31 | 49.9 | 0.002 |
| B | 57 | 0.41 | 51.1 | 0.017 | |
| Sextet | 12 | 0.36 | 46.7 | 0 | |
| γ-Fe2O3-SBA15 | A | 22 | 0.37 | 49.8 | 0.001 |
| B | 37 | 0.36 | 52.3 | 0 | |
| Sextet | 41 | 0.37 | 46.6 | 0 |
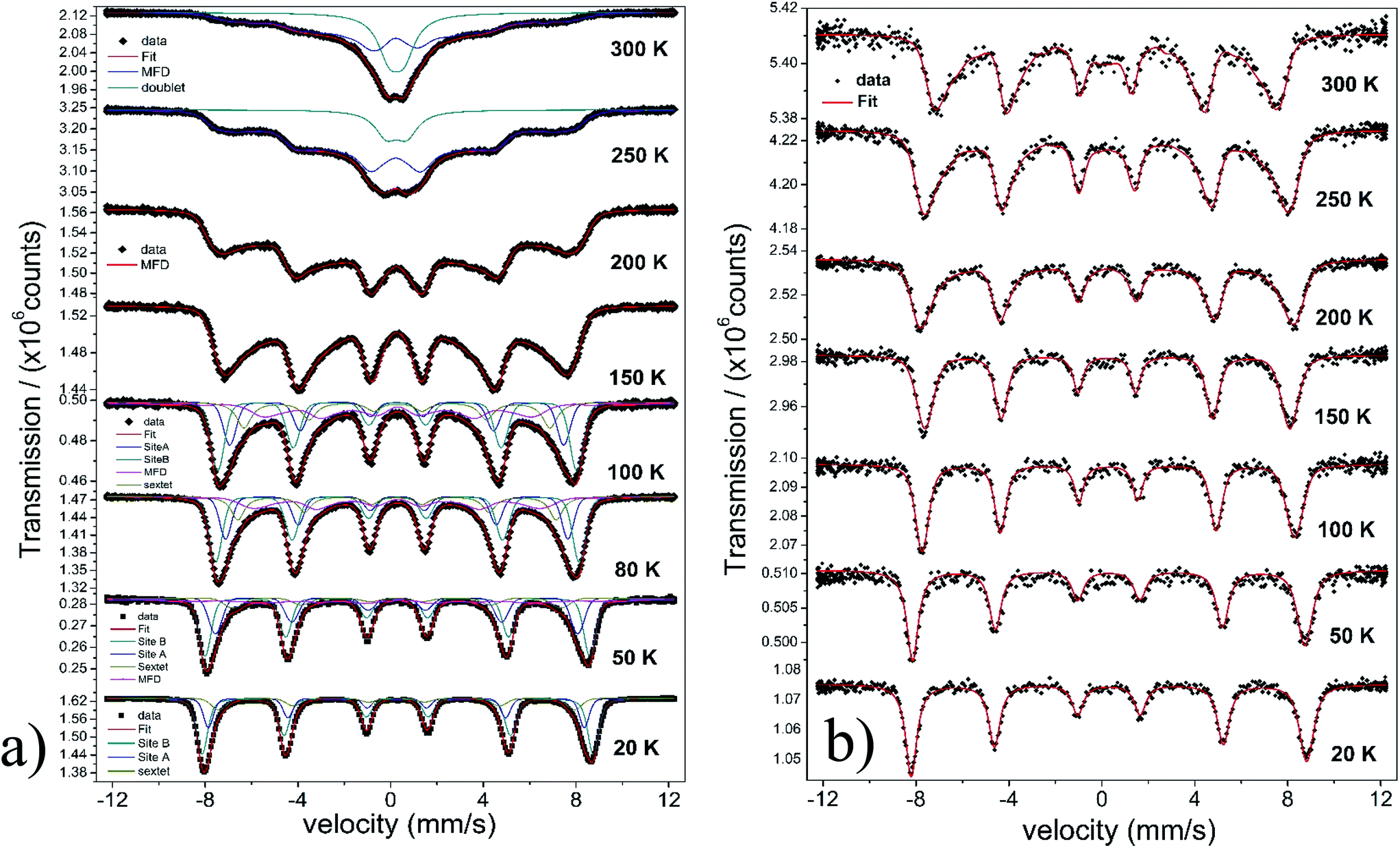
This is in contrast to the γ-Fe2O3@MWCNTs nanohybrids. As seen in Fig. 5b, there can clearly distinguish a superparamagnetic doublet structure at the center of the magnetically split spectrum above ca. 150 K indicating that at least part of the Nps are only weakly magnetically coupled. The doublet area increases with temperature, however, even at 300 K the magnetically split contribution stays dominant. This means that the blocking temperature on the time scale of hyperfine interactions has to be above room temperature. The spectra of γ-Fe2O3-1 and γ-Fe2O3@SiO2 Nps are shown in Fig. 6a and b. The spectra for the γ-Fe2O3@SiO2 Nps phase was fitted using a hyperfine magnetic field distribution (MFD) with hyperfine magnetic fields and isomers shifts close to those found in the spectra for pure for γ-Fe2O3-1 Nps (see Fig. 6a and Table 3). The temperature dependence of spectral shape, however, differs from that of the pure γ-Fe2O3 Nps (Fig. 6b). Up to 250 K no indication of overbarrier fluctuations is found and the Bhf distributions are typical for Nps bigger than 10 nm.8 This result suggests that clustering of particles ocurred during functionalization with SiO2, as also evidenced through the TEM analysis. For uncoated γ-Fe2O3-1 Nps a fitting model containing A and B sites seems to be suitable. However, for measurements performed at temperatures above 80 K a MFD component due overbarrier fluctuations was added, this subspectra is related to the very small particles (Fig. 6a).
 | ||
| Fig. 6 Temperature dependence of Mössbauer spectra for γ-Fe2O3-1 (a) and γ-Fe2O3@SiO2 Nps (b). | ||
The Mössbauer spectra of all samples (exceptγ-Fe2O3@SiO2 Nps) measured at 20 K were fitted using three magnetic components (Fig. 4a, b, 5a, b, 6a and 7), two sextets related to Fe in tetrahedral and octahedral coordination of maghemite, and a third sextet related to a spin disordered surface layer. In fact, the disordered phase in maghemite Nps has been reported in many works. Recently, measurements of polarized small-angle neutron scattering (SANSPOL) and nuclear spin forward (NSF) showed that the presence of spin disordered layer reduced the magnetization to ∼50% of the bulk value.37 In the present paper, above 20 K, the Mossbauer spectra showed the presence of a fourth component that is related to fast relaxing magnetic small nanoparticles. This component is implemented through a MFD. This last component increases with temperature leading a reduced areas values for the A and B iron sites. As one can note, the γ-Fe2O3-SBA15 sample shows the largest paramagnetic component at room temperature, among all the samples considered in this work. This is because the maghemite Nps were grown in the pores of the SBA15 structure. The Fig. 2h shows a SBA15 particle with nicely ordered pores. These pores have an average distance of 9.6 nm, the inset of Fig. 2h shows the gray profile obtained from the yellow reference line, the distance from the first to the eleventh pore was of 105.6 nm, and therefore, the maghemite particles should have particle size smaller than 9.6 nm.
 | ||
| Fig. 7 Temperature dependence of Mössbauer spectra for sample γ-Fe2O3-SBA15. | ||
In short, Mössbauer spectroscopy allows to distinguish various iron oxides from their differing hyperfine parameters. According to our previous works and other literatures, we noticed that the air exposure (time of several days after synthesis) and high chemical reactivity of the magnetite's nanoscopic surface lead inevitably to complete oxidation to maghemite.12,16,17,38 As described by da Costa et al. it seems that once oxidation begins at the surface it will extend to the entire particle volume.38 In our case all isomer shift values determined from fits indicate that the samples are composed of maghemite. We have not found indication for divalent iron that should be present in magnetite. The Relative Absorption Area (RAA) at 20 K for site B was bigger than site A, respectively. The isomer shift (δ) values ranged from 0.31 to 0.40 mm s−1 for sites A and 0.41 to 0.50 mm s−1 for site B. Also, the hyperfine magnetic fields (Bhf) were found to be consistent for sites A and B in inverse cubic spinel maghemite. The third sextet included in the fitting, that corresponds to the outermost layers, have RAA, δ and Bhf ranging from 8 to 24%, δ = 0.35 to 0.42 mm s−1 and Bhf = 47.7 to 49.5 T, respectively.
Table 2 summarizes the saturation magnetization (Ms) obtained from the law of approach to saturation (LAS) by fitting the M–H loops (see Fig. 8) at 300 K with the equation:39
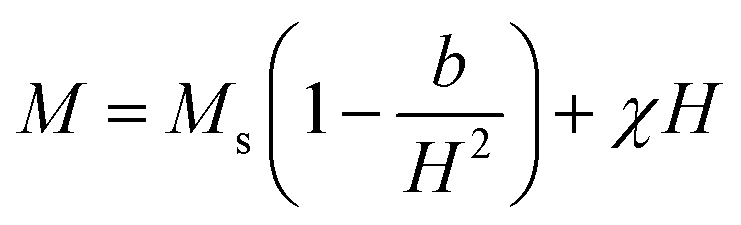
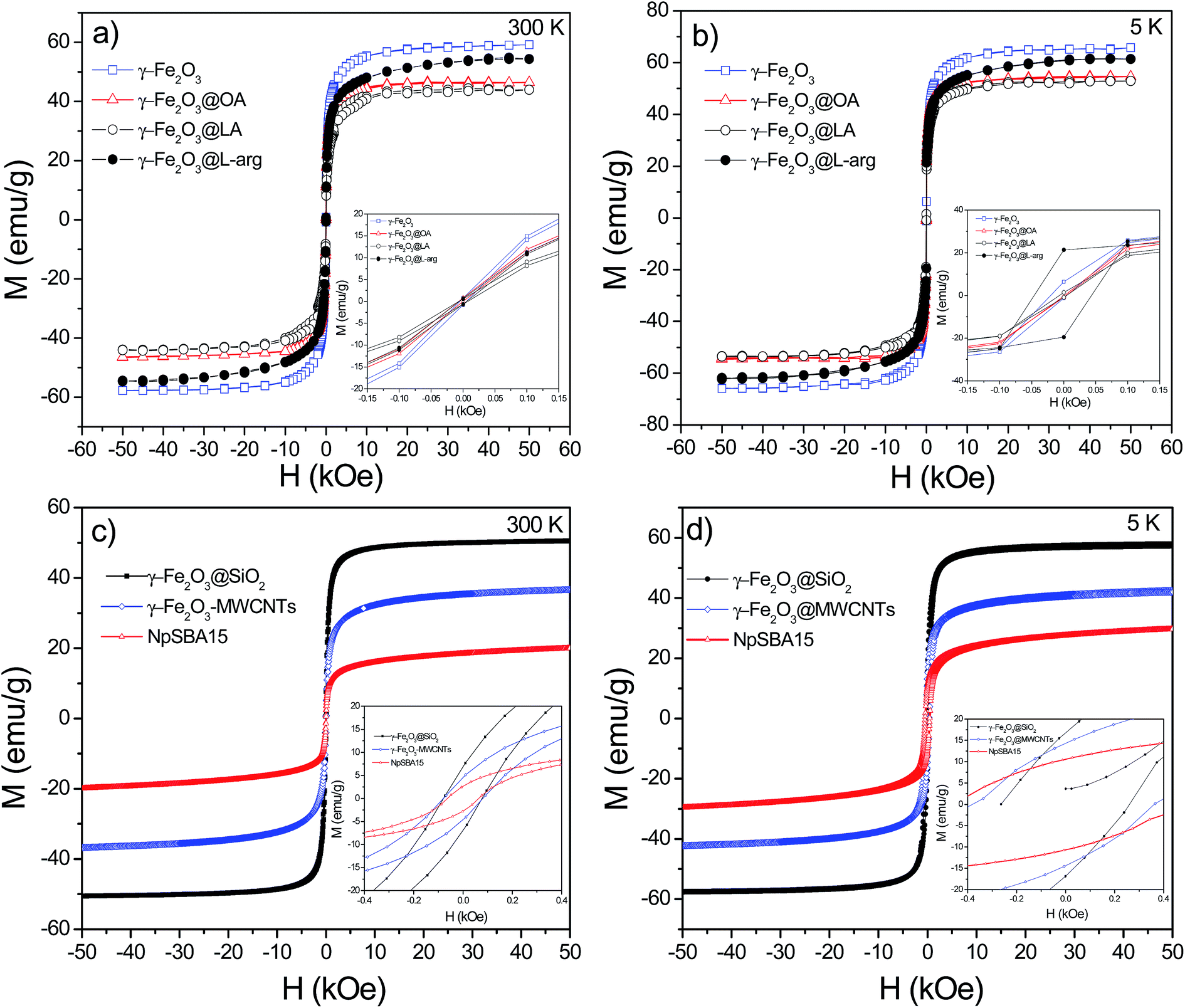 | ||
| Fig. 8 M–H loops at RT and 5 K for γ-Fe2O3-2, γ-Fe2O3@OA, γ-Fe2O3@LA, γ-Fe2O3@L-arg (a and b) and M–H loops at RT and 5 K for γ-Fe2O3@SiO2, γ-Fe2O3@MWCNTs and γ-Fe2O3-SBA15 (c and d). | ||
The Ms values for uncoated γ-Fe2O3 Nps ranged from 59–62 emu g−1. A significant decrease in the Ms value was observed for γ-Fe2O3-1 Nps after coating with a SiO2 shell of 8 nm (average value, see inset in Fig. 2a) according to TEM results. Similarly, for γ-Fe2O3-2 the Ms values decreased after coating with carboxyl OA and LA acids. Nevertheless, the γ-Fe2O3@L-arg Nps exhibit a value of 54 emu g−1 which is higher than the previous functionalized system even to those Nps functionalized with MWCNTs (37 emu g−1).40 Besides, VSM measurements were performed in the as received MWCNTs, before functionalization with γ-Fe2O3 Nps, the measurement showed a ferromagnetic signal (see Fig. S2†). The value obtained from M–H loops was 0.4 emu g−1, which may be assigned to Ni residues from metal catalyst used in the synthesis of carbon nanotubes. This result is consistent with the semi quantitative analysis made by EDX analysis (see Fig. S3†). On the other hand, at 300 K, only samples γ-Fe2O3@MWCNTs and γ-Fe2O3@SiO2 exhibit coercivity field values of ∼80 Oe indicating that these particles are magnetically blocked, in agreement with their Mössbauer spectra recorded at 300 K that showed superparamagnetic and magnetic components with a large RAA for the magnetic component. At 5 K, all samples have a Mr/Ms ratio smaller than 0.5, indicating that these samples are aggregated, magnetic interacting among each other and do not follow the Stoner–Wohlfarth criteria for blocked superparamagnetic particles. At 300 K, the other functionalized samples have coercivity values smaller than 15 Oe and therefore have a superparamagnetic like behavior. These results are in agreement with the smaller RAA for the blocked spectral component.
The temperature dependence of the magnetization is displayed in Fig. 9. In ZFC M–T measurements, all samples showed broad peaks below 300 K. The temperature of the maximum in the ZFC M–T curve is assigned to the blocking like temperature (TB) of Nps. The samples γ-Fe2O3@LA, γ-Fe2O3@OA and γ-Fe2O3-SBA15 showed TB like values smaller than 150 K indicating that these particles are smaller; Mössbauer spectra of these samples showed a strong superparamagnetic contribution at 300 K. The irreversibility temperature where the ZFC and FC M–H curves depart from each other may be used as an indicator of the size distribution width. Thus, sample γ-Fe2O3-SBA15 seems to have a larger size distribution as compared with samples γ-Fe2O3@LA and γ-Fe2O3@OA. The others functionalized samples have a ZFC broad maximum at temperatures close and even above (sample γ-Fe2O3@SiO2) room temperature. Since their particle sizes are similar, the peak position may result from the agglomeration of Nps. In samples γ-Fe2O3@L-arg and γ-Fe2O3@MWCNTs the agglomeration may be promoted by the aminoacid and the carboxyl functional groups decorated on the MWCNTs which can bond to two particles and bring them together. In the case of γ-Fe2O3@SiO2, the difference between the point of zero charge of silica (at pH = 2.5) and γ-Fe2O3 (at pH = 7) may be the reason, at pH in between these values silica has negative charges on its surface and γ-Fe2O3 has positive charges, therefore there is an attractive electric force for clustering.
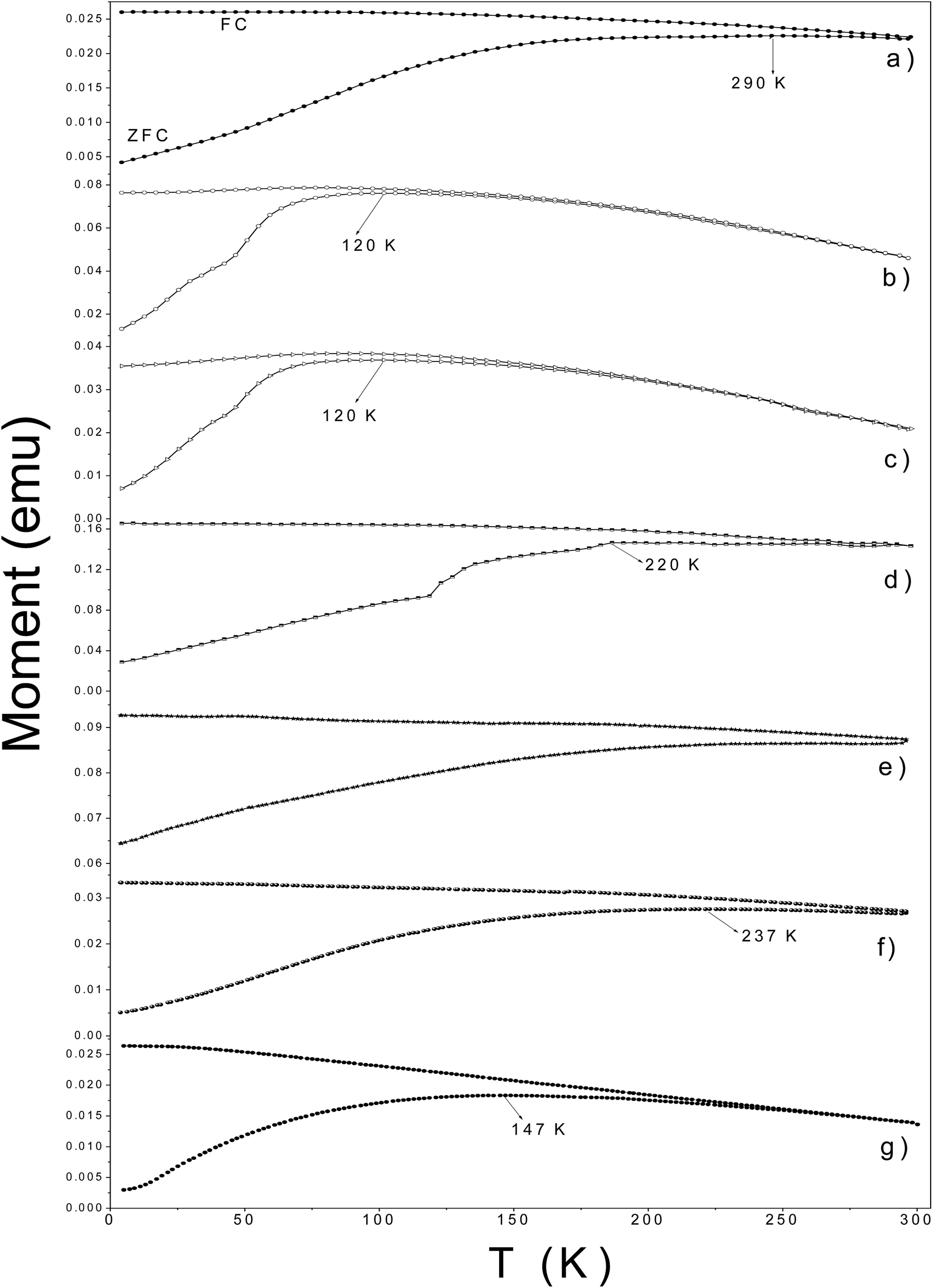 | ||
| Fig. 9 ZFC and FC M–T measurements for γ-Fe2O3-2 (a), γ-Fe2O3@OA (b), γ-Fe2O3@LA (c), γ-Fe2O3@L-arg (d), γ-Fe2O3@SiO2 (e), γ-Fe2O3@MWCNTs (f) and γ-Fe2O3-SBA15 (g). Hext = 80 Oe. | ||
N2 adsorption–desorption isotherms and pore size distributions calculated by the B.J.H. method of pure and functionalized γ-Fe2O3 samples are shown in Fig. 10 and 11. The textural properties including B.E.T. specific surface area, pore volume and pore diameter are given in Table 2. In general, the isotherms are classified as type II according to the IUPAC classification, attributed to slightly porous or macroporous solids.41 As it can be seen the isotherms showed a significant increment along with a hysteresis loop in the relative pressure region between 0.6 and 1, indicating the presence of interparticle porosity probably due to the irregular agglomeration of nanoparticles resulting in a pore size wide distribution of mesopores and macropores (Fig. 11). The estimated pore diameters ranged from 4.7 nm to more than one hundred nanometers which confirmed the presence of mesopores in all the samples. The B.J.H. distributions as well as the isotherms of γ-Fe2O3-1, γ-Fe2O3-EDTA1, γ-Fe2O3@OA and γ-Fe2O3@LA indicated a more regular distribution of the formed pores giving pore sizes uniform values of 8.8, 7.8, 4.7, and 10 nm respectively. The samples obtained magnetic Nps coated with L-arg, HAp, SiO2 and the o-MWCNTs supporting the Nps display wider PSD indicating a more irregular agglomeration.
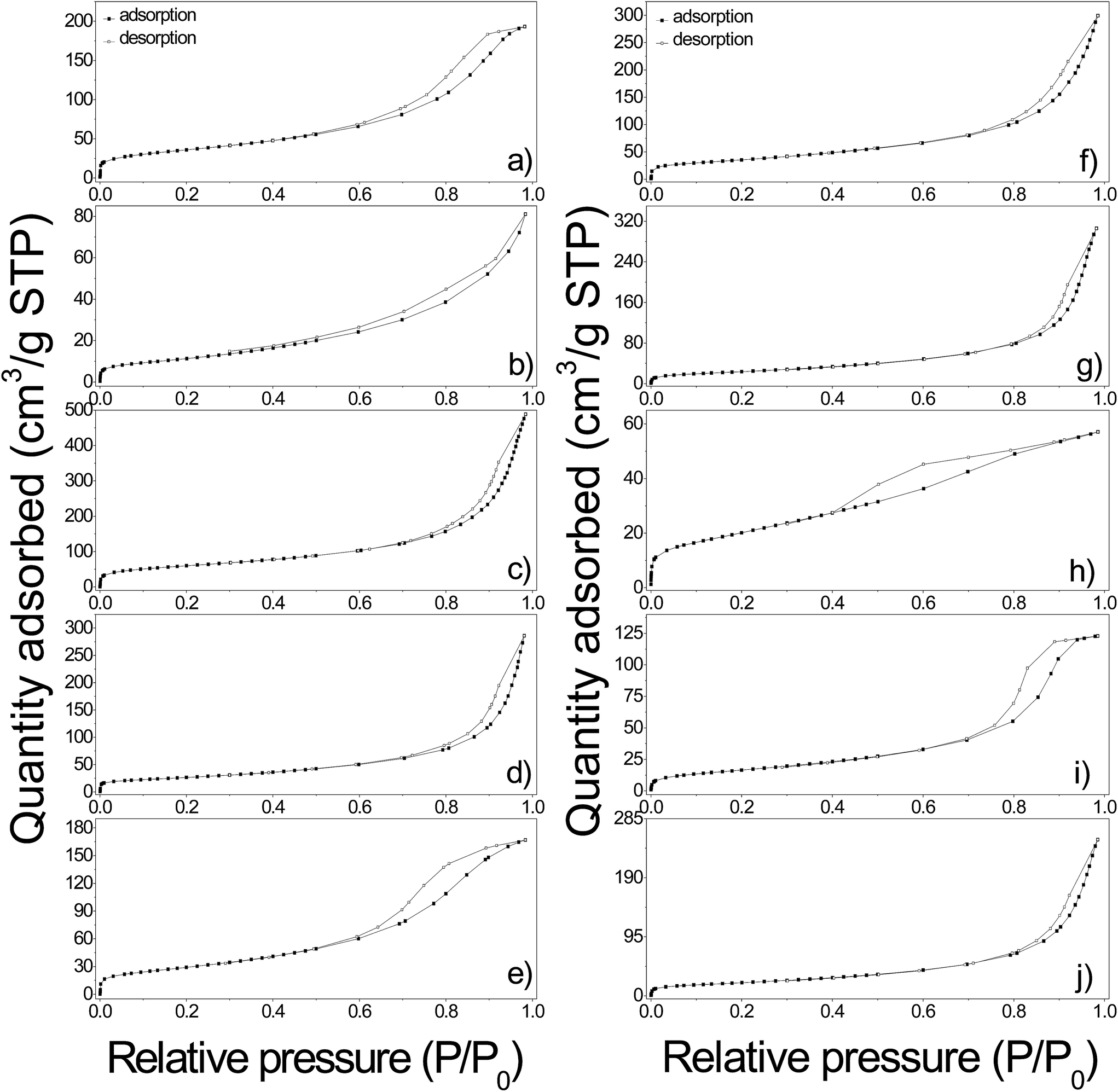 | ||
| Fig. 10 Nitrogen adsorption–desorption isotherms at 77 K of γ-Fe2O3-1 (a), γ-Fe2O3@SiO2 (b), γ-Fe2O3-SBA15 (c), γ-Fe2O3@HAp (d), γ-Fe2O3-EDTA1 (e), γ-Fe2O3@MWCNTs (f), γ-Fe2O3-2 (g), γ-Fe2O3@OA (h), γ-Fe2O3@LA (i) and γ-Fe2O3@L-arg (j). | ||
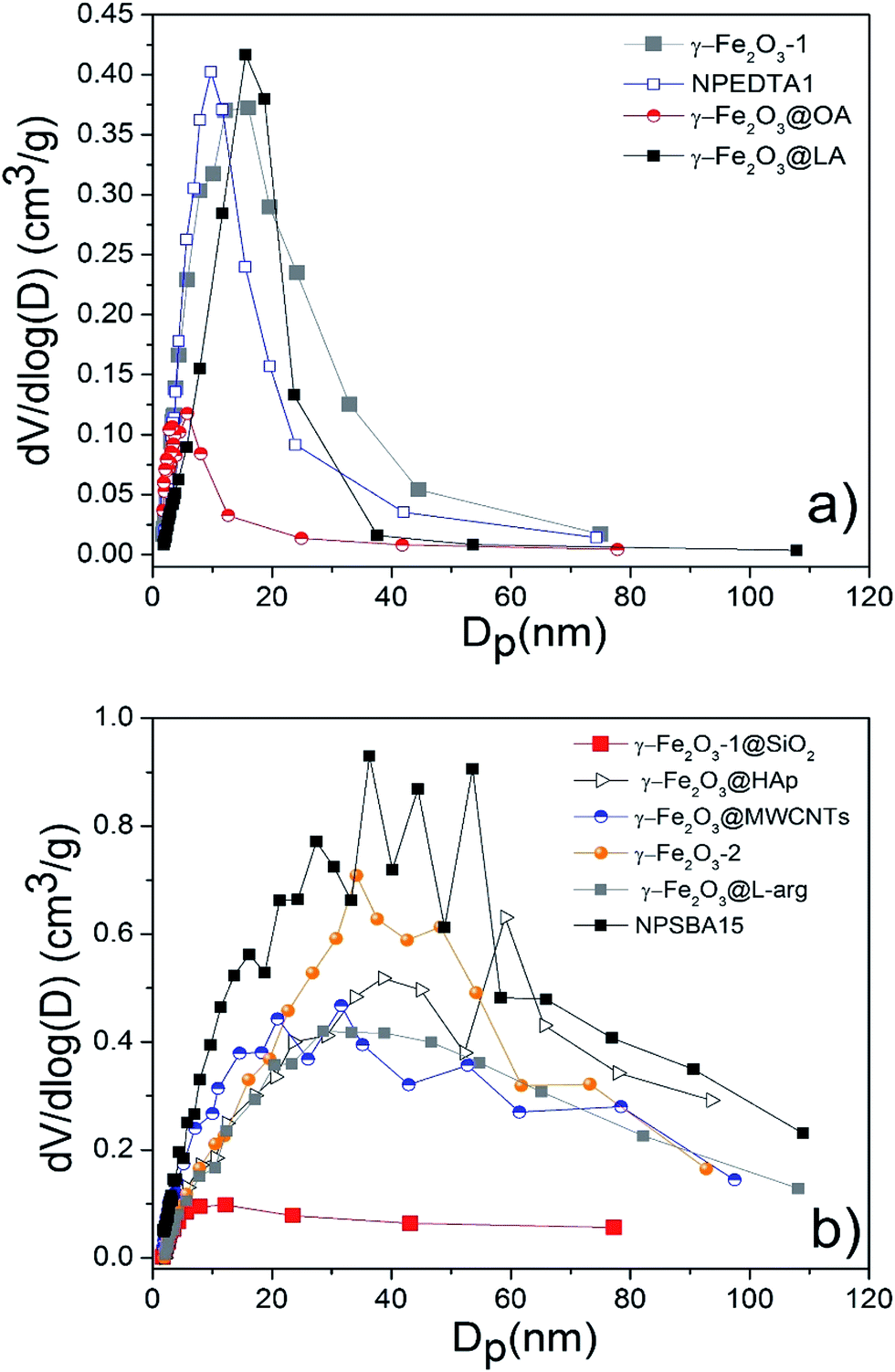 | ||
| Fig. 11 Pore size distribution plots for all indicated samples. Nanoparticles with narrow (a) and broad (b) pore size distribution. | ||
The γ-Fe2O3-1 Nps present a higher B.E.T. specific surface area of 129.7 m2 g−1 compared to γ-Fe2O3-2 Nps (88.3 m2 g−1) synthesized by the same method, but using a different alkaline reagent. This change could be assigned to the difference in the mean particle size since γ-Fe2O3-2 Nps exhibited a larger diameter compared with γ-Fe2O3-1. These values are relatively higher than others found in the literature for magnetic nanoparticles.2 After coating γ-Fe2O3-1 with SiO2 the B.E.T. specific surface area exhibited a noticeable decrease (42.3 m2 g−1) along with a significant reduction in nitrogen adsorption, caused probably by the silica layer (thickness of 8 nm) loaded onto Nps surface. The pure SBA15 sample has a specific BET surface of 790 m2 g−1,13 and it considerably decreased to 214 m2 g−1 after functionalizing with maghemite Nps, this result may be related to the deposition of maghemite Nps in the SBA15 pores that induces a blocking of pore entrance or a simply reduction of the free volume inside pores. The functionalization with fatty acids (oleic and lauric acids) of γ-Fe2O3-2 Nps cause a slightly decrease in the B.E.T. specific surface area since the exposed area is covered by the carboxylic molecules resulting from functionalization. Similarly, the L-arg aminoacid functionalized γ-Fe2O3 Nps exhibited the same behavior with a B.E.T. specific surface area of 77.5 m2 g−1.
On the other side, HAp functionalized γ-Fe2O3 Nps exhibited a B.E.T. specific surface area of 95.5 m2 g−1, which is a higher value in comparison to those found in literature.42 The B.E.T. specific surface area value for γ-Fe2O3-EDTA1 indicates that EDTA strongly favor the increment in the B.E.T. specific surface area as a comparison with fatty and amino acids loaded onto Nps surface. In addition the sample γ-Fe2O3@MWCNTs nanohybrids shows an increment in the B.E.T. specific surface area (129.6 m2 g−1) respect to the MWCNTs without functionalization (110 m2 g−1).
Adsorption experiments were carried out for individual solutions of copper(II) and lead(II) at two different times in order to check the metal adsorption performance of functionalized nanoparticles. After adsorption not iron traces were found for the final Cu(II) and Pb(II) solutions in any case.
Copper was satisfactorily adsorbed onto uncoated and functionalized γ-Fe2O3 Nps. The final Cu(II) concentrations in the solutions and adsorption capacities for periods of 7 and 20 h are shown in Table 4. The given values represent the average values determined for two independent experiments carried out with each sample during 7 and 20 h for the same initial concentration of 40 mg L−1. In general, an increment of the adsorbed amount was observed for larger times. In the comparison of results obtained for γ-Fe2O3-1 and γ-Fe2O3-2, it can be extracted that the mean particle size of uncoated maghemite Nps was not a key factor in the Cu(II) adsorption capacity since adsorbed amount is similar for both times. The samples containing SiO2 Nps and mesoporous SBA15 have comparable Cu(II) adsorption capacities as the bare Nps. As it can be seen, although all the functionalized systems adsorb significant quantities of Cu(II), the samples γ-Fe2O3@HAp and γ-Fe2O3@L-arg Nps showed the best performance achieving a high adsorption capacity of 73 and 88 mg g−1 after 20 h of contact.
| Adsorbent | C7 (mg L−1) | q7 (mg g−1) | C20 (mg L−1) | q20 (mg g−1) |
|---|---|---|---|---|
| γ-Fe2O3-1 | 6 | 61.3 | 6.7 | 78 |
| γ-Fe2O3-2 | 8.3 | 57.1 | 11.5 | 69.3 |
| γ-Fe2O3@SiO2 | 3.9 | 64.6 | 13.7 | 65.4 |
| γ-Fe2O3@OA | 12.7 | 49.3 | 18.2 | 57.3 |
| γ-Fe2O3@LA | 8.1 | 57.5 | 15.2 | 62.7 |
| γ-Fe2O3@L-arg | 3.3 | 66.1 | 9.6 | 72.6 |
| γ-Fe2O3@HAp | 0.3 | 71.5 | 1 | 88.2 |
| γ-Fe2O3-EDTA1 | 9 | 55.8 | 12.7 | 67.2 |
| γ-Fe2O3@MWCNTs | 5.5 | 62.1 | 13.3 | 66.2 |
| γ-Fe2O3-SBA15 | 2.3 | 62.5 | 2.9 | 61.4 |
For Pb(II) batch experiments all the final solutions showed Pb(II) concentrations lower than 0.05 mg L−1 from an initial concentration of 50 mg L−1, which means that all the Pb(II) species were adsorbed for the functionalized Nps. Although some of our samples showed small specific surface area values, their colloidal stability and zeta potential value allow for their adsorption capacity. One can notice that the fatty and amino acids coated Nps which present negative charge at their surface, have strong electrostatic affinity for metal cations through the free carboxylate presented in the surface. Regarding the EDTA functionalized Nps, Dragan et al. reported the complexation of EDTA with metal cations occurs via a tetrahedral tetracoordinate complex with covalent Pb–N bonds and ionic Pb–carboxylate bonds.43 However, it is important to note that in the above configuration only two carboxyl groups of EDTA could participate while the remaining carboxyl groups are bond to Fe core through a monodentate configuration.17 In the case of nanoparticles functionalized with hydroxyapatite, HAp, it was proposed that Pb(II) adsorption occurred through an ion exchange reaction. The next irreversible equation is in general valid for the pH region between pH = 3.0 to 6.0.44
| Ca10(POH4)6(OH)2 + xPb2+ → xCa2+ + Ca10−xPbx(PO4)6(OH)2 |
In our work, we used a pH = 5.5 for the initial solution of Pb(II). The reaction could occur also for other divalent cations such as Cu(II). In the case of silica, it has an isoelectric point (p.z.c.) at pH = 2.5, above this point its surface is coated by OH− groups favoring adsorption via electrostatic interaction with the metal cations.45
Moreover, maghemite Nps are not only used for magnetic separation purposes, they also present significant adsorption capabilities. Liu et al. found that Cd(II) was bound directly to Fe3O4 despite this was coated with humic acid.46 In our case γ-Fe2O3-1 and γ-Fe2O3-2 adsorbed better Cu(II) quantities than silica and MWCNTs bond to the Nps. It is worth mentioning that the surface chemical structure of maghemite presents Fe–OH and ≡Fe–O− sites, which act as a Lewis base.46 These functional groups can interact and coordinate with Pb(II) metal ions depending on the pH and p.z.c. of the adsorbent.47 The pH value of initial solutions is close to 5.5, being the Cu2+ and Pb2+ the predominant species in solution.48 Regarding the p.z.c. of maghemite, it was reported to be 8.6,45 thus particle surface is being likely a protonated surface at pH 5.5, so an electrostatic repulsion is expected. However, the affinity of metal ions to γ-Fe2O3 is higher than that of H+ ions, indicating that metal ions replace the adsorbed H+ ions from the Nps surface through an ion exchange mechanism.47,49
Conclusions
In this study, several γ-Fe2O3 Nps functionalized with organic acids, aminoacid, silica and carbon nanotubes were successfully synthesized by the co-precipitation method, obtaining Nps with particle sizes ranging from 7–16 nm according to Rietveld refinement and TEM images analysis. The zeta potential studies and IR results showed that the functionalization was successful. The magnetic properties such as the saturation magnetization is affected by the surface's modification with carboxyl, aminoacid and silica. The γ-Fe2O3 Nps displayed a superparamagnetic like behavior as proved by Mössbauer spectroscopy and ZFC M–T measurements. The functionalized γ-Fe2O3 Nps exhibited BET specific surface area ranging from 74 to 214 m2 g−1 suitable for metal adsorption. All the nanomaterials present remarkable uptake capacities to adsorb Cu(II) and Pb(II) cations. Under the studied chemical conditions functionalized γ-Fe2O3 Nps can significantly reduce the heavy metal concentration in solution, with Pb(II) being totally adsorbed after 7 h of interaction. Thus, it was concluded that these nanomaterials are promising to be applied for water remediation processes.Acknowledgements
This work was partially supported by The National Council of Science, Technology and Technological Innovation (CONCYTEC/Cienciactiva-Peru). The authors wish to thank Dr Alexandre Mello by allow access to the X-ray photoelectron spectroscopy equipment of the Multiuser Laboratory of Surfaces and Nanostructures at CBPF. M. Morales thanks Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for his fellowship, process number 305748/2015-7. F. J. Litterst and E. Sadrollahi are grateful for partial support by DAAD. This work was also supported by the Ministerio de Economía y Competitividad of Spanish Government through the research projects CTM2015-69246-R.Notes and references
- F. Fernández-Luqueño, F. López-Valdez, P. Gamero-Melo, S. Luna-Suárez, E. N. Aguilera-González, A. I. Martínez, M. García-Guillermo, G. Hernández-Martínez, R. Herrera-Mendoza, M. A. Álvarez-Garza and I. R. Pérez-Velázquez, Afr. J. Environ. Sci. Technol., 2013, 7, 567 Search PubMed
.
- I. Ali, Chem. Rev., 2012, 112, 5073 CrossRef CAS PubMed
- B. I. Kharisov, H. V. R. Dias, O. V. Kharissova, V. M. Jiménez-Pérez, B. O. Pérez and B. M. Flores, RSC Adv., 2012, 2, 9325 RSC
- M. Arbabi and N. Golshani, Int. J. Epidemiol., 2016, 3, 283–293 Search PubMed
- L. J. Holcombe and G. P. Behrens, Environ. Prog., 1987, 6, 74 CrossRef CAS
- M. M. Khin, A. S. Nair, V. J. Babu, R. Murugan and S. Ramakrishna, Energy Environ. Sci., 2012, 5, 8075 CAS
- W. Wu, Q. He and C. Jiang, Nanoscale Res. Lett., 2008, 3, 397 CrossRef CAS PubMed
- S. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204 CrossRef CAS PubMed
- R. A. Sperling and W. J. Parak, Philos. Trans. R. Soc., A, 2010, 368, 1333 CrossRef CAS PubMed
- J. A. R. Guivar, E. G. R. Fernandes and V. Zucolotto, Talanta, 2015, 141, 307 CrossRef CAS PubMed
- Y. S. Kang, S. Risbud, J. F. Rabolt and P. Stroeve, Chem. Mater., 1996, 8, 2209 CrossRef CAS
- J. A. R. Guivar, A. Bustamante, J. Flores, M. M. Santillan, A. M. Osorio, A. I. Martínez, L. Valladares and C. H. W. Barnes, Hyperfine Interact., 2014, 224, 89 CrossRef
- J. Aguado, J. M. Arsuaga, A. Arencibia, M. Lindo and V. Gascón, J. Hazard. Mater., 2009, 163, 213 CrossRef CAS PubMed
- C. L. Chen, X. K. Wang and M. Nagatsu, Environ. Sci. Technol., 2009, 43, 2362 CrossRef CAS PubMed
- C. Luo, Z. Tian, B. Yang, L. Zhang and S. Yan, Chem. Eng. J., 2013, 234, 256 CrossRef CAS
- J. A. R. Guivar, E. A. Sanches, F. Bruns, E. Sadrollahi, M. A. Morales, E. O. López and F. Jochen Litterst, Appl. Surf. Sci., 2016, 389, 721 CrossRef
- J. A. Ramos Guivar, M. A. Morales and F. Jochen Litterst, J. Magn. Magn. Mater., 2016, 420, 324 CrossRef
- EPA, Method 200.7, Trace elements in water, solids and biosolids by inductively coupled plasma atomic emission spectrometry, EPA-821-R-01–010, January 2001 Search PubMed
- T. Zhao, C. Hou, H. Zhang, R. Zhu, S. She, J. Wang, T. Li, Z. Liu and B. Wei, Sci. Rep., 2014, 4, 5619 CrossRef CAS PubMed
- D. Rehana, A. K. Haleel and A. K. Rahiman, J. Chem. Sci., 2015, 127, 1155 CrossRef CAS
- R. Ghosh, L. Pradhan, Y. P. Devi, S. S. Meena, R. Tewari, A. Kumar, S. Sharma, N. S. Gajbhiye, R. K. Vatsa, B. N. Pandey and R. S. Ningthoujam, J. Mater. Chem., 2011, 21, 13388 RSC
- R. G. López, M. G. Pineda, G. Hurtado, R. Díaz de León, S. Fernández, H. Saade and D. Bueno, Int. J. Mol. Sci., 2013, 14, 19636 CrossRef PubMed
- H. P. Yiu, M. A. Keane, Z. A. D. Lethbridge, M. R. Lees, A. J. El Haj and J. Dobson, Nanotechnology, 2008, 19, 255606 CrossRef CAS PubMed
- J. Chen, H. Shen, X. Li, J. Ruan and W. Yuan, Chem. Pap., 2016, 70, 1642 Search PubMed
- W. T. Herrera, J. A. R. Guivar, J. C. Gonzalez and E. M. Baggio-Saitovitch, 7th International Conference on nanomaterials - Research & application, Proceedings NANOCON, TANGER Ltd, Ostrava, Czech Republic, 1st edn, 2015, pp. 511–519, ISBN 978-80-87294-63-5 Search PubMed
- M. Bloemen, W. Brullot, T. T. Luong, N. Geukens, A. Gils and T. Verbiest, J. Nanopart. Res., 2012, 14, 1100 CrossRef PubMed
- N. Baccile, R. Noiville, L. Stievano and I. V. Bogaert, Phys. Chem. Chem. Phys., 2013, 15, 1606 RSC
- Y. Zhang, B. Wang, X. Meng, G. Sun and C. Gao, Ann. Biomed. Eng., 2011, 39, 414 CrossRef PubMed
- J. Y. Park, E. S. Choi, M. J. Baek and G. H. Lee, Mater. Lett., 2009, 63, 379 CrossRef CAS
- A. E. Aliaga, C. Garrido, P. Leyton, G. F. Diaz, J. S. Gomez-Jeria, T. Aguayo, E. Clavijo, M. M. Campos-Vallette and S. Sanchez-Cortes, Spectrochim. Acta, Part A, 2010, 76, 458 CrossRef CAS PubMed
- D. Wilson and M. A. Langell, Appl. Surf. Sci., 2014, 303, 6 CrossRef CAS
- Z. Wang, H. Zhu, X. Wang, F. Yang and X. Yang, Nanotechnology, 2009, 20, 465606 CrossRef PubMed
- J. B. Wu, Y. F. Lin, J. Wang, P. J. Chang, C. P. Tasi, C. C. Lu, H. T. Chiu and Y. W. Yang, Inorg. Chem., 2003, 42, 4516 CrossRef CAS PubMed
- Y. Shan and L. Gao, Nanotechnology, 2005, 16, 625 CrossRef CAS
- C. Chaneac, E. Tronc and J. P. Jolivet, J. Mater. Chem., 1996, 6, 1905 RSC
- X. T. H. Hsieh, K. S. Ho, X. Bi, Y. K. Han, Z. L. Chen, C. H. Hsu and Y. C. Chang, Eur. Polym. J., 2009, 45, 613 CrossRef
- M. Herlitschke, S. Disch, I. Sergueev, K. Schlage, E. Wetterskog, L. Bergström and R. P. Hermann, J. Phys.: Conf. Ser., 2016, 711, 012002 CrossRef
- G. M. da Costa, C. Blanco-Andujar, E. De Grave and Q. A. Pankhurst, J. Phys. Chem. B, 2014, 118, 11738 CrossRef CAS PubMed
- J. A. R. Guivar, A. I. Martínez, A. O. Anaya, L. D. L. S. Valladares, L. L. Félix and A. B. Dominguez, Adv. Nanopart., 2014, 3, 114 CrossRef CAS
- A. Demir, A. Baykal, H. Sözeri and R. Topkaya, Synth. Met., 2014, 187, 75 CrossRef CAS
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051 CrossRef CAS
- L. El Hammari, H. Merroun, T. Coradin, S. Cassaignon and A. Laghzizil, Mater. Chem. Phys., 2007, 104, 448 CrossRef CAS
- S. Dragan and A. Fitch, J. Chem. Educ., 1998, 75, 1018 CrossRef CAS
- Y. Takeuchi, T. Suzuki and H. Arai, J. Chem. Eng. Jpn., 1998, 21, 98 CrossRef
- G. Cao and Y. Wang, Nanostructures and Nanomaterials, Synthesis, properties and applications, ©World Scientific Publishing Co. Pte. Ltd, 2nd edn, 2004, ISBN: 978-981-4322-50-8 Search PubMed.
- J. F. Liu, Z. S. Zhao and G. B. Jiang, Environ. Sci. Technol., 2008, 42, 6949 CrossRef CAS PubMed
- M. Kumari, C. U. Pittman Jr and D. Mohan, J. Colloid Interface Sci., 2015, 442, 120 CrossRef CAS PubMed
- J. Aguado, J. M. Arsuaga and A. Arencibia, Int. J. Environ. Technol. Manage., 2010, 12, 381 CrossRef CAS
- Y. F. Shen, J. Tang, Z. H. Nie, Y. D. Wang, Y. Ren and L. Zuo, Sep. Purif. Technol., 2009, 68, 312 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02750h |
| This journal is © The Royal Society of Chemistry 2017 |