
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence[2 + 2 + 2] cyclotrimerisation as a convenient route to 6N-doped nanographenes: a synthetic introduction to hexaazasuperbenzenes†‡
Lankani P. Wijesinghe a,
Sarath D. Pereraab,
Eugene Larkina,
Gearóid M. Ó Máillea,
Robert Conway-Kennya,
Buddhie S. Lankagea,
Longsheng Wanga and
Sylvia M. Draper*a
a,
Sarath D. Pereraab,
Eugene Larkina,
Gearóid M. Ó Máillea,
Robert Conway-Kennya,
Buddhie S. Lankagea,
Longsheng Wanga and
Sylvia M. Draper*a
aSchool of Chemistry, Trinity College, Dublin, D2, Ireland. E-mail: smdraper@tcd.ie
bChemistry Department, Open University of Sri Lanka, Nugegoda, Sri Lanka
First published on 3rd May 2017
Abstract
6N-containing polyphenylene precursors were generated via [2 + 2 + 2] cyclotrimerisation. On methoxy substitution complete ring-closure can be achieved (via FeCl3-mediated oxidative cyclodehydrogenation) to give asymmetric and C3v symmetric hexaazasuperbenzenes. Investigations using DDQ/H+ mediated conditions reveal this to be a promising alternative and selective route to partial cyclodehydrogenation.
It is now well-documented that the incorporation of heteroatoms such as N,1–3 P,4,5 S,6–8 or B9,10 into the core of polycyclic aromatic hydrocarbon (PAH) derivatives fundamentally affects their electronic and optical properties.11–13 It is one means by which to tailor the HOMO–LUMO gap in such materials and to confer upon them either p- or n-type semiconducting properties as required in organic field-effect transistors and organic light emitting diodes.14–16 Depending on the position of the heteroatom (periphery or core), such substituents can also provide ligand or coordination functionality to PAH-based molecules.17,18 This latter effect has been demonstrated most notably in systems with a coronene skeleton as part of their aromatic core and pyrimidine units in peripheral sites.19,20 The introduction of graphitic nitrogen atoms also opens up the possibility of new applications e.g. as sites for enhancing the electrocatalytic behaviour of carbon-materials.21
Polycyclic heteroaromatics based on hexaphenylbenzene have emerged as desirable synthetic targets on many fronts, not just as a starting unit for bottom-up synthesis but also as well-defined substructures for the surface-mediated formation of heteroatom-doped graphene and nanoribbons.11,22,23 However, the synthetic challenges are magnified by the presence of these heteroatoms, and have led to interesting strategies that differ significantly from their all-carbon analogues.24 In terms of controlling the nature and extent of heteroatom doping, there is still much to achieve in the resulting substitution patterns and the degree of ring fusion. In unfused systems, 6N substitution of hexaphenylbenzene has recently been achieved.25 Fused 2N and 4N systems have been generated through the use of pyrimidyl units,26 while 6N substitution has only been achieved in pyrrole-based systems.3
This work aims to tackle one of the current major challenges, that of increasing the degree of N-doping into the periphery of the aromatic core without compromising the extent of aromaticity in the final product. In the process we have produced hexaazasuperbenzenes with differing anisotropic molecular shapes, structural rigidity, extended π-surfaces, and shed some light on how N-doping influences the outcome of metal and DDQ/H+ mediated cyclodehydrogenation reactions.
[2 + 2 + 2] cycloadditions involving alkynes have been used as an elegant, one-step and potentially one-reagent method for the generation of annulated benzene derivatives.27 Herein, we exploit the [2 + 2 + 2] trimerisation of pyrimidyl containing acetylenes as a convenient route to yield 6N-doped polyphenylenes in high yield. The subsequent oxidative cyclodehydrogenation reactions gave nitrogen-doped nanographenes with N-loadings of up to 11.11 atom% within the aromatic core with differing N-donor arrangements.
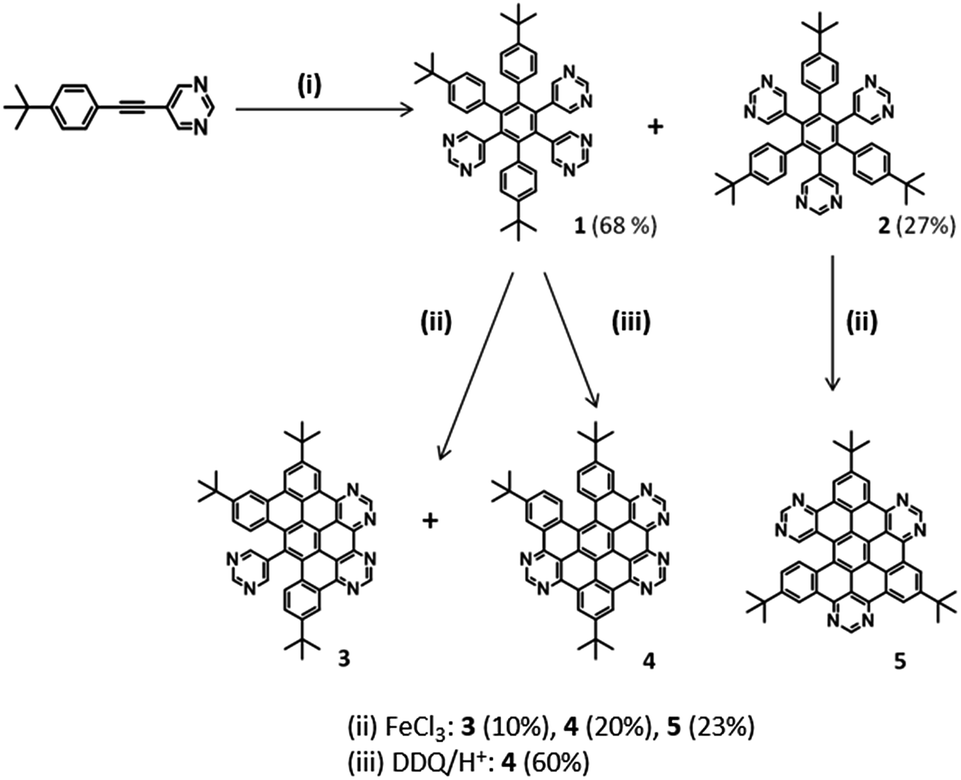
Two series of 6N polyphenylenes were synthesised by varying the functional groups in the monopyrimidyl acetylene precursor (tert-butyl or methoxy – see Schemes 1 and 2) and subjecting them to standard dicobalt octacarbonyl-catalysed cyclotrimerisation conditions.28 Asymmetric 3,5,6-tri-(4-tert-butylphenyl)-1,2,4-tri(5-pyrimidyl)benzene (1), and the symmetric 2,4,6-tri(4-tert-butylphenyl)-1,3,5-tri(5-pyrimidyl)benzene (2) (Scheme 1), were successfully isolated as white crystalline solids in 68% and 27% yields respectively, in the expected statistical ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 [see ESI for single crystal X-ray structure of 2 (S5‡) and the photophysical investigation of 1 and 2 (S6a‡)]. These materials are isomers, differing in terms of their symmetry patterns (C3v or star-shaped, and asymmetric).
:1 [see ESI for single crystal X-ray structure of 2 (S5‡) and the photophysical investigation of 1 and 2 (S6a‡)]. These materials are isomers, differing in terms of their symmetry patterns (C3v or star-shaped, and asymmetric).
 | ||
| Scheme 1 Synthetic route to tert-butyl functionalised 6N-doped hexaazasuperbenzenes (HASB): (i) Co2(CO)8, dioxane, 115 °C, 24 h, under N2, (ii) FeCl3, CH3NO2, CH2Cl2, 298 K, Ar bubbling, 72 h, (iii) DDQ, MeSO3H or CF3SO3H, CH2Cl2. | ||
 | ||
| Scheme 2 Synthetic route to methoxy-functionalised hexaazasuperbenzenes (HASB). (i) Co2(CO)8, dioxane, 115 °C, 24 h, under N2, (ii) FeCl3, CH3NO2, CH2Cl2, 298 K, Ar bubbling, 24 h. | ||
The planarisation of the tert-butyl functionalised 6N polyphenylenes 1 and 2 to obtain 6N nanographenes was studied under a range of common oxidative cyclodehydrogenation conditions: (i) AlCl3/CuCl2/CS2 (ii) FeCl3/CH3NO2/CH2Cl2 and (iii) DDQ/H+ (where H+ = MeSO3H or CF3SO3H). For the asymmetric polyphenylene 1, the AlCl3/CuCl2/CS2 route resulted in a myriad of partially fused products. Although the separation of these was extremely challenging, 5/6th fused (five C–C bonds closed) 4 was isolated in very low yield (5%) after a series of purification attempts.
By comparison, the FeCl3-catalysed procedure proved to be much cleaner: the oxidative cyclodehydrogenation of asymmetric 1 under FeCl3-mediated conditions resulted in the 2/3rd-fused (four C–C bonds formed) 3 and 5/6th fused (five C–C bonds formed) 4 as the two major products (Scheme 1). Preparative thin layer chromatography facilitated the separation of these co-synthesised products 3 (10%) and 4 (20%). In addition, mass spectrometric analysis of the crude mixture showed the presence of 1/2-fused (three C–C bonds formed) and 1/3rd fused (two C–C bonds formed) compounds in trace amounts [see S4(a)‡]. Under the same FeCl3-conditions, the oxidative cyclodehydrogenation of symmetric 2 resulted in the 5/6th fused (five C–C bonds formed) 5 (23%) as the major product. As for 1, the mass spectrometric analysis of the crude reaction material showed the presence of other partially-fused products with varying degrees of fusion [S4(b)‡]. For symmetric 2, the AlCl3 route was not successful and resulted in a mixture of intractable products.
Recently, ring closure via DDQ/H+ oxidation has been explored as a metal-free alternative for the oxidative cyclodehydrogenation of various all-carbon aromatic donors.29 To the best of our knowledge, this route has not been tested for N-containing polyphenylene systems. In this work, the DDQ/H+-mediated oxidative cyclodehydrogenation was investigated for 6N polyphenylene 1, resulting in 5/6th fused 4 as the major product in 60% yield. Since the DDQ/H+ route did not result in other partially fused products, 5/6th fused 4 was isolated in good yield and the purification was relatively straightforward. Interestingly, the five C–C bond closures have taken place at the same positions as in the 5/6th fused product obtained via FeCl3 and AlCl3 (as confirmed by NMR analysis, see S3‡). Furthermore, with extended reaction times (up to 5 days) and the excess addition of FeCl3 (60 equivalents), we were unable to obtain complete ring closure in either tert-butyl functionalised asymmetric 1 or symmetric 2. Given recent literature reports of the efficacy of using stronger acids in the DDQ/H+ process particularly in electron poor polyphenylenes,30 the authors modified their reaction conditions to determine if CF3SO3H would improve the outcome of the reaction. No change was seen either in the nature of the products arising or the yield of 4 produced.
A second series of methoxy-functionalised 6N polyphenylenes was synthesised via cobalt mediated cyclotrimerisation using 5-(3,4,5-trimethoxyphenylethnyl)pyrimidine as the precursor (Scheme 2). The methoxy groups were incorporated to increase the efficiency of the subsequent oxidative cyclodehydrogenation step.31 Both asymmetric 3,5,6-tri-(3,4,5-trimethoxyphenyl)-1,2,4-tri-(5-pyrimidyl)benzene (6) and symmetric 2,4,6-tri-(3,4,5-trimethoxyphenyl)-1,3,5-tri-(5-pyrimidyl)benzene (7) were successfully isolated via column chromatography in 63% and 17% yields respectively (Scheme 2).
Oxidative cyclodehydrogenation of methoxy-substituted asymmetric 6 under FeCl3-mediated oxidative cyclodehydrogenation resulted in fully fused (six C–C bonds formed) 8. Preliminary mass spectral analysis of the crude material indicated the presence of a mixture of products [see S4(c)‡], showing the presence of the fully-fused product 8, along with a 5/6th fused product (five C–C bonds formed). Preparative plate chromatography (SiO2) using toluene:methanol (20:1) as eluent proved to give the best separation. The isolated yield of the pure fully-fused product 8 was low (10%) due to the challenges faced in purification, however 8 is the first example of a fully-fused hexaazasuperbenzene (HASB). Under FeCl3 mediated oxidative cyclodehydrogenation, symmetric 7, resulted in the fully-fused and 5/6th fused compounds according to mass spectrometric evidence of the crude [see S4(d)‡]. In this case the two products could not be separated despite attempting various purification methods.
Fig. 1a compares the absorption spectra of compounds 3, 4, 5 and 8 along with a 4N tetraaza reference compound (N-HSB) in toluene. 4 and 5, with similar degrees of aromaticity, show similar structural features in their absorption spectra, e.g. λabsmax = 325 nm. In comparison the 2/3rd-fused 3 shows a slightly hypsochromically shifted spectrum, due to the decrease in size of its aromatic platform. The methoxy HASB 8 shows the typical structured UV-Vis absorption spectrum of a rigid, fully-fused system centred around λabsmax = 365 nm.
 | ||
| Fig. 1 Normalised (a) UV-Visible absorption spectra and (b) emission spectra of 3, 4, 5, 8 and N-HSB (λexc = 360 nm, ∼10−6 M) in toluene. | ||
Methoxy functionalisation has bathochromically shifted the λmax absorption compared to all carbon HBC32 and N-HSB (Fig. 1), which typically show an absorption maxima band at λmax 355 nm. This band is characteristic of the β band described by Clar,33 which is generally π–π* in character with some n–π* character for N-containing systems. In comparison to the reference N-HSB molecule, the electron-donating methoxy substituents in 8 destabilise the HOMO to a greater extent while the increased N content stabilises the LUMO. A combination of both these effects results in a reduced HOMO–LUMO gap, and the relatively red-shifted absorption spectrum of 8 compared to N-HSB (Fig. 1a). This gap can be calculated directly using the α-band position of the compounds, revealing that the gap is smaller for 8 (2.52 eV) than N-HSB1 (2.58 eV) and its methoxy-substituted diaaza (2N) analogue (2.61 eV).31
The overlaid emission spectra of HASBs 3, 4, 5 and 8 along with reference compound N-HSB are shown in Fig. 1b. A comparison of the spectra of 5/6th fused 4 and 5 shows that they have similar spectral shapes, with λemmax = 495 nm. The 2/3rd-fused 3 exhibits a relatively blue-shifted spectrum in comparison, with λemmax = 463 nm and a second band appearing at λ = 495 nm, due to its reduced aromaticity. It is interesting to note that the emission spectra of the 5/6th-fused products resembles that of N-HSB, with a common λemmax = 494 nm in toluene. Furthermore, comparing diaaza31 (2N) and the all carbon analogue HBC,32 a trend is apparent that upon increasing the number of the N-dopants in the platform, the vibronic structure observed in the emission is lost. This is mainly due to the availability of hydrogen bonding sites on the molecule.
Solvent dependency studies are a useful tool to investigate the nature of the electronic transitions in absorption and emission spectra. The solvent dependent absorption studies of 4 (Fig. 2a) show a bathochromic shift along with a slight broadening of the lower energy bands on increasing the polarity of the solvent. The corresponding solvent dependent emission study is presented in Fig. 2b. The compound is intensely fluorescent, and exhibits prominent vibrational progressions in non-polar solvents (e.g. hexane). As the solvent polarity increases, the vibronic progression is lost and a broad emission results, particularly in methanol. There is a significant bathochromic shift of the λmax (∼2900 cm−1) in going from hexane to methanol. This agrees with the assignment of the first excited state S1 as having 1(π–π*) character.34 The enhanced interaction of the more polar solvent molecules with the chromophore in its excited state (often via H-bond formation), lowers its excited state energy and shifts the emission to longer wavelengths.
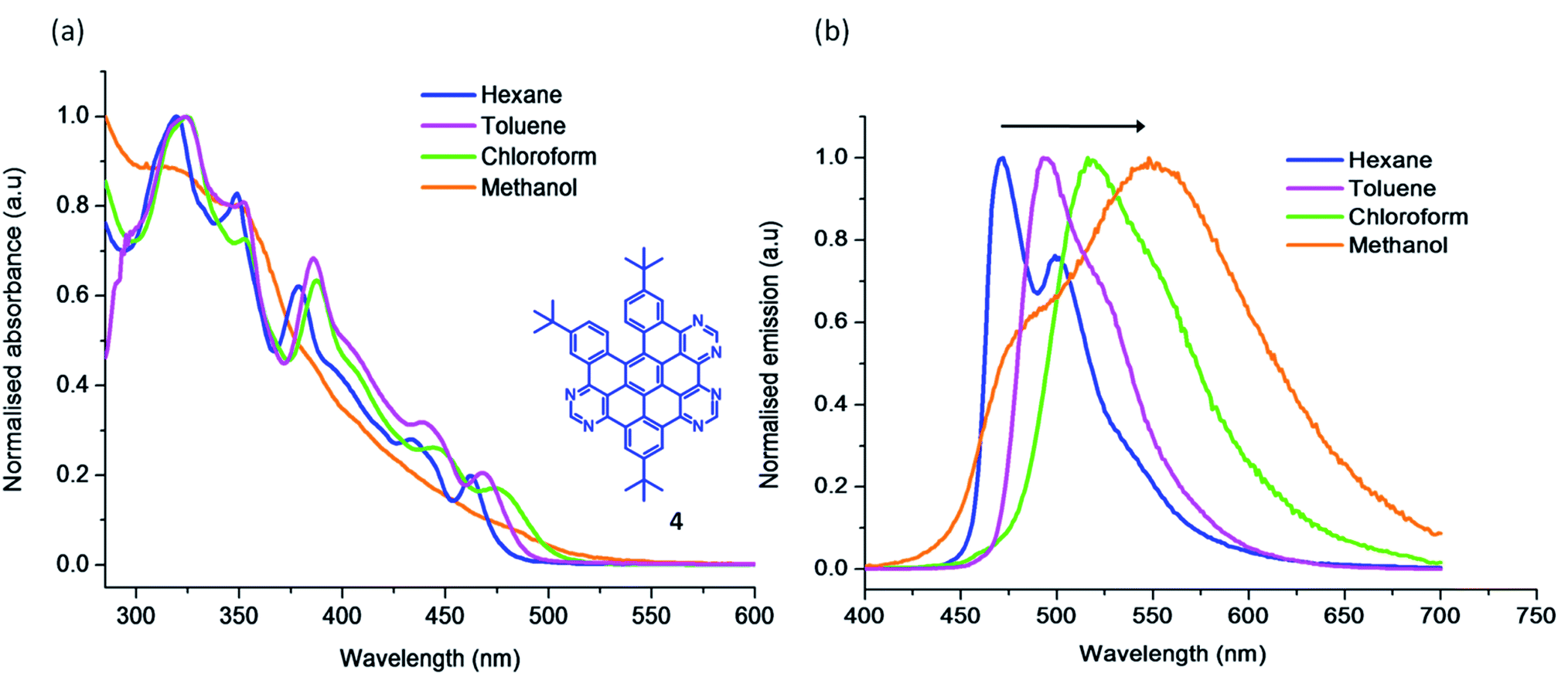 | ||
| Fig. 2 The (a) absorption and (b) emission spectra of 4 in the indicated solvents (∼10−6 M) at RT. | ||
The 6N methoxy substituted 8 shows a broad emission with λemmax = 515 nm and is further red-shifted compared to its tert-butyl counterparts (3, 4 and 5) (see Fig. 1b). Both the inclusion of electron-donating methoxy substituents and the increase in conjugation contribute to the red-shift in emission observed in 8. All of the HASB systems showed comparable lifetimes in the ns range (4–8 ns) at room temperature (Table 1). These nanosecond scale lifetimes further confirm that the emission is mainly from the 1(π–π*) excited state. 4 displays an emission quantum yield of Φem = 0.3 in CHCl3. This slightly reduced quantum yield relative to N-HSB (Φem = 0.4 in toluene) may be a feature of its reduced planarity offering a mechanism to deactivate its excited state via non-radiative pathways.
| Compound | λabs [nm] (ε × 104) [M−1 cm−1]) | λem [nm] (λexc [nm]) | τ/ns (λexc [nm]) (λnm) | Oxidation E1/2/V [ΔEp mV] | Reduction E1/2/V [ΔEp mV] |
|---|---|---|---|---|---|
| a Process is irreversible.b No oxidation in potential window analysed. | |||||
| 3 | 295(1.8), 357(5.0), 405(2.1), 423(2.0), 455(0.8) | 463max, 495 (360) | 7.8(295/463) | +1.3a | −1.56[55], −1.88[59], −2.18[91] |
| 4 | 323(11.3, 352(9.2), 385(7.8), 440(3.5), 468(2.2) | 495(360) | 6.9(340/495) | +1.25a | −1.57[48], −1.91[43], −2.19[45] |
| 5 | 323(11.0), 352(9.1), 385(7.6), 440(3.3), 468(2.1) | 495(360) | 5.3(340/495) | No oxidationb | −1.55[48], −1.91[43], −2.19[45] |
| 8 | 325(6.0), 365(12.1), 386(8.1), 462(2.9), 492(3.3) | 514(360) | 4.6(340/514) | — | — |
| NHSB | 339(5.7), 355(13.7), 376(5.4), 393(3.2), 413(1.5), 451(1.0), 481(1.4) | 494max, 524sh(360) | 13(340/494) | +0.9a | −1.56[60], −2.00[102], −2.32a |
Cyclic voltammetric studies were carried out in dry, degassed dichloromethane using a glassy carbon working electrode (Table 1 and S7‡). 4 shows an irreversible first oxidation wave at Epa = +1.25 V, however the symmetric analogue 5 shows no clear oxidation potential within the spectral window analysed (see S7 in ESI‡). The 2/3-fused 3 (Epa = +1.3 V) showed a slightly higher oxidation potential compared to its 5/6th-fused counterpart 4. The reduced aromaticity in 3 gives rise to a stabilized HOMO making the oxidation in 3 slightly more difficult than 4. The low yields obtained for 8 prevented electrochemical measurements. From the extrapolation of data from previous 2N31 and 4N systems synthesised by Draper et al., the degree of difficulty in oxidation increases with an increasing number of N-dopants. This is due to the reduced electron density in the aromatic platform, leading to stabilisation of the HOMO. As a result, the oxidation of the 6N systems was far more difficult compared to their all-carbon, 2N or 4N counterparts.
The reduction potentials are mainly governed by the electron withdrawing N-atoms present (Table 1 and S7‡). Compound 4 shows three reversible reductions at E1/2 = −1.57 V, −1.94 V and −2.32 with 5 showing similar reduction behaviour (E1/2 = −1.55 V, −1.91 V, −2.19 V). The reduction processes of 5 occur in a broader window than those of 4 which shows a set of clear sharp reduction waves. This supports the observation that 5 undergoes stacking in solution at higher concentrations. The reduction potentials of the 6N systems show that they are more readily reduced compared to the all-carbon, 2N or 4N systems. This is consistent with the increasing electron accepting character of the molecules as a function of the pyrimidyl N-atoms and the stabilisation of the LUMO.
In summary, cobalt octacarbonyl-catalysed [2 + 2 + 2] cyclotrimerisations proved a convenient route to synthesise four 6N containing polyphenylene precursors. The two tert-butyl functionalised 6N polyphenylenes undergo oxidative cyclodehydrogenation using various oxidants to give partially fused systems. The incorporation of methoxy functional groups onto the polyphenylene precursors however facilitates complete ring closure via FeCl3. Cyclodehydrogenation using DDQ/H+ was demonstrated as a promising metal-free alternative route for aromatisation of N-containing polyphenylene systems. It gives rise to significantly increased yields and selectivity of ring closure irrespective of the strength of the acid used. Pending the development of appropriate ring-closure conditions, this cyclotrimerisation approach could be used to access nanographenes with a possible N-loading of 25 atom%. The novel molecular systems generated in this work offer a new opportunity to investigate the influence of N-doping patterns on the supramolecular architectures of the molecules themselves and the nature and properties of their coordination complexes.
Acknowledgements
We thank Drs J. O'Brien, M. Ruether and M. Feeney for spectroscopic and technical assistance.Notes and references
- S. M. Draper, D. J. Gregg and R. Madathil, J. Am. Chem. Soc., 2002, 124, 3486–3487 CrossRef CAS PubMed.
- A. Graczyk, F. A. Murphy, D. Nolan, V. Fernandez-Moreira, N. J. Lundin, C. M. Fitchett and S. M. Draper, Dalton Trans., 2012, 41, 7746–7754 RSC.
- M. Takase, V. Enkelmann, D. Sebastiani, M. Baumgarten and K. Müllen, Angew. Chem., Int. Ed., 2007, 46, 5524–5527 CrossRef CAS PubMed.
- P.-A. Bouit, A. Escande, R. Szűcs, D. Szieberth, C. Lescop, L. Nyulászi, M. Hissler and R. Réau, J. Am. Chem. Soc., 2012, 134, 6524–6527 CrossRef CAS PubMed.
- T. Baumgartner, Acc. Chem. Res., 2014, 47, 1613–1622 CrossRef CAS PubMed.
- C. J. Martin, B. Gil, S. D. Perera and S. M. Draper, Chem. Commun., 2011, 47, 3616–3618 RSC.
- Y.-Z. Tan, S. Osella, Y. Liu, B. Yang, D. Beljonne, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2015, 54, 2927–2931 CrossRef CAS PubMed.
- L. Chen, S. R. Puniredd, Y.-Z. Tan, M. Baumgarten, U. Zschieschang, V. Enkelmann, W. Pisula, X. Feng, H. Klauk and K. Müllen, J. Am. Chem. Soc., 2012, 134, 17869–17872 CrossRef CAS PubMed.
- C. Dou, S. Saito, K. Matsuo, I. Hisaki and S. Yamaguchi, Angew. Chem., Int. Ed., 2012, 51, 12206–12210 CrossRef CAS PubMed.
- F. Miyamoto, S. Nakatsuka, K. Yamada, K.-i. Nakayama and T. Hatakeyama, Org. Lett., 2015, 17, 6158–6161 CrossRef CAS PubMed.
- M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed.
- M. Fan, Z.-Q. Feng, C. Zhu, X. Chen, C. Chen, J. Yang and D. Sun, J. Mater. Sci., 2016, 51, 10323–10349 CrossRef CAS.
- J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Catal., 2015, 5, 5207–5234 CrossRef CAS.
- W. Delaunay, R. Szűcs, S. Pascal, A. Mocanu, P. A. Bouit, L. Nyulaszi and M. Hissler, Dalton Trans., 2016, 45, 1896–1903 RSC.
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed.
- M. Oehzelt, C. Laubschat and D. V. Vyalikh, Nano Lett., 2011, 11, 5401–5407 CrossRef CAS PubMed.
- D. Nolan, B. Gil, F. A. Murphy and S. M. Draper, Eur. J. Inorg. Chem., 2011, 2011, 3248–3256 CrossRef CAS.
- F. A. Murphy and S. M. Draper, J. Org. Chem., 2010, 75, 1862–1870 CrossRef CAS PubMed.
- S. M. Draper, D. J. Gregg, E. R. Schofield, W. R. Browne, M. Duati, J. G. Vos and P. Passaniti, J. Am. Chem. Soc., 2004, 126, 8694–8701 CrossRef CAS PubMed.
- D. J. Gregg, E. Bothe, P. Höfer, P. Passaniti and S. M. Draper, Inorg. Chem., 2005, 44, 5654–5660 CrossRef CAS PubMed.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- A. L. Pinardi, J. I. Martinez, A. Jancarik, I. G. Stara, I. Stary, M. F. Lopez, J. Mendez and J. A. Martin-Gago, Chem. Commun., 2014, 50, 1555–1557 RSC.
- L. Chen, Y. Hernandez, X. Feng and K. Muellen, Angew. Chem., Int. Ed., 2012, 51, 7640–7654 CrossRef CAS PubMed.
- A. Narita, X.-Y. Wang, X. Feng and K. Mullen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- Y. Xiang, Q. Wang, G. Wang, X. Li, D. Zhang and W. Jin, Tetrahedron, 2016, 72, 2574–2580 CrossRef CAS.
- C. Delaney, G. M. Ó. Máille, B. Twamley and S. M. Draper, Org. Lett., 2016, 18, 88–91 CrossRef CAS PubMed.
- C. D. Simpson, J. Wu, M. D. Watson and K. Mullen, J. Mater. Chem., 2004, 14, 494–504 RSC.
- X. Feng, W. Pisula, T. Kudernac, D. Wu, L. Zhi, F. S. De and K. Mullen, J. Am. Chem. Soc., 2009, 131, 4439–4448 CrossRef CAS PubMed.
- L. Zhai, R. Shukla and R. Rathore, Org. Lett., 2009, 11, 3474–3477 CrossRef CAS PubMed.
- D. J. Jones, B. Purushothaman, S. Ji, A. B. Holmes and W. W. H. Wong, Chem. Commun., 2012, 48, 8066–8068 RSC.
- L. P. Wijesinghe, B. S. Lankage, G. M. O. Maille, S. D. Perera, D. Nolan, L. Wang and S. M. Draper, Chem. Commun., 2014, 50, 10637–10640 RSC.
- W. Hendel, Z. H. Khan and W. Schmidt, Tetrahedron, 1986, 42, 1127–1134 CrossRef CAS.
- E. Clar, Polycyclic Hydrocarbons, Academic Press, 1964, vol. 2 Search PubMed.
- R. P. Wayne, Photochemistry, Elsevier, 1971 Search PubMed.
Footnotes |
| † The work was supported financially by Science Foundation Ireland (SFI/IA/3046, SFI/09/IN.I/12974), and Trinity postgraduate awards. |
| ‡ Electronic supplementary information (ESI) available: all experimental procedures, 1H/13C NMR, HRMS, UV-Vis (absorption and emission) for nanographenes 3–5 and 8. Cyclic voltammograms for 3–5. Single crystal X-ray data for 2. CCDC 1507271. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra02648j |
| This journal is © The Royal Society of Chemistry 2017 |