
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring concentration, surface area and surface chemistry effects of colloidal aggregates on fat crystal networks†
Raamanand R. Chauhan *a,
Roel P. A. Dullensa,
Krassimir P. Velikovbc and
Dirk G. A. L. Aarts*a
*a,
Roel P. A. Dullensa,
Krassimir P. Velikovbc and
Dirk G. A. L. Aarts*a
aDepartment of Chemistry, Physical and Theoretical Chemistry Laboratory, University of Oxford, South Parks Road, Oxford, OX1 3QZ, UK. E-mail: raamanand.chauhan@chem.ox.ac.uk; dirk.aarts@chem.ox.ac.uk; Tel: +44 (0)1865 275400
bUnilever R&D Vlaardingen, Olivier van Noortlaan 120, 3133 AT Vlaardingen, The Netherlands
cSoft Condensed Matter, Debye Institute for Nanomaterials Science, Utrecht University, Princetonplein 5, 3584 CC Utrecht, The Netherlands
First published on 31st May 2017
Abstract
We study the effects of concentration, surface area and surface chemistry of fractal fumed silica aggregates on a model fat-structured food system. We use oscillatory stress sweep rheology to determine the storage modulus, the length of the linear viscoelastic region and the rate of network breakdown after the linear region. Differential scanning calorimetry shows that silica is not acting as a seed for nucleation and that the melting profiles for samples with silica are not significantly different. We interpret these results in terms of hydrogen bonding between the silica aggregates and its role in strengthening the fat crystal networks.
1 Introduction
Fat crystal networks are a major component in many foods like butter and chocolate. The microstructure of these three-dimensional crystal networks has been directly linked to the macroscopic properties like mouthfeel and spreadability that are experienced by the consumer.1,2 There have been many different studies on the effects of adding extra components into fat networks, some of which were summarized by Smith et al.3 A common reason for adding new components is to control the crystallization behaviour and the resulting crystal polymorph, which is of paramount importance in the chocolate industry; for example added tripalmitin and monoacylglycerols have been shown to favour the formation of β-crystals over the β′-type in palm oil crystallization.4 Solid sugar and cocoa particles have been used in chocolate model systems to reduce the risk of fat bloom by acting as seeds to create a more homogeneous microstructure.5,6 More recently additives have been introduced into fat crystal networks to provide health benefits, like β-carotene which is an anti-oxidant and a precursor of vitamin A.7Our previous work8 has shown that it is possible to use colloidal silica aggregates to reduce the amount of solid fat whilst maintaining similar rheological and melting properties. In our model system, we begin with a pre-melting step to destroy any fat crystal memory, which leaves only a silica network at high temperatures. This network is relatively weak and so as the fat crystallizes it is able to move and rearrange the silica aggregates. This results in a layer of silica aggregates on the surface of the fat crystal cluster chains forming a composite gel network, which is illustrated in Fig. 1.
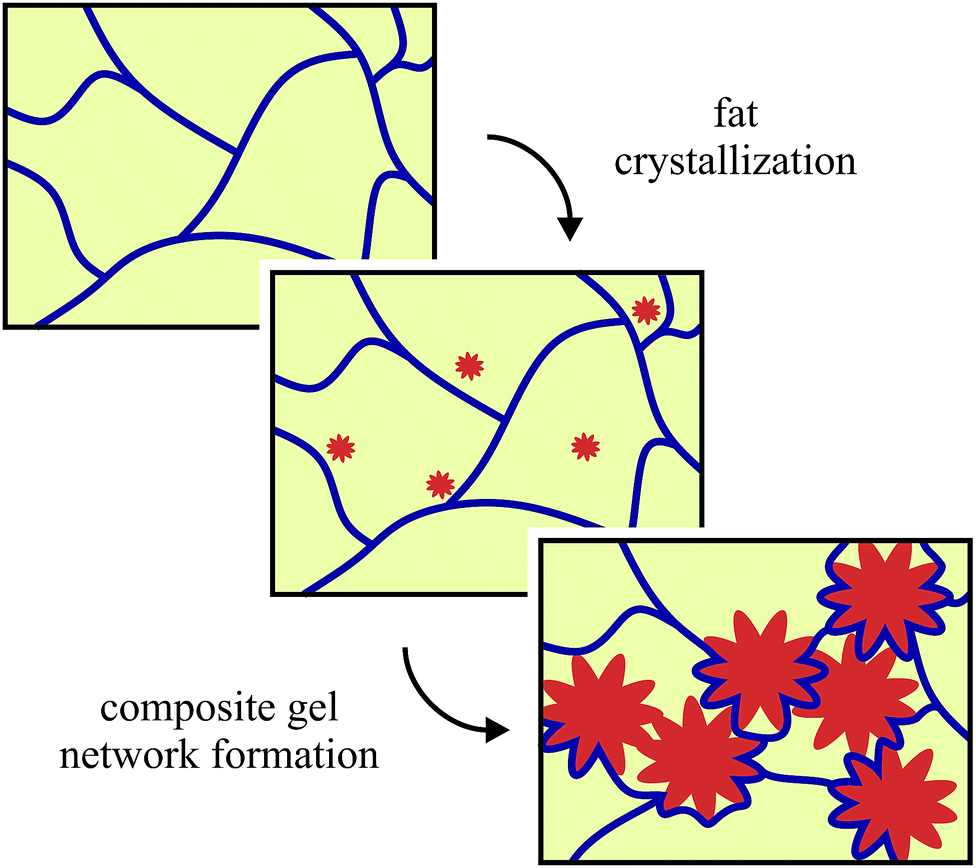 | ||
| Fig. 1 A schematic illustration for the formation of the composite fat-silica networks. The silica network is shown in blue, the oil is yellow and the fat crystals are orange. Not drawn to scale. | ||
Our present study uses the same model system for fat-structured products to further investigate the effects of the fumed silica aggregates. In particular we focus on three different aspects of the silica: (i) concentration, (ii) surface area and (iii) surface chemistry. Our overall aim is to use this model system to understand the role of the added colloidal aggregates on the fat networks and the resulting macroscopic properties. For our previous study,8 we held the silica concentration constant and varied the fat concentration. Here we hold the fat concentration constant and vary the silica concentration, in order to see the effect on the resulting fat crystal network and to determine which properties are silica concentration dependent. Also, from a financial and manufacturing point of view it is important to determine the minimum required amount of colloidal aggregates necessary to have a desired rheological effect.
As the colloidal aggregate we use fumed silica, which is a type of colloidal silicon dioxide used in the food industry as an anti-caking and anti-foaming agent (E551). It is made via flame hydrolysis of silicon tetrachloride (SiCl4) which produces spherical primary particles.9 Whilst they are still hot from the production process, they fuse together irreversibly to form branched, fractal aggregates. By varying the process conditions it is possible to vary the primary particle size, which causes a change in the surface area of the fractal aggregates. Untreated fumed silica is naturally hydrophilic since the surface is covered with silanol (–Si–OH) functional groups. In oil, untreated silica aggregates bind to each other via hydrogen bonding rather than interacting with the non-polar solvent.10 In this study, we conduct experiments with a range of different silicas both with and without solid fat to understand how the silica surface area, and in turn the number of silanol groups available for hydrogen bonding, can affect the resulting fat crystal networks.
Chemical treatment of fumed silica replaces a portion of the surface silanol groups with different chemical functional groups. The chemically treated silicas used in this study are commercially available and the added functional groups are shown in Fig. 2, along with the molecular structure of tripalmitin fat. Typically, in cases where there is a large mismatch between the functional groups on the surface of the particle and in the solvent, the particles will flocculate to form a gel.11,12 The chemically modified silicas have a reduced mismatch with the oil in our model system, compared to the untreated silica, and so we expect to see a difference in the gelation behaviour. It has also been shown by Smith et al.3 that additives with a similar chain length and number of double bonds to the fat are more likely to affect the nucleation and growth processes of the fat crystals and we want to explore how this affects the fat crystal networks that are formed.
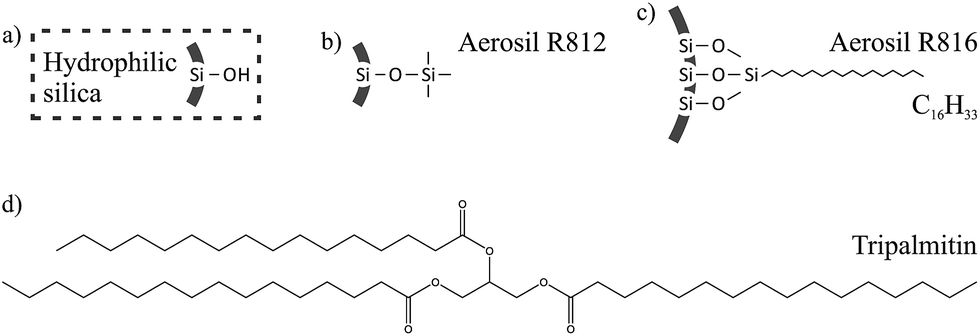 | ||
| Fig. 2 Surface functional groups for (a) untreated hydrophilic silica, (b) R812 hydrophobic silica and (c) R816 hydrophobic silica. (d) The molecular structure of tripalmitin fat. | ||
In the next section we give details on the materials and methods used in these experiments. Section 3 begins with the results for the experiments looking at silica concentration. This is then followed by the results for the surface area and surface chemistry experiments, then finally our conclusions in Section 4.
2 Experimental methods
2.1 Chemicals
Tripalmitin fat (≥85% pure) and soybean oil were purchased from Sigma Aldrich and used without further purification. All fumed silica samples used in this study (A300, A255, 200F, R812 and R816) were provided by Evonik Industries AG.2.2 Sample preparation
Soybean oil and silica were mixed in the required proportions with an Ultra Turrax mixer at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 minutes. Solid tripalmitin was then added and the samples were heated in an oven at 100 °C for 40 minutes with regular shaking by hand to ensure the samples were properly mixed. All samples were then sealed and stored in a refrigerator at 8 °C until use. Before each experiment the bulk samples were melted in an oven at 100 °C until completely liquid.
000 rpm for 5 minutes. Solid tripalmitin was then added and the samples were heated in an oven at 100 °C for 40 minutes with regular shaking by hand to ensure the samples were properly mixed. All samples were then sealed and stored in a refrigerator at 8 °C until use. Before each experiment the bulk samples were melted in an oven at 100 °C until completely liquid.
For the silica concentration experiments, a range of samples was prepared with A300 hydrophilic silica concentrations of 1 wt%, 2 wt%, 3 wt% and 4 wt% both with and without 5 wt% fat. We prepared samples for the surface area and surface chemistry experiments using the five different types of silica which are detailed in Table 1. For each type of silica, two samples were made, one with 5 wt% fat and one without, both containing 2 wt% silica. For all samples in this study, the weight percentages (wt%) are given with respect to the total weight of the sample.
| Name | 1° particle size/nm | Surface area/m2 g−1 |
|---|---|---|
| Hydrophilic silicas | ||
| A300 | 7 | 300 ± 30 |
| A255 | — | 255 ± 30 |
| 200F | 12 | 200 ± 25 |
|
||
| Hydrophobic silicas | ||
| R812 | 7 | 260 ± 30 |
| R816 | 12 | 190 ± 20 |
We have chosen to use mass percentages here since the fractal silica aggregates are able to occlude large amounts of oil which leads to much higher effective volume fractions.10 However it is possible to estimate the volume fractions of the components since their densities are known. The density of soybean oil at 25 °C is reported by Sigma Aldrich as 0.92 g mL−1 and the density of fumed silica is 2.2 g cm−3.9 From our previous work8 we know that the α polymorph of tripalmitin is formed under the cooling conditions used in these experiments. Although the density for the α polymorph of tripalmitin is unknown, the density for the more dense β polymorph is 1.047 g mL−1 and the density of the α polymorph of tristearin is 1.014 g mL−1.13 Therefore, for a typical sample of 5 wt% fat and 2 wt% silica in soybean oil, we have approximately 4.5 vol% fat and 0.9 vol% silica.
2.3 Rheology
We used an AR-G2 controlled stress rheometer from TA Instruments fitted with a Peltier plate. Initially, the sample was melted and approximately 1.5 mL was placed on the rheometer, which was closed to a gap of 500 μm with a 60 mm plate geometry. The sample was then heated to 100 °C and sheared at 50 rad s−1 for 3 minutes in order to break down any fat crystal or silica structures, which was important for minimizing the effects of sample history. Next, the sample was allowed to equilibrate for 2 minutes before a 5 minute oscillatory time sweep, which quantified the amount of structure present in the system.The sample was then cooled down to 10 °C at a rate of 10°C min−1. The fat crystal networks were formed via non-isothermal crystallization since this is a more realistic model for industrial processes. During the cooling ramp the plate geometry was stationary in order to avoid shear-induced crystallization. The samples where subsequently heated up to 25 °C min−1 at a rate of 20 °C min−1 where a second time sweep was conducted for 15 minutes. This time sweep captured the network formation processes and we observed that all samples had stopped evolving before any other measurements were taken.
We used oscillatory stress sweep experiments to measure the samples since, for stresses within the linear viscoelastic region (LVR), we could probe the fat and silica networks without disrupting them. The oscillatory stress amplitudes started from 0.01 Pa and increased up to 1000 Pa, with a frequency of 2π rad s−1. These experiments measured the shear storage and loss moduli, G′ and G′′ respectively, which describe the solid- and liquid-like nature of the samples. Each experiment was repeated at least three times and we present the arithmetic mean with the error bars corresponding to plus minus one standard deviation.
2.4 Differential scanning calorimetry
We used a Q20 heat flux DSC from TA Instruments which was calibrated with an indium sample at 10 °C min−1. Initially, the sample was melted then sheared at 9600 rpm with an Ultra Turrax mixer for 5 minutes. Approximately 10 mg of sample was placed into stainless steel pans, which were subsequently hermitically sealed and placed into the furnace of the DSC.At the beginning of the experiment, the sample was heated at 100 °C for 10 minutes in order to destroy any crystal memory. Next, the sample was cooled at 10 °C min−1 to 10 °C followed immediately by a heating ramp at 20 °C min−1 to bring the sample to 25 °C. The samples were then held isothermally at 25 °C for 15 minutes before the melting measurement was taken. We observed that all samples had stopped evolving during this isothermal step.
The melting transition of the sample was measured by increasing the temperature back up to 100 °C from 25 °C with a rate of 20 °C min−1. The sample was then held at 100 °C for 10 minutes to destroy crystal memory before being crystallized for a second time. The second crystallization went down to −20 °C with the same cooling rate of 10 °C min−1. Preliminary results showed that the crystallization peaks were not fully obtained before 10 °C and so a lower temperature was used in order to observe the entire peak. This heating and cooling profile also allowed us to determine the effects of reheating these samples. For all samples in this study we observed that there was no significant difference between the first and second crystallization curves (data not shown), which confirms that the experiments are not affected by any crystal memory or sample history effects. Each experiment was repeated at least three times and we present the arithmetic mean with the error bands corresponding to plus minus one standard deviation. The crystallization onset temperatures were determined using the TA Universal Analysis software.
3 Results & discussion
3.1 Silica concentration
We use oscillatory stress sweeps to measure G′ and G′′ which describe the solid- and liquid-like properties of the samples respectively. Fig. 3a shows G′ as a function of oscillatory stress (G′′ is not shown), for samples with increasing hydrophilic silica (A300) concentrations both with and without 5 wt% fat present. We note that without fat, 1 wt% silica is not sufficient to form a stress transmitting network (G′ < G′′, G′′ is not shown), see also our previous work.8 For samples with no fat but 2 wt% silica or more we observed stress transmitting networks (G′ > G′′, G′′ is not shown). An interesting result to note here is that although 1 wt% silica without fat (□) is itself not enough to form a silica network, the stress curve for the sample with 1 wt% silica and 5 wt% fat (■) has a similar shape to the other fat-silica samples, suggesting that the same stress-transmitting mechanism is at work. This potentially shows that the colloidal additives alone do not need to be able to form a network in order to strengthen the fat network.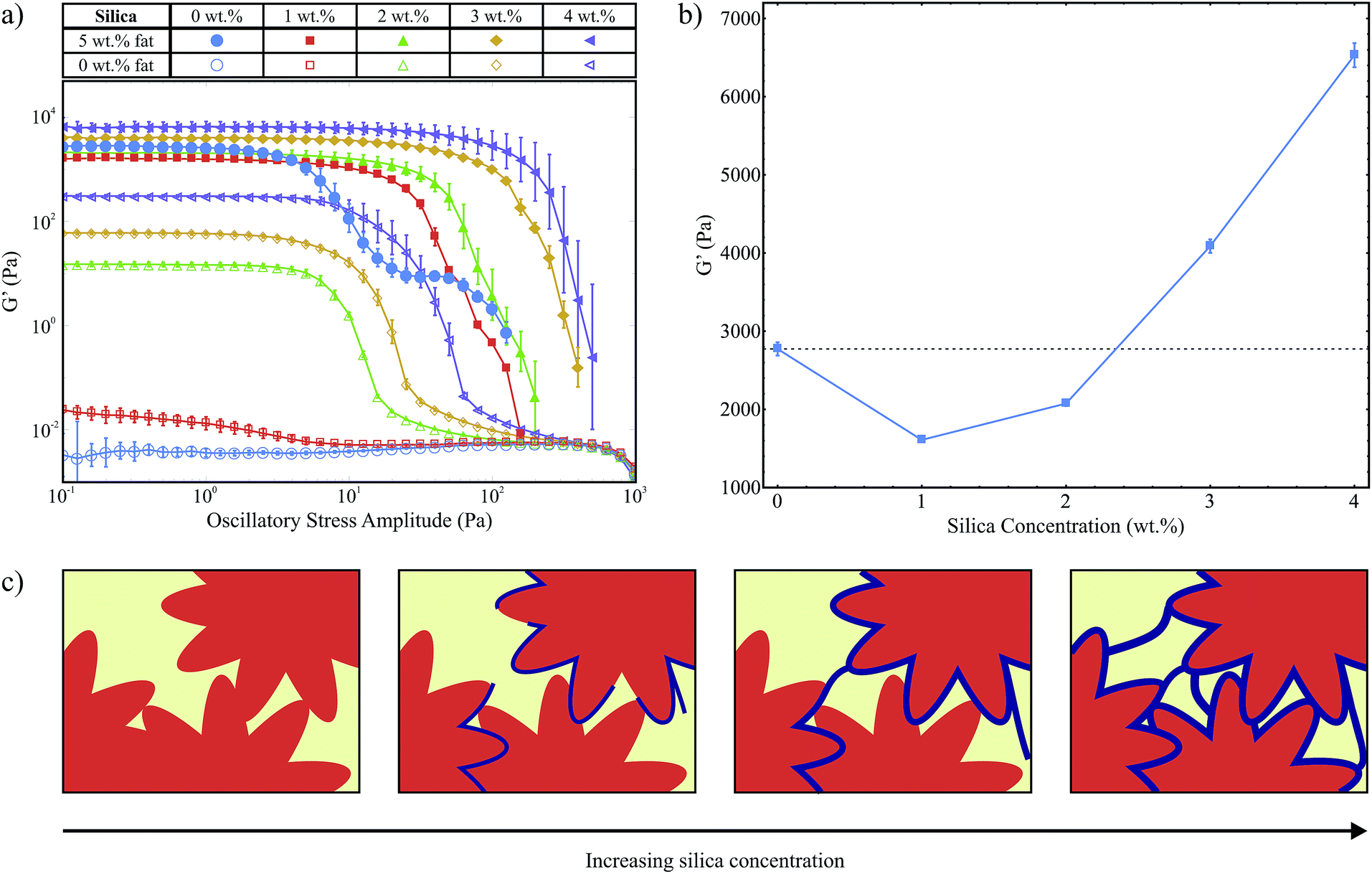 | ||
| Fig. 3 (a) Stress sweep data for samples with a range of silica concentrations, both with and without 5 wt% fat. (b) A plot of the average G′ magnitude within the LVR against silica concentration. (c) A diagram to illustrate the effect of silica concentration on the strength of the fat crystal network. The silica network is shown in blue and the fat crystals are orange. Not drawn to scale. | ||
In Fig. 3a we see a variation in the magnitude of G′ within the linear viscoelastic region (LVR) for the samples containing 5 wt% fat as a function of silica concentration. Fig. 3b is a plot of the average G′ magnitude for samples with 5 wt% fat, between 0.1 Pa and 1 Pa which lies within the LVR for all samples, against silica concentration on a linear scale. We observe an initial decrease followed by an increase which can be explained by the balance of two opposing effects: as the fat crystals grow they will push out the silica aggregates which will lead to a higher concentration of silica around the fat crystals than in the bulk, as illustrated in Fig. 1. As the fat crystals get larger some of the silica aggregates will become trapped between the fat crystals, weakening the fat crystal-fat crystal interactions. However, the silica surrounding the fat crystals will also form a continuous, protective layer which will increase the overall strength of the chains and the network. At lower concentrations, there is less silica to form an effective layer, but there are enough silica aggregates between the crystals to give an overall decrease in the strength of the network. At higher concentrations the second effect becomes dominant and we see an overall increase in G′. This is depicted schematically in Fig. 3c. However, at both low and high silica concentrations we observe a large increase in the length of the LVR with the addition of silica, which means that the silica is able to help the fat network maintain its structure up to much larger applied stress levels.
Polarised light microscopy data (provided as ESI†) showed that the microstructure of the fat crystal network was not significantly affected by silica concentration. This is consistent with our composite gel network model where the fat crystals grow unhindered by the surrounding, weaker silica network.8
Fig. 4a shows the exothermic crystallization peak of the second crystallization process, for samples that contain 5 wt% fat and a range of hydrophilic silica (A300) concentrations. All curves have been translated in the y-direction for clearer interpretation. There is a weak silica concentration dependence on the onset temperature for crystallization. This is a volume exclusion effect where the addition of silica slightly raises the effective fat concentration. We observed in our previous study that higher fat concentrations lead to increased onset temperatures,8 due to an increase in the amount of supercooling which is a driving force for crystallization.
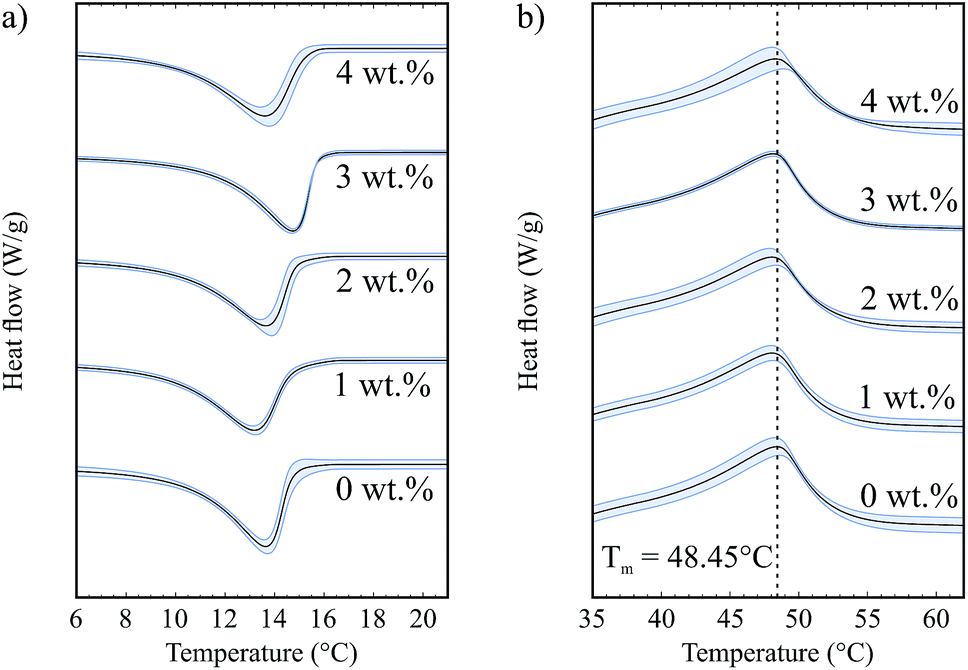 | ||
| Fig. 4 (a) Crystallization curves for samples with different silica concentrations and 5 wt% fat. (b) Melting curves for samples with different silica concentrations and 5 wt% fat. | ||
Fig. 4b shows the endothermic melting curves for the same samples as shown in Fig. 4a. We observe that the curves for samples with and without silica are almost identical, which means that the added colloidal particles do not have a significant effect on the melting of these samples and that we form similar fat networks with comparable fat crystal–fat crystal interactions. The melting temperatures of the different tripalmitin polymorphs are reported by Marangoni as 44.7 °C (α), 55.7 °C (β′) and 65.9 °C (β).14 We see that all of the melting curves shown in Fig. 4b are centred around 48.45 °C, showing that the α polymorph is formed under the conditions of our experiment. This result is corroborated by the data of Himawan et al.15
This study focuses on the crystallization onset temperatures and melting peak temperatures since these are well defined values. However, it is also possible to extract crystallization and melting enthalpies which are related to the area of the peaks. In the ESI,† we provide data for the onset and peak temperatures as well as the enthalpy values for both crystallization and melting processes for each of the datasets presented in this paper.
3.2 Silica surface area
Fig. 5a shows G′ against oscillatory stress for three hydrophilic silicas with different surface areas (SA) both with and without 5 wt% fat and all containing 2 wt% silica. In this set of results all samples showed solid-like behaviour except pure soybean oil (○). For all of the silica samples without fat, the G′ curves are almost identical in shape and magnitude. This indicates that the surface area does not significantly affect the type of pure silica network that forms in oil, with regard to strength or breakdown under an applied stress.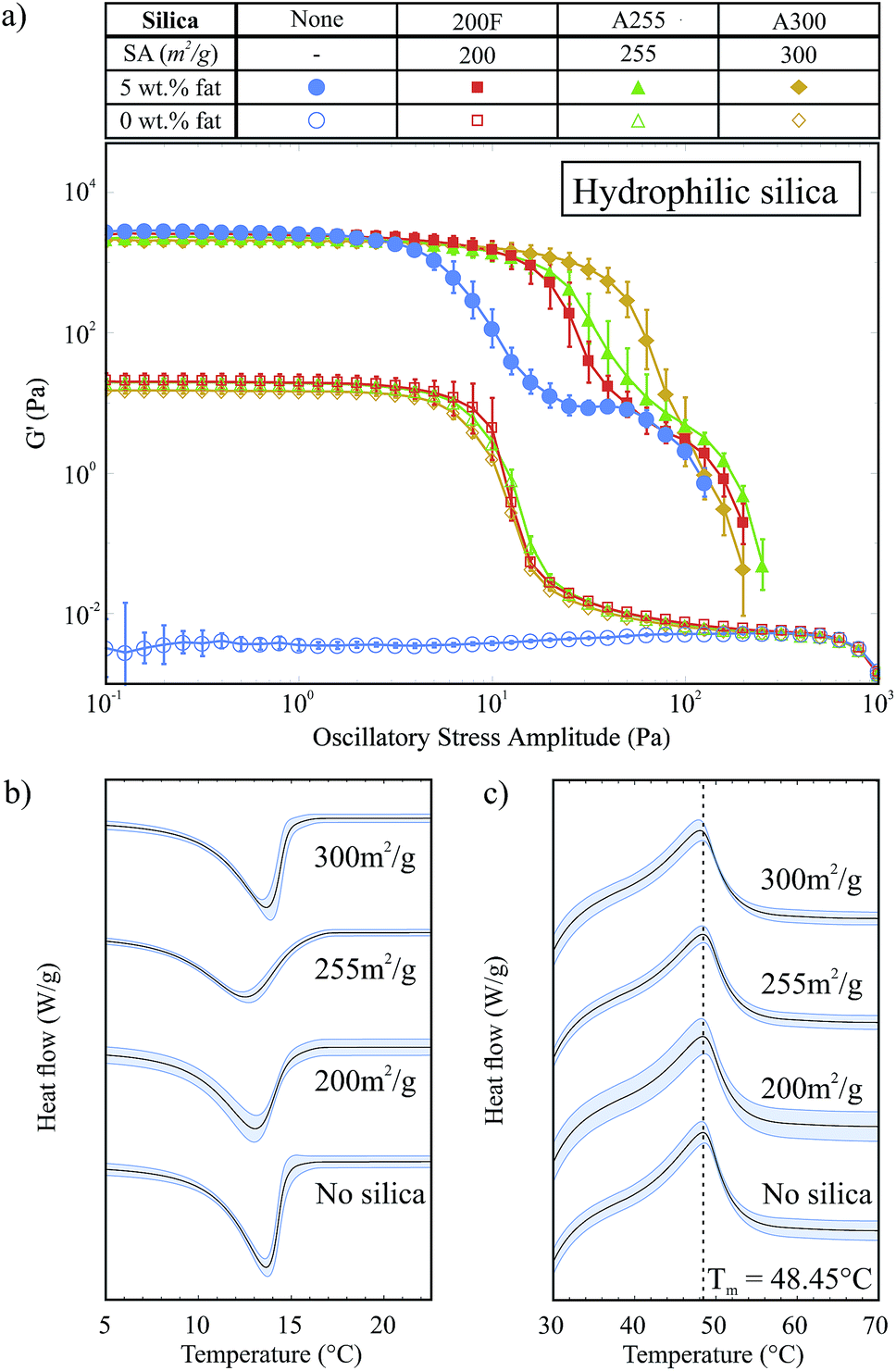 | ||
| Fig. 5 (a) Stress sweep data for samples with 2 wt% hydrophilic silica with different surface areas. (b) Crystallization curves for samples with different silica surface areas and 5 wt% fat. (c) Melting curves for samples with different silica surface areas and 5 wt% fat. | ||
The samples containing 5 wt% fat and 2 wt% silica all have a similar G′ value within the LVR to the 5 wt% fat sample, which indicates that we have the same fat network and that the silica is acting as an auxiliary component supporting the fat network. However, these samples also have an extended LVR compared to the sample with only 5 wt% fat. The length of the LVR does not significantly change with the surface area: instead it is the rate at which G′ decreases after the linear region which varies. Samples containing silica with a higher surface area have G′ curves that decline more slowly after the end of the region of linearity.
This decline in G′ after the LVR describes the structural breakdown of the fat network and specifically the breaking of the connections between fat crystal clusters. As the silica surface area increases, the number of silanol groups available for hydrogen bonding increases and so the silica is able to form more bonds and it becomes easier to rearrange under the applied stress; when one hydrogen bond breaks it is more able to rearrange and form a new bond. This means that the silica is able to help the fat crystal chains to remain connected, or partially connected, and break down more slowly as higher stress levels are applied. The silica network is formed prior to the fat network and is disrupted during the crystallization of the fat (see Fig. 1). This means that the arrangement of the silica around the fat crystals is much more dense compared to that of the open, fractal structure of the pure silica network and possibly explains why we do not see this behaviour in the silica-only samples.
In Fig. 5b we show the exothermic crystallization curves of the second crystallization process for the hydrophilic silica samples with 5 wt% fat. For these experiments, the effective fat concentrations are held constant and so we do not see an effect on the crystallization onset temperature due to increased effective fat concentrations. There is no strong trend of surface area with the crystallization of the fat. The crystallization onset temperatures for the A300, A255, 200F and fat only samples were 15.52 ± 0.73 °C, 15.52 ± 0.08 °C, 14.94 ± 0.08 °C and 14.75 ± 0.21 °C respectively. This result suggests that the hydrophilic silica aggregates are not acting as seeds for nucleation: heterogeneous nucleation is a surface effect and so we would expect to see a clear trend of higher surface areas leading to earlier crystallization. Fig. 5c shows the melting of the fat crystal networks in the presence of the different types of hydrophilic silica. From this data, it is clear that the silica surface area does not affect the melting temperature of the fat.
3.3 Silica surface chemistry
In this section we study two pairs of different silica samples, with the same silica concentrations and closely matched silica surface areas, and so the dominant variable is the surface chemistry. The first pair is the hydrophilic 200F and hydrophobic R816 silicas, which have surface area values of 200 m2 g−1 and 190 m2 g−1 respectively. The second pair is the hydrophilic A255 and hydrophobic R812 silicas which have surface area values of 255 m2 g−1 and 260 m2 g−1 respectively. The hydrophobic silicas have been chemically modified so that a portion of the surface silanol groups were replaced with hydrophobic functional groups. For R816, some silanol groups were replaced with hexadecylsilyl (–SiC16H33) groups and for R812 the trimethylsilyl (–Si(Me)3) group was used, see Fig. 2. Titration with LiAlH4 has been used to determine the fraction of surface silanol groups that have been substituted during the chemical modification of similar fumed silicas.16 Based on these results we estimate that for R812 and R816 approximately 30–50% of the surface silanol groups have been substituted.Fig. 6a shows the stress sweep data for the hydrophilic 200F and hydrophobic R816 silicas and Fig. 6b shows the same data for the hydrophilic A255 and hydrophobic R812 silicas. In both cases, the stress sweep data for the samples without silica present are also shown for reference (♦ and ◊). We note that the hydrophobic silica samples without fat do not form a network within the oil (G′ < G′′, G′′ is not shown). This result is consistent with a combination of factors; less silanol groups available for hydrogen bonding, increased steric hindrance between aggregates and reduced mismatch in chemical nature between the hydrophobic silica and the non-polar soybean oil. Collectively, this means that it is easier for the silica to disperse in the solvent rather than agglomerating to form a gel. However they are not completely liquid-like which can be seen by comparison with the pure soybean oil (◊), which means that there is still a considerable amount of hydrogen bonding present, especially in the R816 case.
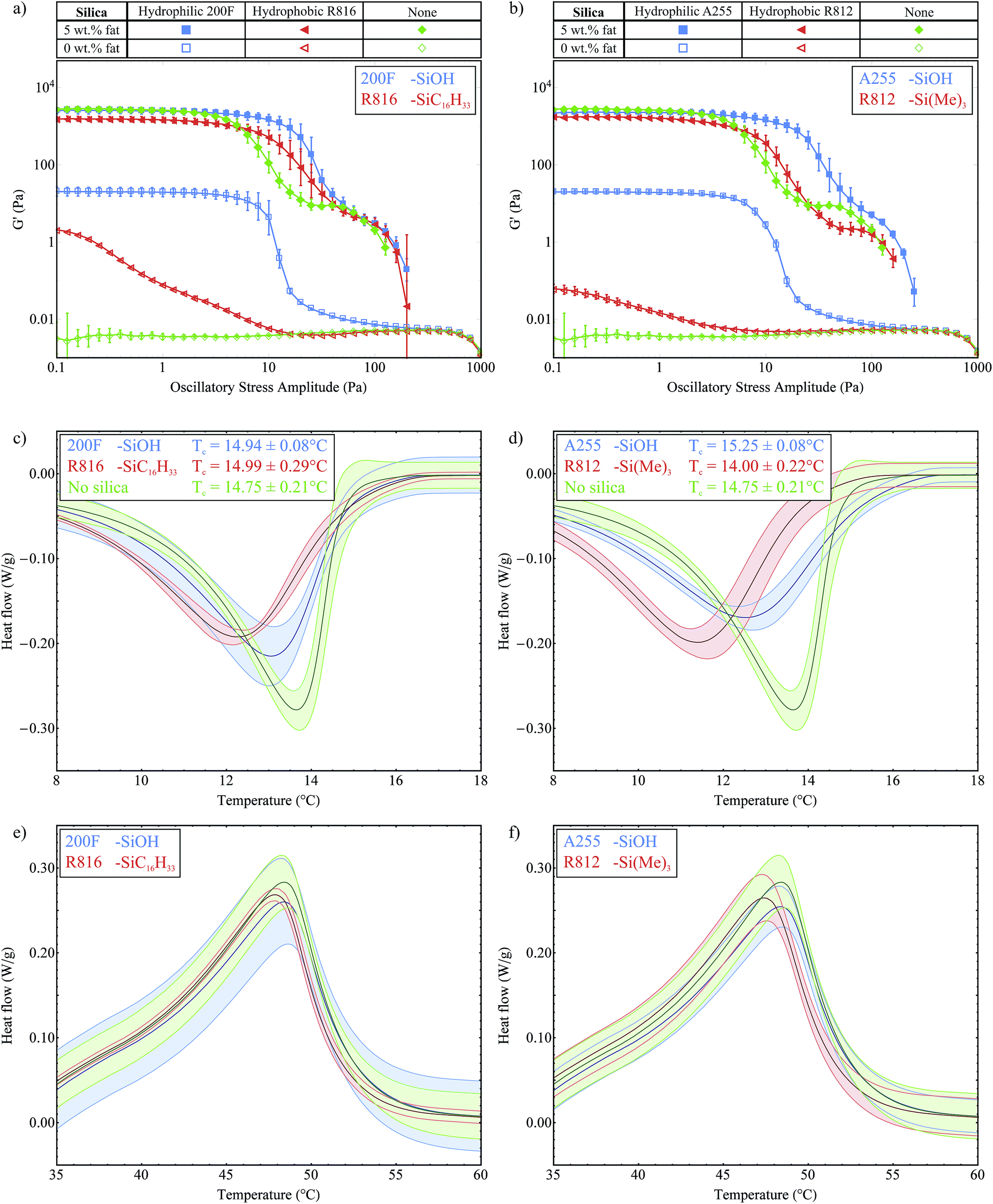 | ||
| Fig. 6 Stress sweep data for (a) hydrophilic 200F and hydrophobic R816 silicas (b) hydrophilic A255 and hydrophobic R812 silicas. Crystallization curves for (c) 200F and R816 silicas (d) A255 and R812 silicas. Melting curves for (e) 200F and R816 silicas (f) A255 and R812 silicas. | ||
With 5 wt% fat present, we see a small but significant drop in the G′ magnitude within the LVR for both of the hydrophobic samples. Without silica present this G′ value was measured at 2773 ± 85 Pa and with R816 this was reduced to 1526 ± 27 Pa and 1721 ± 51 Pa for R812. This could be due to the modified functional groups providing increased steric hindrance and weakening the interactions between the fat crystal crystals in the stress-transmitting chains. The hydrophobic silicas do not greatly increase the length of the LVR; the curves start to decrease at roughly the same point as the fat-only sample (♦), however the gradient of the breakdown is less steep. The hydrophobic silica present is still able to form some hydrogen bonds and to support the fat network as it breaks down, resulting in a slower decline in G′ moving towards higher oscillatory stress levels. These hydrophobic silicas provide another example where the colloidal particles do not form a gel when present on their own in the oil, but they are able to provide some support to the fat crystal network. Overall, these experiments have reiterated the important role of hydrogen bonds between the silica aggregates in reinforcing the fat network.
Polarised light microscopy was used to observe the microstructure of the fat crystal networks in the presence of silicas with different surface chemistries (data provided as ESI†). As with the silica concentration results, the silica surface chemistry did not have a significant effect on the fat crystal microstructure.
Fig. 6c and d show the exothermic crystallization curves of the second crystallization process for the two pairs of silicas all with 5 wt% fat present. From these plots we determined the crystallization onset temperatures to be 14.94 ± 0.08 °C and 14.99 ± 0.29 °C for the 200F and R816 silica respectively, relative to 14.75 ± 0.21 °C for the 5 wt% fat sample. Similarly we measured onset temperatures of 15.25 ± 0.08 °C and 14.00 ± 0.22 °C for the A255 and R812 silicas. This shows that the crystallization of fat is not sensitive to the presence of the different types of silica. Furthermore it shows that the silicas are not acting as seeds for the nucleation process. This experiment was important as we wanted to investigate whether or not the R816 in particular would act as seeds since it has the same carbon chains (C16) as the fatty acid chains of the tripalmitin molecules, see Fig. 2. Although the crystallization onset temperature is not affected by the different silicas, we see that the width of the peaks is broader for the hydrophobic silicas which indicates a slower growth rate. This might be due to disruption in the packing of the fat molecules into lamellae caused by the bulky functional groups on the silica aggregates. Fig. 6e and f show the melting curves for the two pairs of silica. All of these curves lie within the measured variation of each other and so they are not considered to be significantly different.
4 Conclusions
In this study, we have studied in detail three different aspects of fumed silica on a fat-structured model system; concentration, surface area and surface chemistry. We have observed that the silica affects the fat crystal networks response to stress in three distinct ways. The first is that the magnitude of G′ within the LVR varies with both the silica concentration and the surface chemistry. We propose that silica aggregates between the fat crystal clusters can weaken the overall network, but at high enough concentrations a continuous layer of silica can form which is able to strengthen the network.The second way in which the presence of silica affects the fat network is that it extends the linear viscoelastic region. This effect was greatest with higher concentrations of silica and hydrophilic, rather than hydrophobic silica. This effect shows that the silica aggregates, via hydrogen bonding, were able to help the fat crystals maintain their network structure up to higher stress levels. Finally, the silica also changed the breakdown of the fat network after the LVR. Both the surface area and surface chemistry experiments varied the number of available silanol groups, which are used to connect the silica aggregates. With more hydrogen bonds present the silica is able to rearrange and maintain connections for longer, slowing the overall rate of structural breakdown.
This study also observed that the added particles do not need to be able to form a gel in oil in order to strengthen the fat crystal network. This further supports our previously proposed mechanism of a composite network rather than a double network structure comprised of two individual networks.8 We saw two examples of this situation in this study, first with hydrophilic silica at low concentrations (A300 at 1 wt%) and then with both hydrophobic silicas (R816 and R812 at 2 wt%).
The DSC crystallization data from all three experiments show that fumed silica does not act as a seed for the heterogenous nucleation under these conditions. We did however see a weak dependence with silica concentration due to an increased effective fat concentration. The melting data also showed that despite observing large changes in the rheological behaviour upon the addition of silica, the way in which the fat network melts is left unchanged. From a consumers standpoint this is a crucial result because the way fat-structured food products melt is a large part of the consumer experience; for example the way in which chocolate melts in the mouth or butter melts on hot toast.
It is also worth noting here that the 200F hydrophilic silica used in these experiments is food grade and so this is an industrially viable option for the hybrid structuring of fat-structured food products. Food regulations vary across different regions and food categories and so it is difficult to quote the exact allowed amount of silica. However, as an example, 200F silica is currently allowed in grated cheese up to 2 wt% in the USA and up to 3 wt% in soups and condiments in Brazil.17
Acknowledgements
We thank Unilever for partially funding this work.References
- I. Heertje, Food Struct., 2014, 1, 3–23 CrossRef.
- S. S. Narine and A. G. Marangoni, Food Res. Int., 1999, 32, 227–248 CrossRef CAS.
- K. W. Smith, K. Bhaggan, G. Talbot and K. F. van Malssen, J. Am. Oil Chem. Soc., 2011, 88, 1085–1101 CrossRef CAS.
- R. C. Basso, A. P. B. Ribeiro, M. H. Masuchi, L. A. Gioielli, L. A. G. Gonçalves, A. O. dos Santos, L. P. Cardoso and R. Grimaldi, Food Chem., 2010, 122, 1185–1192 CrossRef CAS.
- L. Svanberg, L. Ahrné, N. Lorén and E. Windhab, J. Food Eng., 2011, 104, 70–80 CrossRef CAS.
- L. Svanberg, L. Ahrné, N. Lorén and E. Windhab, Food Res. Int., 2011, 44, 1339–1350 CrossRef CAS.
- S. Calligaris, F. Valoppi, L. Barba, M. Anese and M. C. Nicoli, Food Res. Int., 2014, 66, 257–263 CrossRef CAS.
- R. R. Chauhan, R. P. A. Dullens, K. P. Velikov and D. G. A. L. Aarts, Food Funct., 2017, 8, 352–359 CAS.
- Basic characteristics of AEROSIL® fumed silica: Technical bulletin fine particles 11, Evonik Industries AG technical report.
- S. R. Raghavan, H. J. Walls and S. A. Khan, Langmuir, 2000, 16, 7920–7930 CrossRef CAS.
- S. R. Raghavan, J. Hou, G. L. Baker and S. A. Khan, Langmuir, 2000, 16, 1066–1077 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and surface forces, Academic press, 2nd edn, 1991 Search PubMed.
- D. Small, The Physical Chemistry of Lipids, Plenum Press, New York, 1986, vol. 4, p. 512 Search PubMed.
- A. G. Marangoni and L. H. Wesdorp, Structure and properties of fat crystal networks, CRC Press, 2012 Search PubMed.
- C. Himawan, W. MacNaughtan, I. A. Farhat and A. G. Stapley, Eur. J. Lipid Sci. Technol., 2007, 109, 49–60 CrossRef CAS.
- J. Hou, PhD thesis, Michigan State University, 1997.
- E. Industries AG, AEROSIL® 200F Product Safety Information, 2016.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01803g |
| This journal is © The Royal Society of Chemistry 2017 |