
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectro-ultrafiltration to remove sodium dodecyl sulfate in proteins extracted for proteomics
H. B. Wangabc,
Y. Zhangab,
S. Q. Guiab,
Y. R. Fengab,
H. C. Hanab,
S. H. Maoabc and
F. P. Lu *abc
*abc
aCollege of Biotechnology, Tianjin University of Science and Technology, Tianjin, China. E-mail: lfp@tust.edu.cn
bKey Laboratory of Industrial Fermentation Microbiology, Ministry of Education, Tianjin, China
cNational Engineering Laboratory for Industrial Enzymes, Tianjin, China
First published on 10th May 2017
Abstract
Sodium dodecyl sulfate (SDS) is commonly used to extract membrane proteins in proteomics studies; however, it can reduce the efficiency of tryptic digestion and interfere with the results of liquid chromatography-mass spectrometry (LC-MS) analysis. Available methods for removing surfactants, such as ultrafiltration, acetone precipitation, and gel electrophoresis, are not completely satisfactory. Therefore, in this study, a new method for the depletion of SDS was established, named electro-ultrafiltration, and its performance was compared with other conventional pretreatment methods. Electro-ultrafiltration combines electrophoresis and ultrafiltration to remove SDS from protein samples. This method uses an electric field as the driving force and an ultrafiltration membrane as the separation medium. The performance of the electro-ultrafiltration method in terms of both the signals of LC-MS and the number of proteins identified was superior to that of simple ultrafiltration, but was slightly worse than that of acetone precipitation. These results demonstrate that the electro-ultrafiltration method could help to reduce the influence of SDS on protein digestion and identification, demonstrating its feasibility for application in proteomics.
Introduction
As a powerful technique, shot-gun proteomics has been extensively used across broad applications for the identification of proteins.1,2 It is based on liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis of the peptides digested from protein samples.3,4 One of the main goals of proteomics research is analysis of membrane proteins that are bound strongly to the hydrophobic portion of cell membranes, which requires the use of a surfactant for extraction, such as the most frequently used sodium dodecyl sulfate (SDS). However, this pretreatment has a negative influence on the subsequent trypsin digestion and LC-MS analysis.5,6 Even if only 0.1% SDS remains in the sample, the trypsin digestion efficiency will be substantially suppressed.7 SDS has a strong capacity for absorption in a reversed-phase liquid chromatography (RPLC) column, and therefore can largely interfere with the separation of peptides by RPLC because of their binding with SDS.8 Furthermore, SDS can reduce a peptide's ionization efficiency in MS analysis. Therefore, it is necessary to remove the surfactants after using.The available methods for removing surfactants from proteins include ultrafiltration, acetone precipitation, gel electrophoresis, and use of an ion exchange cartridge.5,9–13 However, these methods are not entirely satisfactory, indicating the need to explore further improvements or develop new methods. Ultrafiltration removes detergents according to a size exclusion principle, and requires relatively expensive centrifugal ultrafiltration tube with a low protein recovery and moderate removal rate of SDS. Acetone precipitation is a convenient method that yields a high removal rate of SDS, but the protein precipitate is difficult to redissolve, leading to a low protein recovery rate.14,15 Gel electrophoresis-based methods result in a high removal rate of SDS approaching 100%, but also show low protein recovery in terms of the harvested peptides from in-gel digestion, and the procedure is time-consuming and laborious.
To overcome these limitations, the aim of our study was to develop a new method and device for the removal of SDS from a protein sample. The principle of the method is based on a combination of electrophoresis and ultrafiltration, and is so-named electro-ultrafiltration. Proteins and SDS molecules move in a conductive solution by exposure to an electric field, and are then separated on an ultrafiltration membrane according to their different molecular sizes. To our knowledge, there has been no similar report to remove detergents from protein samples for use in proteomics research. In this study, the effects of electro-ultrafiltration for the depletion of SDS on the subsequent LC-MS analysis and protein identification were investigated and compared with those of other conventional methods to test its feasibility in application.
Experimental
Material
Sequencing-grade modified trypsin (Promega, Madison, WI, USA), proteinase inhibitor and iodoacetamide (Roche, Basel, Switzerland), HPLC-grade acetonitrile and formic acid (Fisher Scientific, Waltham, MA, USA), ammonium bicarbonate (Beijing Chemical Company, Beijing, China), and all other chemicals (Sigma, St. louis, MO, USA). Water was prepared by a Milli-Q system (Millipore, Billerica, MA, USA).The device made for electro-ultrafiltration
The device for electro-ultrafiltration was made up of two 0.5 mL 10 K centrifugal ultrafiltration tubes (Millipore). The schematic of the electro-ultrafiltration device is shown in Fig. 1. Both centrifugal ultrafiltration tubes were conglutinated into a whole in a mouth-to-mouth orientation, and three parts were separated by the two ultrafiltration membranes. Samples were added to the middle part, while the electrophoresis buffer (0.05 M Tris–HCl, pH 8.0) was added to the other two parts. Two platinum electrodes were inserted into both ends of the electro-ultrafiltration device, with the power source of an electrophoresis instrument (Liuyi, Beijing, China) as a stabilized voltage power supply.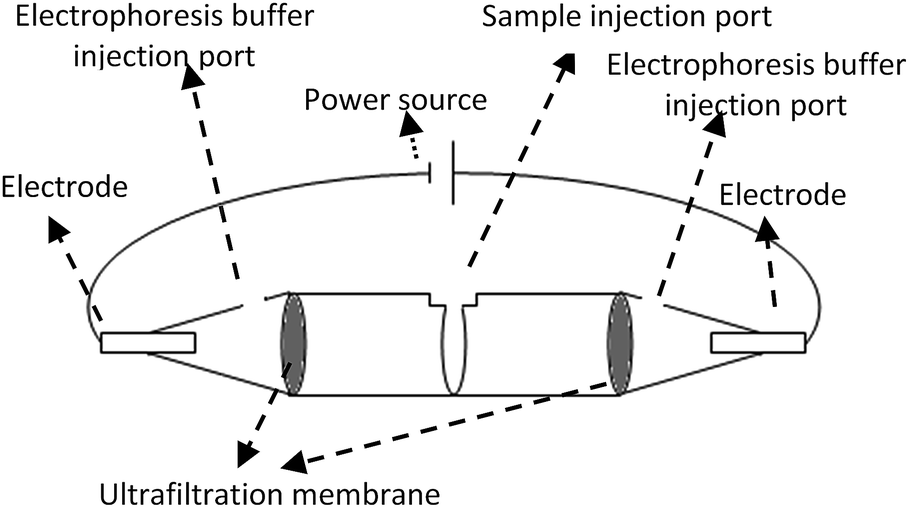 | ||
| Fig. 1 Sketch map of electro-ultrafiltration device constructed by two centrifugal ultrafiltration tubes. | ||
Protein sample preparation
A protein mixture was extracted from cultured SH-SY5Y cells (iCell Bioscience Inc, Shanghai, China) by ultrasonic disruption with 50 mM Tris–HCl buffer (pH 7.5) containing protease inhibitors. SDS (0.5%) was added to the mixture, followed by different pretreatment methods for its removal, including centrifugal ultrafiltration, acetone precipitation, and electro-ultrafiltration. Furthermore, two different control samples were prepared based on the protein mixture extracted from the SH-SY5Y cells. One was tested without the addition of SDS, and the other was tested with the addition of SDS but with no subsequent pretreatment for its removal.Pretreatment methods
Digestion, LC-MS/MS analysis, and protein identification
The dried samples were denatured and reduced in a solution containing 8 M urea, 10 mM dithiothreitol, and 50 mM NH4HCO3 at 37 °C for 4 h. Alkylation was performed in a 50 mM iodoacetamide solution at room temperature for 1 h in the dark, followed by dilution with 50 mM NH4HCO3 buffer to decrease the urea concentration to 1 M. Then, the samples were digested by trypsin at a concentration ratio of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (total protein:trypsin, w/w).
:1 (total protein:trypsin, w/w).
The digested samples were analyzed on an 1100 series HPLC system (Agilent, Santa Clara, CA, USA) coupled with an ion trap mass spectrometer (MSD Trap SL, Agilent) with a C18 column (300 Å, 2.1 × 150 mm, Grace Vydac, USA) at a flow rate of 0.2 mL min−1. The injection amount was 20 μg. Gradient elution of the peptide samples was achieved with mobile phase A (0.1% (v/v) formic acid in H2O) and mobile phase B (0.1% (v/v) formic acid in acetonitrile). The gradient program of mobile phase B consisted of 3% from 0–5 min, 3–40% from 5–55 min, 40–97% from 55–65 min, 97% from 65–68 min and 97–3% from 68–70 min, and 3% from 70–80 min. Electrospray ionization (ESI) trap MS was performed under dry gas at 10 L min−1 at a temperature of 350 °C. The nebulizer pressure was 35 psi, and the capillary voltage was 3500 V. The MS mass window was 300–1800 amu.
The data produced by the RPLC/ESI-Trap MS/MS analysis were then searched against the Swissprot protein database by the MASCOT server (version 2.1; Matrix Science, Boston, MA, USA). The identification conditions were as follows: species option, human; protease, trypsin; missed cleavages, no more than 1; fixed modification, cysteine carbamidomethylation.
Results and discussion
In electro-ultrafiltration, proteins and SDS molecules are driven by an electric field. Although there have been some reports using electric field in the ultrafiltration for the separation of fermented or enzymatic products, the electric field was not used as a driving force for separation, but just an assistant means to reduce concentration polarization.17,18 In some sense, the proposed principle of electro-ultrafiltration is similar to that of gel electrophoresis. Both methods use an electric field as the driving force and size exclusion as a separation approach. The key difference is that gel electrophoresis requires a gel as the separation medium, which makes the procedure more time-consuming and laborious, whereas in electro-ultrafiltration, the separation is carried out in free solution without requiring the support of a solid medium.The effects and efficiency of SDS removal based on different methods have often been compared and evaluated through assessment of the signals of LC-MS or the number of proteins identified.11,19–21 A stronger signal of LC-MS and a better separation effect would indicate that more SDS had been efficiently removed from the pretreatment. Fig. 2 shows the LC-MS total ion chromatograms (TICs) from the two different control samples with and without SDS. A larger and wider peak was observed at the retention time of 60–70 min in the TIC from the control sample with the addition of SDS than in that from the other control sample without the addition of SDS. This result clearly demonstrated that the large and wide peak was due to incomplete tryptic digestion by SDS, demonstrating the negative influence of SDS on RPLC separation, which is in accordance with the results of previous studies.6,22
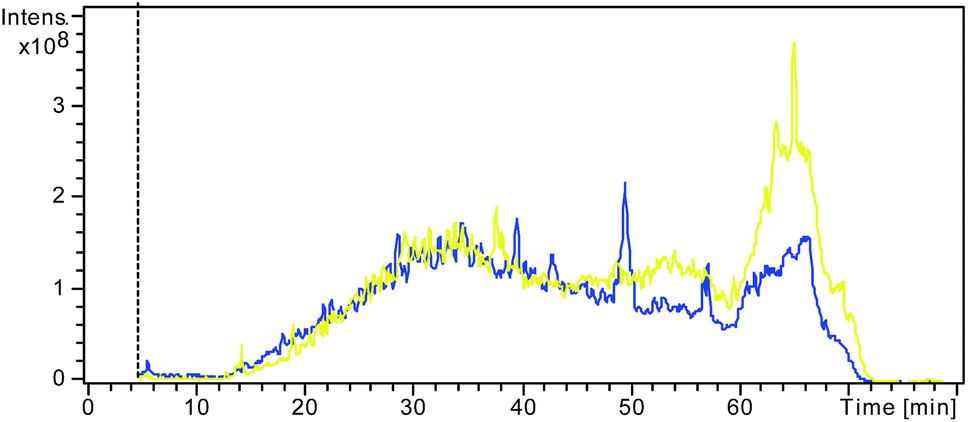 | ||
| Fig. 2 Overlap of the LC/MS TIC from the control sample without addition of SDS (blue) and the other control sample with addition of SDS but no pretreatment to remove SDS (yellow). | ||
Fig. 3 compares the TICs obtained from the samples subjected to the various pretreatments for SDS removal. As shown in Fig. 3(a), the large and wide peak at the retention time of 60–70 min observed in the TIC from the control sample with SDS was diminished in the TIC from the sample pretreated by centrifugal ultrafiltration, suggesting that this treatment method improved the tryptic digestion and RPLC by effectively removing SDS to some extent. Fig. 3(b) shows that acetone precipitation induced further improvement in the tryptic digestion and RPLC compared to centrifugal ultrafiltration. Indeed, besides reduction in the peak at the retention time of 60–70 min, the peaks during the retention times before 60 min were obviously higher. As shown in Fig. 3(c), the newly developed electro-ultrafiltration could also diminish the peak at the retention time of 60–70 min, suggesting that electro-ultrafiltration could also improve tryptic digestion and RPLC by removing SDS to some degree. Fig. 4 compares the extracted ion chromatograms (EICs) of a long retaining peptide with m/z 979.1 from different treated samples. The peptide was subsequently identified as VGAGAPVYMAAVLEYLTAEILELAGNAAR from Histone H2A type. As can be seen, it is large and similar in size that the EIC peaks from the sample without SDS and the samples pretreated by acetone precipitation and electro-ultrafiltration, while the EIC peak from the sample pretreated by centrifugal ultrafiltration is smaller and no EIC peaks was observed from the sample with addition of SDS. The comparisons reflect the strong influence of SDS and the better effect of electro-ultrafiltration on removal of SDS than that of centrifugal ultrafiltration.
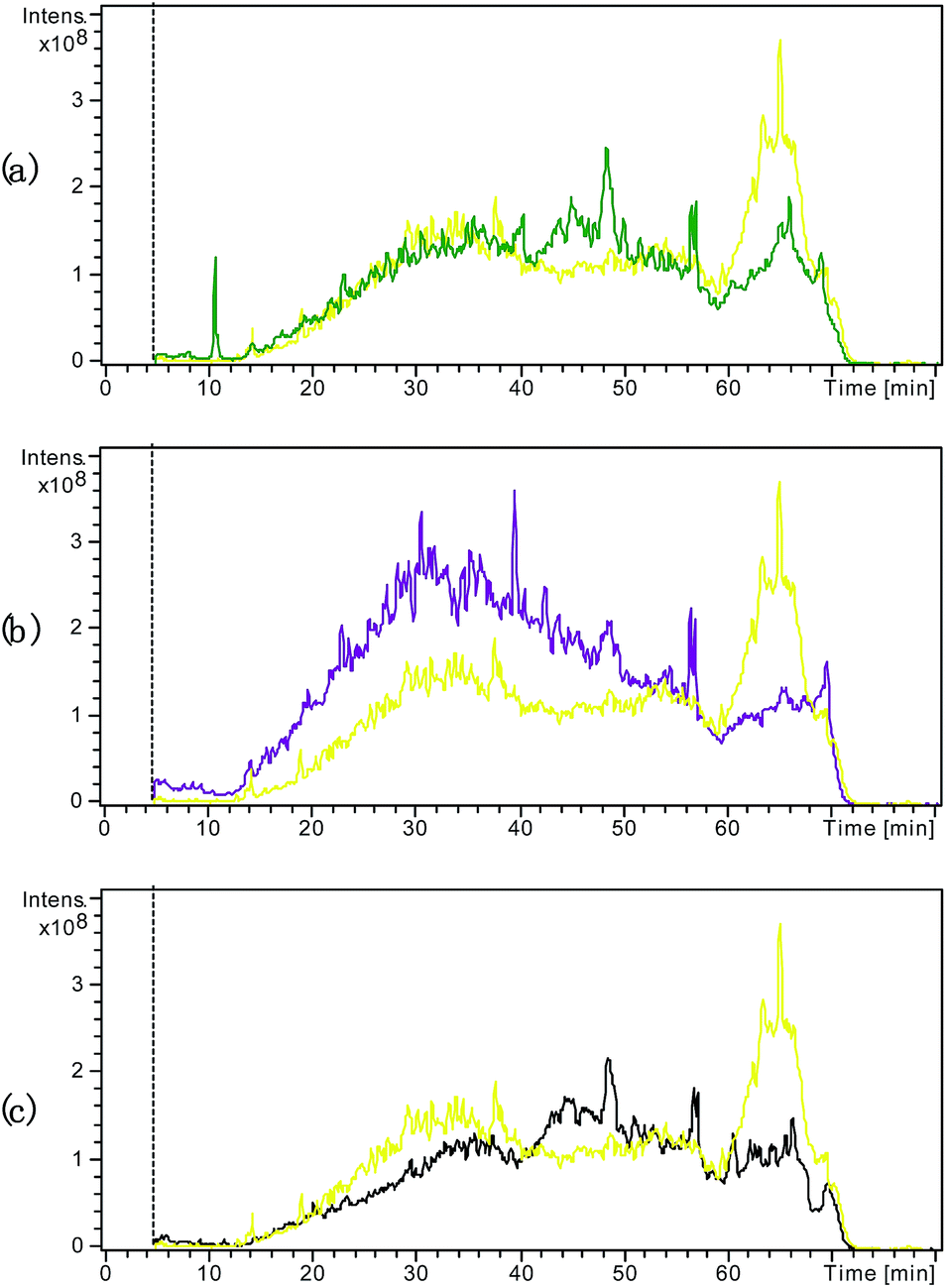 | ||
| Fig. 3 Overlap of the LC/MS TIC from the control sample with addition of SDS but no pretreatment to remove SDS (yellow) and the different pretreated samples by centrifugal ultrafiltration (a), acetone precipitation (b) and electro-ultrafiltration (c). | ||
 | ||
| Fig. 4 The EICs of a long retaining peptide with m/z 979.1 from different pretreated samples. The peptide was identified as “VGAGAPVYMAAVLEYLTAEILELAGN-AAR” belonging to Histone H2A type. | ||
The data sets produced by the RPLC/ESI-Trap MS/MS analysis with the three types of samples were then searched against the Swissprot protein database by MASCOT. On average, 85 ± 9 proteins were identified from the analysis using the samples subjected to ultrafiltration, whereas 120 ± 7 and 108 ± 7 proteins were identified from samples subjected to acetone precipitation and electro-ultrafiltration, respectively. By contrast, only 54 ± 6 proteins were identified from the control samples without pretreatment for the removal of SDS, which suggested that ultrafiltration, acetone precipitation, and electro-ultrafiltration all improved the LC-MS/MS-based protein identification by removal of SDS to some degree. Although electro-ultrafiltration did not show the best identification result, its feasibility for improving LC-MS/MS-based proteomics research was nevertheless revealed. Moreover, the present work represents a preliminary study of electro-ultrafiltration, and therefore many improvements are expected to strengthen its SDS-removal function.
Ultrafiltration depends on the centrifugal force or other aspects of fluid pressure to drive solutions through the membrane and achieve the desired concentration and separation. However, a major operating problem for membrane filtration is concentration polarization, resulting in the buildup of solutes on the membrane surface, thereby reducing the separation efficiency.23–26 In electro-ultrafiltration, the power of filtration is derived from an electric field, which drives charged molecules to move through the solution. Moreover, the driving speed and directions differ for different molecules, depending on their charges, sizes, and masses. Therefore, SDS molecules would theoretically be expected to move faster toward the membrane surface than proteins in electro-ultrafiltration, and the variation in the rates of movement of different proteins would reduce the concentration polarization on the membrane surface. Furthermore, centrifugal ultrafiltration is highly dependent on the tenacity of the membrane due to the high fluid pressure, while electro-ultrafiltration is not. For electro-ultrafiltration, an ordinary dialysis membrane is sufficient, and further membrane support is not needed; thus, its manufacturing costs will be lower than those required for centrifugal ultrafiltration.
The electro-ultrafiltration method combines electrophoresis and ultrafiltration to remove SDS from a protein sample, but it cannot concentrate samples as well as ultrafiltration and acetone precipitation. However, some other convenient methods such as lyophilization can be used after electro-ultrafiltration to improve the concentration effect. Theoretically, electro-ultrafiltration should also be able to remove other small charged ions and surfactants from protein samples besides SDS, thereby reducing their interferences on peptide signals in the subsequent MS analysis.
In summary, a new method termed electro-ultrafiltration was demonstrated to be able to improve protein digestion and LC-MS-based protein identification by the efficient removal of SDS, indicating its feasibility for application in proteomics research for membrane proteins. This study provides a foundation for the application of electro-ultrafiltration in proteomics in the future. However, as this is a preliminary analysis of electro-ultrafiltration, further improvements are needed to strengthen its function before it can be adopted widely in proteomics research.
Acknowledgements
This study was supported by the National Natural Science Foundation [grant No. 21306140], the Key Technology R&D Program of Tianjin [grant No. 14ZCZDSY00012].Notes and references
- R. Aebersold and M. Mann, Nature, 2003, 422, 198–207 CrossRef CAS PubMed.
- S. D. Patterson and R. H. Aebersold, Nat. Genet., 2003, 33, 311–323 CrossRef CAS PubMed.
- B. Domon and R. Aebersold, Science, 2006, 312, 212–217 CrossRef CAS PubMed.
- P. Feist and A. B. Hummon, Int. J. Mol. Sci., 2015, 16, 3537–3563 CrossRef CAS PubMed.
- H. Kawasaki and K. Suzuki, Anal. Biochem., 1990, 186, 264–268 CrossRef CAS PubMed.
- J. P. Vissers, J. P. Chervet and J. P. Salzmann, J. Mass Spectrom., 1996, 31, 1021–1027 CrossRef CAS.
- J. Arribas and J. G. Castano, J. Biol. Chem., 1990, 265, 13969–13973 CAS.
- D. Botelho, M. J. Wall, D. B. Vieira, S. Fitzsimmons, F. Liu and A. Doucette, J. Proteome Res., 2010, 9, 2863–2870 CrossRef CAS PubMed.
- K. Majewska-Nowak, I. Kowalska and M. Kabsch-Korbutowicz, Desalination, 2006, 198, 149–157 CrossRef CAS.
- U. Carraro, D. Doria, C. Rizzi and M. Sandri, Biochem. Biophys. Res. Commun., 1994, 200, 916–924 CrossRef CAS PubMed.
- Y. Liu, Y. Lin, Y. Yan, J. Li, Q. He, P. Chen, X. Wang and S. Liang, Electrophoresis, 2012, 33, 316–324 CrossRef CAS PubMed.
- D. Sun, N. Wang and L. Li, J. Proteome Res., 2012, 11, 818–828 CrossRef CAS PubMed.
- C. Kachuk, K. Stephen and A. Doucette, J. Chromatogr. A, 2015, 1418, 158–166 CrossRef CAS PubMed.
- A. M. Crowell, M. J. Wall and A. A. Doucette, Anal. Chim. Acta, 2013, 796, 48–54 CrossRef CAS PubMed.
- M. Puchades, A. Westman, K. Blennow and P. Davidsson, Rapid Commun. Mass Spectrom., 1999, 13, 344–349 CrossRef CAS PubMed.
- K. Majewska-Nowak, I. Kowalska and M. Kabsch-Korbutowicz, Desalination, 2005, 184, 415–422 CrossRef CAS.
- J. Hong and C. K. Lee, Ann. N. Y. Acad. Sci., 1986, 469, 131–144 CrossRef CAS.
- C. K. Lee and J. Hong, Biotechnol. Bioeng., 1988, 32, 647–654 CrossRef CAS PubMed.
- R. Sharma, B. D. Dill, K. Chourey, M. Shah, N. C. VerBerkmoes and R. L. Hettich, J. Proteome Res., 2012, 11, 6008–6018 CrossRef CAS PubMed.
- S. Xia, H. Yuan, Y. Chen, Z. Liang, L. Zhang and Y. Zhang, Talanta, 2015, 141, 235–238 CrossRef CAS PubMed.
- J. L. Proc, M. A. Kuzyk, D. B. Hardie, J. Yang, D. S. Smith, A. M. Jackson, C. E. Parker and C. H. Borchers, J. Proteome Res., 2010, 9, 5422–5437 CrossRef CAS PubMed.
- N. Zhang, N. Li and L. Li, J. Proteome Res., 2004, 3, 719–727 CrossRef CAS PubMed.
- B. Kwon, J. Molek and A. L. Zydney, J. Membr. Sci., 2008, 319, 206–213 CrossRef CAS.
- J. Leberknight, B. Wielenga, A. Lee-Jewett and T. J. Menkhaus, J. Membr. Sci., 2011, 366, 405–412 CrossRef CAS.
- Q. Xu, Y. Ye, V. Chen and X. Wen, Water Res., 2015, 68, 182–193 CrossRef CAS PubMed.
- M.-C. Vincent-Vela, B. Cuartas-Uribe, S. Álvarez-Blanco and J. Lora-García, Chem. Eng. Process., 2011, 50, 404–408 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |