
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile one-pot fabrication of cellulose nanocrystals and enzymatic synthesis of its esterified derivative in mixed ionic liquids
Guanglei Zhao,
Fengli Wang,
Xiongfei Lang,
Beihai He,
Junrong Li and
Xiaofeng Li *
*
State Key Laboratory of Pulp and Paper Engineering, South China University of Technology, Wushan Road 381, Guangzhou 510641, China. E-mail: xflibio@scut.edu.cn; Tel: +86 20 87111770
First published on 22nd May 2017
Abstract
As an important cellulose derivative, esterified cellulose nanocrystals (E-CNCs) could be applied in biomedical and chemical industries. A facile one-pot enzymatic method was proposed for the preparation of esterified cellulose nanocrystals with methyl laurate by using native cellulose as raw material in a binary ionic liquid (IL) system. 1-Butyl-3-methylimidazolium hydrogen sulfate ([BMIm]HSO4), a hydrophilic IL, acted as catalyst and solvent to produce cellulose nanocrystals (CNCs). 1-Butyl-3-methyl-imidazolium tetrafluoroborate ([BMIm][BF4]), which is hydrophobic, was used as a solvent for improving the lipase performance. The key reaction conditions, including mass ratio of [BMIm]HSO4/[BMIm][BF4], molar ratio of methyl laurate/anhydroglucose unit (AGU) in cellulose, mass ratio of lipase/cellulose, and reaction time, showed significant influences on the enzymatic transesterification of CNCs. The ester products were confirmed by FT-IR and 13C NMR analysis. The crystalline structure of E-CNCs was revealed by XRD. Compared with native cellulose, the E-CNCs showed improved crystallinity. TEM data showed the width of rod-shaped E-CNCs in the range of 10–30 nm. Compared to native cellulose, the thermal stability of E-CNCs was decreased due to the higher surface areas exposed to heat. This is the first successful preparation of E-CNCs with long-chain fatty acids from cheap native cellulose in a one-pot method.
1 Introduction
As a renewable material, cellulose nanocrystals (CNCs) have drawn extensive attention due to their large specific surface area, excellent mechanical properties, low density, high strength and transparency, and electrical and magnetic properties.1–3 The most significant efforts focused on the use of CNCs as reinforcement materials in nanocomposites, but recently, their use in films, membranes, catalyst support materials, and functionalized drug carriers is being explored.4,5 The methods to produce CNCs include acid hydrolysis, enzyme treatment, high-pressure homogenization, or their combinations. Among these methods, acid hydrolysis is the most common.6 However, these methods still have some drawbacks, such as low reaction rate, equipment corrosion, harsh reaction conditions and degradation of cellulose, which limit the availability of CNCs.Currently, the use of ionic liquids (ILs) as a type of green solvent has received extensive attention in the production of CNCs due to their low toxicity, non-flammability, very low vapor pressures and recyclability.7,8 According to Man et al., CNCs could be prepared by treating microcrystalline cellulose with 1-butyl-3-methylimidazolium hydrogen sulphate ionic liquid.6 Recently, Tan et al. had prepared high-crystallinity CNCs by ionic liquid, with diameters ranging from 15 to 20 nm and lengths ranging from 70 to 80 nm.4 In the reaction condition, [BMIm]HSO4 dissociated to individual [BMIm]+ cations and [HSO4]− anions, which form electron donor–electron acceptor (EDA) complexes with the charged species between the hydrogen and oxygen atoms of the cellulose molecules. Then, different cellulose chains separate, resulting from the breakage of the hydroxyl groups. Furthermore, the amorphous regions in native cellulose are more accessible for [BMIm]+ and [HSO4]−, which leads to the selective removal of the amorphous regions by [BMIm]HSO4, leaving behind the crystalline regions.
CNCs have a high surface area-to-volume ratio, which means that they have high activity and are difficult to disperse in apolar solvents.9,10 To overcome these difficulties, various modification approaches have been developed for CNCs to make functional cellulose nanocrystal particles or to improve their dispersion in apolar solvents.2,5,11 According to Kaboorani and Riedl, CNCs could be surface modified by a cationic surfactant of hexadecyltrimethylammonium (HDTMA) bromide, and the modified CNCs had different suspension stabilities and dispersibilities compared to unmodified CNCs.1 Yin et al. had focused on modifying the thermostability and compatibility of CNCs by graft polymerization with styrene.2 Zaman et al. described nanocrystalline cellulose that was rendered cationic by grafting with glycidyltrimethylammonium chloride (GTMAC), and the products were well dispersed and stable in aqueous media due to enhanced cationic surface charge density.12 However, the previous studies on surface-modified CNCs focused on the modification of isolated CNCs mainly by chemical methods.
Recently, there has been an increasing trend towards developing environmentally friendly processes. One major aspect of these processes is the use of biocatalysts (isolated enzymes or whole cells).13 Compared to traditional chemical methods, biocatalysis provides a greener and simpler route to performing chemical transformations for its significant advantages, such as high selectivity, high product purities, moderate reaction conditions, etc.14,15 Over the past thirty years, biocatalysis has been successfully applied in various types of reactions, including hydrolysis, hydroxylation, epoxidation, transamination, and transesterification.13,16,17 In most cases, the use of biocatalysts improved the efficiency of the entire process, reduced the consumption of chemical reagents and simplified the purification processes of the reaction products.13,16,17 Herein, biocatalysis has been considered as an attractive tool for organic synthesis.
Among biocatalysts, lipases (triacylglycerol hydrolases, EC 3.1.1.3) have attracted great interest in recent years.18,19 The natural function of lipases is to catalyze hydrolysis of fatty acid esters in aqueous media. In non-aqueous media with very low water content, they are capable of catalyzing the reverse reaction of hydrolysis, including esterification and transesterification. In non-aqueous media, lipases prefer hydrophobic solvents such as hexane, t-amyl alcohol, [BMIm]BF4 or [BMIm]PF6, in which they can well maintain the active site conformation of their proteins.20,21 In many polar solvents, however, these enzymes may have decreased catalytic activities or even be deactivated.22–24 Herein, for the biocatalytic reactions that polar substrates are involved in, there is a conflict between maintaining the enzyme activity and dissolving the polar substrates when choosing a reaction solvent. Generally, hydrophobic solvents should be chosen for better lipase performance, in which, however, the hydrophilic substrates cannot be dissolved.8,16,25 This conflict greatly hampers the application of lipases in catalyzing the modification of polar compounds such as cellulose.
In this research, we proposed a simple, efficient and green approach to prepare esterified CNCs (E-CNCs) in one pot via enzymatic transesterification in a binary IL media containing a hydrophilic IL ([BMIm]HSO4) and a hydrophobic one ([BMIm]BF4). A two-step cascade reaction composed of the decomposition of cellulose (step 1) followed by enzymatic transesterification of CNCs (step 2) is investigated. In the first step, the IL [BMIm]HSO4 serves as a moderate catalyst, with which the large-scale celluloses can be hydrolyzed to form nanocelluloses (CNCs). Compared to the large-scale celluloses, the nanosized cellulose dispersed in the ILs have reduced steric hindrance and increased specific surface area, which favors the subsequent enzymatic function. The other IL [BMIm][BF4] was used as a good solvent for both the enzyme and the acyl donor fatty acid methyl ester. In [BMIm][BF4], lipase can well maintain its catalytic activity, catalyzing the transesterification of CNCs with the methyl ester to form E-CNCs. This approach combined the significant advantages of (1) hydrophilic IL (catalytic capability for glycosidic bond cleavage, low toxicity, nonflammability, and recyclability), (2) hydrophobic IL (good solvent for biocatalytic process, low toxicity, nonflammability, and recyclability) and (3) enzymatic esterification (green catalyst, high selectivity, mild reaction conditions) in a tandem manner in one pot (simplified procedure, shortened reaction time).
Since the lipase was used as catalyst and its natural function is towards the hydrolysis of esters with the existence of water, to avoid the hydrolysis of the E-CNCs, we adopt methyl laurate, a fatty acid ester, as the acyl donor. In addition, modification conditions, including the mass of [BMIm]HSO4/[BMIm][BF4], the mass ratio of lipase/cellulose, the molar ratio of methyl laurate/anhydroglucose unit (AGU) in cellulose, and the reaction time, were investigated. E-CNCs were further characterized by Fourier transform infrared (FT-IR) spectroscopy, 13C NMR spectroscopy, X-ray diffractometry (XRD), transmission electron microscopy (TEM), and thermogravimetric analysis (TGA).
2 Experimental
2.1. Materials
Microcrystalline cellulose (MCC) was purchased from Aladdin Chemistry Co. Ltd. (Shanghai, China) and dried at 50 °C for 48 h before use. 1-Butyl-3-methylimidazolium hydrogen sulfate ([BMIm]HSO4, > 97%) and 1-butyl-3-methyl-imidazolium tetrafluoroborate ([BMIm]BF4, > 99%) were obtained from Shanghai Cheng Jie Chemical Co. Ltd. (Shanghai, China). Methyl laurate (>97%) was purchased from Aladdin Chemistry Co. Ltd. (Shanghai, China). The lipase from Candida rugosa, type VII (nominal activity: >700 U mg−1 enzyme) was obtained from Sigma-Aldrich (Shanghai, China). All other chemicals were of analytical grade.2.2. General procedure for preparation of E-CNCs and CNCs
Dried cellulose (0.3 g) was added into [BMIm]HSO4 (3 g) in a three-necked round flask. The mixture was stirred until homogeneous and heated in an oil bath at 80 °C for 3 h.4 After the mixture was cooled to reaction temperature, [BMIm]BF4 and methyl laurate were added according to the conditions required by the experimental design. Then, the lipase was added with the lipase/cellulose mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 to 5:10, and the reaction was carried out under stirring at 50 °C for 3 h. After completion of the reaction, the reaction mixture was cooled to room temperature and then washed thoroughly with sufficient anhydrous ethanol to eliminate the residual ILs and unreacted reagents. Finally, the solid was filtered and dried in vacuum at 50 °C for 24 h.
:10 to 5:10, and the reaction was carried out under stirring at 50 °C for 3 h. After completion of the reaction, the reaction mixture was cooled to room temperature and then washed thoroughly with sufficient anhydrous ethanol to eliminate the residual ILs and unreacted reagents. Finally, the solid was filtered and dried in vacuum at 50 °C for 24 h.
Dried cellulose (0.3 g) was added into [BMIm]HSO4 (3 g) in a three-necked round flask. The mixture was stirred until homogeneous and heated in an oil bath at 80 °C for 3 h. After adding the ethanol into the mixture, off-white precipitates of cellulose formed. Consequently, the mixture was sonicated at room temperature for 15 min. The suspension was washed with deionized water using repeated centrifugation to isolate the CNCs, and the precipitates were freeze dried.4 The yield of CNCs was measured according to the previous literature.26
2.3. Determination of DS
The degree of substitution (DS) value of modified cellulose is defined as the average number of hydroxyl group substituents per D-glucopyranosyl structural unit of the biopolymer.27,28 The DS values of cellulose esters were determined according to the reported titration method. About 0.05 g (accuracy of weighing ± 0.01 mg) of the sample was dissolved in 20 mL of 0.05 mol L−1 NaOH, followed by vigorous stirring at 75 °C for 30 min. The excess NaOH was back-titrated with standard 0.05 mol L−1 HCl using phenolphthalein as the indicator. The titration was repeated three times, and the average value of the HCl volume was used for the calculations. A blank was also titrated. The amount of COOH was calculated using the following equation:| n = (V0 − V)c, | (1) |
The DS was calculated using the following equation:
| DS = 162n/(m − M′n), | (2) |
2.4. Characterization of the E-CNCs
The crystallinity index (CrI) was calculated from the X-ray diffraction patterns using eqn (3):
| CrI = (I002 − Iam)/I002, | (3) |
3 Results and discussions
3.1. Effects of reaction conditions on DS of the products
A binary IL system ([BMIm]HSO4–[BMIm][BF4]) was adopted for E-CNC preparation in one pot. [BMIm]HSO4 acted as a catalyst for the hydronium ions to penetrate the cellulose chains in the amorphous domains, promoting the hydrolytic cleavage of the glycosidic bonds and releasing individual crystallites. [BMIm]BF4 performed as a suitable co-solvent, in favor of the lipase-catalyzed transesterification. The results of negative controls also confirmed that CNCs can form in the existence of [BMIm]HSO4. No E-CNCs were formed in absence of lipase.From both the theoretical and practical viewpoints, it is of considerable importance to understand the effects of key reaction factors on the reaction efficiency.
As shown in Fig. 1a, DS values of E-CNCs gradually increased from 0.07 to 0.129 along with the increase in mass ratio of [BMIm]HSO4/[BMIm][BF4] from 3:1 to 3:5. Further extending the mass ratio of [BMIm]HSO4/[BMIm][BF4] above 3:5 showed a slight effect on the DS value of E-CNCs. The reason for this phenomenon is that high concentration of the hydrophilic IL [BMIm]HSO4 may deactivate the enzyme proteins by stripping essential water from the protein molecules. Furthermore, the increase in the proportion of the hydrophobic [BMIm][BF4] made the entire reaction media more suitable for enzymatic reaction, leading to an increase in the DS values.
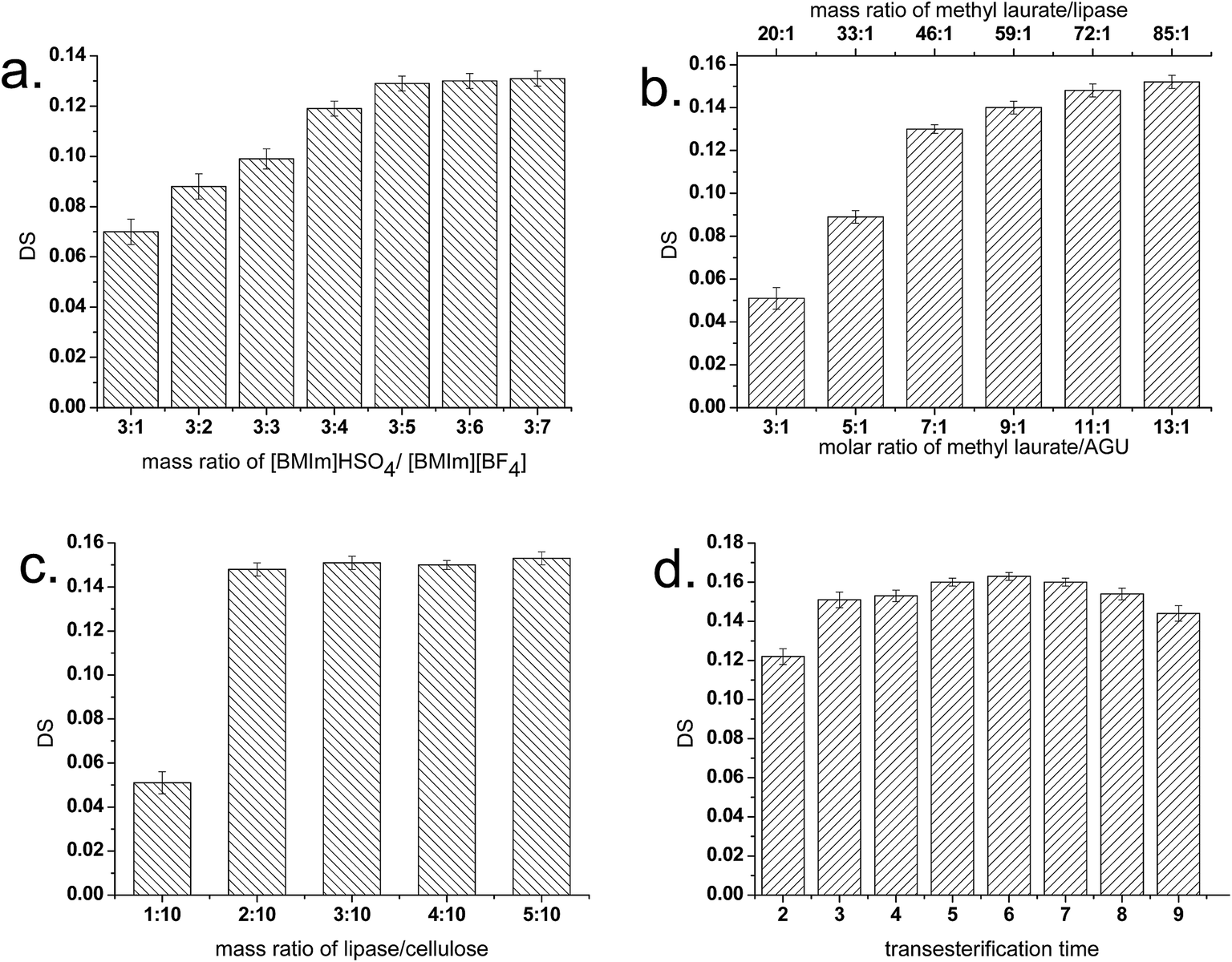 | ||
| Fig. 1 Effects of (a) mass ratio of [BMIm]HSO4/[BMIm][BF4], (b) molar ratio of methyl laurate/AGU, (c) mass ratio of lipase/cellulose, and (d) transesterification time on the DS. Reaction conditions: (a) molar ratio of methyl laurate/AGU, 11:1; mass ratio of lipase/cellulose, 2:10; reaction time, 3 h; temperature, 50 °C; (b) [BMIm]HSO4/[BMIm][BF4], 3:5; mass ratio of lipase/cellulose, 2:10; reaction time, 3 h; temperature, 50 °C; (c) [BMIm]HSO4/[BMIm][BF4], 3:5; molar ratio of methyl laurate/AGU, 11:1; reaction time, 3 h; temperature, 50 °C; (d) [BMIm]HSO4/[BMIm][BF4], 3:5; molar ratio of methyl laurate/AGU, 11:1; mass ratio of lipase/cellulose, 2:10; temperature, 50 °C. | ||
The effect of molar ratio of methyl laurate/AGU and mass ratio of methyl laurate/lipase on DS is presented in Fig. 1b. When the molar ratio of methyl laurate/AGU and mass ratio of methyl laurate/lipase were increased, the DS value of E-CNCs rose rapidly. Lipases could catalyze the transesterification reaction by forming a highly reactive lipase-acyl donor intermediate, which could then react with the hydroxyl group of the anhydroglucose units of cellulose.8 In this experiment, with the amount of the acyl donor methyl laurate increasing and with the dosages of lipase and cellulose being fixed, the DS of products rose. It was also found that when the molar ratio of methyl laurate/AGU was increased higher than 13:1, an obvious precipitation of CNCs may occur, which made this system become heterogeneous. A similar trend was also observed by Barthel and Heinze, who attempted to acylate cellulose with fatty acid chloride in ionic liquids.31 The precipitation of CNCs should be mainly due to the excess methyl laurate, which has been proven by our complementary experiments. In the control experiments, precipitation of CNCs was also found when the molar ratio of methyl laurate/AGU was increased beyond 11:1 in the same reaction system without the addition of lipases. Since no enzymatic transesterification occurred, the precipitation of CNCs was ascribed to the increase of methyl ester concentration. As a hydrophobic solvent, the increase of ethyl laurate may increase the hydrophobicity of the entire reaction medium and thus reduce the solubility of CNCs in the reaction system.
Fig. 1c shows the effect of mass ratio of lipase/cellulose on the DS values. Although the lipase has high specific activity, a mass ratio of lipase to cellulose below than 1:10 is still insufficient for E-CNC preparation. An increase of the DS of E-CNCs from 0.051 to 0.145 with mass ratio increase of lipase/cellulose from 1:10 to 2:10 was observed. Further increase in mass ratio of lipase beyond 2:10, however, led to a slight increase in the DS value of E-CNCs. The results indicate that beyond this limit, the lipase tends to be saturated with the substrate, and thus the DS of the reaction increases rather slowly to a constant value.
Fig. 1d shows that DS increased with transesterification time of 2 to 6 h, then decreased to 0.144 as the transesterification time was extended to 9 h. One reason for this phenomenon may be that hydrolysis of E-CNCs occurred. Another reason is the partial deactivation of lipase in the reaction due to the extended time. A similar phenomenon was also observed in previous literature.8
3.2. FTIR analysis
FTIR spectra of native cellulose and E-CNCs with different DS values are presented in Fig. 2. Compared to native cellulose, a new peak in the carbonyl area around 1734 cm−1 appeared, which indicates the success of the reaction. With the increase of DS, the intensities of the peak at 1734 cm−1 increased.2,10,11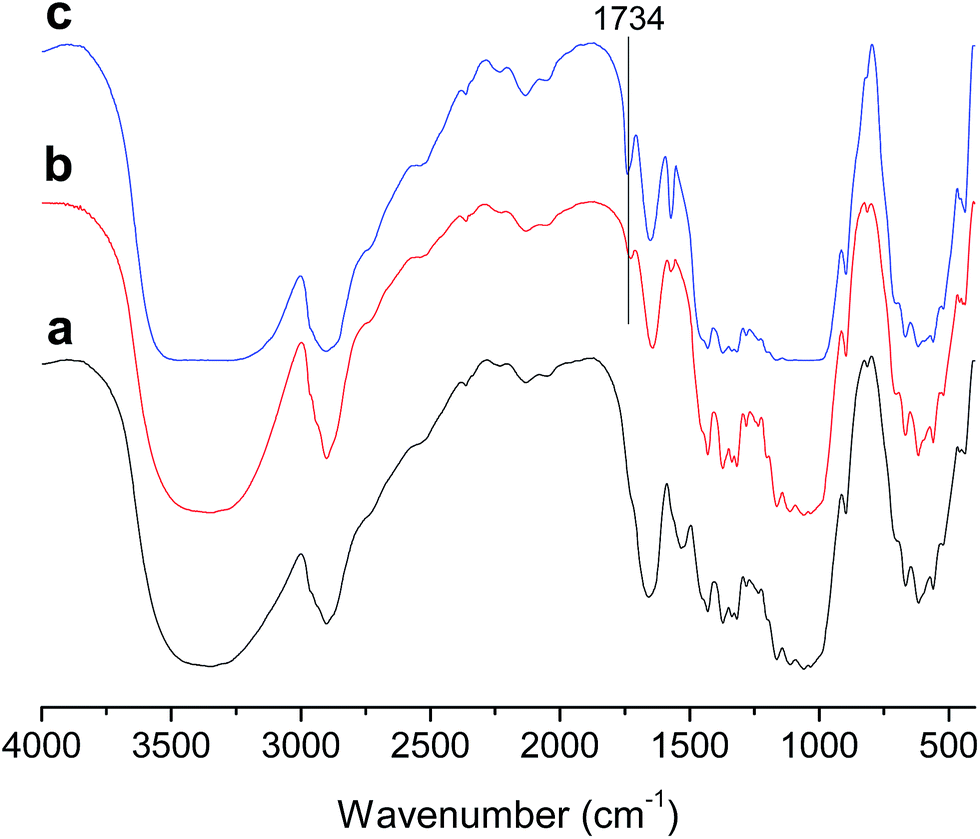 | ||
| Fig. 2 FTIR spectra of (a) native cellulose and E-CNCs of (b) DS = 0.045, (c) DS = 0.125. | ||
3.3. Solid-state 13C NMR measurements
Fig. 3 shows the solid-state 13C NMR spectra of cellulose (a) and E-CNCs (b). The signals at δ = 102.85, 70.14, 72.55, 86.55 and 59.68 ppm were assigned to the carbon atoms on the glucose ring at C1, C2, C3, C4, C5 and C6, respectively.32 Compared with native cellulose, characteristic signals emerged on the spectra after transesterification. The signals arising at 171.70 and 10–57 ppm were typical of the carbons of carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), methyl (–CH3) and ethyl groups (–CH2), respectively.33 The peak at 57.29 ppm was assigned to the C6′ carbon bearing a substituted acyl group. Both C6′ and C6 were observed, which indicates partial substitution at C6. The above results further confirm the successful transesterification. A similar phenomenon was also observed in previous literature.34–36 The shift of the peak confirmed that the main reactive site of the hydroxyl group in cellulose is at C-6 positions.
O), methyl (–CH3) and ethyl groups (–CH2), respectively.33 The peak at 57.29 ppm was assigned to the C6′ carbon bearing a substituted acyl group. Both C6′ and C6 were observed, which indicates partial substitution at C6. The above results further confirm the successful transesterification. A similar phenomenon was also observed in previous literature.34–36 The shift of the peak confirmed that the main reactive site of the hydroxyl group in cellulose is at C-6 positions.
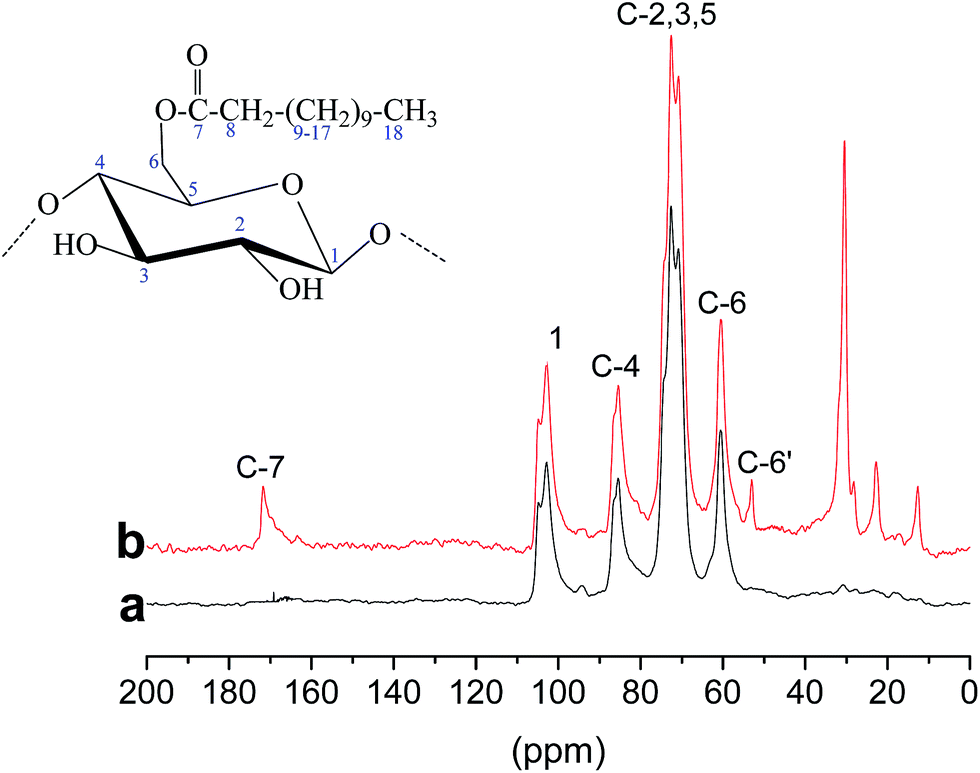 | ||
| Fig. 3 13C NMR spectra: (a) cellulose, (b) E-CNCs (DS 0.140). | ||
3.4. X-ray diffraction
As shown in Fig. 4, the X-ray diffraction of native cellulose shows main diffraction peaks at around 2θ = 14.5, 16.3 and 23.5°, which are in line with the characteristics of cellulose I.37,38 The diffraction patterns of CNCs were similar to native cellulose. The diffraction patterns of E-CNCs do not significantly differ from native cellulose and CNCs, which indicates that the reaction took place mainly at the surface, without affecting the inner crystalline structure of cellulose.39 From Table 1, it is observed that the crystallinity indexes of CNCs and E-CNCs are higher than that of native cellulose. The diffraction peak at 22.5° is more acute, and the relative peak intensity is significantly increased.38 As both catalyst and solvent, using [BMIm]HSO4 is an effective way to selectively remove the amorphous phase of cellulose.4 With the increase of DS, the crystallinity index decreased; this is because the acyl group destroyed the regular arrangement of the cellulose molecules and weakened the hydrogen bond.40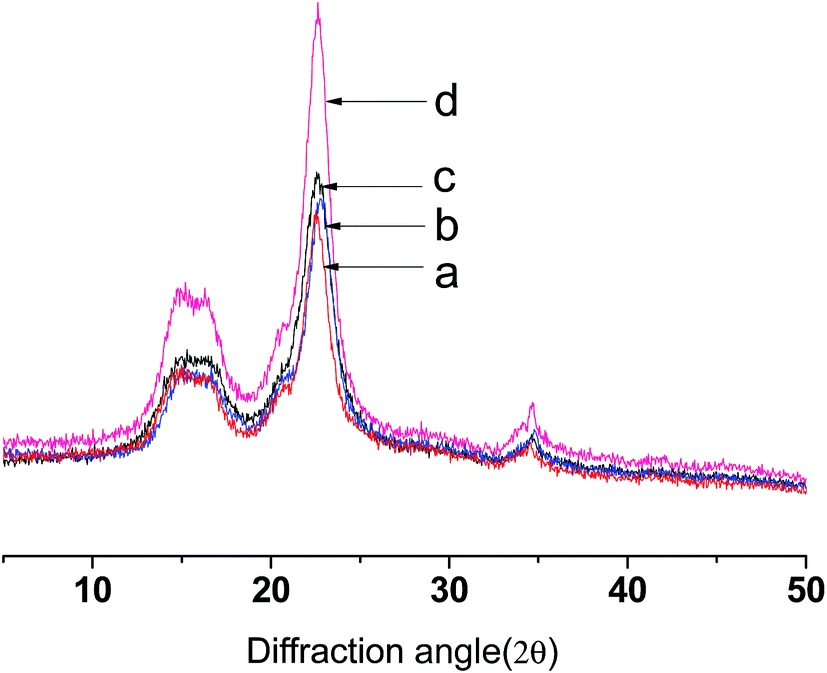 | ||
| Fig. 4 XRD of (a) native cellulose, E-CNCs of (b) DS = 0.125 and (c) DS = 0.065, and (d) CNCs. | ||
| Sample | Crystallinity index | |
|---|---|---|
| a | Native cellulose | 69.70% |
| b | E-CNCs (DS = 0.125) | 75.66% |
| c | E-CNCs (DS = 0.065) | 79.89% |
| d | CNCs | 85.66% |
3.5. Morphology of E-CNCs and CNCs
Fig. 5 shows the TEM images of CNCs and E-CNCs (DS = 0.125). The morphology of CNCs and E-CNCs are all rod-like shapes with the width range of 10–30 nm. Besides, compared with CNCs, E-CNC rod clusters could be observed in some places, while they were separated in others. This is mainly because [BMIm]HSO4 is an effective way to selectively remove the amorphous phase of cellulose, with the native cellulose reduced from micron to nanometer scale. On the other hand, during the reaction, the O–H of cellulose was partial substituted, which reduced the agglomeration, and thus, the E-CNCs could disperse better. | ||
| Fig. 5 TEM of CNCs and E-CNCs of DS = 0.125. | ||
As observed in the image of CNCs, the rods were approximately uniform with the same length. The yield of CNCs was over 78%, which is much higher than the previous studies.26,41,41 The uniform shape was prepared in one step, without dialysis and filtration or other procedures.
3.6. TG analysis
TG and DTG curves of native cellulose, CNCs and E-CNCs are shown in Fig. 6. From the TG curves, native cellulose, CNCs and E-CNCs all have an initial small weight loss at the temperature below 100 °C, which is due to the evaporation of water.42,43 This finding was in good accordance with the characteristic peak of 1640 cm−1 in the FTIR spectra.4 Native cellulose decomposed within a narrow temperature range (280–360 °C), while the decomposition of CNCs and E-CNCs took place within a relatively wider temperature range (155–340 °C). The decomposition temperatures of CNCs and E-CNCs were lower than that of native cellulose. From the DTG curves, the decomposition temperature of CNCs was similar to that of native cellulose. However, there were two peaks in the curves of E-CNCs, which correspond to the decomposition of CNCs and E-CNCs, respectively.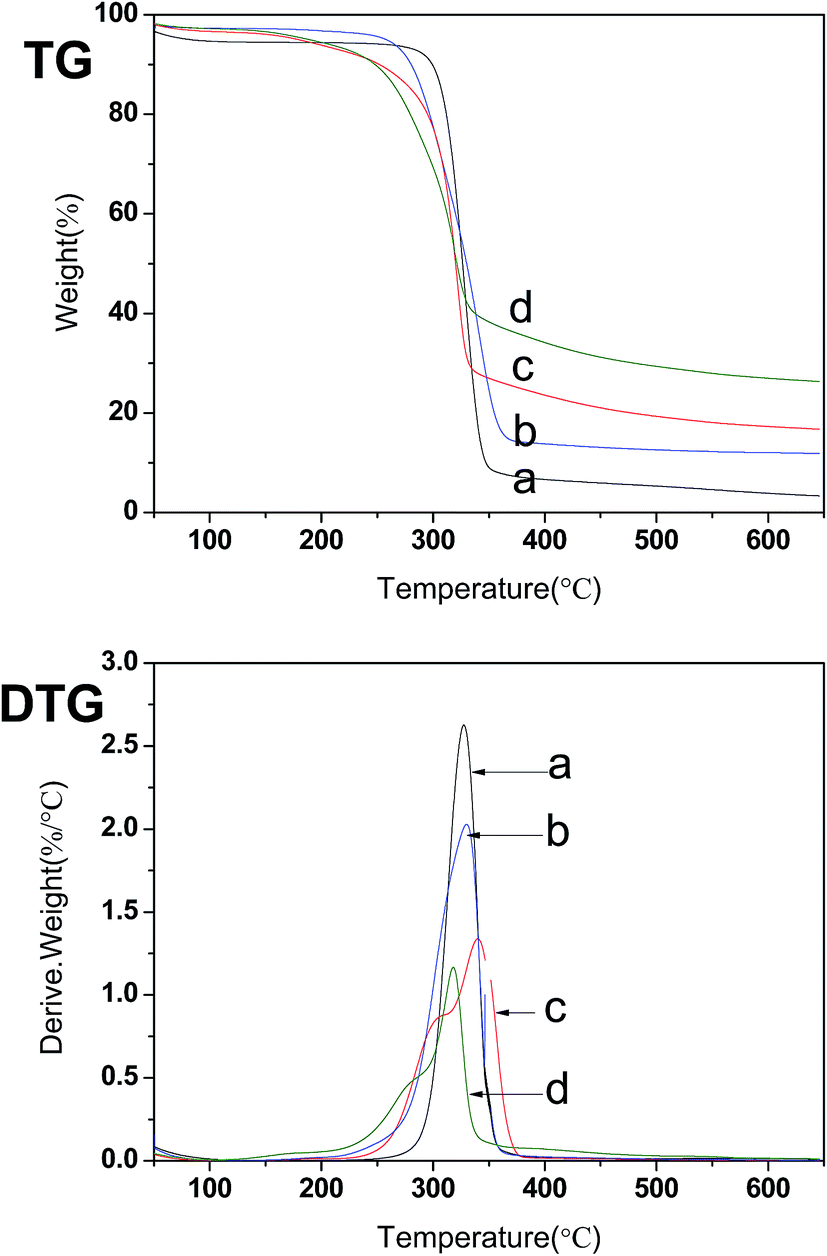 | ||
| Fig. 6 TG and DTG of (a) native cellulose, (b) CNCs, and E-CNCs of (c) DS = 0.065 and (d) DS = 0.125. | ||
A decreasing trend of decomposition temperature suggests that the thermal stability of CNCs and of E-CNCs are lower than that of native cellulose. Reduction in the thermal stability of CNCs and E-CNCs could be due to several reasons. First, the molecular weights of CNCs and E-CNCs are much lower than that of native cellulose.6,44 Second, lowered degradation temperatures of CNCs and E-CNCs could be due to the smaller fiber dimensions as compared to native cellulose, which lead to greater surface area exposed to heat.4,45 With transesterification, the acyl group destroyed the regular arrangement of the cellulose molecules and weakened the hydrogen bonds, thus decreasing the degree of crystallinity, so the thermal stability of E-CNCs was lower than that of the CNCs.40
4 Conclusions
A cost-efficient two-step cascade for the enzymatic preparation of esterified cellulose nanocrystals with long-chain fatty acids (E-CNCs) was established in one pot by using mixed ionic liquids. Highly valued E-CNCs with different DS values can be easily obtained by controlling the reaction conditions and using cheap, bulk native cellulose as raw materials. Compared with the previously reported acid hydrolysis methods, this approach was more efficient and environmentally friendly, producing a yield of E-CNCs higher than 78%. Moreover, this approach greatly simplified the experimental steps, reduced the consumption of chemical regents, allowed the reaction to be carried out under mild conditions and thus reduced the corrosion of the equipment. Compared with native cellulose, the E-CNCs have an improved crystallinity and reduced thermal stability, showing wider application prospects. Furthermore, the biocatalysts are renewable/degradable, and the ILs can be effectively recycled, making this strategy a more promising alternative to traditional chemical approaches.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 31270636, 21676105), Program for New Century Excellent Talents in University (NCET-12-0192) and Self Determined Research Fund of SCUT from the College Basic Research and Operation of MOE (No. 2015ZZ111).Notes and references
- A. Kaboorani and B. Riedl, Ind. Crops Prod., 2015, 65, 45–55 CrossRef CAS.
- Y. Yin, X. Tian, X. Jiang, H. Wang and W. Gao, Carbohydr. Polym., 2016, 142, 206–212 CrossRef CAS PubMed.
- Q. Xu, Y. Gao, M. Qin, K. Wu, Y. Fu and J. Zhao, Int. J. Biol. Macromol., 2013, 60, 241–247 CrossRef CAS PubMed.
- X. Y. Tan, S. B. Abd Hamid and C. W. Lai, Biomass Bioenergy, 2015, 81, 584–591 CrossRef CAS.
- S. E. A. W. Thielemans, Nanoscale, 2014, 6, 7764–7779 RSC.
- Z. Man, N. Muhammad, A. Sarwono, M. A. Bustam, M. Vignesh Kumar and S. Rafiq, J. Polym. Environ., 2011, 19, 726–731 CrossRef CAS.
- B. Jastorff, R. Störmann, J. Ranke, K. Mölter, F. Stock, B. Oberheitmann, W. Hoffmann, J. Hoffmann, M. Nüchter, B. Ondruschka and J. Filser, Green Chem., 2003, 5, 136–142 RSC.
- X. X. Lu, Z. G. Luo, S. J. Yu and X. Fu, J. Agric. Food Chem., 2012, 60, 9273–9279 CrossRef CAS PubMed.
- L. Tang, B. Huang, Q. Lu, S. Wang, W. Ou, W. Lin and X. Chen, Bioresour. Technol., 2013, 127, 100–105 CrossRef CAS PubMed.
- G. Frisoni, M. Baiardo, M. Scandola, D. Lednická, M. C. Cnockaert, J. Mergaert and J. Swings, Biomacromolecules, 2001, 2, 476–482 CrossRef CAS PubMed.
- G. R. G. Rodionova, B. H. B. Hoff, M. L. M. Lenes, O. E. O. Eriksen and O. G. O. Gregersen, Cellulose, 2013, 20, 1167–1174 CrossRef CAS.
- M. Zaman, H. Xiao, F. Chibante and Y. Ni, Carbohydr. Polym., 2012, 89, 163–170 CrossRef CAS PubMed.
- G. Torrelo, U. Hanefeld and F. Hollmann, Catal. Lett., 2015, 145, 309–345 CrossRef CAS.
- S. P. A. R. Kazlauskas, Curr. Opin. Biotechnol., 2003, 14, 432–437 CrossRef.
- C. Wong, Adv. Synth. Catal., 2007, 349, 1287 CrossRef CAS.
- X. Li, Z. Lu, G. Zhao, H. Wu and Y. Yu, Bioresour. Technol., 2012, 114, 6–11 CrossRef CAS PubMed.
- X. F. Li, M. H. Zong and G. L. Zhao, Appl. Microbiol. Biotechnol., 2010, 88, 57–63 CrossRef CAS PubMed.
- A. M. Iribarren and L. E. Iglesias, RSC Adv., 2016, 6, 16358–16386 RSC.
- B. Norjannah, H. C. Ong, H. H. Masjuki, J. C. Juan and W. T. Chong, RSC Adv., 2016, 6, 60034–60055 RSC.
- R. M. L. Fred Van Rantwijk, Trends Biotechnol., 2003, 21, 131–138 CrossRef.
- J. A. Arcos, C. G. Hill and C. Otero, Biotechnol. Bioeng., 2001, 73, 104–110 CrossRef CAS PubMed.
- M. D. Romero, L. Calvo, C. Alba, A. Daneshfar and H. S. Ghaziaskar, Enzyme Microb. Technol., 2005, 37, 42–48 CrossRef CAS.
- M. Ferrer, M. A. Cruces, M. Bernabe, A. Ballesteros and F. J. Plou, Biotechnol. Bioeng., 1999, 65, 10–16 CrossRef CAS PubMed.
- S. Gremos, D. Zarafeta, D. Kekos and F. Kolisis, Bioresour. Technol., 2011, 102, 1378–1382 CrossRef CAS PubMed.
- G. Feng, H. Wu, X. Li, F. Lai, G. Zhao, M. Yang and H. Liu, Biochem. Eng. J., 2015, 95, 56–61 CrossRef CAS.
- J. Ling-ling, C. Xiao-quan and L. Zong-ren, Chem. Bioeng., 2008, 12, 63–66 Search PubMed.
- E. Stojanovi, K. Jeremi, S. Jovanovi and M. D. Lechner, Starch – Stärke, 2005, 57, 79–83 CrossRef.
- C. F. Liu, A. P. Zhang, W. Y. Li, F. X. Yue and R. C. Sun, J. Agric. Food Chem., 2009, 57, 1814–1820 CrossRef CAS PubMed.
- J. Han, C. Zhou, A. D. French, G. Han and Q. Wu, Carbohydr. Polym., 2013, 94, 773–781 CrossRef CAS PubMed.
- C. Li, B. Knierim, C. Manisseri, R. Arora, H. V. Scheller, M. Auer, K. P. Vogel, B. A. Simmons and S. Singh, Bioresour. Technol., 2010, 101, 4900–4906 CrossRef CAS PubMed.
- S. Barthel and T. Heinze, Green Chem., 2006, 8, 301–306 RSC.
- K. Zhang, E. Brendler, A. Geissler and S. Fischer, Polymer, 2011, 52, 26–32 CrossRef CAS.
- S. Berlioz, S. Molina-Boisseau, Y. Nishiyama and L. Heux, Biomacromolecules, 2009, 10, 2144–2151 CrossRef CAS PubMed.
- Y. L. Yang, H. B. Xie and E. H. Liu, Green Chem., 2014, 16, 3018–3023 RSC.
- T. F. L. K. Thomas Heinze, Cellulose, 2003, 10, 283–296 CrossRef.
- M. Fumagalli, D. Ouhab, S. M. Boisseau and L. Heux, Biomacromolecules, 2013, 14, 3246–3255 CrossRef CAS PubMed.
- F. X. E. D. Lihua Liu, Polym. Mater.: Sci. Eng., 2014, 30, 95–99 Search PubMed.
- R. M. Sheltami, I. Abdullah, I. Ahmad, A. Dufresne and H. Kargarzadeh, Carbohydr. Polym., 2012, 88, 772–779 CrossRef CAS.
- P. Tingaut, T. Zimmermann and F. Lopez-Suevos, Biomacromolecules, 2010, 11, 454–464 CrossRef CAS PubMed.
- J. S. A. H. Chanzy, Cellulose, 1995, 2, 111–127 CrossRef.
- M. Daizhong, Enzymolysis Preparation of Pure Nano-Crystalline Cellulose, South China University of Technology, Guangzhoy, China, 2016 Search PubMed.
- Z. Liu, H. Wang, Z. Li, X. Lu, X. Zhang, S. Zhang and K. Zhou, Mater. Chem. Phys., 2011, 128, 220–227 CrossRef CAS.
- M. I. Voronova, A. G. Zakharov, O. Y. Kuznetsov and O. V. Surov, Mater. Lett., 2012, 68, 164–167 CrossRef CAS.
- N. Johar, I. Ahmad and A. Dufresne, Ind. Crops Prod., 2012, 37, 93–99 CrossRef CAS.
- F. Jiang and Y. Hsieh, Carbohydr. Polym., 2013, 95, 32–40 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |