DOI:
10.1039/C7RA02702H
(Paper)
RSC Adv., 2017,
7, 19439-19447
Addressing the mid-point of polymer chains for multiple functionalization purposes through sequential thiol–epoxy ‘click’ and esterification reactions†
Received
6th March 2017
, Accepted 22nd March 2017
First published on 3rd April 2017
Abstract
A synthetic strategy is devised for the preparation of mid-chain multifunctional polymers. In this strategy, small molecules carrying one or two epoxide units along with two polymerization-initiating sites were prepared in two to three synthetic steps. Atom transfer radical polymerization (ATRP) of methylmethacrylate (MMA) monomer using the newly designed initiators gave rise to epoxide-mid-reactive polymers. A careful chain cleavage study confirmed the midpoint location of the reactive site(s) on the polymer chain. Dual-modification of these reactive epoxide unit(s) through sequential thiol–epoxy and esterification reactions then afforded the midpoint multiply functionalized polymeric structures. Due to compatibility of the ATRP process with the reactive epoxide unit(s) and the cascade nature of the functionalization processes, the present strategy is free from the protective-group requirement of organic synthesis. Moreover, it allows for varying the number and nature of functionalities at the midpoint of a polymer chain.
Introduction
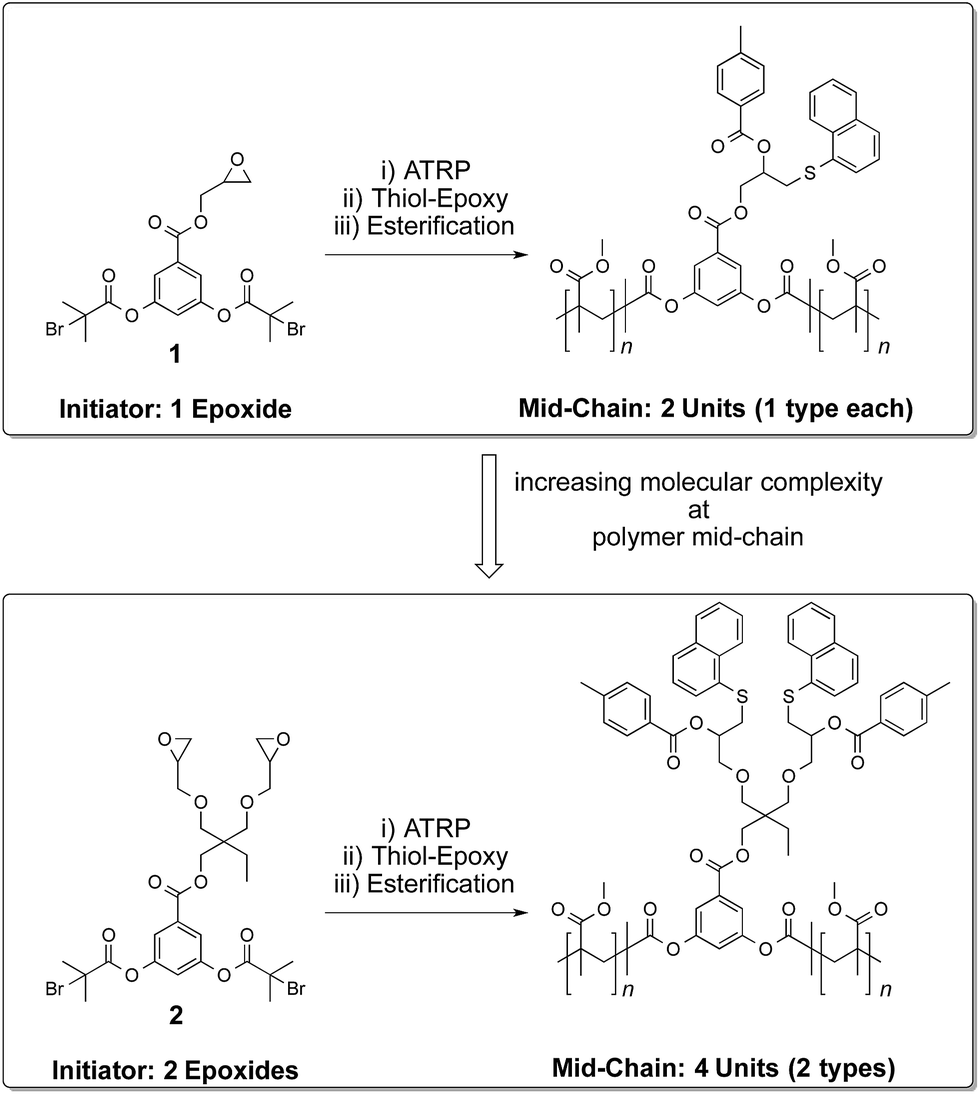
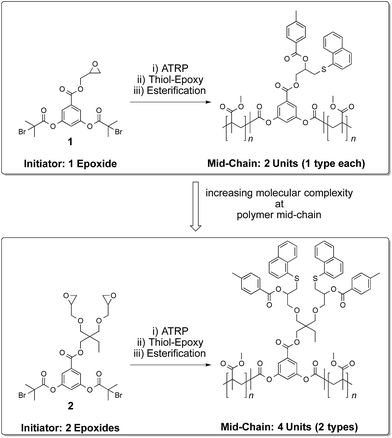
Post-polymerization modification of polymers is a valuable tool in generating functional soft materials.1–8 In this regard, side-chain1–5 and end-chain6–8 polymer functionalization has received enormous attention. General methods for addressing the polymer mid-chain for functionalization processes, however, are relatively few.9–12 Strategies seeking further increase in the molecular complexity at the midpoint of a polymer chain by carrying out more than one protective-group-free functionalization step remains, to the best of our knowledge, unknown. Towards this end, we demonstrate a modular strategy for multiple functionalization of the polymer mid-chain through sequential application13 of thiol–epoxy ‘click’14 and esterification reactions. The devised approach is free from protective-group requirements and allows for adjusting the number and nature of functional groups at the middle of a polymer chain (Scheme 1).
 |
| | Scheme 1 Application of orthogonal and sequential processes to address polymer mid-chain for multiple functionalizations. | |
Our approach relies on the functional group tolerance of the ATRP process15–20 that allows for the preparation of polymers substituted with reactive epoxide groups21 at the centre of a polymer chain. These epoxide units can undergo a ring opening reaction with a thiol molecule to form a thio-ether linkage.14,20,22–24 This base-catalyzed chemistry is established to be an efficient process that is capable of transforming thousands of epoxide repeat units into functionalized thio-ether linkages. The process is practically simple and proceeds under ambient conditions. An added advantage of this chemistry is that a reactive hydroxyl group is produced upon completion of the coupling reaction. This secondary hydroxyl group, although less reactive than the primary hydroxyl group, can be converted into an ester moiety upon reaction with an acid chloride molecule.25 This sequential functionalization processes gives rise to doubly functionalized structures in a protective-group-free fashion. Commercial availability of a variety of functional thiol and acid chloride molecules further enhances the scope of the present strategy in the preparation of functional materials.
Experimental details
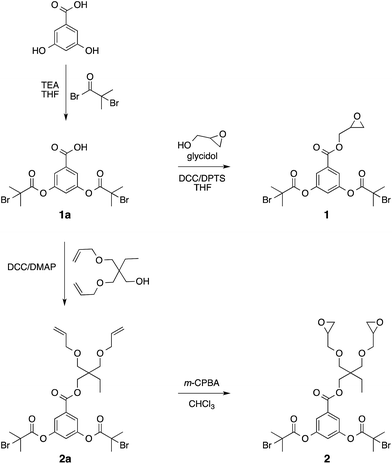
Synthesis of 1a
To a stirring solution of bromoisobutyryl bromide (10.44 g, 45.41 mmol) in THF (40 mL) was added triethylamine (TEA) (8.66 g, 85.64 mmol) drop wise at 5 °C and stirring was continued for another 20 min. To this reaction mixture was added 3,5-dihydroxybenzoic acid (2.0 g, 12.97 mmol) in THF (25 mL) drop wise through a syringe pump (0.86 mL min−1) at −5 °C. The reaction mixture was stirred for 2 hours at −5 °C and then warmed up to 0 °C. After this, the triethylamine salts formed during the reaction were removed by filtration. The filtrate was concentrated under reduced pressure and the crude material obtained was dissolved in DCM (50 mL) and washed with water (2 × 50 mL) and brine (20 mL). The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The crude product was then purified by silica gel column chromatography using a DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH solvent gradient (98:2 to 96:4). The resulting brown solid was further purified by crystallization from a DCM:hexane mixture to afford 2.12 g of the product as a white solid (yield = 36%). 1H-NMR (δ, ppm, 300 MHz, DMSO-d6): 13.59 (br s, 1H), 7.67 (d, J = 2.24 Hz, 2H), 7.41 (t, J = 2.24 Hz, 1H), 2.05 (s, 12H); 13C-NMR (δ, ppm, 75 MHz, DMSO-d6): 169.29, 165.45, 150.81, 133.39, 120.10, 119.50, 56.83, 29.89; ESI-MS m/z = 472.92 [M + Na]+ (calcd 472.93 for C15H16Br2O6Na); IR (cm−1): 2939, 1754, 1696, 1586, 1462, 1443, 1406, 1387, 1370, 1299, 1255, 1133, 1095, 1007, 986, 925, 770, 732, 654, 602, 572.
:MeOH solvent gradient (98:2 to 96:4). The resulting brown solid was further purified by crystallization from a DCM:hexane mixture to afford 2.12 g of the product as a white solid (yield = 36%). 1H-NMR (δ, ppm, 300 MHz, DMSO-d6): 13.59 (br s, 1H), 7.67 (d, J = 2.24 Hz, 2H), 7.41 (t, J = 2.24 Hz, 1H), 2.05 (s, 12H); 13C-NMR (δ, ppm, 75 MHz, DMSO-d6): 169.29, 165.45, 150.81, 133.39, 120.10, 119.50, 56.83, 29.89; ESI-MS m/z = 472.92 [M + Na]+ (calcd 472.93 for C15H16Br2O6Na); IR (cm−1): 2939, 1754, 1696, 1586, 1462, 1443, 1406, 1387, 1370, 1299, 1255, 1133, 1095, 1007, 986, 925, 770, 732, 654, 602, 572.
Synthesis of 1
To a stirring solution of 1a (1.9 g, 4.2 mmol), glycidol (0.59 g, 7.98 mmol), and 4-(N,N′-dimethylamino)pyridinium-4-toluenesulfonate (DPTS) (0.23 g, 0.84 mmol) in DCM (30 mL), was added dicyclohexylcarbodiimide (DCC) (1.04 g, 5.04 mmol) in several portions at 0 °C. The resulting reaction mixture was then stirred at room temperature for 12 hours. After this time, the reaction mixture was cooled to 0 °C. This resulted in precipitation of dicyclohexylurea (DCU) that was removed through filtration. The filtrate was concentrated under reduced pressure and the crude thus obtained was purified by silica gel column chromatography using heptane:EtOAc solvent gradient (95:5 to 85:15) to afford 0.94 g of the product as a white solid (yield = 44%). 1H-NMR (δ, ppm, 300 MHz, CDCl3): 7.76 (d, J = 2.26 Hz, 2H), 7.24 (t, J = 2.26 Hz, 1H), 4.68 (dd, J = 3.04, 12.23 Hz, 1H), 4.16 (dd, J = 6.42, 12.23 Hz, 1H), 3.35 (m, 1H), 2.91 (t, J = 4.40 Hz, 1H), 2.72 (dd, J = 2.67, 4.92 Hz, 1H), 2.07 (s, 12H); 13C-NMR (δ, ppm, 75 MHz, CDCl3): 169.81, 164.51, 151.25, 132.17, 120.57, 119.77, 66.46, 54.87, 49.42, 44.92, 30.69; ESI-MS m/z = 528.94 [M + Na]+ (calcd 528.96 for C18H20Br2O7Na); IR (cm−1): 2961, 1754, 1724, 1596, 1465, 1440, 1389, 1368, 1346, 1298, 1258, 1220, 1137, 1096, 997, 927, 906, 865, 790, 767, 745, 682, 652.
Synthesis of 2a
To a stirring solution of 1a (2.0 g, 4.42 mmol), trimethylolpropane diallyl ether (1.09 g, 5.08 mmol), and dimethylaminopyridine (DMAP) (0.108 g, 0.884 mmol) in THF (20 mL) was added DCC (1.09 g, 5.30 mmol) in several portions at 0 °C. The resulting reaction mixture was then stirred at room temperature for 12 hours. After this time, the reaction mixture was cooled to 0 °C. This resulted in the precipitation of dicyclohexylurea (DCU) that was removed through filtration. The filtrate was concentrated under reduced pressure and the crude thus obtained was purified by silica gel column chromatography using heptane:EtOAc solvent gradient (98:2 to 95:5) to afford 1.6 g of the product as a colourless viscous liquid (yield = 56%). 1H-NMR (δ, ppm, 300 MHz, CDCl3): 7.69 (d, J = 2.21 Hz, 2H), 7.21 (t, J = 2.21 Hz, 1H), 5.86 (m, 2H), 5.18 (m, 4H), 4.32 (s, 2H), 3.95 (d, J = 5.53 Hz, 4H), 3.40 (s, 4H), 2.07 (s, 12H), 1.53 (q, J = 7.47 Hz, 2H), 0.91 (t, J = 7.47 Hz, 3H); 13C-NMR (δ, ppm, 75 MHz, CDCl3): 169.81, 164.62, 151.21, 135.06, 133.15, 120.24, 119.34, 116.79, 72.48, 70.49, 66.56, 54.97, 42.77, 30.67, 23.33, 7.80; ESI-MS m/z = 647.08 [M + H]+ (calcd 647.08 for C27H37Br2O8); IR (cm−1): 2933, 1758, 1726, 1596, 1462, 1443, 1391, 1370, 1350, 1299, 1256, 1224, 1134, 1095, 990, 925, 764, 681, 654.
Synthesis of 2
To a stirring solution of 2a (1.6 g, 2.46 mmol) in CHCl3 (10 mL), was added a solution of 70% m-chloroperoxybenzoic acid (3.4 g, 19.74 mmol) in CHCl3 (25 mL) at 0 °C in a drop wise fashion. The resulting reaction mixture was stirred at room temperature for 20 hours, and then washed with 0.3 M NaOH (2 × 25 mL) and water (25 mL), dried over Na2SO4, and concentrated under reduced pressure to give a colourless liquid. This crude product was purified by silica gel column chromatography using a DCM:EtOAc solvent gradient (99:1 to 95:5) to afford 0.79 g of the product as a colourless viscous liquid. (Yield = 47%). 1H-NMR (δ, ppm, 300 MHz, CDCl3): 7.71 (d, J = 2.24 Hz, 2H), 7.21 (t, J = 2.24 Hz, 1H), 4.31 (s, 2H), 3.72 (m, 2H), 3.48 (m, 4H), 3.36 (m, 2H), 3.10 (m, 2H), 2.75 (t, J = 4.85 Hz, 2H), 2.56 (dd, J = 2.79, 4.97 Hz, 2H), 2.07 (s, 12H), 1.54 (q, J = 7.58 Hz, 2H), 0.91 (t, J = 7.58 Hz, 3H); 13C-NMR (δ, ppm, 75 MHz, CDCl3): 169.68, 164.49, 151.11, 132.89, 120.15, 119.25, 72.11, 72.09, 71.51, 66.17, 54.87, 50.89, 44.02, 42.82, 30.52, 23.07, 7.62; ESI-MS m/z = 696.10 [M + NH4]+ (calcd 696.07 for C27H40Br2NO8); IR (cm−1): 2924, 1756, 1722, 1595, 1461, 1442, 1388, 1371, 1300, 1258, 1225, 1133, 1097, 997, 902, 848, 764, 684, 654.
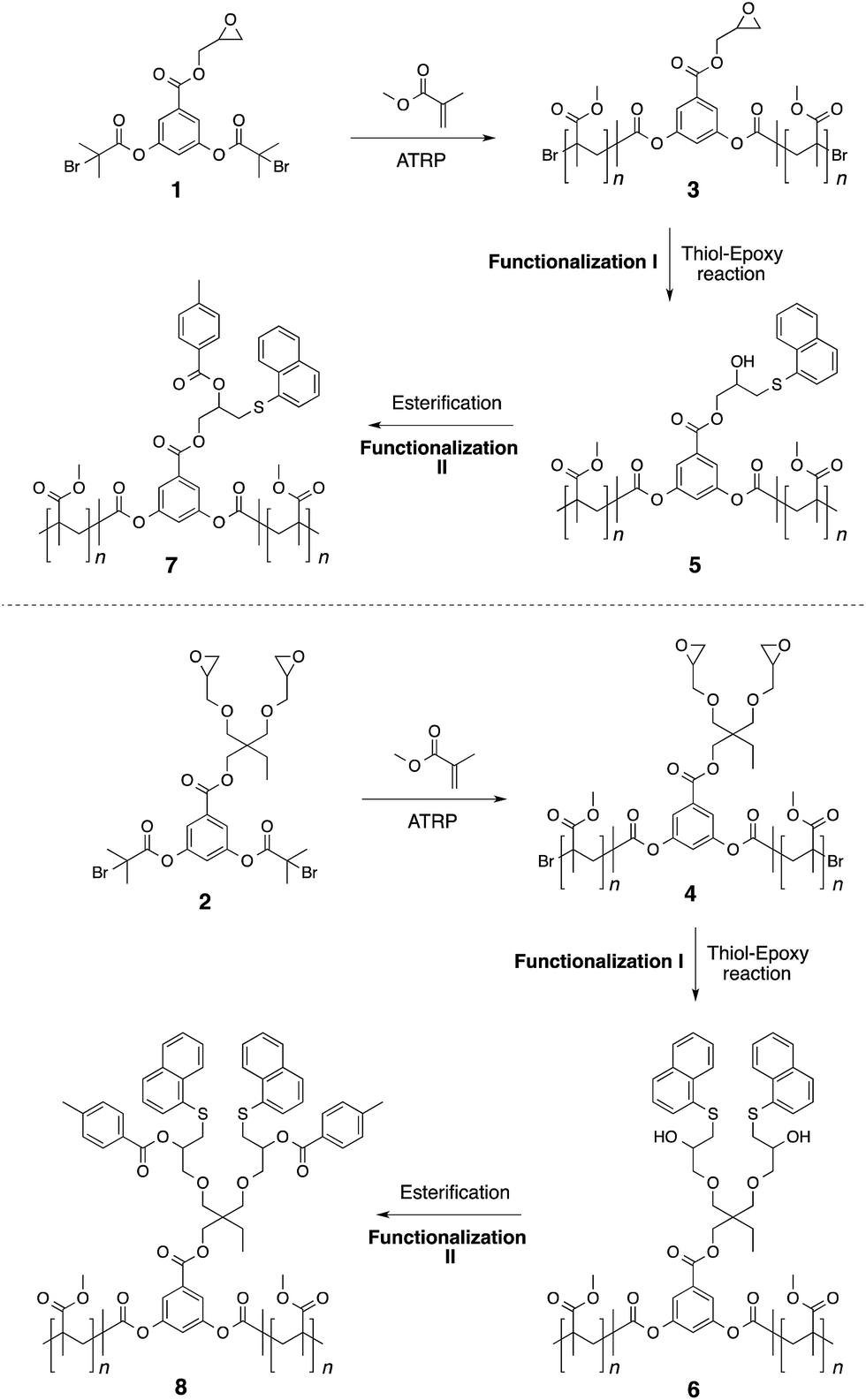
Epoxy functionalized polymer 3
Atom transfer radical polymerization (ATRP) initiator 1 (0.03 g, 0.059 mmol), methyl methacrylate (5.08 g, 50.77 mmol), pentamethyldiethylenetriamine (PMDETA) (0.032 g, 0.188 mmol), and anisole (7.0 mL) were taken in a Schlenk tube and degassed by two freeze–pump–thaw cycles. Cu(I)Br (0.013 g, 0.94 mmol) was added to the frozen solution and degassed again by one freeze–pump–thaw cycle. The reaction mixture was then stirred under nitrogen atmosphere in a pre-heated oil bath at 35 °C for 90 minutes. After this time, the reaction vessel was opened to air, cooled to room temperature, and precipitated into 400 mL of methanol. The precipitated polymer was isolated by filtration and purified by passing through a small plug of silica gel using DCM (150 mL) as eluent. The collected fractions were concentrated and precipitated into 300 mL of methanol. The polymer precipitate was collected by filtration and dried to give 0.64 g of the product as a white powder (monomer conversion 13%). 1H-NMR (δ, ppm, 300 MHz, CDCl3): 7.61 (br s, 2H), 7.08 (br s, 1H), 4.67 (d, J = 12.21 Hz, 1H), 4.16 (m, 1H), 3.84–3.35 (br s, backbone, –OCH3), 2.91 (t, J = 4.53 Hz, 1H), 2.72 (m, 1H), 2.13–0.63 (br m, backbone); GPC: Mn = 15000, Mw = 19500, PDI (Mw/Mn) = 1.3; IR (cm−1): 2955, 1723, 1481, 1441, 1387, 1268, 1240, 1193, 1145, 987, 965, 840, 750.
Epoxy functionalized polymer 4
Atom transfer radical polymerization (ATRP) initiator 2 (0.03 g, 0.044 mmol), methyl methacrylate (5.07 g, 50.70 mmol), PMDETA (0.032 g, 0.185 mmol), and anisole (7.0 mL) were taken in a Schlenk tube and degassed by two freeze–pump–thaw cycles. Cu(I)Br (0.013 g, 0.92 mmol) was added to the frozen solution and degassed again by one freeze–pump–thaw cycle. The reaction mixture was then stirred under nitrogen atmosphere in a pre-heated oil bath at 35 °C for 90 minutes. After this time, the reaction vessel was opened to air, cooled to room temperature, and precipitated into 400 mL of methanol. The precipitated polymer was isolated by filtration and purified by passing through a small plug of silica gel using DCM (150 mL) as eluent. The collected fractions were concentrated and precipitated into 300 mL of methanol. The polymer precipitate was collected by filtration and dried to give 0.77 g of the product as a white powder (monomer conversion 15%). 1H-NMR (δ, ppm, 300 MHz, CDCl3): 7.57 (br s, 2H), 7.03 (br s, 1H), 4.29 (s, 2H), 3.84–3.35 (br s, backbone, –OCH3), 3.10 (s, 2H), 2.76 (t, J = 4.83 Hz, 2H), 2.57 (m, 2H), 2.07–0.63 (br m, backbone); GPC: Mn = 26300, Mw = 33200, PDI (Mw/Mn) = 1.2; IR (cm−1): 2961, 1723, 1447, 1270, 1239, 1189, 1142, 993, 966, 839, 746.
Thiol functionalized polymer 5 (using LiOH as a base)
To a stirring solution of polymer 3 (0.13 g, 0.0081 mmol) and 1-thionaphthol (0.019 g, 0.12 mmol) in THF (1.5 mL), was added LiOH (0.204 mg, 0.0048 mmol) in 0.040 mL of water (a stock solution was prepared before) at 0 °C. The resulting reaction mixture was stirred for 18 hours at ambient temperature. THF was evaporated and the crude polymer was dissolved in DCM (20 mL) and washed with water (10 mL). The organic layer was dried, concentrated, and the polymer obtained was dissolved in a minimum quantity of DCM and precipitated into MeOH (50 mL), then filtered and dried to give 0.12 g of the product (yield = 91%). 1H-NMR (δ, ppm, 300 MHz, CDCl3): 8.44 (d, J = 8.27 Hz, 1H), 7.85 (d, J = 7.64 Hz, 1H), 7.77 (d, J = 8.01 Hz, 1H), 7.70 (d, J = 7.27 Hz, 1H), 7.62–7.38 (br m, 5H), 7.08 (br s, 1H), 4.42 (m, 2H), 4.08 (br s, 1H), 3.84–3.34 (br s, backbone –OCH3), 3.20 (br s, 2H), 2.07–0.63 (br m, backbone); GPC: Mn = 15800, Mw = 20400, PDI (Mw/Mn) = 1.3; IR (cm−1): 2955, 1723, 1481, 1441, 1387, 1268, 1240, 1193, 1145, 987, 965, 840, 750.
Thiol functionalized polymer 5 (using TEA as a base)
To a stirring solution of polymer 3 (0.030 g, 0.00187 mmol) and 1-thionaphthol (0.014 g, 0.09 mmol) in THF (0.3 mL), was added TEA (0.113 g, 0.112 mmol) at ambient temperature. The resulting reaction mixture was stirred for 16 hours. The solvent was reduced under low pressure and the crude polymer was dissolved in DCM (4 mL) and washed with water. The organic layer was dried, concentrated, and the polymer obtained was dissolved in a minimum quantity of DCM and precipitated into MeOH (30 mL), then filtered and dried to give 0.025 g of the product in 82% yield.
Thiol functionalized polymer 6 (using LiOH as a base)
To a stirring solution of polymer 4 (0.1 g, 0.005 mmol) and 1-thionaphthol (0.024 g, 0.15 mmol) in THF (1.5 mL), was added LiOH (0.188 mg, 0.0045 mmol) in 0.037 mL of water (a stock solution was prepared before) at 0 °C. The resulting reaction mixture was stirred for 18 hours at ambient temperature. THF was evaporated and the crude polymer was dissolved in DCM (20 mL) and washed with water (10 mL). The organic layer was dried, concentrated, and the polymer obtained was dissolved in a minimum quantity of DCM and precipitated into MeOH (50 mL), then filtered and dried to give 0.092 g of the product (yield = 91%). 1H-NMR (δ, ppm, 300 MHz, CDCl3): 8.39 (d, J = 8.26 Hz, 2H), 7.83 (d, J = 7.54 Hz, 2H), 7.73 (d, J = 7.71 Hz, 2H), 7.61 (d, J = 7.20 Hz, 2H), 7.53 (m, 6H), 7.38 (t, J = 7.94 Hz, 2H), 7.03 (br s, 1H), 4.23 (s, 2H), 3.84–3.35 (br s, backbone –OCH3), 3.09 (m, 4H), 2.07–0.63 (br m, backbone); GPC: Mn = 25500, Mw = 33700, PDI (Mw/Mn) = 1.3; IR (cm−1): 2961, 1723, 1447, 1270, 1239, 1189, 1142, 993, 966, 839, 746.
Thiol functionalized polymer 6 (using TEA as a base)
To a stirring solution of polymer 4 (0.020 g, 0.001 mmol) and 1-thionaphthol (0.020 g, 0.128 mmol) in THF (0.4 mL), was added TEA (0.0161 g, 0.16 mmol) at ambient temperature. The resulting reaction mixture was stirred for 16 hours. The solvent was removed and the crude polymer was dissolved in DCM (4 mL) and washed with water. The organic layer was dried, concentrated, and the polymer obtained was dissolved in a minimum quantity of DCM and precipitated into MeOH (30 mL), then filtered and dried to give 0.016 g of the product in 78% yield.
Bifunctional polymer 7
To a stirring solution of polymer 5 (0.060 g) in pyridine (1 mL), was added p-toluoyl chloride (0.1 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 1 hour and then at room temperature for 18 hours. Pyridine was removed under reduced pressure and the crude polymer thus obtained was dissolved in DCM (10 mL) and washed with water (10 mL), saturated aqueous solution of NaHCO3 (10 mL), and 0.5 M HCl solution (10 mL). The organic layer was dried, concentrated, and the polymer thus obtained was dissolved in a minimum amount of DCM and precipitated into MeOH (40 mL). The precipitate was collected by filtration, washed with MeOH, and dried to give 0.05 g of the product (yield = 82%). 1H-NMR (δ, ppm, 300 MHz, CDCl3): 8.42 (d, J = 8.30 Hz, 1H), 7.77 (m, 5H), 7.55–7.38 (m, 5H), 7.18 (d, J = 8.03 Hz, 2H), 7.04 (br s, 1H), 5.52 (m, 1H), 4.65 (m, 2H), 3.84–3.34 (br m, backbone –OCH3), 2.40 (s, 3H), 2.07–0.63 (br m, backbone); GPC: Mn = 14900, Mw = 19500, PDI (Mw/Mn) = 1.3 IR (cm−1): 2955, 1723, 1481, 1441, 1387, 1268, 1240, 1193, 1145, 987, 965, 840, 750.
Bifunctional polymer 8
To a stirring solution of polymer 6 (0.050 g) in pyridine (1 mL), was added p-toluoyl chloride (0.08 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 1 hour and then at room temperature for 18 hours. Pyridine was removed under reduced pressure and the crude polymer thus obtained was dissolved in DCM (10 mL) and washed with water (10 mL), saturated solution of NaHCO3 (10 mL), and 0.5 M HCl solution (10 mL). The organic layer was dried, concentrated, and the polymer thus obtained was dissolved in a minimum amount of DCM and precipitated into MeOH (40 mL). The precipitate was collected by filtration, washed with MeOH, and dried to give 0.042 g of the product (yield = 83%). 1H-NMR (δ, ppm, 300 MHz, CDCl3): 8.36 (d, J = 8.14 Hz, 2H), 7.72 (m, 10H), 7.50 (m, 6H), 7.35 (t, J = 7.72 Hz, 2H), 7.12 (d, J = 7.76 Hz, 4H), 7.02 (br s, 1H), 5.28 (m, 2H), 4.16 (s, 2H), 3.84–3.33 (br m, backbone –OCH3), 2.35 (s, 6H), 2.07–0.64 (br m, backbone); GPC: Mn = 27300, Mw = 35900, PDI (Mw/Mn) = 1.3; IR (cm−1): 2961, 1723, 1447, 1270, 1239, 1189, 1142, 993, 966, 839, 746.
Hydrolysis of 3
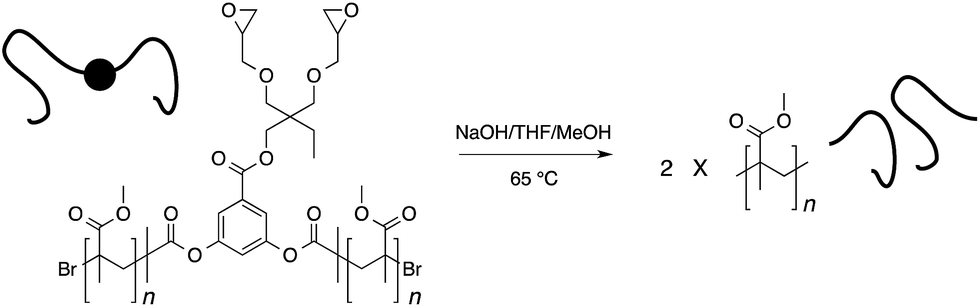
To a solution of polymer 3 (50 mg, 0.0031 mmol) in THF (2 mL) and MeOH (2 mL), NaOH (51 mg, 1.281 mmol) was added and the reaction mixture was refluxed at 65 °C for 20 hr. The solvent was then removed under reduced pressure and the crude thus obtained was suspended in water and then filtered. The solid was then washed with MeOH. Same procedure was employed for the hydrolysis of polymer 4.
Results and discussion
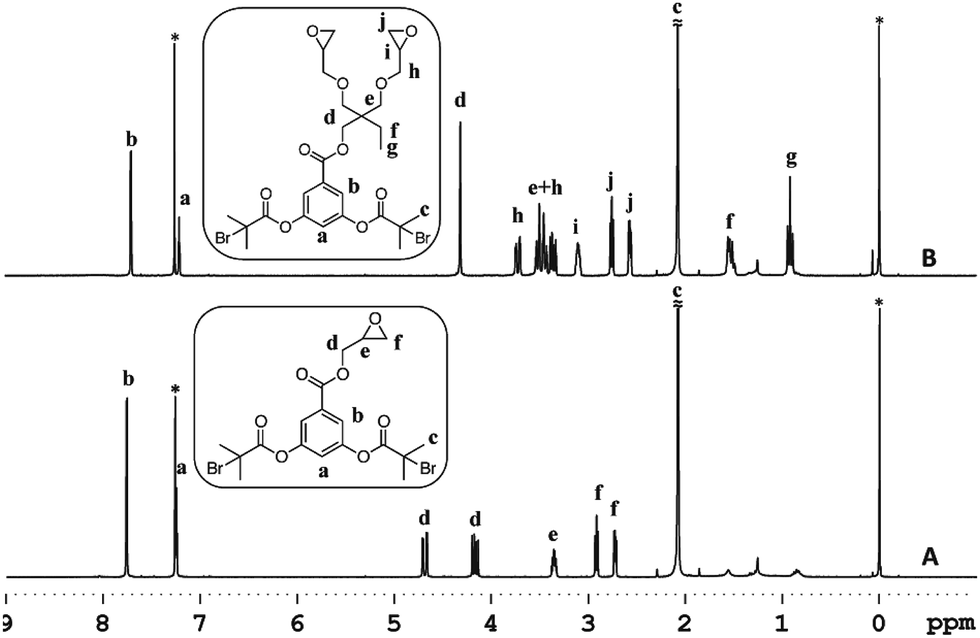
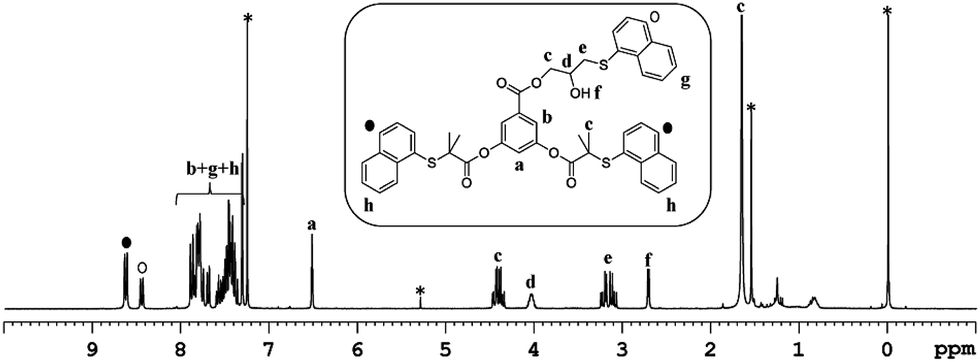
To investigate the feasibility of the aforementioned concept, ATRP-based polymerization initiators carrying one (1) and two (2) epoxide groups along with two initiating sites were designed (Scheme 2). It was envisaged that polymerization from such dual initiating species would give rise to polymer chains having reactive epoxide unit(s) at the midpoint of a polymer chain. To achieve synthesis of these initiators, 3,5-dihydroxybenzoic acid was subjected to an esterification reaction with bromoisobutyryl bromide (Scheme 2). The resulting benzoic acid derivative 1a was further subjected to an esterification reaction with glycidol molecule to give initiator 1. To obtain initiator 2, 1a was converted into the olefin derivative 2a through an esterification reaction and then epoxidized using m-perchlorobenzoic acid in chloroform to furnish initiator 2. 1H-NMR spectroscopy confirmed the structure of these initiators as epoxy proton resonances could be observed in the range of 2.5–3.5 ppm and the aromatic proton resonances could be located in the range of 7–8 ppm (Fig. 1). This aromatic unit is referred to as the core unit in the forthcoming discussion. Initially, initiators 1 and 2 were subjected to a reaction with 1-naphthalenethiol using LiOH or triethylamine as a base. From this study, it became clear that thiol–halide reaction also occurred along with the thiol–epoxy reaction at the initiator level as two sets of aromatic resonances could be assigned to two different types of naphthalene substitutions (Fig. 2 and S4†).
 |
| | Scheme 2 Synthesis of polymerization initiators. | |
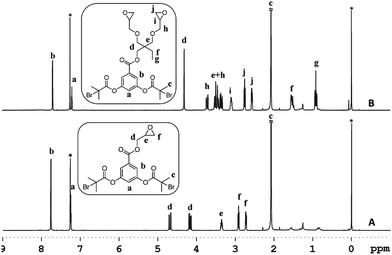 |
| | Fig. 1 1H-NMR of the ATRP initiators 1 (A) and 2 (B) in CDCl3. Solvent signals are shown with an asterisk. Tetramethylsilane was used as an internal standard. | |
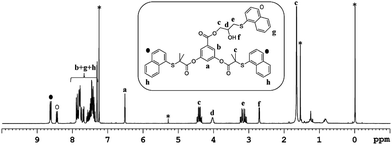 |
| | Fig. 2 1H-NMR (CDCl3) of the product formed upon reaction of initiator 1 with 1-naphthalenethiol in the presence of LiOH as a base. Solvent signals are shown with an asterisk. | |
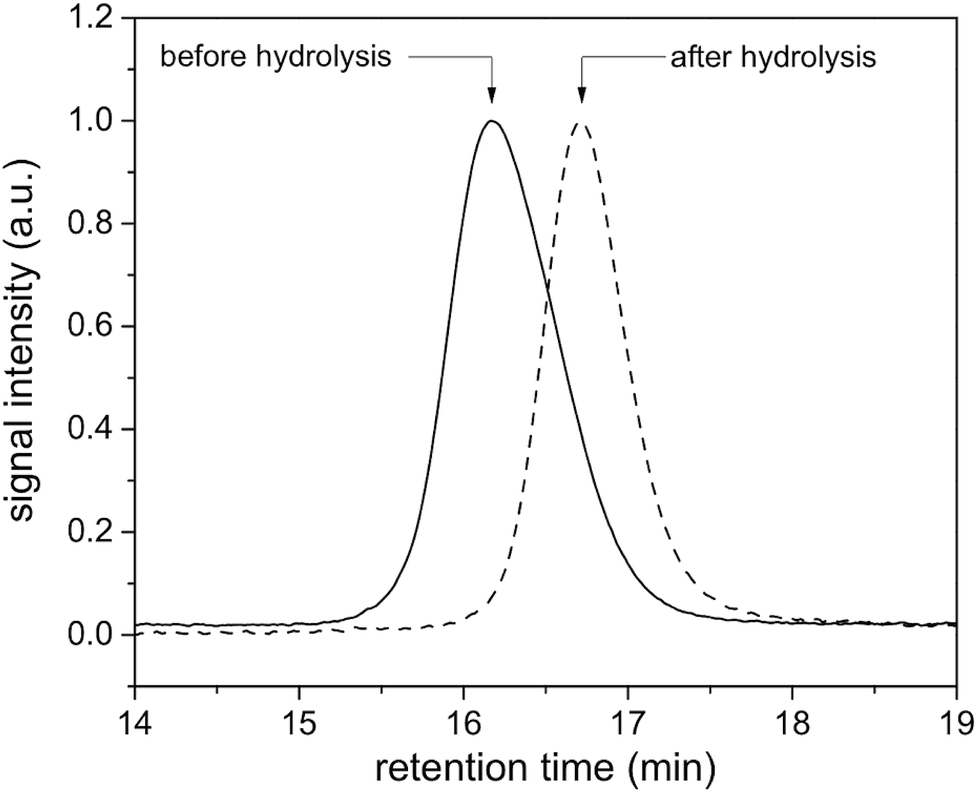
Initiators 1 and 2 were then used for polymerization of methyl methacrylate monomer to give epoxide mid-functional poly(methyl methacrylate) (PMMA) polymers 3 (Mn = 15 kDa, Mw/Mn = 1.3) and 4 (Mn = 26 kDa, Mw/Mn = 1.2), respectively (Scheme 3). To confirm that the reactive centre was located at the centre of the polymer chain, the ester bonds that connect the polymer chain to the aromatic-core, in polymer 4, were hydrolysed under basic conditions (Scheme 4). This resulted in cleavage of the polymer chain into two fragments of nearly equal molecular weight (Mn = 13 kDa, Mw/Mn = 1.2) (Fig. 3 and S5†). A similar result was obtained in the case of polymer 3. These experiments suggested that the reactive sites were located on the centre of the polymer chain.
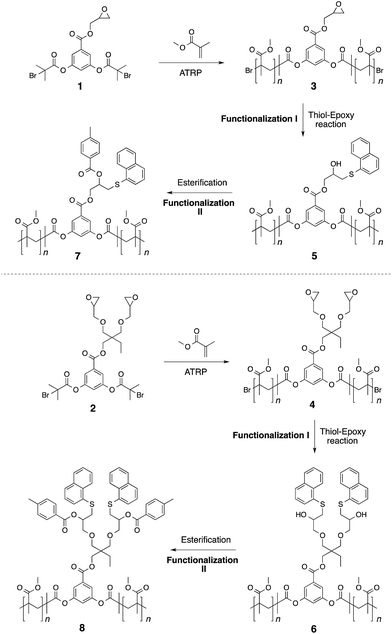 |
| | Scheme 3 Synthesis of polymers 7 and 8. | |
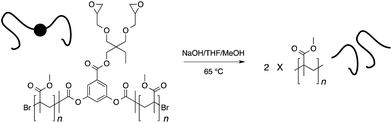 |
| | Scheme 4 Cleavage of the polymer chain to ascertain location of the reactive sites. | |
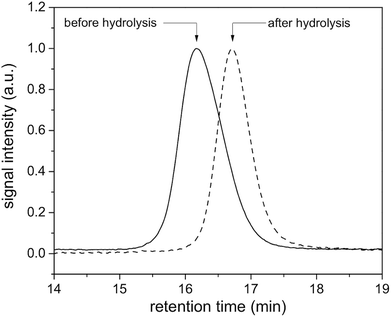 |
| | Fig. 3 GPC traces of the polymer 4 before (solid line) and after hydrolysis (dash line) in DMF. | |
To further investigate the cleavage chemistry, the polymer sample resulting from the hydrolysis reaction was purified and analysed by 1H-NMR spectroscopy (Fig. 4 and S6†). In this analysis, it became clear that the ester bonds that connect the polymer chains to the aromatic core were hydrolysed as the resonances from the aromatic core (7–8 ppm) disappeared along with the methylene (4.2 ppm) and epoxide proton (2.5–3.5 ppm) signals in the 1H-NMR spectrum of the hydrolysed sample. The signals from the PMMA backbone (0.6–2.2 ppm), however, remained unchanged. More importantly, the integration of the methoxy-side chains of the polymer (3.3–3.9 ppm) matched with the integration of the polymer backbone suggesting that the side chain ester groups remained intact. This is in accordance with the previous polymer chain cleavage studies carried out under similar conditions on PMMA-based star polymers.26
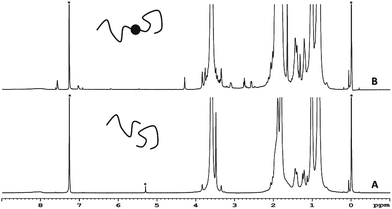 |
| | Fig. 4 1H-NMR of the polymer 4 before (B) and after hydrolysis (A) in CDCl3. Solvent signals are shown with an asterisk. Tetramethylsilane was used as an internal standard. | |
Having established the central location of the reactive site(s), the first modification of the epoxide unit(s) in polymers 3 and 4 was achieved using 1-naphthalenethiol (functionalization I in Scheme 3). This reaction was carried out in the presence of LiOH at room temperature. To ensure complete conversion of the sterically hindered epoxy units, an excess of the thiol reagent was utilized (epoxy:thiol/1:15). This resulted in the formation of the thio-ether bond and generation of the hydroxyl unit. Use of triethylamine as a base (instead of LiOH) also produced the same results. However, in case of LiOH, a catalytic amount was sufficient for thio-ether formation, whereas high excess of triethylamine is required for complete transformation of the epoxide group into the corresponding functionalized structure. Interestingly, only one set of aromatic proton resonances could be observed. Indicating that the halide-chain-end of the polymers did not participate in the substitution reaction as observed in the case of initiators. Modification of the hydroxyl unit in polymers 5–6 with toluoyl chloride led to the formation of the targeted midpoint multifunctionalized structures 7 and 8 (functionalization II in Scheme 3). Once again, high excess of the acid chloride reagent was utilized due to the steric demand and relatively low reactivity of the secondary hydroxyl units (hydroxyl group:acid chloride/1:240).
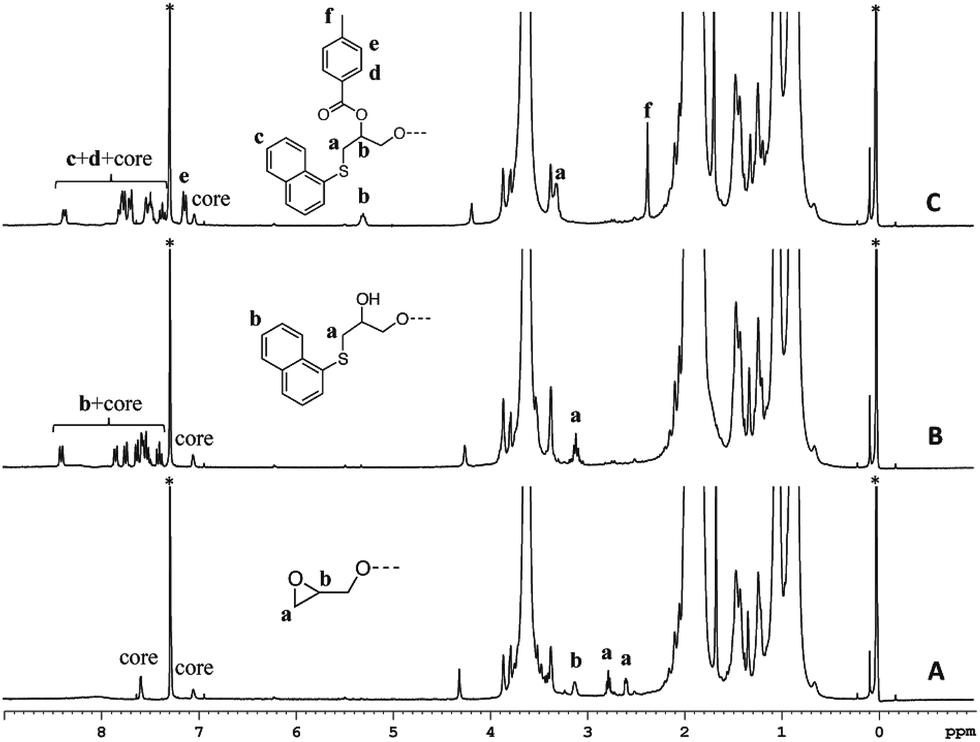
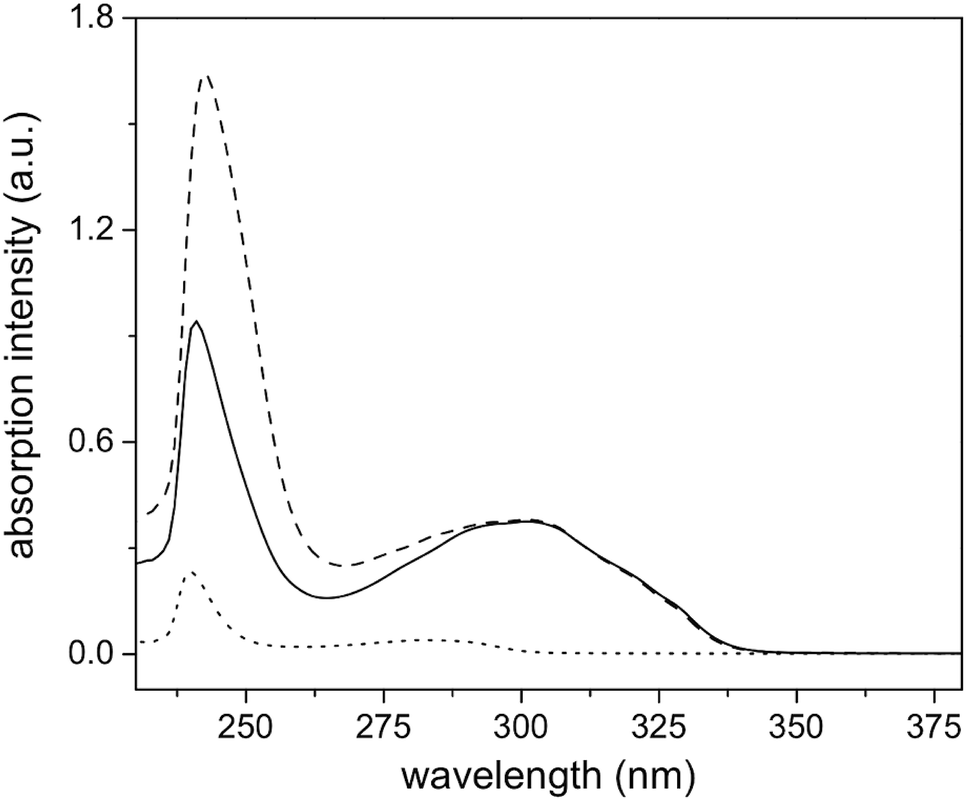
The 1H-NMR spectrum of polymer 4 (A), and functionalized polymers 6 (B), and 8 (C) are shown in Fig. 5. Polymer 4 exhibited the characteristic proton resonances of the PMMA backbone in the range of 0.6–2.2 and 3.3–3.9 ppm. The epoxy proton signals could be detected at 2.57, 2.76, and 3.1 ppm. The first functionalization resulted in the disappearance of the epoxy proton resonances and emergence of the aromatic proton signals from the naphthalene group (7.3–8.4 ppm). Based on the small molecule study, this set of aromatic proton resonances could be assigned to the thioether formed upon the thiol–epoxy reaction. Absence of the other set of the aromatic resonances suggested that the thiol–bromo reaction did not occur in the present set of materials. This may be due to the steric hindrance created by the methyl group of poly(methyl methacrylate). Besides absence of a second set of aromatic signals, as seen at the initiator level, area integration analysis also suggested that the thiol–halide reaction did not occur (see Fig. S7† for area integration). UV-Vis spectroscopy also suggested a successful functionalization step as the absorption band centred on 300 nm could be assigned to the naphthalene chromophore (Fig. 6 and S8†). For the second functionalization, toluoyl chloride was chosen as a reactant. This choice was made because the signal from the methyl group of the toluoyl unit emerges in a region of the 1H-NMR spectrum that is free from proton resonance signals of the polymer backbone and mid-chain groups. Moreover, this signal is high in intensity and resolution when compared to the other signals. This allowed us to determine the extent of hydroxyl group conversion with the help of 1H-NMR spectroscopy. The esterification of polymer 6 gave rise to polymer 8 and area integration analysis between the proton resonances of the toluoyl unit (designated ‘e’ and ‘f’ in Fig. 5) and the naphthalene group suggested complete conversion of the hydroxyl units into the desired toluoyl esters (see Fig. S9† for area integrations). The proton resonance signal at 5.28 ppm could be ascribed to a proton located at the carbon adjacent to the newly formed ester group. In UV-Vis spectroscopy, the absorption band at 245 nm further increased in intensity after the esterification reaction (Fig. 6), most likely due to the incorporation of the toluoyl units in the molecular structure, once again suggesting successful functionalization of the polymer mid-chain through a sequential thiol–epoxy and esterification reaction.
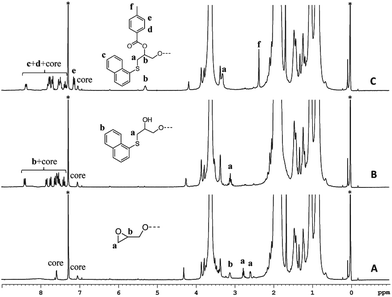 |
| | Fig. 5 1H-NMR of polymer 4 (A), and end-functionalized polymers 6 (B) and 8 (C) in CDCl3. Solvent signals are shown with an asterisk. Tetramethylsilane was used as an internal standard. | |
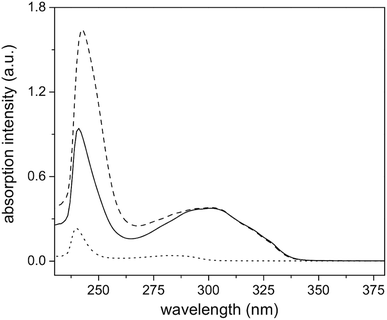 |
| | Fig. 6 UV-Vis absorption spectra of polymers 4 (dot line), 6 (solid line), and 8 (dash line) in chloroform. | |
To examine the effect of the functionalization reactions upon the hydrodynamic volume of the polymers, gel permeation chromatography (GPC) studies were undertaken. These studies established that the hydrodynamic volume of the precursor polymers and the functionalized structures remained the same as no changes in the retention time of the precursors and their functionalized analogues could be detected in the GPC chromatograms (Fig. S10 and S11†).
Conclusions
To summarize, a general synthetic strategy for midpoint multifunctionalization of a polymer chain is established. For this, ATRP polymerization initiators appended with one and two epoxide unit(s) along with two polymerization initiation sites were prepared. Polymerization of methyl methacrylate monomer by using these initiators gave access to polymers with epoxide unit(s) at the centre of the polymer chain. The hydrolysis study confirmed the midpoint location of the reactive site(s) on the polymer chain. The first mid-chain functionalization was achieved through the thiol–epoxy reaction that instated optically active naphthalene functionality and gave rise to a reactive hydroxyl group at the centre of the polymer chain. Further functionalization of the hydroxyl unit with toluoyl chloride then led to the formation of the targeted midpoint multifunctional materials. The number of the mid-chain functionalities could be adjusted by adjusting the number of epoxide unit(s) on the polymerization initiator. The functional group compatibility of the ATRP process and the cascade nature of the functionalization processes delivered the established strategy free from the protection/deprotection requirement of organic synthesis and allowed for facile preparation of complex yet well-defined multifunctional polymeric structures.
Acknowledgements
This work was by National Research Foundation of Korea grant funded by the Korea government (MSIP) (NRF-2015R1D1A1A01057796 and 2015R1A2A2A01006008) and Global Frontier R&D Program (No. 2013M3A6B1078869).
Notes and references
- P. Theato and H.-A. Klok, Functional Polymers by Post- Polymerization Modification, Wiley, Weinheim, Germany, 2013 Search PubMed.
- For excellent review articles, please see:
(a) C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200–1205 CrossRef CAS PubMed;
(b) J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS PubMed;
(c) R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed;
(d) C. Barner-Kowollik and A. J. Inglis, Macromol. Chem. Phys., 2009, 210, 987–992 CrossRef CAS;
(e) A. Sanyal, Macromol. Chem. Phys., 2010, 211, 1417–1425 CrossRef CAS;
(f) U. Mansfeld, C. Pietsch, R. Hoogenboom, R. Becer and U. S. Schubert, Polym. Chem., 2010, 1, 1560–1598 RSC;
(g) C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS PubMed;
(h) B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1–13 CrossRef CAS;
(i) H. Durmaz, A. Sanyal, G. Hizal and U. Tunca, Polym. Chem., 2012, 3, 825–835 RSC;
(j) M. A. Harvison and A. B. Lowe, Macromol. Rapid Commun., 2011, 32, 779–800 CrossRef CAS PubMed;
(k) P. J. Roth, C. Boyer, A. B. Lowe and T. P. Davis, Macromol. Rapid Commun., 2011, 32, 1123–1143 CrossRef CAS PubMed;
(l) A. B. Lowe and M. A. Harvison, Aust. J. Chem., 2010, 63, 1251–1266 CrossRef CAS;
(m) A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745–4750 RSC;
(n) W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS;
(o) K. A. Gunay, P. Theato and H. A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2012, 51, 1–28 CrossRef.
- For selected examples, please see:
(a) M. Malkoch, R. J. Thibault, E. Drockenmuller, M. Messerschmidt, B. Voit, T. P. Russell and C. J. Hawker, J. Am. Chem. Soc., 2005, 127, 14942–14949 CrossRef CAS PubMed;
(b) L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS;
(c) S. Ghosh, S. Basu and S. Thayumanavan, Macromolecules, 2006, 39, 5595–5597 CrossRef CAS;
(d) R. C. Li, J. Hwang and H. D. Maynard, Chem. Commun., 2007, 3631–3633 RSC;
(e) S. K. Yang and M. Weck, Macromolecules, 2008, 41, 346–351 CrossRef CAS;
(f) A. V. Ambade, C. Burd, M. N. Higley, K. P. Nair and M. Weck, Chem.–Eur. J., 2009, 15, 11904–11911 CrossRef CAS PubMed;
(g) N. T. Brummelhuis and M. Weck, ACS Macro Lett., 2012, 1, 1216–1218 CrossRef;
(h) E. M. Kolonko, J. K. Pontrello, S. L. Mangold and L. L. Kiessling, J. Am. Chem. Soc., 2009, 131, 7327–7333 CrossRef CAS PubMed;
(i) N. Cengiz, H. Kabadayioglu and R. Sanyal, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4737–4746 CrossRef CAS;
(j) J. B. Uprety, J. F. Reuther and B. M. Novak, Macromolecules, 2012, 45, 8155–8165 CrossRef;
(k) K. A. McEwan, S. Slavin, E. Tunnah and D. M. Haddleton, Polym. Chem., 2013, 4, 2608–2614 RSC;
(l) X. X. Deng, L. Li, Z. L. Li, A. Lv, F. S. Du and Z. C. Li, ACS Macro Lett., 2012, 1, 1300–1303 CrossRef CAS;
(m) A. Lv, X. X. Deng, L. Li, Z. L. Li, Y. Z. Wang, F. S. Du and Z. C. Li, Polym. Chem., 2013, 4, 3659 RSC;
(n) R. Kakuchi and P. Theato, Polym. Chem., 2014, 5, 2320–2326 RSC;
(o) R. Kakuchi and P. Theato, ACS Macro Lett., 2013, 2, 419–422 CrossRef CAS.
- For protective-group-free multifunctionalization of polymers, please see: A. Saha, S. De, M. C. Stuparu and A. Khan, J. Am. Chem. Soc., 2012, 134, 17291–17297 CrossRef CAS PubMed.
-
(a) C. Ott, R. Hoogenbomm and U. S. Schubert, Chem. Commun., 2008, 3516–3518 RSC;
(b) E. M. Kolonko and L. L. Kiessling, J. Am. Chem. Soc., 2008, 130, 5626–5627 CrossRef CAS PubMed;
(c) I. Kosif, E. J. Park, R. Sanyal and A. Sanyal, Macromolecules, 2010, 43, 4140–4148 CrossRef CAS;
(d) S. Onbulak, S. Tempelaar, R. J. Pounder, O. Gok, R. Sanyal, A. P. Dove and A. Sanyal, Macromolecules, 2012, 45, 1715–1722 CrossRef CAS;
(e) L. Billiet, D. Fournier and F. D. Prez, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6552–6564 CrossRef CAS;
(f) D. Fournier and F. D. Prez, Macromolecules, 2008, 41, 4622–4630 CrossRef CAS;
(g) L. Billiet, D. Fournier and F. D. Prez, Polymer, 2009, 50, 3877–3886 CrossRef CAS;
(h) P. Espeel, F. Goethals, F. Driessen, L.-T. T. Nguyen and F. D. Prez, Polym. Chem., 2013, 4, 2449–2456 RSC;
(i) M. V. Walter, P. Lundberg, A. Hult and M. Malkoch, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2990–2995 CrossRef CAS;
(j) Y. Y. Durmaz, M. Sangermano and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2862–2868 CrossRef CAS;
(k) G. Yilmaz, H. Toiserkani, D. O. Demirkol, S. Sakarya, S. Timur, Y. Yagci and L. Torun, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 110–117 CrossRef CAS;
(l) M. Stemmelen, F. Pessel, V. Lapinte, S. Caillol, J.-P. Habas and J.-J. Robin, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2434–2444 CrossRef CAS;
(m) M. E. Buck and D. M. Lynn, Polym. Chem., 2012, 3, 66–80 RSC.
- For excellent review articles, please see:
(a) R. P. Quirk and D. L. Pickel, Polym. Sci., 2012, 6, 351–412 Search PubMed;
(b) M. A. Tasdelen, M. U. Kahveci and Y. Yagci, Prog. Polym. Sci., 2005, 36, 455–567 CrossRef;
(c) F. L. Verso and C. N. Likos, Polymer, 2008, 49, 1425–1434 CrossRef;
(d) S. Hilf and A. F. M. Kilbinger, Nat. Chem., 2009, 1, 537–546 CrossRef CAS PubMed;
(e) M. A. Harvison, P. J. Roth, T. P. Davis and A. B. Lowe, Aust. J. Chem., 2011, 64, 992–1006 CrossRef CAS.
- For protective-group-free multifunctionalization of polymer chain-end, please see:
(a) I. Gadwal and A. Khan, Polym. Chem., 2013, 4, 2440–2444 RSC;
(b) Y. Kohsaka, K. Yamamoto and T. Kitayama, Polym. Chem., 2015, 6, 3601–3607 RSC.
- For selected examples, please see:
(a) M. P. Robin, M. W. Jones, D. M. Haddleton and R. K. O'Reilly, ACS Macro Lett., 2012, 1, 222–226 CrossRef CAS;
(b) S. Hilf, R. H. Grubbs and A. F. M. Kilbinger, J. Am. Chem. Soc., 2008, 130, 11040–11048 CrossRef CAS PubMed;
(c) S. Hilf and A. F. M. Kilbinger, Macromolecules, 2009, 42, 4127–4133 CrossRef CAS;
(d) S. Hilf and A. F. M. Kilbinger, Macromolecules, 2010, 43, 208–212 CrossRef CAS;
(e) A. A. Nagarkar, A. Crochet, K. M. Fromm and A. F. M. Kilbinger, Macromolecules, 2012, 45, 4447–4453 CrossRef CAS;
(f) G. N. Grover, S. N. S. Alconcel, N. M. Matsumoto and H. D. Maynard, Macromolecules, 2009, 42, 7657–7663 CrossRef CAS PubMed;
(g) L. Tao, C. S. Kaddis, R. R. Ogorzalek Loo, G. N. Grover, J. A. Loo and H. D. Maynard, Macromolecules, 2009, 42, 8028–8033 CrossRef CAS PubMed;
(h) K. L. Heredia, Z. P. Tolstyka and H. D. Maynard, Macromolecules, 2007, 40, 4772–4779 CrossRef CAS;
(i) A. P. Narrainen, L. R. Hutchings, I. Ansari, R. L. Thompson and N. Clarke, Macromolecules, 2007, 40, 1969–1980 CrossRef CAS;
(j) I. A. Ansari, N. Clarke, L. R. Hutchings, A. P. Narrainen, A. E. Terry, R. L. Thompson and J. R. P. Webster, Langmuir, 2007, 23, 4405–4413 CrossRef CAS PubMed;
(k) L. R. Hutchings, A. P. Narrianen, R. L. Thompson, N. Clarke and I. Ansari, Polym. Int., 2008, 57, 163–170 CrossRef CAS.
-
(a) D. Colak, I. Cianga, A. E. Muftuoglu and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 727–743 CrossRef CAS;
(b) B. Iskin, G. Yilmaz and Y. Yagci, Polym. Chem., 2011, 2, 2865–2871 RSC.
-
(a) E. Gungor, G. Cote, T. Erdogan, H. Durmaz, A. L. Demirel, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1055–1065 CrossRef CAS;
(b) O. Altintas, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1218–1228 CrossRef CAS;
(c) O. Altintas, A. L. Demirel, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5916–5928 CrossRef CAS;
(d) A. Gozgen, A. Dag, H. Durmaz, O. Sirkecioglu, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 497–504 CrossRef CAS.
-
(a) L. Tao, J. Xu, D. Gell and T. P. Davis, Macromolecules, 2010, 43, 3721–3727 CrossRef CAS;
(b) L. Tao, J. Liu and T. P. Davis, Biomacromolecules, 2009, 10, 2847–2851 CrossRef CAS PubMed;
(c) A. Debuigne, M. Hurtgen, C. Jérôme and C. Detrembleur, ACS Symp. Ser., 2012, 217–230 CrossRef CAS;
(d) M. Degirmenci, A. Acikses and N. Genli, J. Appl. Polym. Sci., 2012, 123, 2567–2573 CrossRef CAS;
(e) C. Li, J. Hu, J. Yin and S. Liu, Macromolecules, 2009, 42, 5007–5016 CrossRef CAS;
(f) A. Hanisch, H. Schmalz and A. H. E. Müller, Macromolecules, 2012, 45, 8300–8309 CrossRef CAS;
(g) K. Yue, J. He, C. Liu, M. Huang, X.-H. Dong, K. Guo, P. Ni, C. Wesdemiotis, R. P. Quirk, S. Z. D. Cheng and W.-B. Zhang, Chin. J. Polym. Sci., 2013, 31, 71–82 CrossRef CAS.
- M. Beija, M.-T. Charreyre and J. M. G. Martinho, Prog. Polym. Sci., 2011, 36, 568–602 CrossRef CAS.
- Multi-Component and Sequential Reactions in Polymer Synthesis, ed. P. Theato, Springer International Publishing, Heidelberg, 2015, vol. 269 Search PubMed.
- M. C. Stuparu and A. Khan, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3057–3070 CrossRef CAS.
-
(a) T. E. Patten, J. Xia, T. Abernathy and K. Matyjaszewski, Science, 1996, 272, 866–868 CAS;
(b) J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS;
(c) K. Matyjaszewski, T. E. Patten and J. Xia, J. Am. Chem. Soc., 1997, 119, 674–680 CrossRef CAS.
-
(a) K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed;
(b) N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS PubMed.
- P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338–1354 RSC.
- For synthesis of polymers with reactive chain-ends via ATRP, please see:
(a) K. Matyjaszewski, Y. Nakagawa and S. G. Gaynor, Macromol. Rapid Commun., 1997, 18, 1057–1066 CrossRef CAS;
(b) Y. Nakagawa and K. Matyjaszewski, Polym. J., 1998, 30, 138–141 CrossRef CAS.
- For synthesis of polymers with an epoxy chain-end via ATRP, please see:
(a) N. V. Tsarevsky, S. A. Bencherif and K. Matyjaszewski, Macromolecules, 2007, 40, 4439–4445 CrossRef CAS;
(b) X. Zhang, J. Xia and K. Matyjaszewski, Macromolecules, 2000, 33, 2340–2345 CrossRef CAS;
(c) M. Degirmenci, O. Izgin, A. Acikses and N. Genli, React. Funct. Polym., 2010, 70, 28–34 CrossRef CAS.
- For synthesis of random copolymers and block copolymers carrying epoxide units via ATRP, please see: S. De, C. Stelzer and A. Khan, Polym. Chem., 2012, 3, 2342–2345 RSC.
- For synthesis of well-defined polyglycidyl methacrylates, please see:
(a) M. Rodlert, E. Harth, I. Rees and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4749–4763 CrossRef CAS;
(b) D. Damiron, J. Mazzolini, F. Cousin, C. Boisson, F. D'Agosto and E. Drockenmuller, Polym. Chem., 2012, 3, 1838–1845 RSC;
(c) A. Moayeri, B. Lessard and M. Maric, Polym. Chem., 2011, 2, 2084–2092 RSC;
(d) P. F. Caňamero, J. L. de la Fuente, E. L. Madruga and M. Ferngauenter. Pa, Macromol. Chem. Phys., 2004, 205, 2221–2228 CrossRef;
(e) C. S. Gudipati, M. B. H. Tan, H. Hussain, Y. Liu, C. He and T. P. Davis, Macromol. Rapid Commun., 2008, 29, 1902–1907 CrossRef CAS;
(f) H. Gao, M. Elsabahy, E. V. Giger, D. Li, R. E. Prud'homme and J.-C. Leroux, Biomacromolecules, 2010, 11, 889–895 CrossRef CAS PubMed;
(g) Q. Zhang, S. Slavin, M. W. Jones, A. J. Haddleton and D. M. Haddleton, Polym. Chem., 2012, 3, 1016–1023 RSC;
(h) A. Hayek, Y. Xu, T. Okada, S. Barlow, X. Zhu, J. H. Moon, S. R. Marder and S. Yang, J. Mater. Chem., 2008, 18, 3316–3318 RSC;
(i) H. Zhu, Q. Liu and Y. Chen, Langmuir, 2007, 23, 790–794 CrossRef CAS PubMed;
(j) M. Ma, F. Li, F.-J. Chen, S.-X. Cheng and R.-X. Zhuo, Macromol. Biosci., 2010, 10, 183–191 CrossRef CAS PubMed;
(k) R. París, M. Liras and I. Quijada-Garrido, Macromol. Chem. Phys., 2011, 212, 1859–1868 Search PubMed.
- S. De and A. Khan, Chem. Commun., 2012, 48, 3130–3132 RSC.
-
(a) A. Brändle and A. Khan, Polym. Chem., 2012, 3, 3224–3227 RSC;
(b) I. Gadwal, J. Rao, J. Baettig and A. Khan, Macromolecules, 2014, 47, 35–40 CrossRef CAS;
(c) I. Gadwal, M. C. Stuparu and A. Khan, Polym. Chem., 2015, 6, 1393–1404 RSC;
(d) S. Binder, I. Gadwal, A. Bielmann and A. Khan, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2040–2046 CrossRef CAS;
(e) I. Gadwal and A. Khan, RSC Adv., 2015, 5, 43961–43964 RSC;
(f) C. Buerkli, S. H. Lee, E. Moroz, M. C. Stuparu, J. C. Leroux and A. Khan, Biomacromolecules, 2014, 15, 1707–1715 CrossRef CAS PubMed;
(g) D. Zhang, C. Liu, S. Chen, J. Zhang, J. Cheng and M. Miao, Prog. Org. Coat., 2016, 101, 178–185 CrossRef CAS.
-
(a) S. Li, J. Han and C. Gao, Polym. Chem., 2013, 4, 1774–1787 RSC;
(b) J. S. Basuki, L. Esser, H. T. T. Duong, Q. Zhang, P. Wilson, M. R. Whittaker, D. M. Haddleton, C. Boyer and T. P. Davis, Chem. Sci., 2014, 5, 715–726 RSC.
-
(a) F. S. Gungor and B. Kiskan, React. Funct. Polym., 2014, 75, 51–55 CrossRef CAS;
(b) B. Hanbeyoglu, B. Kiskan and Y. Yagci, Macromolecules, 2013, 46, 8434–8440 CrossRef CAS;
(c) A. Musa, B. Kiskan and Y. Yagci, Polymer, 2014, 55, 5550–5556 CrossRef CAS.
- L. Xue, U. S. Agarwal, M. Zhang, B. B. P. Staal, A. H. E. Müller, C. M. E. Bailly and P. J. Lemstra, Macromolecules, 2005, 38, 2093–2100 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization details are provided. See DOI: 10.1039/c7ra02702h |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
JiHyeon Hwanga,
Youngson Choeb,
Joona Banga and
Anzar Khan
a,
JiHyeon Hwanga,
Youngson Choeb,
Joona Banga and
Anzar Khan