DOI:
10.1039/C7RA02700A
(Paper)
RSC Adv., 2017,
7, 24168-24176
Synergistic activating effect of promoter and oxidant in single step conversion of methane into methanol over a tailored polymer-Ag coordination complex†
Received
6th March 2017
, Accepted 27th April 2017
First published on 3rd May 2017
Abstract
Single-step conversion of methane to its oxygenated derivatives, such as methanol, is a challenging topic in C1 chemistry. The presence of Brønsted-acidic sites, N- and O-type chelating ligands, and noble metals are demonstrated to be essential criteria for effective catalysis of this reaction. Considering these criteria, a catalytic complex was tailored herein. Poly-D-glucosamine (Ch) was used as chelating ligand for Ag, to incorporate the robust redox properties of Ag(I). The prepared AgCh complex was characterized by techniques including solid-state 1H-NMR, FE-TEM, XANES, and XPS. Besides highlighting the utility of chelate complexation for providing new materials, this study elucidates the effects of the oxidant and promoters on the methane oxidation. The catalytic activity was tested for different oxidant combinations, including hydrogen peroxide, oxygen, and carbon dioxide. Of all of them, a mixture of hydrogen peroxide and oxygen showed the highest selectivity for oxidation of methane to methanol. Further, it was observed that the addition of 1-butyl-3-methylimidazolium chloride [BMIM]+Cl− as a promoter to the hydrogen peroxide and oxygen-containing AgCh system could enhance methanol production. The methanol yield reached up to 3166 μmol, representing an 18-fold yield increase and an 8-fold methane conversion increase when compared to the results (175 μmol) without a promoter.
Introduction
Global climate change is an increasingly serious problem resulting from increasing global industrialization and the associated output of greenhouse gases, such as methane (CH4). While carbon dioxide (CO2) is the major contributor to the greenhouse effect by volume, CH4 is a much more potent greenhouse gas. Thus, strategies to increase the use of waste CH4 gas as a feedstock for the production of industrially important chemicals, such as methanol and acetic acid, are an effective means to reduce harmful CH4 emissions and may reduce the greenhouse effect. Many attempts have previously been made to utilize CH4 in the preparation of syngas,1–3 which is a major feedstock for methanol production. However, the formation of the final methanol product requires multiple high-temperature steps and the use of different catalysts of type Cu/ZnO/Al2O3.4,5 Consequently, the direct single-step conversion of CH4 to its oxygenates, such as methanol and acetic acid, remains a challenge. The inertness of CH4, caused by its high C–H bond dissociation energy (ΔHC–H = 439.57 kJ mol−1), is a key problem that has prevented the use of soft reaction conditions for CH4 conversion, such as low acidity, low temperature, and low pressure. Therefore, catalysts that are tailored to increase the C–H bond activity and thereby enable faster reaction kinetics for CH4 conversion, especially to oxygenates, are urgently required.
It is important to understand which properties of catalysts are advantageous for the conversion of CH4 to its oxygenates, as previously investigated by several research groups.6,7 Metal complexes have been extensively tested as catalysts for the conversion of CH4 because of their ability to undergo inorganic nucleophilic reactions. Kirillova et al.6 tested vanadium-based metal complexes as catalysts in the homogeneous catalytic conversion of CH4 to acetic acid, and found that complexes with more N,O- or O,O-polydentate groups in their ligands showed the best catalytic activity. The use of these ligands enabled a 50% improvement in the yield of acetic acid. Their report was consistent with previous reports of alkane oxidation reactions in which the N,O-type ligands (N-heterocyclic carboxylic acids) were recognized for their involvement in the proton transfer steps.6 Therefore, catalysts with these favorable ligand environments are suitable for further tailoring. The role of the noble metal atoms in these catalyst complexes may be understood by considering Pt(bpym)Cl2 and HAuCl4 ref. 7 and 8 complexes, which were previously employed for the production of methanol from CH4 in homogeneous systems with a highly acidic solvent medium. While these complexes showed the best catalytic activity for CH4 conversion (TON > 500), the conversion reaction did not reach completion, yielding methyl bisulfate (CH3OSO3H). The harsh conditions employed in these reaction systems meant they were not investigated further. Since then, trends in the field have shifted towards mimicry of the CH4 monooxygenase (MMO) enzyme,9 which oxidizes CH4 to methanol in nature, and the replacement of hard solvents by hydrogen peroxide (H2O2). As an example, the complex [CuICuICuI(7-N-Etppz)]1+, where 7-N-Etppz stands for the organic ligand 3,3′-(1,4-diazepane-1,4-diyl)bis[1-(4-ethylpiperazine-1-yl)propan-2-ol], was reported by Liu et al. for methanol production.10 Based on previous reports of increased yields from zeolite-supported Cu, Fe catalysts,11 the [CuICuICuI(7-N-Etppz)]1+ complex was supported on silica nanoparticles resulting in an increased yield of methanol. However, while the harsh conditions of the earlier reaction systems were successfully replaced, the central aim of achieving a higher CH4 conversion was not addressed. Noble metal complexes that function under milder conditions are yet to be tested and understood. The few reported examples of heterogeneous catalyst systems for CH4 conversion also reveal specific properties of the catalysts. For example, Sun et al. replaced the acidic solvent (sulfuric acid) used in a homogeneous reaction system with a solid acidic catalyst, i.e., a heteropolyacid chemisorbed on silica.12 Their study demonstrated and generalized the requirement for catalysts with acidic properties by showing that acidic catalysts convert CH4 to acetic acid under mild conditions. Brønsted-acidic groups also play a considerable role in the proton exchange steps involved in producing methanol and acetic acid.13 Thus, to reduce the high bond dissociation energy of CH4 and thereby achieve a higher conversion of CH4 to its oxygenates, catalysts should have several N,O-type ligands, high acidity, Brønsted-acidic sites, and be heterogeneously supported. Further, previous reports7,14 have shown that the use of inorganic chloride salts as promoters can increase the CH4 conversion. Though activity increased after using promotor, the total conversion of CH4 by mass did not meet the expectations. Weak attraction between the catalyst and the added salt promoter still remains as a drawback in this case. Thus making use of promoting salts that interact strongly with the catalyst, forming an intermediate structure which boosts the kinetics of CH4 conversion is preferable.
Our strategy is to use a biopolymer, poly-D-glucosamine (Ch), to ligate Ag ions with its N- and O-based chelating groups. This biopolymer also contains highly acidic Brønsted acid sites, and the regular arrangement of the chelating atoms in Ch enables a systematic arrangement of active Ag sites for the CH4 conversion reaction. Therefore Ag coordinated Ch (AgCh) complex meets all the criteria mentioned above for producing a much higher yield of methanol and is competitive with the performance of other zeolite-based catalysts. We highlight that, unlike with zeolite-supported catalysts, the AgCh catalyst's competitive performance results solely from the active Ag sites, with no contribution from the support. We show that the total TON can be directly correlated to the number of moles of Ag contained in the catalyst. These observations prove that our tailored catalyst is highly active for the CH4 conversion reaction. We also investigated the effect of halide ion-containing ionic liquids on the reaction, finding that the inclusion of these ionic liquids created a stable intermediate structure that gave ∼13 times greater CH4 conversion. The interaction between the ionic liquids and the catalyst is discussed later in this article. We also prepared CuCh and FeCh using the same method and compared their performance to that of AgCh to prove the effect of the noble metal in the AgCh complex.
Experimental section
Materials and methods
Silver nitrate (AgNO3, >99%), copper(II) nitrate pentahydrate (CuNO3·5H2O, 98%), iron(III) nitrate nonahydrate (FeNO3·9H2O), poly-D-glucosamine, and the ionic liquids 1-butyl-3-methylimidazolium chloride (>98%) and 1-butyl-3-methylimidazolium bromide (>97%) were purchased from Sigma Aldrich. Sodium hydroxide (NaOH) and hydrogen peroxide (H2O2) (30%) were purchased from Sanchun chemicals, Korea. High purity methane (99.95%) and carbon dioxide (99.999%) were used for reactions, and high purity hydrogen (99.99%) and oxygen (99.999%) were used as standards for gas chromatography (GC) analysis. Double-distilled water was used in all reactions.
Catalyst preparation
Poly-D-glucosamine (500 mg) was dispersed in double-distilled water (250 mL) contained in a beaker (500 mL capacity). The dispersion was heated to 80 °C on a hot plate and stirred for 20 min at 600 rpm. Then, the required metal nitrate (Ag, Fe, or Cu) (5 mmol) was dissolved in water (100 mL) in a separate beaker. This solution was added dropwise to the poly-D-glucosamine dispersion. After a further 10 min, the pH of the solution was adjusted to 9.3 by the addition of sodium hydroxide with careful stirring, forming the poly-D-glucosamine-metal complex as a solid precipitate. The complex was filtered, washed several times with water, and air-dried. Catalyst with Ag, Fe and Cu are abbreviated as AgCh, FeCh and CuCh respectively.
Sample characterization
Fourier transform infrared (FTIR) spectroscopy was recorded using a Varian 2000 Fourier transforms infrared spectrophotometer by accumulating 32 scans between 2000 and 4000 cm−1. The samples were pelletized with KBr and put in an FTIR chamber for analysis. X-ray photoelectron spectroscopy (XPS) analyses (ESCALAB 250, UK) were performed under a vacuum of ∼1.3 × 10−7 Pa. A monochromated X-ray Al Kα source (energy: 15 kV, 150 W) with hγ of 1486.6 eV was used for all measurements. The analyzed area was 400 μm2. The binding energy values were charge-corrected to the C 1s signal (284.5 eV). Peak deconvolution was performed with XPS peaks 41 software. X-ray diffraction (XRD) spectra were recorded between 2θ of 10 and 90°. A Cu anode was used, and continuous scans were taken for each sample. Solid-state 1H magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance II+ 400 MHz spectrometer. The instrument was operated at a rate of 400 MHz with a spin rate of 13 kHz. X-ray absorption spectroscopy (XAS) was used to collect Ag, Fe, and Cu K-edge X-ray absorption near edge structure (XANES) data, using the lab-source R-XAS Looper (Rigaku R-XAS) with a 3 kW X-ray generator and a Mo–LaB6 target-filament combination. A Johannson-type curved Ge (220) crystal with a Rowland circle of radius of 320 mm was used in the monochromator section. The incident X-ray intensity (I0) was monitored using a semi-transmission type proportional counter filled with Ar gas, with Be windows on both sides. Transmitted X-ray intensity (It) was measured with a high-speed scintillation counter. Thin layered samples (sandwiched between Kapton tape) were mounted between the I0 and It detectors, in the transmission geometry. The Fe and Cu K-edge energies were calibrated using standard Fe and Cu foils, that were measured under the above conditions. Thermogravimetric analysis (TGA), using a TGA N 1000 (SCINCO) thermogravimetric analyser was used to study the degradation patterns of the catalysts. The samples were heated to 600 °C with a ramping rate of 5 °C min−1. Elemental analysis was carried out using a Flash EA 1112 elemental analyzer. Samples were dissolved in acetic acid (0.1 M) for analysis. Transition electron microscopy (TEM) images were recorded using a JEOL-200CX. Ion chromatography (IC) was used to analyse products in solution. The method specifications used were: column: Aminex 87H (Bio-rad), high performance liquid chromatography (HPLC): Ultimate 3000 (Dionex, USA), eluent: H2SO4 (0.01N), flow: 0.5 mL min−1, oven temperature: 40 °C, and detector: refractive index (RI) (ERC refractoMAX520).
Catalytic activity test
The catalyst was added to an ice-cooled reactor vessel (150 mL capacity) containing double-distilled water (15 mL) and H2O2 (5 mL, 30%). Thus charged, the vessel's lid was fitted tightly and the vessel was degassed by purging with N2 for 5 min. Then, the reactor was pressurized to the required pressure with CH4 (g). The system was heated at 60 °C and electrically stirred at a rate of 900 rpm. After attaining the correct temperature, the reaction was allowed to proceed for the required time. After this time, the system was simply allowed to cooldown to room temperature and then cooled with ice to condense the gaseous oxygenated products. The liquid was filtered and the products were analysed by IC to determine the product contents in units of milligrams, from which amount of the product in units of moles was calculated. All results were compared to standards for confirmation. The gas-phase products were analysed by GC (Agilent 7890A TCD/FID). The number of moles of CO2 was calculated from a calibration curve. The CO2 content (mol) was calculated by eqn (1) and (2),| | |
Concentration of CO2 = peak area × Rf
| (1) |
| | |
Amount of CO2 (mol) = concentration of CO2 × volume of reaction vessel (mL),
| (2) |
where Rf is the response factor.
Results
Characterization of poly-D-glucosamine-based metal complexes
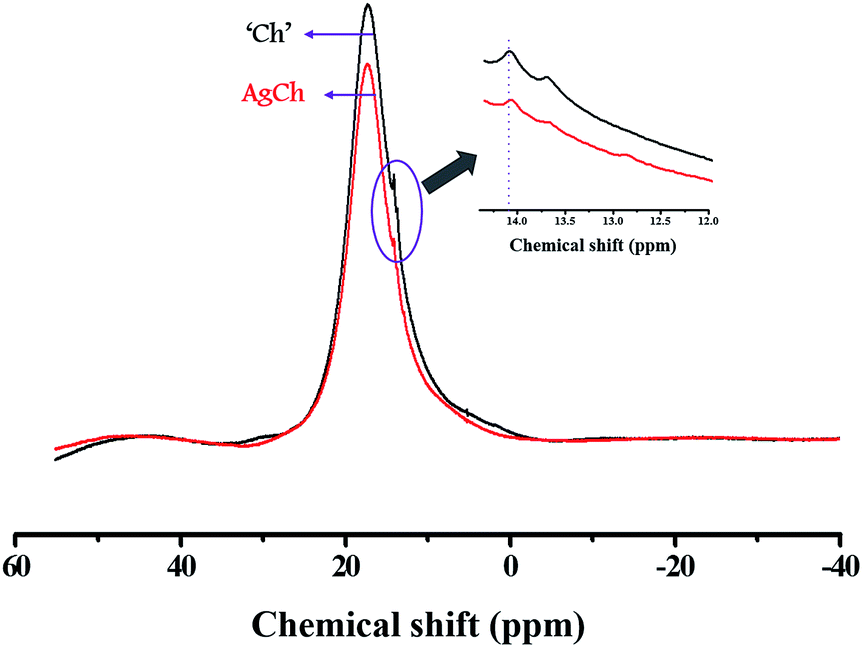
The solid-state 1H MAS NMR spectra for the poly-D-glucosamine catalyst and its Ag coordination complex, AgCh, are shown in Fig. 1. A wide and broad peak was observed from 0 to 20 ppm, which clearly indicates the presence of highly acidic Brønsted acid groups with strong hydrogen bonding. The large width of the peak also confirms the presence of several hydrogen atom environments with different electropositivity, which affects the degree of hydrogen bonding. The hydrogen atoms of amine groups also contribute to the peak broadening as the ppm range for N–H coupling lies within the range of observed broad peak. After the formation of the AgCh complex, a small shift to lower ppm values over the entire peak range and a decrease in peak intensity were observed.
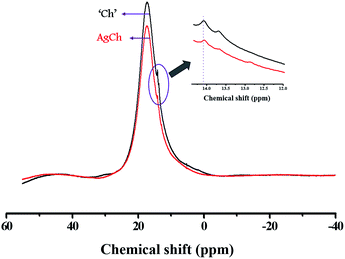 |
| | Fig. 1 Solid-state 1H MAS NMR spectra for Ch (black line) and AgCh (red line). | |
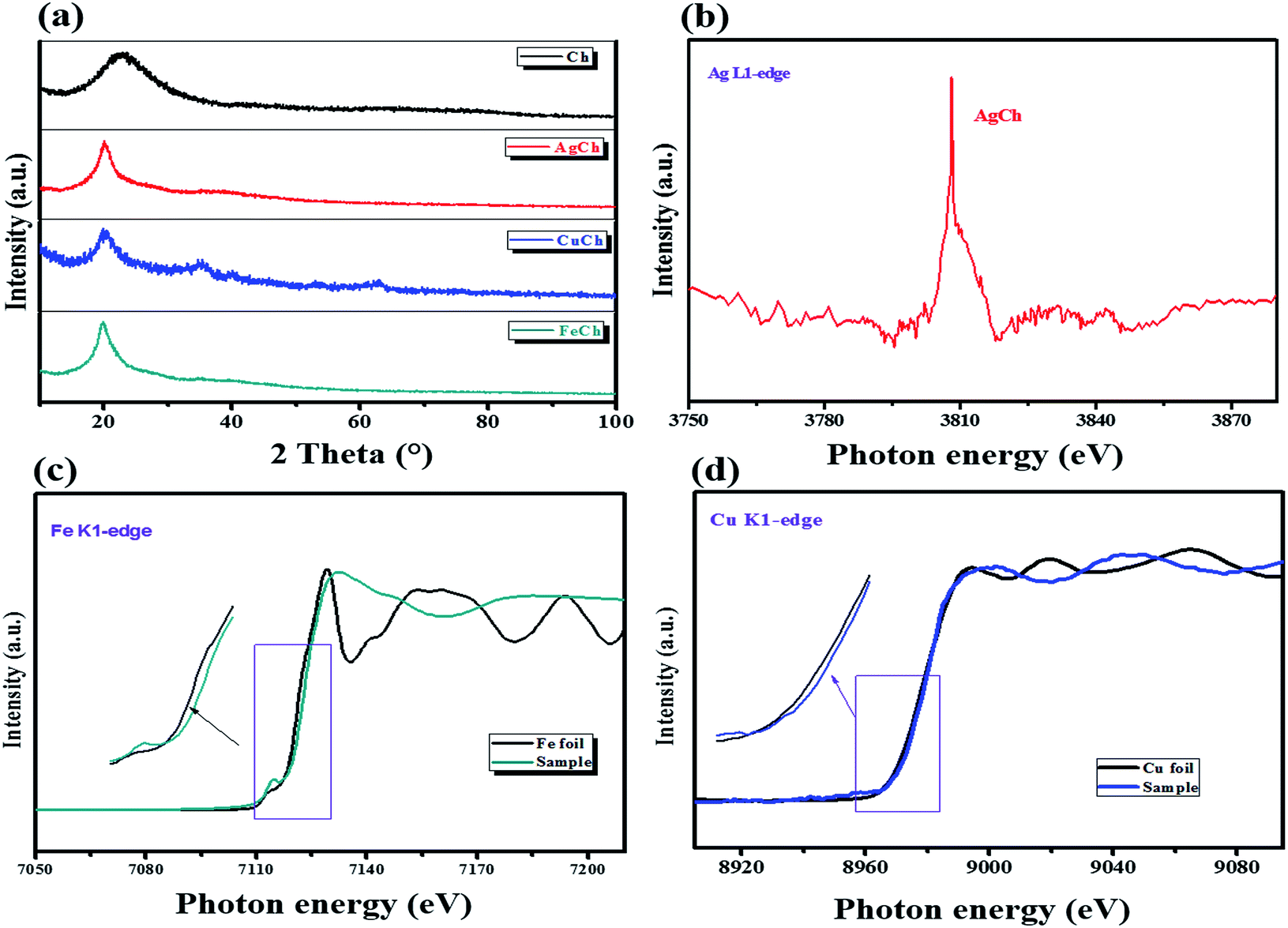
FTIR spectra were recorded for both Ch and the AgCh, CuCh, and FeCh complexes (Fig. 2). The characteristic broad band between 2900 and 3600 cm−1 corresponds to the combined absorptions of N–H and O–H vibrations.15 The peaks in the range 1567–1651 cm−1 are assigned to N–H deformational vibrations. After the addition of the metals to Ch, a decrease in the intensity of the absorbance between 3400 and 3600 cm−1, assigned to N–H vibrations, was observed. This decrease indicates the complex of the metal since the donation of electrons from nitrogen to metal weakens the N–H bonds. Complexation of the metal is further confirmed by the disappearance of the weak band at 1567 cm−1, associated with N–H vibrations, and a shift in the peak at 1651 cm−1. A sharp peak at 524 cm−1, usually assigned to ligand-to-metal bonding, was also observed in all metal complexes, further confirming the complexation of Ch with metals. No marked changes in the absorbance of O–H bonds were observed. The crystallization behavior of Ch and the prepared catalysts was studied using XRD, and the spectra are shown in Fig. 3(a). The pristine Ch showed a broad reflection at 2θ = 22.9°, which implies that pristine Ch is amorphous. After the complexation, this broad reflection sharpened and shifted to 2θ = 20°, indicating higher crystallinity after complexation of Ch with metals. No reflections corresponding to the metals were observed in the complexed Ch, suggesting that the metal atoms are only present as chelates, rather than metal particles. The crystalline structure varied between each of the three complexes, with the FeCh complex showing the sharpest reflection, with the sharpness decreasing in the order of AgCh followed by CuCh. Fig. 3(b) shows the Ag L1-edge XANES of the AgCh catalyst. The peak at ∼3808.7 eV corresponds to the transition from core 2s orbital to the unoccupied p-type orbitals, which occurs at a higher energy in the AgCh than the corresponding peak in metallic Ag foil (∼2.6 eV). Based on this difference in energy, we tentatively suggest that the complexed Ag is present in the +1 oxidation state, as Ag+ ions, although the low Ag content in the AgCh sample and low spectral resolution introduce noise into the presented spectra which may slightly affect the reported energies. This XAS evidence for the presence of Ag+ in the AgCh complex is complemented by the XPS results for Ag (see below), which also suggest the presence of Ag(I) in the sample. Fig. 3(c) shows the Fe K-edge XANES spectra for both the reference foil and FeCh. The pre-edge feature, the energy shoulder at 7122 eV, and the white line intensity of the FeCh spectrum indicate that the electronic structure of FeCh is different to that of the Fe foil. The diverse spectral features of FeCh are similar to those previously reported for iron oxide, especially Fe(II) oxide,16,17 indicating the presence of Fe2+ ions in the FeCh samples. The Cu K-edge XANES spectra for both CuCh and Cu foil are shown in Fig. 3(d). The CuCh XANES spectrum exhibits pre-edge spectral features indicative of 1s–3d transition.18 The spectral features of the CuCh sample match previously reported spectra of CuO and CuS compounds,19 suggesting that Cu2+ ions are present in the sample. Each of our samples showed a shift in the white line to a slightly higher energy, compared to their respective metallic foils, which previous reports attribute to the fractional oxidation of Fe20 and Cu21 atoms in the samples. Fractional oxidation is expected to occur in the complexes prepared in the present study because of the substantial presence of both hydroxyl ions and different ligating functional groups in the chelating polymer.
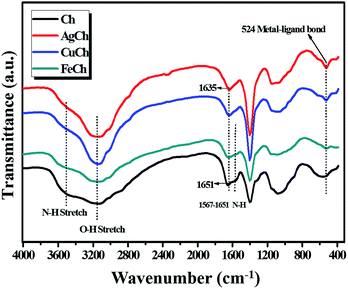 |
| | Fig. 2 FTIR spectra for Ch and the different catalyst species. | |
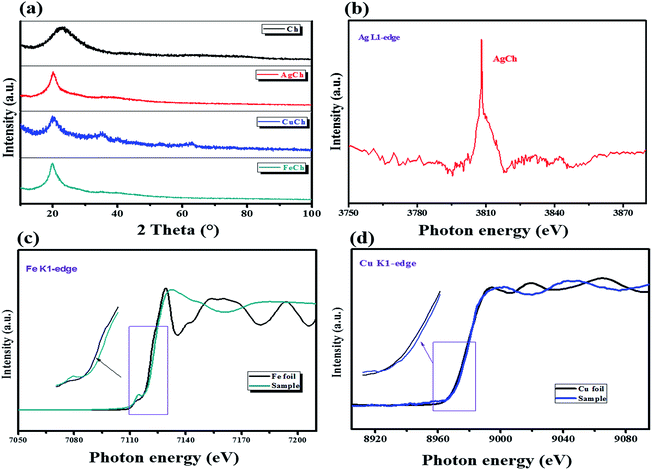 |
| | Fig. 3 (a) XRD spectra and (b–d) XANES spectra for AgCh, FeCh, and CuCh respectively. | |
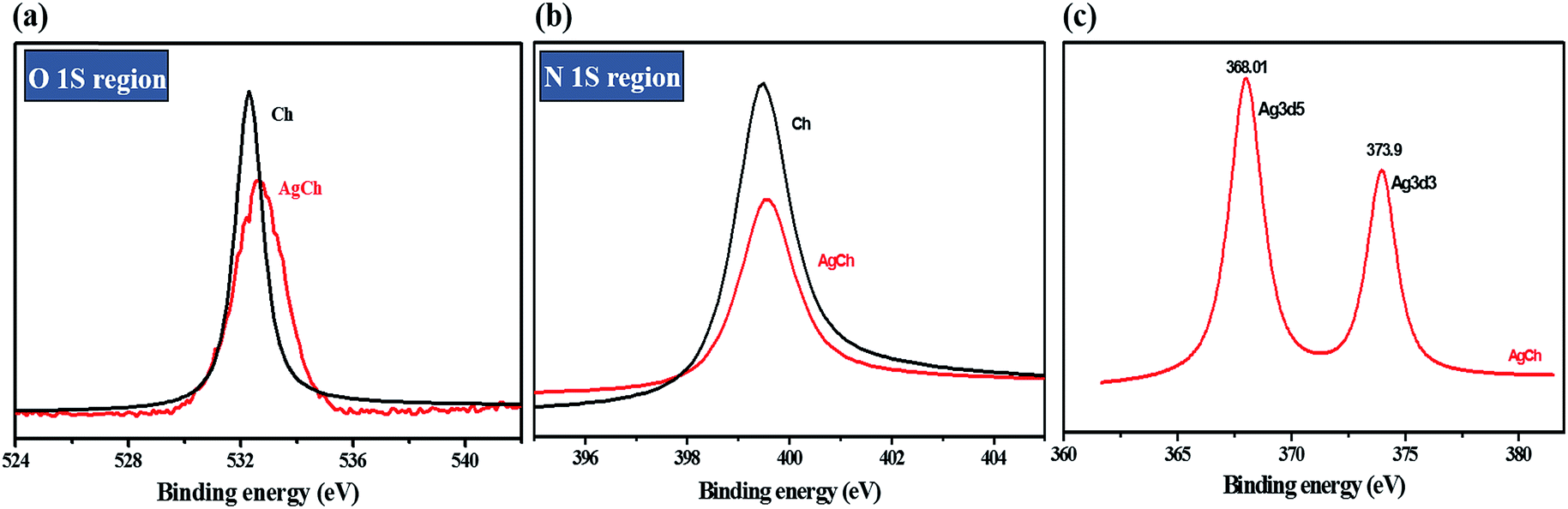
The surface composition of the catalyst complexes was analysed by XPS. The changes observed in the spectrum of the Ch polymer after the formation of complexes can be seen for AgCh in Fig. 4. XPS before and after complexation comparison for Fe and Cu catalysts is shown in ESI, Fig. S2.† The intensity of the O 1s and N 1s peak of Ch decreased after AgCh formation. In both regions, the peak shifted to slightly higher values of binding energy, which clearly indicates the transfer of electron density from Ch to Ag. As in the FTIR spectra, where the strong N–H absorbance diminishes after complexation, the XPS spectrum reveals that the lone pair of electrons from the nitrogen atom of amine groups contributes to coordination of Ag. Fig. 4(c) confirms the presence of Ag, and the two characteristic peaks at 368 and 373.9 eV confirm the oxidation state of Ag to be +1.
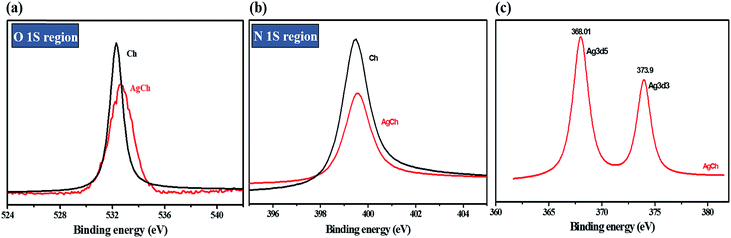 |
| | Fig. 4 High-resolution XPS spectra for (a) O 1s, (b) N 1s, and (c) Ag regions of the AgCh complex. | |
The TEM images of the Ch polymer presented in Fig. 5(a) show that the pristine polymer has a loose, fibrillar structure, indicating the presence of free polymer chains. The formation of the AgCh complex [Fig. 5(b) and (c)] induces a change in the morphology, from fibrillar to uniform cube-like structures. These cubes with varying size between 100 to 200 nm, are observed for AgCh only; no cube-like structures are observed for AgCh only; no cube-like structures are observed for the other metal–Ch complexes (Fig. S3, ESI†). The cubes are suggested to form because of the tendency of the polymer to chelate metal atom in three-dimensional configurations.
 |
| | Fig. 5 TEM images of (a) Ch and (b–d) AgCh at different magnification. | |
Discussion
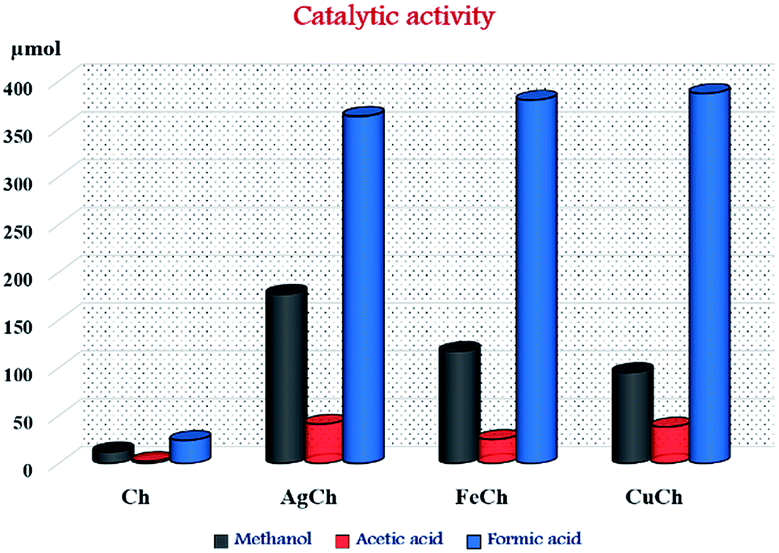
The activity of all the prepared catalysts is shown in Fig. 6 and Table 1. Zero conversion of the CH4 was observed when reaction was carried out without catalyst. Calculated moles, as present in their respective complex catalyst, of metal (Ag, Fe, Cu) nitrates employed as catalysts were able to show a moderate activity. However, no methanol was produced showing complete selectivity towards formic acid. The pristine Ch produced a negligible amount of methanol (12 μmol) indicating possibility of methanol formation with this material when employed as catalyst. Adding AgNO3 (which showed best activity among the tested metal nitrates) and Ch into the reaction mixture produced formic acid, surprisingly, but not methanol. This observation confirms metal nitrates dominates the reaction path way to formic acid. After metal complexation, there was an abrupt increase in methanol formation, along with other byproducts including formic acid and acetic acid. These results imply that the metal–Ch complex synergistically makes an active component and thus, an active catalyst for the conversion of CH4 to its oxygenates. Among the prepared AgCh, FeCh, and CuCh catalysts, AgCh showed the best performance, producing 175 μmol of methanol and 227 μmol of total oxygenated products for 0.15 g of the catalyst at a CH4 pressure of 3 × 106 Pa. CuCh was more active than FeCh, nevertheless both these catalysts showed activity that was ∼20 times lower than that of AgCh when the turnover number (TON) was considered. The concentration of over-oxidized products was higher than the concentration of methanol in all cases. Higher stability of Ag-complexes in peroxide medium might be responsible for best activity shown by AgCh compared to FeCh and CuCh. Ag-complexes reported to form a strong ionized intermediates where charge is normally a radical.22 These radical species, which are generated by homolytic cleavage nature of peroxide's O–O bond, are known to interact strongly with organic species to form chemicals. High activity of AgCh in this study also can be explained under similar mechanism in which radical species formed by AgCh complex in H2O2 medium can interact with hydrocarbon CH4 to form organic molecules like methanol, formic acid and etc. Though the formic acid and acetic acid are over oxidized products of methanol in this reaction, the AgCh complex, which shows high stability of radical species, was able to retain higher concentration of methanol in given time. Cu shows different oxidation states (CuI and CuII) and so do Fe (FeII and FeIII). Interconversion of these oxidation states reacting with OH. Produced by H2O2 during the reaction,23 causes easy over oxidation of methanol thereby giving more selectivity towards formic and acetic acid in case of these two metal complexes.
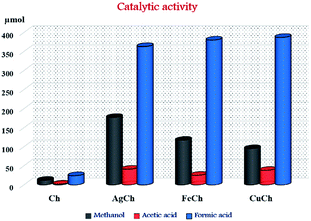 |
| | Fig. 6 CH4 oxidation activity comparison of AgCh, FeCh and CuCh catalysts. | |
Table 1 Product quantification and percentage of CH4 conversion for different catalysts used in this worka
| Entry |
Catalyst |
Oxidant |
Product formed (μmol) |
CH4 conversionb (%) |
| CH3OH |
HCOOH |
CH3COOH |
CO2 |
| Reaction conditions – catalyst: 0.15 g, solvent: 15 mL DI H2O + 5 mL 30% H2O2, P(CH4): 3 × 106 Pa, temperature: 60 °C, time: 3 h. % conversion calculated using formula − total moles of oxygenates formed/initial moles of CH4 × 100. |
| 1 |
No |
H2O2 |
— |
— |
— |
— |
— |
| 2 |
AgNO3 + Ch |
H2O2 |
— |
65 |
— |
— |
— |
| 3 |
Ch |
H2O2 |
12 |
24 |
2 |
0 |
0.03 |
| 4 |
AgCh |
H2O2 |
175 |
363 |
41 |
47.57 |
0.43 |
| 5 |
CuCh |
H2O2 |
95 |
387 |
38 |
27.86 |
0.37 |
| 6 |
FeCh |
H2O2 |
117 |
380 |
25 |
24.49 |
0.37 |
| 7 |
AgCh |
H2O2 + O2 |
156 |
77 |
50 |
19.52 |
0.21 |
| 8 |
CuCh |
H2O2 + O2 |
87 |
64 |
30 |
13.31 |
0.13 |
| 9 |
FeCh |
H2O2 + O2 |
97 |
133 |
44 |
16.5 |
0.19 |
| 10 |
AgCh + [BMIM]+Cl− |
H2O2 |
0 |
6152 |
414 |
47.92 |
4.50 |
| 11 |
AgCh + [BMIM]+Cl− |
H2O2 + O2 |
3166 |
2370 |
287 |
37.27 |
4.00 |
| 12 |
AgCh + [BMIM]+Br− |
H2O2 |
0 |
2710 |
436 |
38.40 |
2.16 |
| 13 |
AgCh + [BMIM]+Br− |
H2O2 + O2 |
952 |
1680 |
312 |
33.55 |
2.02 |
The activity of the AgCh catalyst was further studied by varying the amount catalyst from 0.03 to 0.15 g (Fig. 7(a)). Increasing the amount of the catalyst resulted in increased methanol formation. A catalyst loading of 0.03 g produced 48 μmol of methanol, which increased to 175 μmol at a loading of 0.15 g. When a larger amount of AgCh was used, the products contained more formic acid than methanol. Higher amount of catalysts produces more methanol which may over oxidised to formic acid with the time and hence the more formic acid production suggesting that the catalyst was highly active. Investigations of the effect of reaction time, shown in Fig. 7(b), found that the maximum yield of liquid-phase products was obtained after 3 h, after which the production of liquid-phase products decreased. These results suggest that reaction times longer than 3 h lead to deactivation of the catalyst. The over-oxidation of CH4 to produce gaseous products may also cause the observed decrease in the content of liquid products at reaction times longer than 3 h.
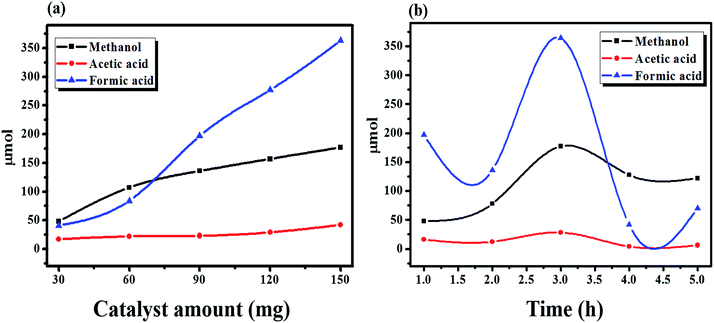 |
| | Fig. 7 The effect of (a) catalyst amount and (b) reaction time on the conversion of CH4 to its different oxygenate products. | |
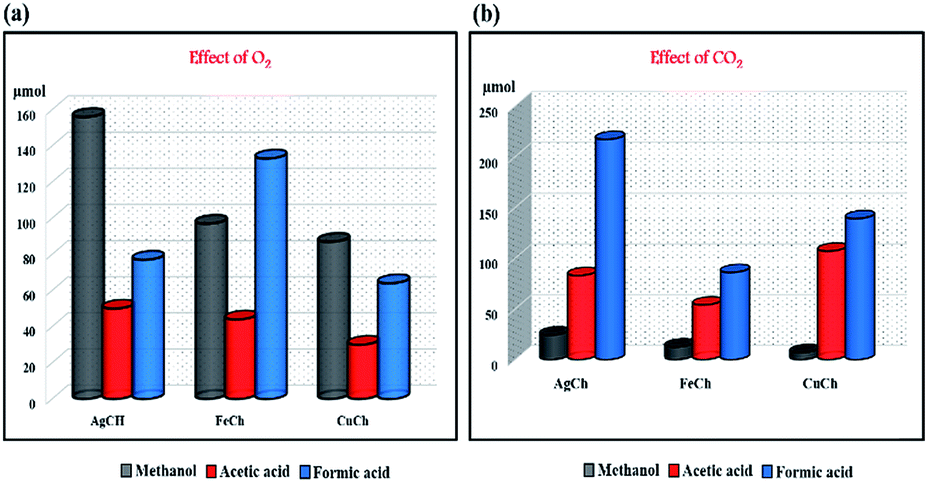
Effects of O2 and CO2 on CH4 oxidation
We studied in detail the effect of an additional oxidant, O2 (in the presence and absence of H2O2), on the single-step conversion of CH4 to methanol by our Ch catalysts. The results of the activity comparisons for each of the different catalysts are shown in Fig. 8(a) and Table 1, and showed trends that followed our earlier results. The AgCh performed well in the presence of O2, compared to CuCh and FeCh. Interestingly, the yield of oxygenates was comparatively less than in reactions conducted without O2. This finding suggests that when the reactor was purged with O2, the primary source of O atoms for use in the conversion of CH4 changes from H2O2 to O2. The observation of H2O2 consumption in both systems confirms that when H2O2 alone was used, it acts as an oxidant for CH4 conversion and when O2 was purged into reactor, H2O2 involves in the reaction as O atom active exchanger with O2 thereby minimizing its participation in over oxidizing side-reaction. This explanation was further supported by investigations of the amount of over-oxygenated products generated compared to the amount of methanol, which showed that more methanol was produced in the O2-containing systems. The lower selectivity towards over-oxidized products observed for systems that contained O2 certainly suggests that there is a shift in source of O atoms from H2O2 to O2 in activating the catalytic system for the CH4 conversion reaction. When the reaction was carried out with O2 and no H2O2, a low amount of methanol was obtained. Hence, a combination of O2 and H2O2 is the optimal choice for higher selectivity towards methanol.
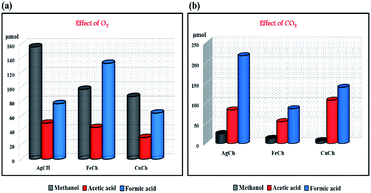 |
| | Fig. 8 The effect of (a) O2 and (b) CO2 on the conversion of CH4 to its different oxygenated products. | |
The effect of CO2 on the reaction is shown graphically in Fig. 8(b), which shows the amount of methanol obtained was low in all cases. These results are as expected since CO2 is a carboxylating agent. The acetic acid concentration was high when CO2 was present, suggesting that the direct carboxylation of methanol to acetic acid can occur. A comparison of the activity of the different catalysts again shows AgCh to be the best catalyst in all cases.
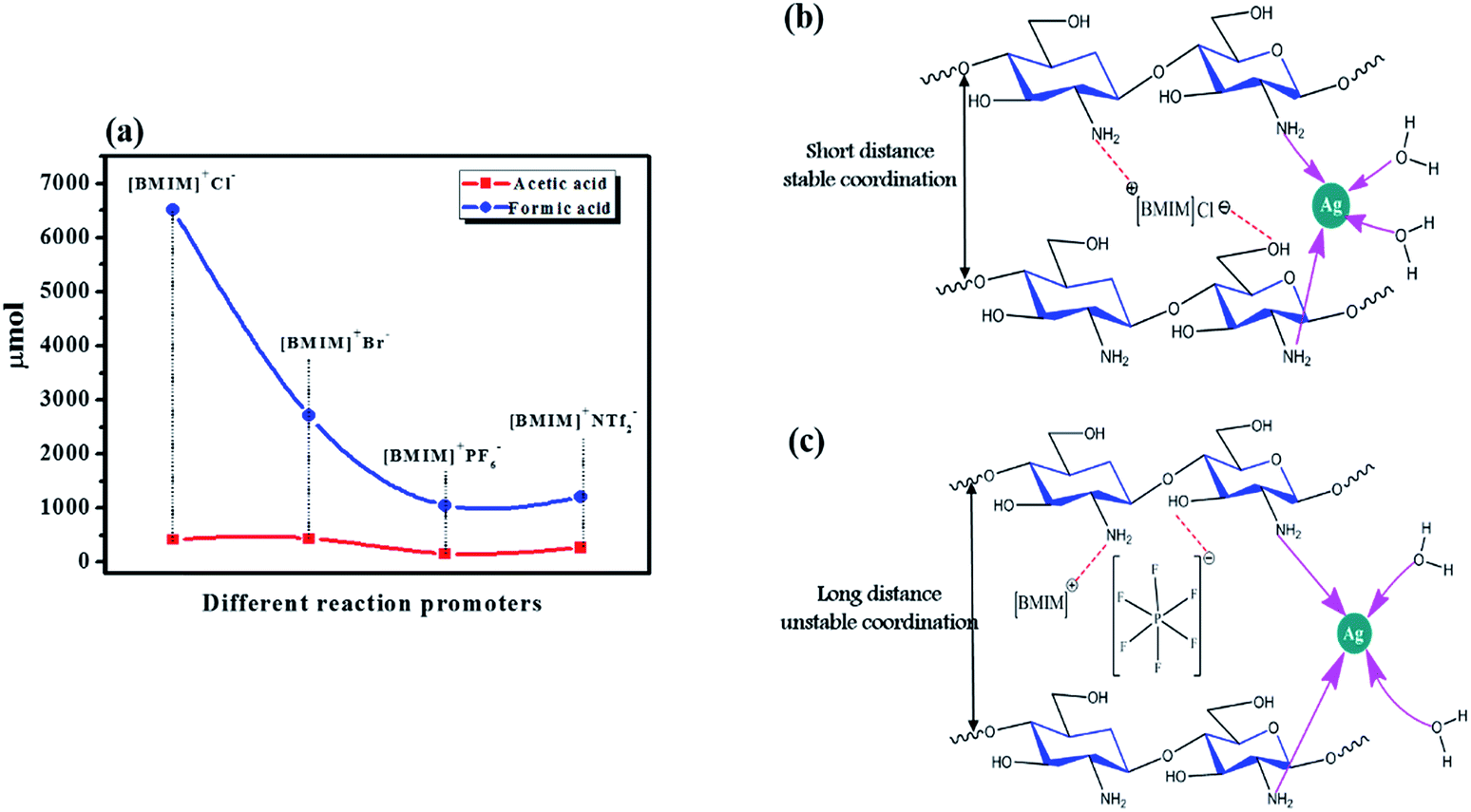
Halide ion-containing ionic liquids as reaction promoters
A recent study sought a suitable reaction promoter for the conversion of CH4 to its oxygenates.11 A previous report demonstrates that the addition of Cu to an Au–Pd/TiO2 catalyst (AuPdCu/TiO2) led to a 5-fold increase in the CH4 conversion productivity, in line with other reports.24 The promoter's role is to activate CH4 by reducing the bond dissociation energy of the C–H bond. Halide anions are well known as CH4 activators, and scientists have made use of alkyl halides and halide salts for this purpose. Although the final CH4 conversion was increased when halide salts were used, it was still either relatively low or showed low selectivity towards oxygenates.8,11 This lower performance was possibly because of a weaker attraction between the formed halide ions (from the halide salt) in the system and the catalyst. Halide ions remain in solution rather than associating strongly with the catalyst and therefore do not act efficiently during the catalytic cycle involved in CH4 activation. Hence, we suggest that the promoter's halide anion and the associated cations must bind strongly to the catalyst by means of a chemical bond. Thus, the formation of a suitable intermediate structure (as shown in Fig. 9(b and c)) can occur, providing a nearby source of promoter ions during the conversion reaction.
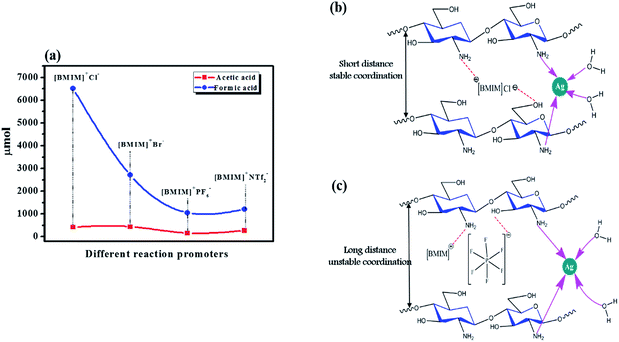 |
| | Fig. 9 (a) The effect of the different ionic liquids used as reaction promoters. (b and c) Possible structures of AgCh after interacting with ionic liquids of different size. | |
CH4 monooxygenase (MMO) is an enzyme that converts CH4 to methanol and oxidizes C1 to C5 hydrocarbons in nature.9 This enzyme is also highly active towards the oxidation of halogenated alkanes.25 Its structure includes Fe centers, which are coordinated with the N atoms of histidine and the O atoms of carboxylates and water. Our catalyst provides a similar environment to MMO, and the use of imidazolium-based halide ion-containing ionic liquids as promoters provides easily accessible halide ions for triggering the formation of halogenated CH4. By analogy to the high activity of MMO for halogenated alkane oxidation and the similarity in catalyst environment between our Ch-based catalysts and MMO, we expect that the halogenated CH4 will be immediately oxidized. Another advantage of this system is the presence of [BMIM]+, which can act similarly to a reductase in some organic reactions.26 We suggest that this reductase action may provide a similar driving force for activation of CH4 as provided for MMO by nicotinamide adenine dinucleotide (NADH). Our experiments showed that 11-fold improvement in the conversion of CH4 to its oxygenates was achieved using these promoters. [BMIM]+Cl− and [BMIM]+Br− were employed as reaction promoters with AgCh, which had the best performance in earlier tests compared to the other prepared catalyst complexes (entry no. 10 in Table 1). The CH4 conversion increased abruptly, with 11-fold and 5.5-fold increases for [BMIM]+Cl− and [BMIM]+Br− respectively. This finding suggests that Cl− ions are better halide promoters for the reaction with the AgCh catalyst. To confirm the improvement in activity was a result of the presence of halide anions, we also compared the performance of ionic liquid promoters with PF6− and NTf2− anions (shown in Fig. 9(a)), which are extensively used in many gas phase reactions.27,28 Even though these anions were effective promoters, their performance was considerably lower than that of the halide ions. These results also demonstrate the high possibility of cation activity in the conversion reaction. The PF6− and NTf2− anions are larger than the halide anions, which affects the coordination structure and stability of catalyst, as shown in Fig. 9(b and c). A control experiment was done using only [[BMIM]+Cl−. The only observed product was formic acid (2085 μmol), which indicated that the [BMIM]+Cl− was three times less effective when used alone than when it was used as a promoter with the catalyst. These results provide evidence that the ionic liquids form a structure with the catalyst that favors higher CH4 conversion. Hence, our attempt to establish an intermediate structure between catalysts and promoter was effective in achieving a higher conversion of CH4. The maximum conversion of CH4 observed after using ionic liquids as promotors is 4.5%. When compared to traditional catalysis, the achieved conversion is very low. However for CH4 conversion into its oxygenates via one-step, the 4.5% conversion is still noteworthy. A tabulated comparison of CH4 conversion and methanol selectivity from the literature is shown in Table S3.† It can be seen that the performance of our catalyst at lower temperature is comparable to other reports which are done at higher temperatures.
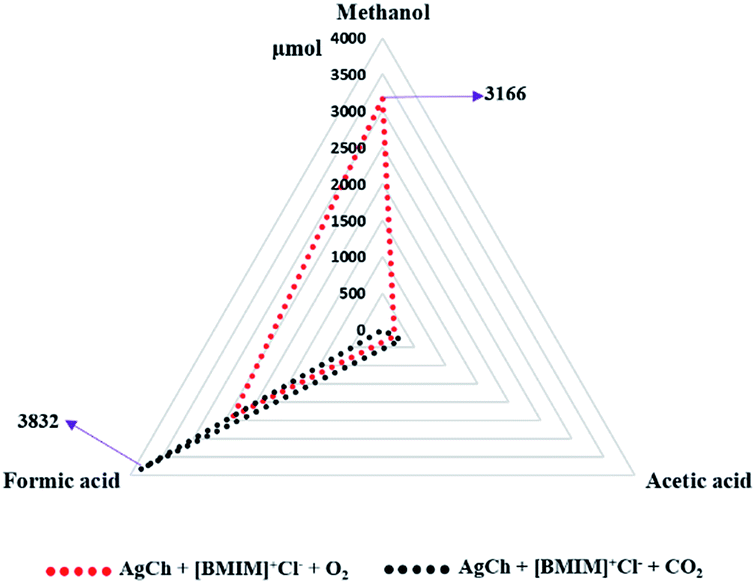
Finally, we combined the findings of our optimizations by testing the influence of oxygen and CO2 on the CH4 conversion reaction with the AgCh catalyst, the [BMIM]+Cl− promoter and the H2O2 co-oxidant. The results, shown in Fig. 10, indicate that the total conversion was lower when this catalyst system was used without O2 or CO2. However, the addition of O2 resulted in the production of 3166 μmol of methanol, (entry no. 9 in Table 1) which is ∼18 times higher than the yield from AgCh alone. This comparison reveals that the use of O2 reduces the over-oxidation of methanol, and hence a combination of H2O2 and O2 is the best oxidant for methanol synthesis. However, the ionic liquid cation was responsible for the production of ∼1500 μmol of formic acid. The selectivity towards methanol was found to be 54% in this case as highest selectivity for methanol detected in this work. The methanol selectivity comparison with the literature is shown in Table S3† which explains the 54% selectivity was worthy when compared previous reports. When CO2 was added, no methanol was observed. Instead, 3832 μmol of formic acid was produced, which is more than double compared to the amount of formic acid formed in O2 purged system (∼1500 μmol). This finding suggests that the combination of catalyst, promoter and CO2 presents a strong CH4 carbonylation environment.
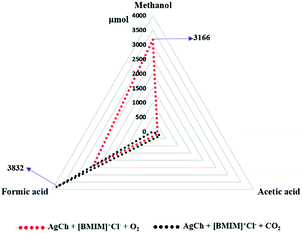 |
| | Fig. 10 CH4 conversion activity of AgCh and [BMIM]+Cl− combinations with different gaseous oxidants (O2 and CO2). | |
Conclusions
A catalytic AgCh complex for conversion of CH4 to its oxygenates was prepared and fully characterized. The complex contained Ag(I), which provides the redox properties necessary to undergo the inorganic nucleophilic reactions involved in the conversion of CH4 to methanol. The suitability of Ag for this conversion was demonstrated by comparing AgCh with catalysts prepared using various metal atoms (Fe & Cu). Efforts to identify the best oxidant revealed that H2O2 alone led to a higher CH4 conversion percentage than a combination of O2 and H2O2. This result was suggested to occur because of the shifting of O utilization source from H2O2 to O2, which renders rate of reaction. We determined the best environment to maximize methanol selectivity in our study of ionic liquids with halide anions as promoters. We found that a combination of AgCh with [BMIM]+Cl− and H2O2 increased the CH4 conversion 11-fold, but only over oxidized products were observed. The increased conversion confirmed the utility of our strategy to establish an interaction between the catalysts and the promoter. Finally, a system using a combination of H2O2, O2, AgCh and [BMIM]+Cl− showed the best selectivity for methanol and a 13-fold improvement in the yield of methanol compared to the system with H2O2 alone, producing 3166 μmol of methanol. As an extended work, experiments are ongoing in our laboratory to establish a cyclic radicle mechanism among the added halide anions and CH4 in order to invent a new green feasible method for this oxidation reaction.
Acknowledgements
This work was supported by the Energy Efficiency & Resources (No. 20163010092210) of the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korea government Ministry of Trade, Industry & Energy and this work was supported by Nano-Material Fundamental Technology Development (2016M3A7B4909370) through National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning.
Notes and references
- J. Galuszka, R. Pandey and S. Ahmed, Catal. Today, 1998, 46, 83–89 CrossRef CAS.
- J. G. Seo, M. H. Youn, Y. Bang and I. K. Song, Int. J. Hydrogen Energy, 2011, 36, 3505–3514 CrossRef CAS.
- M. Rezaei, S. M. Alavi, S. Sahebdelfar and Z.-F. Yan, J. Nat. Gas Chem., 2006, 15, 327–334 CrossRef CAS.
- K. C. Waugh, Catal. Today, 1992, 15, 51–75 CrossRef CAS.
- M. Behrens, F. Studt, I. Kasatkin, S. Kuhl, M. Havercker, F. Abild-pedersen, S. Zander, F. Girgsdies, P. Kurr, B. Kniep, M. Tovar, R. W. Fischer, J. K. Norskow and R. Schlogl, Science, 2012, 336, 893–898 CrossRef CAS PubMed.
- M. V. Kirillova, M. L. Kuznetsov, P. M. Reis, J. A. L. Da Silva, J. J. R. Fraústo Da Silva and A. J. L. Pombeiro, J. Am. Chem. Soc., 2007, 129, 10531–10545 CrossRef CAS PubMed.
- R. A. Periana, Science, 1998, 280, 560–564 CrossRef CAS PubMed.
- Q. Yuan, W. Deng, Q. Zhang and Y. Wang, Adv. Synth. Catal., 2007, 349, 1199–1209 CrossRef CAS.
- M. H. Ab Rahim, R. D. Armstrong, C. Hammond, N. Dimitratos, S. J. Freakley, M. M. Forde, D. J. Morgan, G. Lalev, R. L. Jenkins, J. A. Lopez-Sanchez, S. H. Taylor and G. J. Hutchings, Catal. Sci. Technol., 2016, 6, 3410–3418 CAS.
- C.-C. Liu, C.-Y. Mou, S. S.-F. Yu and S. I. Chan, Energy Environ. Sci., 2016, 9, 1361–1374 CAS.
- C. Hammond, M. M. Forde, M. H. Ab Rahim, A. Thetford, Q. He, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer, D. M. Murphy, A. F. Carley, S. H. Taylor, D. J. Willock, E. E. Stangland, J. Kang, H. Hagen, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2012, 51, 5129–5133 CrossRef CAS PubMed.
- M. Sun, E. Abou-Hamad, A. J. Rossini, J. Zhang, A. Lesage, H. Zhu, J. Pelletier, L. Emsley, V. Caps and J.-M. Basset, J. Am. Chem. Soc., 2013, 135, 804–810 CrossRef CAS PubMed.
- K. Narsimhan, V. K. Michaelis, G. Mathies, W. R. Gunther, R. G. Griffin and Y. Román-Leshkov, J. Am. Chem. Soc., 2015, 137, 1825–1832 CrossRef CAS PubMed.
- K. Otsuka, M. Hatano and T. Komatsu, Catal. Today, 1989, 4, 409–419 CrossRef CAS.
- X. Wang, Y. Du and H. Liu, Carbohydr. Polym., 2004, 56, 21–26 CrossRef CAS.
- Z. Guo, L. L. Henry, V. Palshin and E. J. Podlaha, J. Mater. Chem., 2006, 16, 1772–1777 RSC.
- Y. L. Wei, K. W. Chen and H. P. Wang, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct., 2009, 27, 1385 CrossRef CAS.
- J. N. Nian, S. A. Chen, C. C. Tsai and H. Teng, J. Phys. Chem. B, 2006, 110, 25817–25824 CrossRef CAS PubMed.
- P. Kumar, R. Nagarajan and R. Sarangi, J. Mater. Chem. C, 2013, 1, 2448 RSC.
- M. Wilke, F. Farges, P. E. Petit, G. E. Brown and F. Martin Jr, Am. Mineral., 2001, 86, 714–730 CrossRef CAS.
- C. Lamberti, S. Bordiga, F. Bonino, C. Prestipino, G. Berlier, L. Capello, F. D. Acapito, F. X. Llabres i Xamena and A. Zecchina, Phys. Chem. Chem. Phys., 2003, 5, 4502–4509 RSC.
- H. Yin, D. L. Hachey and N. A. Porter, J. Am. Soc. Mass Spectrom., 2001, 12, 449–455 CrossRef CAS PubMed.
- E. Vidrio, H. Jung and C. Anastasio, Atmos. Environ., 2008, 42(18), 4369–4379 CrossRef CAS PubMed.
- M. D. Pham, Y. P. Lin, Q. Van Vuong, P. Nagababu, B. T. A. Chang, K. Y. Ng, C. H. Chen, C. C. Han, C. H. Chen, M. S. Li, S. S. F. Yu and S. I. Chan, Biochim. Biophys. Acta, Proteins Proteomics, 2015, 1854, 1842–1852 CrossRef CAS PubMed.
- R. L. Lieberman and A. C. Rosenzweig, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 147–164 CrossRef CAS PubMed.
- Y. Oh and X. Hu, Chem. Commun., 2015, 51, 13698–13701 RSC.
- A. A. Chaugule, A. H. Tamboli, F. A. Sheikh, W. J. Chung and H. Kim, J. Mol. Liq., 2015, 208, 314–321 CrossRef CAS.
- A. A. Chaugule, A. H. Tamboli and H. Kim, RSC Adv., 2016, 6, 42279–42287 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02700a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Sung Ok Wonb and
Jeong Gil Seo
b,
Sung Ok Wonb and
Jeong Gil Seo