
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of nanocrystallized zirconium carbide based on an aqueous solution-derived precursor
Jing-Xiao Wangabc,
De-Wei Ni *bc,
Shao-Ming Dongbc,
Guang Yanga,
Yan-Feng Gaoa,
Yan-Mei Kanbc,
Xiao-Wu Chenbc,
Yan-Peng Caoabc and
Xiang-Yu Zhang*bc
*bc,
Shao-Ming Dongbc,
Guang Yanga,
Yan-Feng Gaoa,
Yan-Mei Kanbc,
Xiao-Wu Chenbc,
Yan-Peng Caoabc and
Xiang-Yu Zhang*bc
aSchool of Materials Science and Engineering, Shanghai University, Shanghai 200444, China
bState Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, China. E-mail: deweini@mail.sic.ac.cn; xyzhang@mail.sic.ac.cn; Fax: +86 21 69906085; Tel: +86 21 69906033
cStructural Ceramics and Composites Engineering Research Center, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, China
First published on 25th April 2017
Abstract
Nanocrystallized zirconium carbide (ZrC) powder was synthesized by an aqueous solution-based process using zirconium acetate and sucrose as starting reagents. Polyvinyl pyrrolidone (PVP) was used to combine the reactants to form a suitable precursor for ZrC. Through such a novel aqueous solution-based process, fine-scale mixing of the reactants is achieved in an environmentally friendly manner. The formed precursor can be converted to ZrC by carbothermal reduction reaction at 1600–1650 °C. Thanks to a limited amount of residual free carbon, the as-synthesized ZrC powder had an ultra-fine particle size (50–100 nm) and a low oxygen content less than 1.0 wt%. The conversion mechanisms from as-synthesized pre-ceramic precursor to ZrC powder were investigated systematically and revealed by means of FTIR, TG-DSC, XRD, Raman, SEM and TEM.
1. Introduction
Zirconium carbide (ZrC) has been considered as a strategic material in many applications, such as field emitters, a coating for nuclear reactor particle fuels, and cutting tools etc., because of its outstanding properties (high melting point, high elastic modulus and hardness, and small neutron absorption cross-section etc.).1–3 However, due to its strong covalent bonding nature and low self-diffusion coefficient, ZrC shows very poor sinterability, which limits its applications. It has been widely realized that smaller particles can lower the sintering temperature and facilitate densification processes due to the larger surface area and higher reactivity of the smaller particles.4–6Conventionally, ZrC is synthesized by solid state reactions between carbon and Zr, ZrH2, or ZrO2. In many cases, high temperatures are required for the carbide-forming reactions because the powders are mixed together on a relatively coarse scale, e.g., micrometer-scale, which also leads to metal carbide products with relatively large particle sizes.7 Studies have also shown that ZrC can be synthesized by using precursors via solution-based processing methods. This becomes increasingly popular due to the advantages of homogenous distribution of all reactants at molecular level, which can lower the reaction temperature and shorten the reaction time, thus leading to fine particle size. Sacks et al. synthesized nano ZrC powders (∼50–130 nm) using mixed solutions of zirconium n-propoxide/n-propanol (as a zirconia source) and either phenolic resin or glycerol (as a carbon source).8 Investigation by Yan et al. revealed that carbon sources and various ligands chelation showed a remarkable influence on morphology and particle size of the synthesized ZrC powders.9–11 With an additional foaming process, Li et al. synthesized ZrC nanopowder (175 nm) using zirconium oxychloride octahydrate and sucrose as Zr and C sources respectively.12
In most of the reported cases for ZrC synthesis by solution-based processing, organic reactants and organic solvent were usually used, which is expensive, complex and not environmentally friendly. In this work, ZrC was synthesized by carbothermal reduction via an aqueous solution-derived precursor, in which zirconium acetate and sucrose were used as Zr and C sources respectively. Polyvinyl pyrrolidone (PVP) was used to combine the reactants to form a proper precursor for ZrC. The conversion mechanisms from as-synthesized pre-ceramic precursor to ZrC powder were investigated in detail. As well, the effect of residual free carbon on the synthesized ZrC was investigated.
2. Experimental procedures
2.1. Materials and processing
Zirconium acetate (ZrO(CH3COO)2, purity 99%, Yixing Xinxing Zirconium Co., Ltd, Yixing, China), sucrose (C12H22O11, AR grade, Aladdin industrial Corporation, Shanghai, China), and polyvinyl pyrrolidone (PVP, Beijing HWRK Chem Co., Ltd, Beijing, China) were used as starting materials. According to reaction ZrO2 + 3C = ZrC + 2CO (g), molar ratio between ZrO(CH3COO)2 and C12H22O11 was adjusted in a range of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 4:1.5. The PVP addition was controlled at around 2 wt% in the precursor solution. The starting materials were mixed with deionized water under constant stirring for 24 h to form a uniform aqueous solution-derived precursor.
:1 to 4:1.5. The PVP addition was controlled at around 2 wt% in the precursor solution. The starting materials were mixed with deionized water under constant stirring for 24 h to form a uniform aqueous solution-derived precursor.
The obtained precursor was cross-linked and cured at 85 °C for 12 h in drying oven. And then it was pyrolyzed at 600 °C for 2 h under flowing argon atmosphere with a heating rate of 1 °C min−1. The pyrolyzed precursor was subsequently heat treated at 1400–1650 °C for 2 h in vacuum for carbothermal reduction with a heating rate of 5 °C min−1. The as-obtained products after carbothermal reduction could be ground into fine powders easily for further characterization.
2.2. Characterization
Fourier transform infrared (FTIR) spectra of both the starting materials and precursors were measured by NICOLET Is 10 spectrometer (Thermo Fisher Scientific, Massachusetts, USA). Thermal gravimetric analysis (TG) and differential scanning calorimetry (DSC) were performed on a Netzsch STA 499 F3 Jupiter® in argon flow at a heating rate of 10 °C min−1 over the range of 40–1400 °C.The morphologies of the as-synthesized powders were observed using a field emission scanning electron microscope (FESEM, Merlin Compact, Zeiss, Germany). As well, the characterizations of morphologies were performed using a 200 kV transmission electron microscope (TEM, JEOL JEM-2100F, Japan) along with selected area electron diffraction (SAED). The particle size of the synthesized ZrC powder were determined by post processing of the SEM images, using freeware software ImageJ®. To reduce the error in the determination, at least ten images with hundreds of crystallites were taken in random regions of the samples and used for the statistical analysis. X-ray diffraction (XRD) patterns were collected on a Rigaku Ultima IV diffractometer with Cu target (40 kV, 40 mA) over the angle range of 10–85°. The Rietveld refinement of the XRD profile was carried out to determine the influence of residual carbon on crystal structure of the as-synthesized powder. Raman spectroscopy (Thermo Nicolet, USA) was also carried out for the synthesised powders. The oxygen content of the as-synthesized ZrC powder was determined by a nitrogen/oxygen determinator (TC600, LECO, St. Joseph, MI). The carbon content of the as-synthesized powders was detected by chemical analysis.
3. Results and discussion
Using ZrO(CH3COO)2 and C12H22O11 as starting reagents, PVP was used to combine the reactants to form the precursor for ZrC. Infrared spectroscopy was used to investigate the structural evolution and principal interactions of the precursor during heat treatment (Fig. 1). The FTIR spectra of the starting materials are also shown here for comparison. The respective assignments of the absorption peaks are 3440 (v(O–H) + v(NH2)), 2940 (v(C–H)), 1654 (v(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)), 1566 (v(CO)), 1435 (v(CO) + δ(CH)), 1295 (δ(C–H) + v(C–CH3)), 1052 (v(OOC)), 925 (v(C–CH3) + v(CO)), and 650 (π(C–CH3) + v(Zr–O) + ring distortion) and 400–500 (v(Zr–O)) cm−1.13 For the precursor after cross-linking and curing at 85 °C, the broad peak at 3440 cm−1 assigned to v(O–H) and v(NH2) is attributed to the stretching vibration of –NH2 and –OH groups. It shifts to lower wavenumber, indicating –NH2 and –OH groups take part in the complexation reaction. The peak at 1654 cm−1 from FTIR spectra of PVP is assigned to v(CO), which is easily chelated to zirconium, thus largely reducing the tendency of zirconium to form bulk gels. Furthermore, the peaks at around 3400 and 1650 cm−1 of the precursor prior to pyrolysis are strong indication of the presence of absorbed water, which is also confirmed in the following discussion on DSC and TG results. The two peaks located at 650, 490 cm−1, which are attributed ring distortion + v(Zr–O) of the cyclic structure after zirconium chelation and v(Zr–O) + v(Zr–N), respectively,14 appear in the FTIR curve of the precursor. This is very important for achievement of the homogeneous and intimate mixing of all the components for the followed carbothermal reduction reaction. After heat treatment at 600 °C, almost all the absorption peaks disappear, indicating pyrolysis processes complete and nearly no residual organics exist.
O)), 1566 (v(CO)), 1435 (v(CO) + δ(CH)), 1295 (δ(C–H) + v(C–CH3)), 1052 (v(OOC)), 925 (v(C–CH3) + v(CO)), and 650 (π(C–CH3) + v(Zr–O) + ring distortion) and 400–500 (v(Zr–O)) cm−1.13 For the precursor after cross-linking and curing at 85 °C, the broad peak at 3440 cm−1 assigned to v(O–H) and v(NH2) is attributed to the stretching vibration of –NH2 and –OH groups. It shifts to lower wavenumber, indicating –NH2 and –OH groups take part in the complexation reaction. The peak at 1654 cm−1 from FTIR spectra of PVP is assigned to v(CO), which is easily chelated to zirconium, thus largely reducing the tendency of zirconium to form bulk gels. Furthermore, the peaks at around 3400 and 1650 cm−1 of the precursor prior to pyrolysis are strong indication of the presence of absorbed water, which is also confirmed in the following discussion on DSC and TG results. The two peaks located at 650, 490 cm−1, which are attributed ring distortion + v(Zr–O) of the cyclic structure after zirconium chelation and v(Zr–O) + v(Zr–N), respectively,14 appear in the FTIR curve of the precursor. This is very important for achievement of the homogeneous and intimate mixing of all the components for the followed carbothermal reduction reaction. After heat treatment at 600 °C, almost all the absorption peaks disappear, indicating pyrolysis processes complete and nearly no residual organics exist.
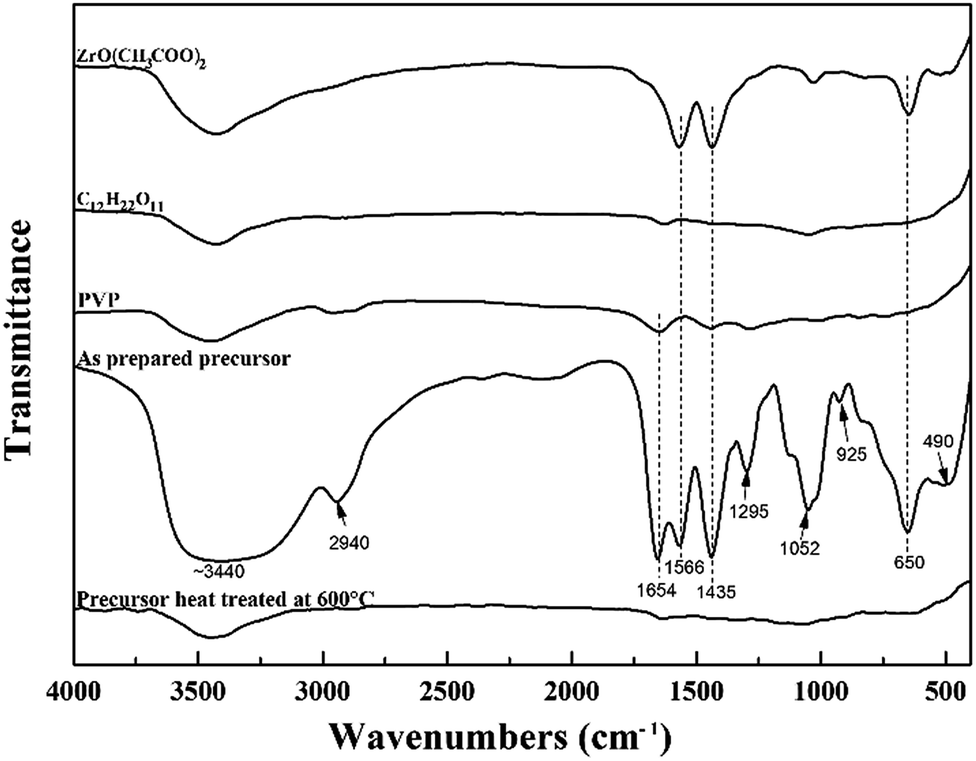 | ||
| Fig. 1 FTIR spectra of the starting materials and precursors. | ||
The pyrolysis process of the precursor material was studied by simultaneous TG and DSC analysis, which are illustrated in Fig. 2. The pyrolysis processes of the precursor can be divided into four stages. Due to the evaporation of free water and separation of adsorbed water, there is a considerable weight loss occurring below 160 °C, which results in an apparent endothermic peak on the DSC curve. The weight loss at this stage is around 12.23%. Next, the second stage is located between 160 and 240 °C, corresponding to the evaporation of constitution water and organic by products, where the weight loss is around 12.24%. This is consistent with a shoulder endothermic peak on the DSC curve. Then, the third stage between 240 and 500 °C is attributed to the decomposition of ZrO(CH3COO)2, C12H22O11 and PVP, leading to a weight loss of around 27.12%. After 500 °C, the weight loss tends to be very slow, where the TG curve is almost flat. This stage is probably corresponding to the continuous carbonization of sucrose and the transformation of ZrO2 crystalline phase.15,16 Subsequently, carbothermal reduction reaction started at temperature around 1400 °C. Based on the above discussion of TG-DSC results, the pyrolysis temperature of the precursor is settled at 600 °C to reduce the volatile components, and the carbothermal reduction temperature is set to be 1400–1600 °C. Based on TG analysis of individual raw materials (not shown here), a molar ratio of 4:1.5 for ZrO(CH3COO)2:C12H22O11 was first used in the precursor according to carbothermal reduction reaction ZrO2 + 3C = ZrC + 2CO (g).
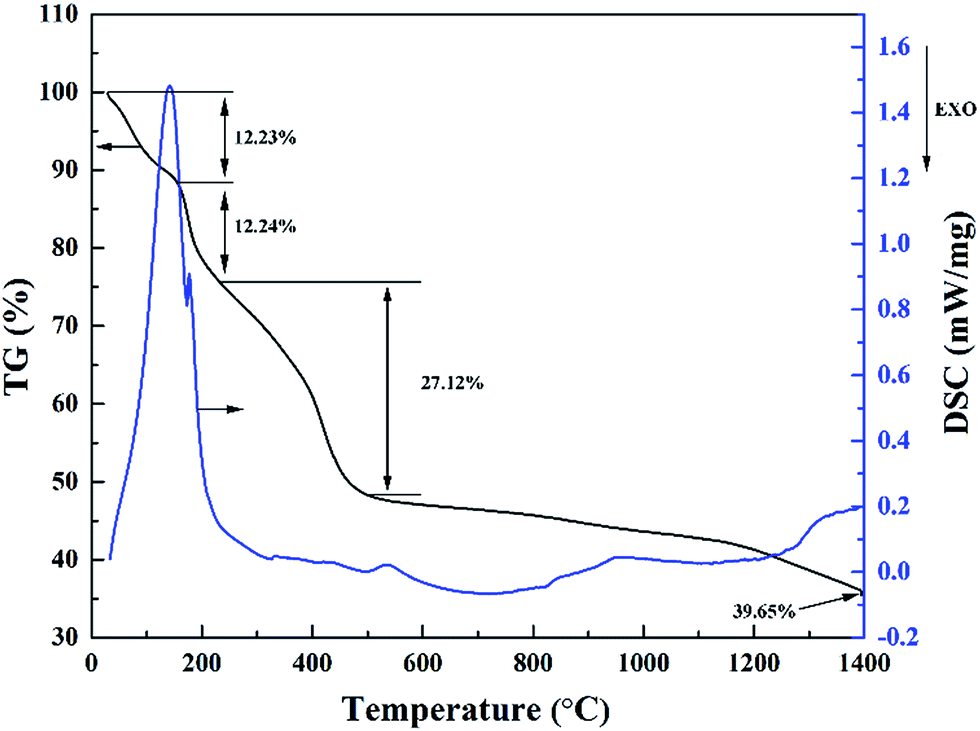 | ||
| Fig. 2 TG-DSC curves of the as-obtained ZrC precursor dried at 85 °C. | ||
XRD was used to study the conversion of phase composition during heat treatment. Fig. 3 shows the XRD patterns of ZrC precursor after heat treatment at temperatures from 1400 to 1600 °C, where the molar ratio of ZrO(CH3COO)2:C12H22O11 was 4:1.5 in the precursor. After heat treatment at 1400 °C, the initial formation of ZrC is observed in the sample but t-ZrO2 is the predominant phase. This result is in good agreement with the TG analysis discussed above. As well, it was observed that the vacuum degree of the furnace chamber started to decrease at about 1350 °C, which was a sign of the carbothermal reduction reaction beginning. On the other hand, it is well known that tetragonal phase of zirconia can be stabilized to room temperature by carbon and tiny particle size.17,18 Therefore, it's t-ZrO2 rather than m-ZrO2 observed in the sample. ZrC becomes the predominant phase in the sample heat treated at 1500 °C, but small amount of t-ZrO2 still existed. After heat treatment at 1600 °C, pure ZrC can be detected and no evidence of ZrO2 is observed on the XRD pattern. However, measurement indicated that oxygen content of the as synthesized ZrC powder at 1600 °C is as high as 1.27 wt%. Therefore, a further heat treatment at a higher temperature (1650 °C) had been carried out, where the oxygen content was reduced to 1.0 wt%.
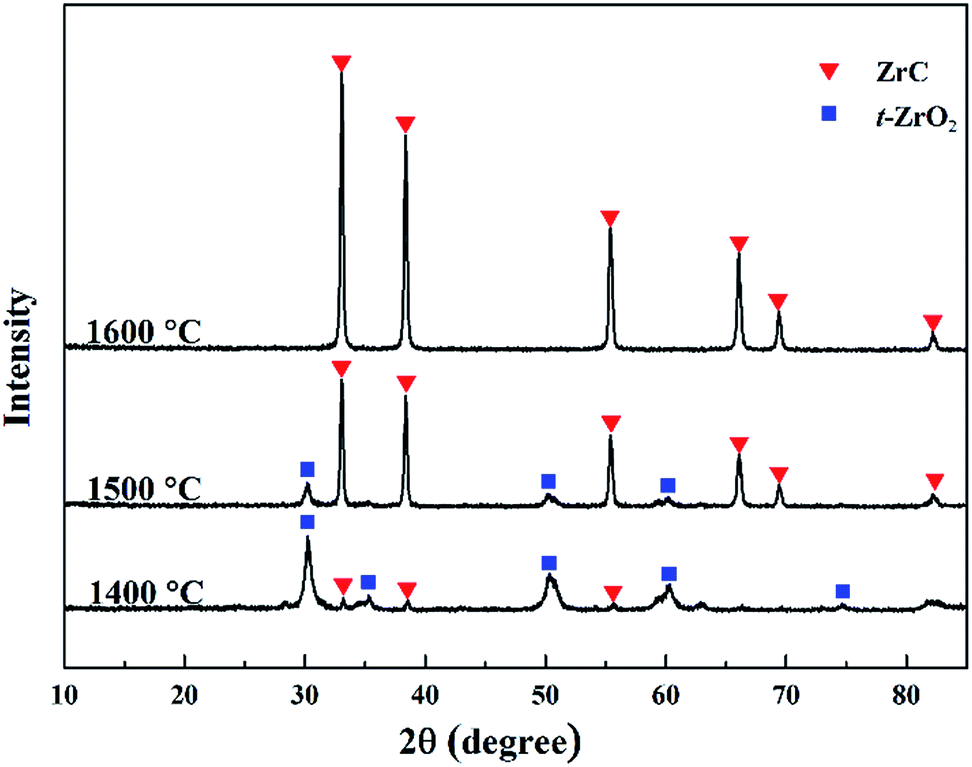 | ||
| Fig. 3 XRD patterns of the ZrC precursor (ZrO(CH3COO)2:C12H22O11 = 4:1.5) after heat treatment at temperatures from 1400 to 1600 °C. | ||
SEM images of the as-synthesized ZrC powders are presented in Fig. 4. It is clear that there are two completely different morphologies in both powders. In addition to the equiaxial ZrC particles with some extent of agglomeration, a flocculent material also presents surrounding the ZrC particles. EDS analysis (not shown here) indicates that the flocculent material is residual carbon. Chemical analysis shows that the residual free carbon in the ZrC powder synthesized at 1600 °C is around 3.7 wt%. Compared with the ZrC powder synthesized at 1600 °C, less residual carbon (1.4 wt%) is achieved at 1650 °C, as also shown in Fig. 4. Particle size of the ZrC powders are mainly distributed in the range of 50–100 nm with a mean size of 56 nm, which is much smaller than ZrC powders synthesized through traditional solid state reactions or sol–gel methods.19,20 No evident particle coarsening is observed at the higher temperature (1650 °C). It is known that grain growth is mostly determined by mass transfer and mainly occurs among particles via surface diffusion.21 Previously, it has been reported that carbon black could limit HfO2 growth by physically separating the oxide particles.22 Thus, it is believed that the existence of the residual carbon in this work reduces the particle mutual contact, thus preventing the particle coarsening and leading to ultra-fine particle size.
 | ||
| Fig. 4 SEM images of ZrC powders obtained at (a) 1600 °C and (b) 1650 °C. | ||
To further investigate the effect of residual free carbon on the synthesized ZrC powders, ZrC powders from precursors with different ratio of ZrO(CH3COO)2:C12H22O11 were prepared. Corresponding Rietveld refinement for XRD patterns of the synthesized ZrC powders are shown in Fig. 5. ZrC is the only phase detected for all the three ZrC powders which is nicely refined using cubic Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group in the region of 10–85 degrees. According to the Rietveld refinement, lattice parameter of the cubic ZrC phase was calculated, which is 4.6595 Å, 4.6872 Å and 4.6880 Å, respectively.
m space group in the region of 10–85 degrees. According to the Rietveld refinement, lattice parameter of the cubic ZrC phase was calculated, which is 4.6595 Å, 4.6872 Å and 4.6880 Å, respectively.
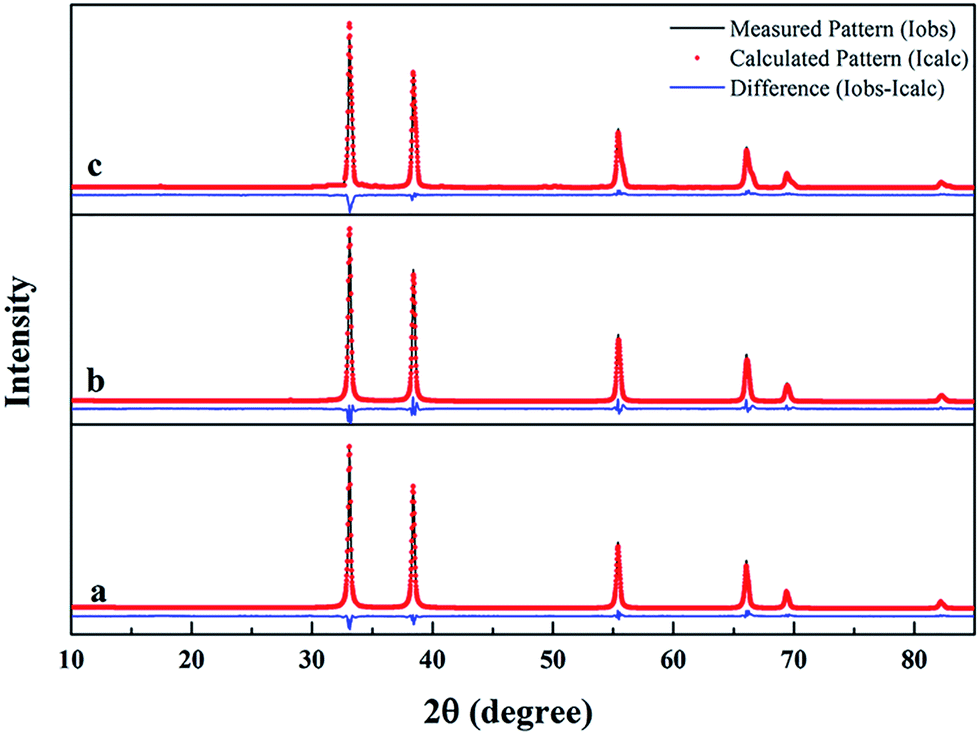 | ||
| Fig. 5 Rietveld refinement for XRD patterns of synthesized ZrC powders from precursors with different ratio of ZrO(CH3COO)2:C12H22O11: (a) 4:1.5; (b) 4:1.2; (c) 4:1. | ||
It has been reported in literature that non-stoichiometric ZrC1−x could be formed in the Zr–C binary system.23,24 However, it was thought in the present case that the residual carbon would react with ZrC lattice to form non-stoichiometric Zr1−xC according to the following reaction:
| (1 − x)ZrC + xC → Zr1−xC | (1) |
The lattice parameter a of stoichiometric ZrC has been reported to be 4.693 Å (PDF#350784). The decrease in lattice parameter of ZrC phase in this work implies the formation of non-stoichiometric Zr1−xC according to the above reaction. As the increase of residual free carbon (x value), more Zr vacancies/non-stoichiometric Zr1−xC could be formed, leading to smaller lattice parameter. Furthermore, vacancies/non-stoichiometry also results in microstrains in the lattice, which is 0.441, 0.341, 0.158, respectively, as indicated by the Rietveld refinement results of XRD.
Chemical analysis indicates that the contents of residual free carbon in the synthesized ZrC powders are around 1.4 wt%, 0.8 wt% and 0.5 wt% respectively. Raman spectra of the synthesized ZrC powders are shown in Fig. 6. Two typical peaks centered at ∼1330 and ∼1590 cm−1 are detected in all the three samples, corresponding to the D and G peaks of free carbon, revealing the presence of residual free carbon in the powders.25 Moreover, the intensity of D and G peaks decrease significantly with the increase of ZrO(CH3COO)2:C12H22O11 ratio, indicating the decrease of residual carbon in the powders. This is in good agreement with the chemical analysis.
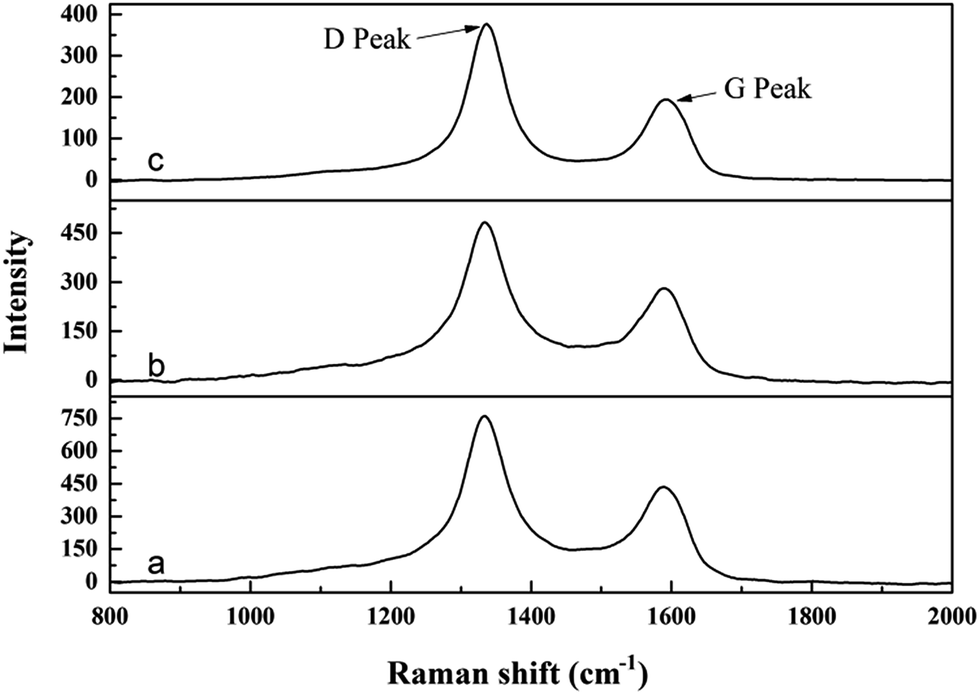 | ||
| Fig. 6 Raman spectra of synthesized ZrC powders from precursors with different ratio of ZrO(CH3COO)2:C12H22O11: (a) 4:1.5; (b) 4:1.2; (c) 4:1. | ||
Fig. 7 exhibits the TEM and HRTEM images of synthesized ZrC powders from precursors with different ratio of ZrO(CH3COO)2:C12H22O11. SAED images (insets in Fig. 7a–c) reveal that the equiaxial particles are cubic ZrC. It can be found that the three powders show similar morphology with comparable particle size (56 nm, 60 nm and 61 nm respectively). With the decrease of C12H22O11 in the starting materials, the content of amorphous free carbon decreases remarkably, showing good agreement with the chemical analysis discussed above. It appears that a limited amount (0.5 wt%) of residual free carbon is enough in preventing grain coarsening of ZrC. Further decrease of C12H22O11 in the starting materials can lead to serious grain coarsening or ZrO2 impurity. As can be seen in the HRTEM images (Fig. 7d–f), the particles have lattice fringes with the interlayer distance of 0.23–0.27 nm (as indicated in the images), which corresponds to the (200) or (111) planes of ZrC crystal. The calculated lattice parameter based on the lattice fringes show very good consistent with the values from XRD results. This further confirms the formation of non-stoichiometric Zr1−xC in the synthesized ZrC powders. It is believed that the non-stoichiometric Zr1−xC powders with nano size would be beneficial for the subsequent sinter and densification of the materials.23
 | ||
| Fig. 7 TEM and HRTEM images of synthesized ZrC powders from precursors with different ratio of ZrO(CH3COO)2:C12H22O11: (a and d) 4:1.5; (b and e) 4:1.2; (c and f) 4:1. The selected area electron diffraction (SAED) patterns of ZrC particles are inserted in (a), (b) and (c) at the * position respectively. | ||
4. Conclusions
Nanocrystallized ZrC powders were synthesized by an environmentally friendly aqueous solution-based processing using zirconium acetate and sucrose, combined with polyvinyl pyrrolidone (PVP) to form a precursor for ZrC with high stability. The precursor can be transformed into cubic ZrC by carbothermal reduction reaction at 1600–1650 °C. It is revealed that the limited amount of residual free carbon can react with ZrC and lead to the formation of non-stoichiometric Zr1−xC. Furthermore, the limited amount (∼0.5 wt%) of residual free carbon can also reduce the mutual contact of ZrC particles and prevents the particle coarsening, leading to the ultra-fine particle size (∼50–100 nm) in the synthesized ZrC powders. It is believed that the non-stoichiometric Zr1−xC powders with ultra-fine particle size would be beneficial for the subsequent densification of the materials.Acknowledgements
The financial support from the “The National Key Research and Development Program of China” (No. 2016YFB070020X), Chinese Academy of Sciences Innovative Funding (CXJJ-17-M169), “CAS Pioneer Hundred Talents Program” and Shanghai Key Projects of Basic Research (Grant No. 14JC1406200) are greatly acknowledged.References
- K. Minato, T. Ogawa, K. Sawa, A. Ishikawa, T. Tomita, S. Iida and H. Sekino, Nucl. Technol., 2000, 130, 272–281 CAS.
- H. O. Pierson, Handbook of refractory carbides and nitrides: Properties, characteristics, processing and applications, Noyes Publication, New York, NY, United States, 1997 Search PubMed.
- X. Zhang, Q. Qu, J. Han, W. Han and C. Hong, Scr. Mater., 2008, 59, 753–756 CrossRef CAS.
- R. Chaim, M. Levin, A. Shlayer and C. Estournès, Adv. Appl. Ceram., 2013, 107, 159–169 CrossRef.
- X. Kuang, G. Carotenuto and L. Nicolais, Adv. Perform. Mater., 1997, 4, 257–274 CrossRef CAS.
- M. J. Mayo, Int. Mater. Rev., 1996, 41, 85–115 CrossRef CAS.
- M. Zhang, Q. Hu, B. Huang and J. Li, J. Alloys Compd., 2011, 509, 8120–8125 CrossRef CAS.
- M. D. Sacks, C.-A. Wang, Z. Yang and A. Jain, J. Mater. Sci., 2004, 39, 6057–6066 CrossRef CAS.
- C. Xie, M. Chen, X. Wei, M. Ge and W. Zhang, J. Am. Ceram. Soc., 2012, 95, 866–869 CAS.
- C. Yan, R. Liu, Y. Cao, C. Zhang and D. Zhang, J. Sol-Gel Sci. Technol., 2012, 64, 251–256 CrossRef CAS.
- C. Yan, R. Liu, Y. Cao, C. Zhang and D. Zhang, Ceram. Int., 2013, 39, 3409–3412 CrossRef CAS.
- F. Li, X. Huang and G.-J. Zhang, Ceram. Int., 2015, 41, 3335–3338 CrossRef CAS.
- H. Y. Liu, X. Q. Hou, X. Q. Wang, Y. L. Wang, D. Xu, C. Wang, W. Du, M. K. Lü and D. R. Yuan, J. Am. Ceram. Soc., 2004, 87, 2237–2241 CrossRef CAS.
- X. Tao, W. Qiu, H. Li and T. Zhao, Polym. Adv. Technol., 2010, 21, 300–304 CAS.
- Z. Dong, X. Zhang, Q. Huang, J. Zhang, X. Zuo, W. Li, G. Yuan and X. Li, Ceram. Int., 2015, 41, 7359–7365 CrossRef CAS.
- F. Li, M. Liang, X.-F. Ma, X. Huang and G.-J. Zhang, J. Porous Mater., 2015, 22, 493–500 CrossRef CAS.
- Y. Li, C. Tian, L. Zhao, Y. Li, S. Jin and S. Li, J. Chin. Ceram. Soc., 2009, 37, 1273–1276 CAS.
- R. C. Garvie and M. F. Goss, J. Mater. Sci., 1986, 21, 1253–1257 CrossRef CAS.
- M. Dolle, D. Gosset, C. Bogicevic, F. Karolak, D. Simeone and G. Baldinozzi, J. Eur. Ceram. Soc., 2007, 27, 2061–2067 CrossRef CAS.
- C. Yan, R. Liu, Y. Cao, C. Zhang and D. Zhang, J. Am. Ceram. Soc., 2012, 95, 3366–3369 CrossRef CAS.
- D.-W. Ni, J.-X. Liu and G.-J. Zhang, J. Eur. Ceram. Soc., 2012, 32, 2557–2563 CrossRef CAS.
- J. X. Liu, Y. M. Kan and G. J. Zhang, J. Am. Ceram. Soc., 2010, 93, 980–986 CrossRef CAS.
- X.-G. Wang, W.-M. Guo, Y.-M. Kan, G.-J. Zhang and P.-L. Wang, J. Eur. Ceram. Soc., 2011, 31, 1103–1111 CrossRef.
- O. M. Barabash and Y. N. Koval, Crystal Structure of Metals and Alloys, Kiew, Naukova Dumka, 1986, pp. 215–216, in Russian Search PubMed.
- A. Sadezky, H. Muckenhuber, H. Grothe and R. Niessner, Carbon, 2005, 43, 1731–1742 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |