
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient water oxidation with amorphous transition metal boride catalysts synthesized by chemical reduction of metal nitrate salts at room temperature†
Yisu Yang‡
a,
Linzhou Zhuang‡a,
Thomas E. Rufforda,
Shaobin Wangb and
Zhonghua Zhu *a
*a
aSchool of Chemical Engineering, The University of Queensland, Brisbane 4072, Australia. E-mail: z.zhu@uq.edu.au; Fax: +61 733654199; Tel: +61 733653528
bDepartment of Chemical Engineering, Curtin University, Perth, WA 6845, Australia
First published on 28th June 2017
Abstract
We present a variety of amorphous transition-metal borides prepared at room temperature by a chemical reduction method as highly active catalysts for the oxygen evolution reaction (OER). The amorphous borides exhibit activities much higher than the corresponding crystalline (spinel, layered double hydroxide and perovskite) metal oxides containing the identical metal compositions, which have already been regarded as promising OER catalysts. The amorphous Ni/Fe borides showed the best mass normalized OER current density of 50 A g−1 at an overpotential of 0.35 V, transcending the performance of the state-of-the-art OER catalyst, RuO2. Amorphous transition-metal borides demonstrated extremely high active OER catalytic activity. The outstanding catalytic activity can be attributed to the amorphous structure, the large specific surface areas (above 110 m2 g−1) and the electron-enriched transition metal sites stemming from boron doping. The stoichiometry of the catalysts can be controlled precisely even for the synthesis of quaternary metal boride catalysts, which made it feasible to further optimize the catalytic activity. These results indicated that it is facile to prepare highly active OER catalysts by the one-step chemical reduction process at room temperature.
Introduction
The oxygen evolution reaction (OER) has become the focus of research interest and efforts due to its potential application in energy storage. When coupled with a cathodic reaction (e.g. carbon dioxide reduction, hydrogen reduction) it provides a viable route to store intermittent renewable energy such as wind or solar, in the form of a chemical fuel.1–3 However, the sluggish kinetics due to the four-electron transfer result in a high overpotential being required for OER.4 Although RuO2 and IrO2 exhibit outstanding OER activities,5,6 the scarcity and high cost of these noble metal oxides significantly constrain their commercial viability. Therefore, it is greatly desired to develop cost-effective and robust electrocatalysts with remarkable OER activities.Recently, the materials derived from earth-abundant 3d-orbital transition metals7–10 (e.g. Fe, Co, Ni, Mn) has aroused research interest due to their low cost and excellent performance in electrochemical water oxidation, and they efficiently enhance the OER process and reduce the cost of chemical-fuel production. To this day, varieties of crystalline 3d-metal materials including spinels, perovskites and layered double hydroxides (LDH) have been systematically studied as OER catalysts,9,11–14 some of which even show superior activities to the noble metal catalysts. Song et al.11 reported the exfoliated monolayer nanosheet of Ni–Fe LDH as highly electrocatalysts for OER, competing favorably against the costly IrO2 catalyst. Suntivich et al.9 identified that the intrinsic OER activity of Ba0.5Sr0.5Co0.8Fe0.2O3−δ perovskite with an eg-orbital occupancy close to unity is more than one order of magnitude higher than the state-of-the-art IrO2 catalyst. Importantly, May et al.15 found that the activity of Ba0.5Sr0.5Co0.8Fe0.2O3−δ catalyst increased with electrochemical cycling, possibly due to the amorphization of surface layer, as shed new light to the fabrication of amorphous 3d-metal OER catalysts. The latest research demonstrated that amorphous quaternary Ba–Sr–Co–Fe oxide showed higher OER current density to Ba0.5Sr0.5Co0.8Fe0.2O3−δ perovskite.4 In our previous research,8 it is also reported that the amorphous Ni-containing materials exhibit OER activities higher than the state-of-the-art RuO2 catalysts, comparing favorably against their crystalline counterparts. Although amorphous materials being excellent candidate catalysts for OER, to date no efficient route to prepare highly active OER catalysts has been identified.
The latest developed photochemical decomposition method has been successfully applied in fabricating amorphous metal oxides as highly active OER catalysts.3,4,16,17 However, this method required the use of specific organic compounds (metal 2-ethylhexanoate precursors) and deep UV-light irradiation, which made the synthesis process costly and difficult to scale up. Alternatively, a facile route to prepare 3d-metal catalysts is the chemical reduction method with sodium borohydride as reducing agent. Since it is an extremely simple way to synthesize amorphous transition-metal borides with controlled compositions at room temperature, this method is extensively used in the fabrications of catalysts for various energy storage applications (e.g. hydrogen generation from hydrazine decomposition,18 hydrogen evolution reaction,19 oxygen reduction reaction20).
Herein, we put forward the chemical reduction method to construct various amorphous borides based on Fe, Co and Ni, the main compositions for most active OER catalysts reported to date.2,21–23 Three typical crystalline structures with high OER activity were selected as references, namely spinel, LDH and perovskite.9,11,12,14 All the amorphous materials exhibited significantly higher activity towards OER compared to the corresponding crystalline metal oxides. The binary metal catalysts (Ni–Fe and Ni–Co) prepared by chemical reduction method exhibited superior OER activity compared to the state-of-the-art RuO2 catalyst, highlighting the chemical reduction method as a facile, rapid and universal approach to prepare elaborate OER catalysts. Further research into the synthesis of multi-component catalysts using this method showed that the elemental compositions of the products were strictly controlled even for quaternary metal boride catalysts (Ba–Sr–Co–Fe and La–Sr–Co–Fe).
Experimental
Materials
Deionized water was used in all experiments. Sodium borohydride (NaBH4, 98%), nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O, 97%), iron(II) sulfate heptahydrate (FeSO4·7H2O, 98%), cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, 98%), barium nitrate (Ba(NO3)2, 99%), strontium nitrate (Sr(NO3)2, 99%) and triethanolamine (TEA, 98%) were purchased from Aldrich Chemical. Lanthanum(III) nitrate hexahydrate (La(NO3)3·6H2O, 99%) was purchased from Alfa Aesar Chemical. Ethylenediaminetetraacetic acid (EDTA) and potassium hydroxide (KOH, 97%) were purchased from Ajax Finechem. Urea, citric acid monohydrate and ammonia solution (30 wt%) were purchased from Chem-Supply Pty. Ltd. Two commercial catalysts were used in this work: ruthenium(IV) oxide catalyst (RuO2, 99%) from Alfa Aesar Chemical and La0.6Sr0.4Co0.2Fe0.8O3−δ perovskite (labelled here as La6Sr4Co2Fe8-PERO) from Fuelcellmaterials Pty. Ltd.Preparation of crystalline metal oxides
Preparation of amorphous metal borides
Amorphous metal boride catalysts with similar stoichiometric ratios of metals to the spinel, LDH and perovskite metal oxides were prepared by reduction of 50 mL metal nitrate solutions with 0.2 g NaBH4 in 30 mL water. The NaBH4 was added dropwise to the metal nitrate solutions and stirred for 5 minutes or until the production of hydrogen bubbles stopped. The amorphous products were collected by centrifugation, washed three times with water and ethanol, then dried in a vacuum dryer at room temperature.The 50 mL metal nitrate solutions contained: 0.3842 g Co(NO3)2·6H2O and 1.067 g Fe(NO3)3·9H2O for Co1Fe2-AMOR; 0.872 g Co(NO3)2·6H2O and 0.404 g Fe(NO3)3·9H2O for Co3Fe1-AMOR; 0.3839 g Ni(NO3)2·6H2O and 1.067 g Fe(NO3)3·9H2O for Ni1Fe2-AMOR; 0.8724 g Ni(NO3)2·6H2O and 0.404 g Fe(NO3)3·9H2O for Ni3Fe1-AMOR; 0.2614 g Ba(NO3)2, 0.2118 g Sr(NO3)2, 0.4656 g Co(NO3)2·6H2O and 0.1616 g Fe(NO3)3·9H2O for Ba5Sr5Co8Fe2-AMOR; and 0.5196 g La(NO3)2·6H2O, 0.1694 g Sr(NO3)2, 0.1164 g Co(NO3)2·6H2O and 0.6464 g Fe(NO3)3·9H2O for La6Sr4Co2Fe8-AMOR.
Characterization
Elemental compositions of metals and boron were measured with a Perkin Elmer Optima 8300DV Inductively-Coupled Plasma Optical Emission Spectrometer (ICP-OES). X-ray diffraction (XRD) patterns were recorded on a Bruker D8-Advanced X-ray diffractometer using nickel-filtered Cu-Kα radiation. Scanning electron microscopy (SEM) images were collected on a JEOL JSM-7001F at an acceleration voltage of 10 kV. The energy dispersive X-ray spectroscopy (EDS) was recorded on a Tecnai F20 transmission electron microscope. X-ray photoelectron spectra were acquired on a Kratos Axis ULTRA X-ray photoelectron spectrometer (XPS) incorporating a 165 mm hemispherical electron energy analyzer and a monochromatic Al Kα (1486.6 eV) radiation at 150 W (15 kV, 10 mA). The binding energies were determined using the C 1s line at 284.5 eV from adventitious carbon as a reference. N2 sorption isotherms were measured at 77 K on a Micromeritics TriStar II 3020 after degassing the samples for 12 h at 80 °C. Specific surface areas (SBET) were determined from the N2 isotherms with the Brunauer Emmett Teller method.Electrochemical measurements of thin film electrodes
Thin film electrodes were fabricated by sonication of 10 mg active catalyst and 10 mg carbon black (Super C65, TIMCAL C'NERGY) for 30 minutes in 1 mL ethanol with 100 μL 5 wt%. Nafion solution. A 5 μL aliquot of this suspension was drop-cast onto a glassy carbon disk electrode (4 mm diameter, 0.126 cm2 area) and left to dry under a glass jar. The typical catalyst loading was 0.36 mg cm−2.Rotating disk electrode (RDE) voltammograms were measured in an O2 (99.999%) saturated 0.1 M KOH solution at room temperature with a Biologic VMP2/Z multichannel potentiostat. The electrochemical cell had a Pt-chain counter electrode and a Ag|AgCl (3 M NaCl) reference electrode. The reference electrode was calibrated with respect to a reversible hydrogen electrode (RHE) in a hydrogen saturated electrolyte with a Pt-chain working electrode. Cyclic voltammograms were measured at a scan rate of 10 mV s−1 and the thermodynamic potential for the hydrogen electrode reactions was determined from the mean of the two potentials at which the current crossed zero.
All potentials reported in this study are iR-corrected potentials calculated by the following equation (eqn (1)):
| ERHE(iR-corrected) = EAg|AgCl + 0.937 − iR | (1) |
Results and discussions
The molar ratios of the elements determined by ICP-OES and the specific surface areas determined by N2 adsorption of the samples are listed in Table 1. Obviously, the amorphous samples exhibited almost the same relative metal compositions as the corresponding crystalline metal oxides. The slight differentials between amorphous and crystalline samples indicated the metal compositions of the amorphous samples were strictly controlled by using the chemical reduction method. More importantly, all the metal elements were homogeneously distributed in the catalysts as confirmed by the EDS mapping (see Fig. S1–S3†) spectra. During the reduction process, borohydride undergoes hydrolyzation according to the reaction below (eqn (2)–(6)),25| NaBH4 + 2H2O → 4H2↑ + NaBO2 | (2) |
| Co2+ + 2Fe3+ + 5OH− → CoFe2O4 + H2O + 3H+ | (3) |
| Co2+ + 2BH4− → CoB2 + 2H2 | (4) |
| Fe3+ + 3BH4− → FeB3 + 6H2 | (5) |
CoFe2O4 + xBH4− → CoFe2BxO4−x + xOH− + ![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) xH2 xH2
| (6) |
| Sample | Atomic ratio of element (in total elements at%) | Specific surface area (m2 g−1) | ||||||
|---|---|---|---|---|---|---|---|---|
| Co | Ni | Fe | Ba | Sr | La | B | ||
| Co1Fe2-SPIN | 32.7 | 67.3 | 33.8 | |||||
| Co1Fe2-AMOR | 23.1 | 46.7 | 30.2 | 232.0 | ||||
| Co3Fe1-LDH | 75.2 | 24.8 | 49 | |||||
| Co3Fe1-AMOR | 54.1 | 18.5 | 27.4 | 139.6 | ||||
| Ni1Fe2-SPIN | 33.6 | 66.4 | 45.2 | |||||
| Ni1Fe2-AMOR | 19.9 | 40.0 | 40.1 | 244.2 | ||||
| Ni3Fe1-LDH | 74.8 | 25.2 | 124.6 | |||||
| Ni3Fe1-AMOR | 45.3 | 16.2 | 38.5 | 110.2 | ||||
| Ba5Sr5Co8Fe2-PERO | 40.1 | 10.0 | 25.2 | 24.7 | 0.5 | |||
| Ba5Sr5Co8Fe2-AMOR | 13.1 | 3.3 | 8.0 | 8.3 | 67.3 | 20.5 | ||
| La6Sr4Co2Fe8-PERO | 10.0 | 40.2 | 19.8 | 30.0 | 6.8 | |||
| La6Sr4Co2Fe8-AMOR | 4.2 | 16.7 | 6.6 | 10.2 | 62.3 | 144.8 | ||
Interestingly, the quaternary samples showed a boron content (atomic ratio ∼ 65%) much higher than that of the binary samples (∼27–38%). Shih et al.26 found that the alkali earth metals (especially barium) are the most efficient precipitants to chemisorb boron species in the aqueous solution in the form of metal perborate (MeB2O4, B content 66%). On the other hand, alkali earth metals hexaborides (MeB6, B content 85.7%) could also be formed,27 leading to a high B level in the final products. Meanwhile, the amorphous transition metal (Fe, Co, Ni) borides can be formed in a wide compositional (relatively low) range19,28,29 due to the formation of a variety of polymorphs (MeB, Me2B, Me3B, Me4B3, etc., maximum B content 50% corresponding to MeB). As a result, the quaternary samples containing alkali earth metals (Ba, Sr) showed a much higher boron content.
It can be also seen from Table 1 that all the amorphous samples except Ni3Fe1-AMOR exhibited much larger specific surface areas than the corresponding crystalline metal oxides. Usually, the crystallization of a stable crystalline structure occurs at a high temperature. For example, the synthesis of Ba5Sr5Co8Fe2-PERO and La6Sr4Co2Fe8-PERO required a calcination step at 900 °C and 1250 °C respectively.13,30 However, the high synthesis temperature required for the perovskites leads to the sintering of the samples, resulting in a relatively low specific surface area (Ba5Sr5Co8Fe2-PERO: 0.5 m2 g−1, La6Sr4Co2Fe8-PERO: 6.0 m2 g−1) and a restrained catalytic performance. In contrast, the amorphous samples were prepared at room temperature with no sintering step, showing much larger specific surface areas (Ba5Sr5Co8Fe2-AMOR: 20.5 m2 g−1, La6Sr4Co2Fe8-AMOR: 144.8 m2 g−1). Among all the amorphous samples, Ni1Fe2-AMOR has the largest specific surface area of 244.2 m2 g−1. Due to the layered structure, Ni3Fe1-LDH is the only crystalline sample with a specific surface area larger than 100 m2 g−1 (124.6 m2 g−1). The corresponding amorphous sample Ni3Fe1-AMOR exhibited a comparable specific surface area of 110.2 m2 g−1, which is only slightly lower but still at the same level with Ni3Fe1-LDH. Notably, considering no surfactants or structure directing agents were used and the amorphous samples were prepared at room temperature, these results highlighted the chemical reduction method as a facile and efficient approach to fabricate amorphous materials with homogeneous elemental distributions and high specific surface areas.
The XRD patterns of the amorphous borides and the corresponding metal oxides were shown in Fig. 1. It can be seen that all the materials prepared by chemical reduction method exhibited amorphous structures. The total number of six metals (Fe, Co, Ni, Ba, Sr, La) involved in the synthesis highlighted the chemical reduction method as a facile and effective approach to construct amorphous metal-based materials. It is reported recently that the amorphous materials containing Ni, Fe and Co prepared by various methods (e.g. electrochemical deposition,22 photochemical decomposition3 and vapor induced precipitation8) exhibited superior OER activities to the noble metal oxides catalysts such as IrO2 and RuO2. Since Fe, Co and Ni, three most active transition metals, were all included in the synthesis, the fabrication of highly-active OER catalyst can be expected using the chemical reduction method. The other samples showed typical XRD patterns corresponding to crystalline materials, namely spinel, LDH and perovskite, which were reported to show promising activity towards water oxidation.
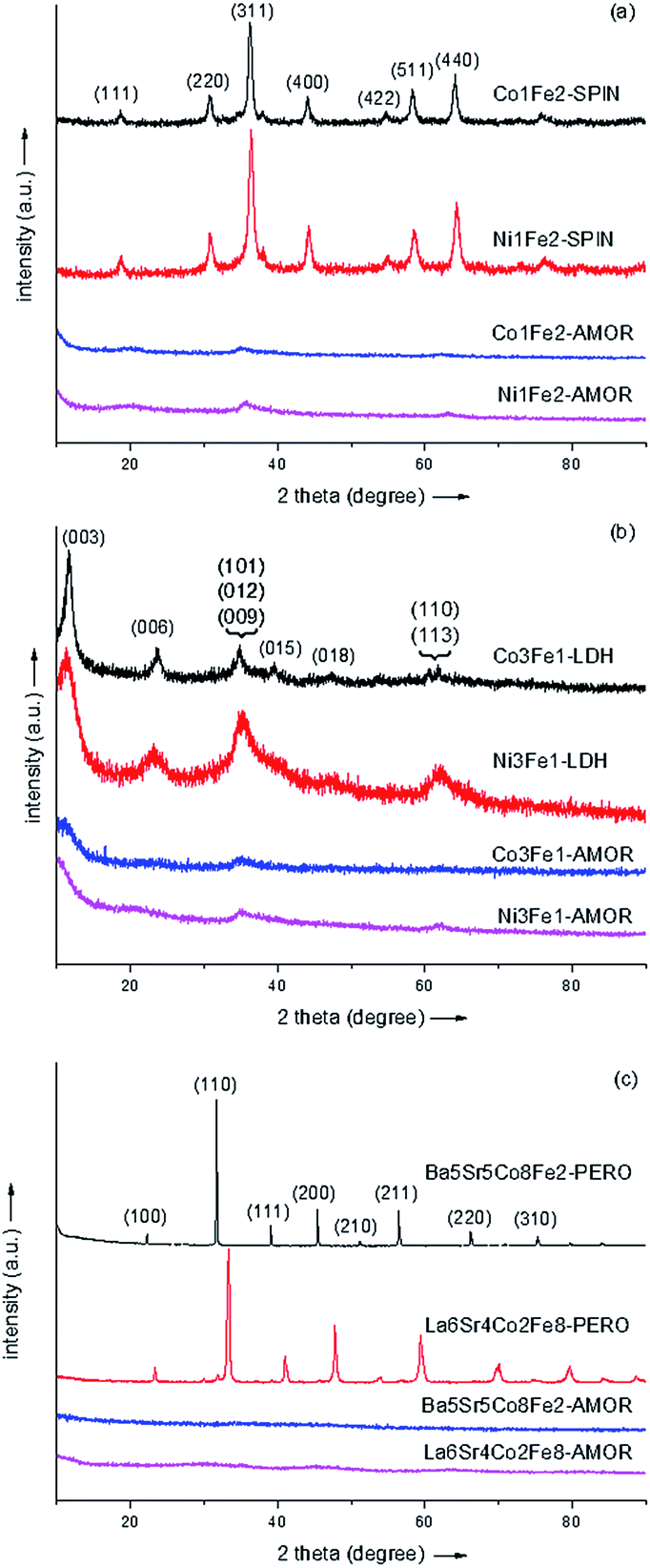 | ||
| Fig. 1 XRD patterns of the transition-metal materials with crystalline structures and the corresponding amorphous metal borides: spinels (a), layered double hydroxides (b) and perovskites (c). | ||
The SEM images of the amorphous catalysts are shown in Fig. 2 and the SEM images of the crystalline catalysts are included in Fig. S4 of the ESI.† The images in Fig. 2a and b showed that the Co3Fe1-AMOR and Ni3Fe1-AMOR precipitated from solutions with the lower Fe concentrations were constituted by nanoparticles. These nanoparticles were closely accumulated, leading to a porous structure (Fig. S1 and S2†) consistent with the surface areas of 139.6 m2 g−1 and 110.2 m2 g−1 determined from N2 sorption analyses. With the increase of Fe contents, Co1Fe2-AMOR (Fig. 2c) and Ni1Fe2-AMOR (Fig. 2d) exhibited nano-sheet like structures (Fig. 2c and d). These nano-sheets were loosely stacked to form sponge-like structures, leading to even higher specific surface areas (232.0 and 244.2 m2 g−1 respectively). The texture of the amorphous samples with higher Fe content looks more like iron boride nanoparticles sponge morphologies that have been reported.31 The quaternary metal borides catalysts (Fig. 2e and f) were also precipitated as nanoparticle aggregates with similar morphologies to the Co:Fe and Ni:Fe amorphous metal borides; these morphologies are very different to the non-porous morphologies of the corresponding crystalline perovskites Ba5Sr5Co8Fe2-PERO and La6Sr4Co2Fe8-PERO shown (Fig. S4e and f†). The SEM results are consistent with the specific surface area data shown in Table 1. All the amorphous samples were enriched by pore systems, showing large specific surface areas (see Table 1). Although Ni1Fe2-SPIN and Co1Fe2-SPIN (see Fig. S4a and b†) with spinel phase showed morphologies of nanoparticles similar to Ni1Fe2-AMOR and Co1Fe2-AMOR, the specific surface areas (see Table 1) of these two samples were much lower compared with the amorphous samples, indicating the agglomeration of the nanoparticles of the crystalline spinels during the calcination process. Similarly, due to sintering at high temperature, the perovskite catalysts (Ba5Sr5Co8Fe2-PERO, La6Sr4Co2Fe8-PERO) showed a non-porous bulk phase (Fig. S4e and f†), while the corresponding amorphous borides exhibited highly porous structures (Fig. 2e and f). Ni3Fe1-LDH and Co3Fe1-LDH exhibited layered structure (see Fig. S4e and f†), which is typical for layered double hydroxides, showing the largest specific surface areas among the crystalline samples. The obtained SEM results corresponded well with the textural properties shown in Table 1, again demonstrating that porous materials with high specific surface areas can be achieved by using chemical reduction method.
 | ||
| Fig. 2 SEM images of the amorphous borides catalysts: Co3Fe1-AMOR (a), Ni3Fe1-AMOR (b), Co1Fe2-AMOR (c), Ni1Fe2-AMOR (d), Ba5Sr5Co8Fe2-AMOR (e) and La6Sr4Co2Fe8-AMOR (f). | ||
The XPS spectra of the amorphous samples were displayed in Fig. 3. It can be seen from Fig. 3a that in the Co 2p level, Co3Fe1-AMOR showed a peak at 779.4 eV, as can be attributed to the Co(II) species. It is reported that metallic Co can be identified at ca. 778.2 eV for Co–B catalysts. In this study, no elemental Co peak was observed, indicating that the catalyst was severely oxidized when exposed in the air. Notably, compared with the binding energy of Co(II)O (779.7 eV), the slight negative shift of 0.3 eV of the Co(II) in the catalyst suggested an electron-enriched Co structure, which is further evidenced by the B 1s scan. The B 1s signal at 189.0 eV was ascribed to elemental B in the amorphous borides, which was positively shifted by 1.9 eV compared with pure B. This suggested an electron transfer from alloying B to vacant d-orbital of Co, leading to electron-enriched Co sites. These electron-enriched Co sites were identified as highly active for various catalytic reactions, including hydrogenation,32,33 hydrogen evolution19 and hydrolysis reactions.34,35 The Fe 2p band showed a peak at 710.6 eV, corresponding to Fe(III) species. Since there was considerable amount of B (27.3 at%) in this sample, this peak was also affected by the electron transfer from B, and showed a negative shift of 0.2 eV compared with pure Fe2O3. Similar negative shift of 0.2 eV for Ni was also observed for the sample Ni3Fe1-AMOR. Since Fe 2p signal overlaps strongly with Ni LMM Auger peaks, the Fe 2p level of Ni3Fe1-AMOR was not shown here. These results suggested that in the amorphous transition metal borides catalysts, the transition metals were enriched by electrons due to the electron transfer from boron. Similar electron transfer was also reported by several other groups.19
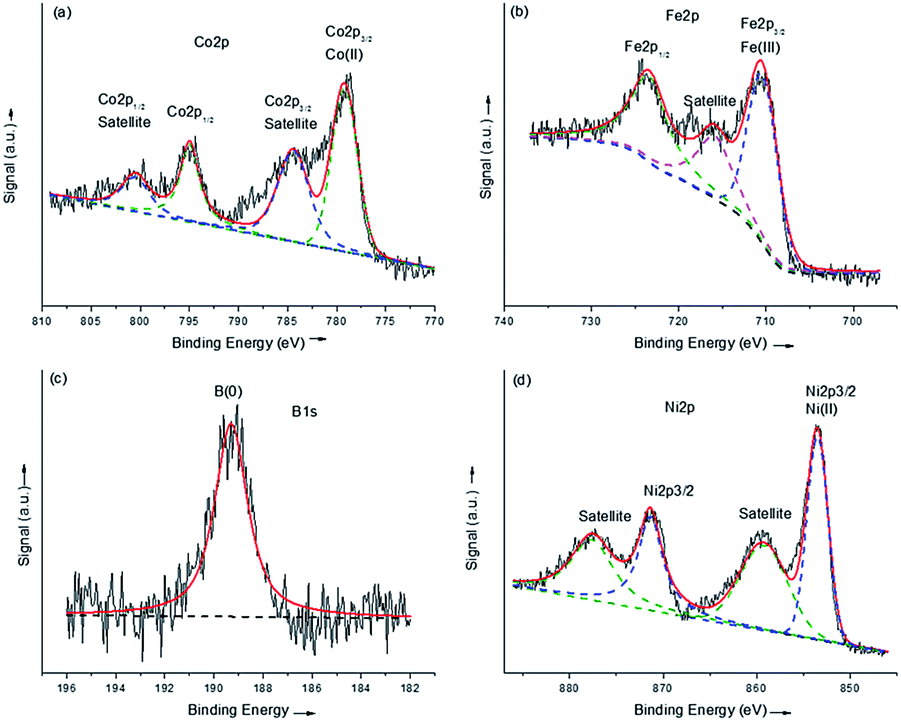 | ||
| Fig. 3 XPS spectra of the amorphous samples synthesized via chemical reduction method: Co 2p (a), Fe 2p (b) and B 1s (c) region of Co3Fe1-AMOR and Ni 2p level of Ni3Fe1-AMOR. | ||
For the amorphous quaternary sample Ba5Sr5Co8Fe2-AMOR, a considerable amount of SrCO3 can be identified. In the Sr 3d level two distinct peaks were observed for the amorphous samples, suggesting the existence of SrCO3 species (see Fig. S5a†). Correspondingly, the O 1s region of the amorphous catalysts exhibited a peak at 532.5 eV which is typical for metal carbonates (see Fig. S5b†). Since the alkaline earth metals are highly active elements, they will undergo hydrolyzation during the reduction process to form hydroxyl groups which will then react with the CO2 in air, leading to the formation of metal carbonates. In this context, BaCO3 would also exist in the amorphous catalyst. However, due to the strong overlap of BaO (780.0 eV) and BaCO3 (780.1 eV), neither of them can be clearly identified in the Ba 3d region shown in Fig. S5c.† The existence of SrCO3 for the other quaternary sample La6Sr4Co2Fe8-AMOR is also confirmed from its Sr region (see Fig. S5d†). The metal carbonates were also observed by Zhang et al. while preparing amorphous Ba–Sr–Co–Fe metal oxides catalysts using the photochemical degradation method, indicating that it is difficult to avoid the formation of metal carbonates in the amorphous catalysts.
The outstanding OER catalytic performances of amorphous borides catalysts were justified by the activities of the binary samples in this study, which were displayed in Fig. 4a–d. The activity of the state-of-the-art RuO2 catalyst was also shown as reference (red curve). It can be seen from Fig. 4a that the two amorphous Co–Fe catalyst exhibited much higher OER activities than the samples with spinel and LDH structure. This result was in accordance with our previous finding8 that transition-metal catalyst with amorphous structure can compare favorably against the corresponding crystalline metal oxides for room-temperature water splitting. It should be emphasized that these amorphous transition-metal materials exhibited OER activities superior to that of the state-of-the-art RuO2 catalyst,3,8 which can be seen from Fig. 4a. Interestingly, although the elemental compositions of Co3Fe1-AMOR and Co1Fe2-AMOR varies, they exhibited quite similar Tafel slopes of 86 and 84 mV dec−1, respectively, indicating the same active phase evolved under OER condition for these two samples. This is in accordance with the finding reported by Burke et al.36 that a highly active FexCo1−xOOH phase with moderate high Fe level (0.33 < x < 0.79) emerged under OER condition. Masa et al.23 found the strain exerted on the crystal lattice of cobalt by boron directly arises from their chemical interaction involving electron transfer and hybridization of B 2p states with the metal d orbitals. The lattice strain weakens the Co–Co bonds, diminishing the energy barrier for formation of the OOH intermediate during OER. Furthermore, the electron-enriched transition-metal sites as confirmed by XPS results might also contribute to improve the OER activities of the samples. As a result, the sample Co1Fe2-AMOR with a larger specific surface area of 232 m2 g−1 (Co3Fe1-AMOR: 139.6 m2 g−1) exhibited a better catalytic performance as shown in Fig. 4a.
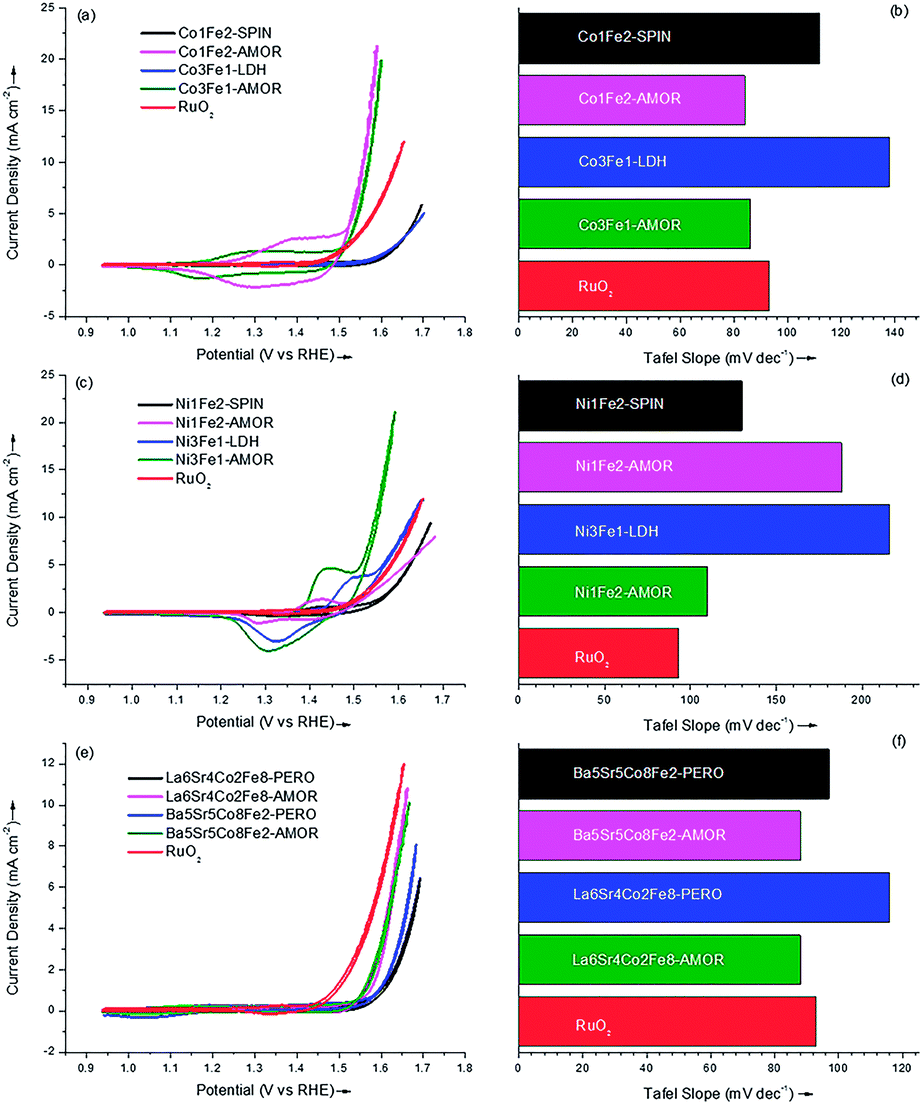 | ||
| Fig. 4 OER activities and corresponding Tafel slopes of the amorphous samples and the corresponding crystalline metal oxides: activities (a) and Tafel slopes (b) of Co–Fe binary catalysts; activities (c) and Tafel slopes (d) of Ni–Fe binary catalysts; activities (e) and Tafel slopes (f) of quaternary catalysts. | ||
The OER current densities of the amorphous Ni–Fe catalysts and the corresponding crystalline metal oxides were displayed in Fig. 4c and d. Again, the amorphous samples showed better activities toward OER, with the sample Ni3Fe1-AMOR giving the best catalytic performance, transcending that of RuO2 catalyst. Different from the Co–Fe catalysts, the Tafel slopes of the amorphous Ni–Fe samples were significantly affected by their elemental compositions. Ni3Fe1-AMOR exhibited the lowest Tafel slope of 110 mV dec−1, while Ni1Fe2-AMOR showed a relatively high value of 188 mV dec−1, suggesting the active sites changed with the increasing Fe content in the catalysts. This is different from Co-based AMOR. In a Co oxide compound, valence transition between Co2+ and Co3+ is easy and the redox potential of Co3+/Co2+ is lower than that of Ni2+/Ni0. Thus, the Tafel slopes is much less dependent on Co loading and elemental composition. Friebel et al.37 found the Ni–Fe catalysts undergo oxidation to form γ-Ni1−xFexOOH phase, where Fe3+ shows a significantly lower overpotential than do Ni3+ in either γ-Ni1−xFexOOH or γ-NiOOH, being identified as highly active Fe sites for OER. However, as the Fe level rises above 25 at%, a γ-FeOOH phase containing either no or <3 at% Ni nucleates, which is quite inefficient for water oxidation. Therefore, it is reasonable that the amorphous sample Ni3Fe1-AMOR with a Fe level of 25 at% showing the lowest Tafel slope while Ni1Fe2-AMOR exhibiting increased Tafel slope and decreased OER activity due to the formation of γ-FeOOH phase.
Fig. 4e and f show the catalytic performance of the quaternary samples. Although the amorphous Ba5Sr5Co8Fe2-AMOR and La6Sr4Co2Fe8-AMOR samples did not reach the level of RuO2 catalyst, they still showed higher activities than those of the corresponding crystalline metal oxides. Considering that these two samples exhibited Tafel slopes (88 mV dec−1) which were similar to the amorphous Co–Fe binary catalysts, it can be speculated that the active phase was composed of Co and Fe. Since the quaternary samples contained large amounts of B and metal carbonates as confirmed by ICP-OES and XPS test, the overall catalytic activity can be reduced due to doping of these non-active compositions. It should be emphasized recent studies show that multi-component metal materials served as promising OER catalysts. Smith et al.3 studied the amorphous metal oxide catalysts fabricated by photochemical degradation method for OER, and found the combination of 33% Fe, 33% Co and 33% Ni showed the best catalytic performance. Gerken et al.38 found that Ni–Fe oxides containing a third metal (e.g. Al, Ga, Cr) emerged as highly effective OER catalysts. The latest high-throughput screening revealed that Ce doping may further increase the OER activity of the catalysts composed of Fe, Co and Ni.39,40 On the other hand, very recently, the amorphous cobalt boride prepared by chemical reduction method has been identified as promising OER catalysts which is superior to RuO2 and IrO2.23 This study focused on finding a facile route to fabricate and screen multi-component materials. Although the quaternary samples were less active compared with the binary ones, the homogeneous and strictly controlled elemental distribution of the quaternary samples highlighted the unique benefit of chemical reduction method in preparing elaborate OER catalysts affords.
The catalytic parameters of all the samples were summarized in Table 2. Importantly, all the amorphous samples prepared via chemical reduction method compared favorably against the corresponding crystalline metal oxides, namely spinel, LDH and perovskite, all of which are promising structures for OER reported to date. This demonstrates chemical reduction method can be a simple and efficient approach to fabricate highly active OER catalysts. The sample Ni3Fe1-AMOR showed the best catalytic performance with an OER current density of 50.9 A g−1 at an overpotential of 0.35 V. Co3Fe1-AMOR (34.9 A g−1) and Co1Fe2-AMOR (47.1 A g−1) also exhibited outstanding OER activities which transcended that of the noble metal RuO2 (14.2 A g−1) catalyst. Fig. S6† showed 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s stability tests performed on the two most active samples Co1Fe2-AMOR and Ni3Fe1-AMOR under harsh anodic conditions (0.7 V vs. Ag|AgCl in 0.1 M KOH solution), which indicated excellent stabilities of transition metal borides as OER catalysts. It is worthy to notify that the elemental compositions of the amorphous samples were strictly controlled as identical as those of the corresponding crystalline metal oxides, however, in practical application the compositions can be controlled arbitrarily and further optimized to improve their OER performance. In this study, we have demonstrated chemical reduction method as a simple and efficient approach to prepare highly active amorphous transition metal borides catalysts for OER with strictly controlled compositions. Therefore, it can be expected that new combinations of transition metal borides for electro water oxidation can be discovered via chemical reduction method.
000 s stability tests performed on the two most active samples Co1Fe2-AMOR and Ni3Fe1-AMOR under harsh anodic conditions (0.7 V vs. Ag|AgCl in 0.1 M KOH solution), which indicated excellent stabilities of transition metal borides as OER catalysts. It is worthy to notify that the elemental compositions of the amorphous samples were strictly controlled as identical as those of the corresponding crystalline metal oxides, however, in practical application the compositions can be controlled arbitrarily and further optimized to improve their OER performance. In this study, we have demonstrated chemical reduction method as a simple and efficient approach to prepare highly active amorphous transition metal borides catalysts for OER with strictly controlled compositions. Therefore, it can be expected that new combinations of transition metal borides for electro water oxidation can be discovered via chemical reduction method.
| Sample | Mass activity (A g−1) | Tafel slope (mV dec−1) |
|---|---|---|
| Co1Fe2-SPIN | 1.5 | 112 |
| Co1Fe2-AMOR | 47.1 | 84 |
| Co3Fe1-LDH | 1.9 | 138 |
| Co3Fe1-AMOR | 34.9 | 86 |
| Ni1Fe2-SPIN | 6.5 | 130 |
| Ni1Fe2-AMOR | 10.1 | 188 |
| Ni3Fe1-LDH | 16.9 | 216 |
| Ni3Fe1-AMOR | 50.9 | 110 |
| Ba5Sr5Co8Fe2-PERO | 2.2 | 97 |
| Ba5Sr5Co8Fe2-AMOR | 4.7 | 88 |
| La6Sr4Co2Fe8-PERO | 1.9 | 116 |
| La6Sr4Co2Fe8-AMOR | 3.7 | 88 |
| RuO2 | 14.2 | 93 |
Conclusions
In conclusion, we demonstrated chemical reduction method as a facile and efficient approach to prepare amorphous transition-metal borides materials with strictly controlled elemental compositions for efficient water oxidation. The amorphous borides catalysts are featured by their large specific surface areas and electron-enriched transition-metal sites, and compared favorably against the corresponding crystalline metal oxides, namely spinel, LDH and perovskite, which have been reported as promising structures for OER. More importantly, three amorphous samples, Co1Fe2-AMOR, Co3Fe1-AMOR and Ni3Fe1-AMOR exhibited outstanding OER activities, which transcended that of the state-of-the-art RuO2 catalysts. This indicates that efficient OER catalysts can be developed by using chemical reduction method. The homogeneous and strictly controlled elemental distributions of the amorphous borides were justified by further research into the synthesis of quaternary samples (Ba–Sr–Co–Fe and La–Sr–Co–Fe). Since the elemental compositions of the amorphous borides can be tuned arbitrarily during the synthesis process, the catalytic performance can be further optimized and the discovery of new combinations of transition-metal borides as efficient OER catalysts can be expected.Acknowledgements
This research was funded by an Australian Research Council Discovery Project (DP130102151). Yang acknowledges financial support from a University of Queensland (UQ) International Postgraduate Scholarships. The authors acknowledge the facilities, scientific and technical assistance of the Australian Microscopy and Microanalysis Research Facility at the UQ Centre for Microscopy and Microanalysis.Notes and references
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. X. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2011, 111, 5815 CrossRef CAS.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- R. D. L. Smith, M. S. Prevot, R. D. Fagan, Z. P. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS PubMed.
- C. J. Zhang, C. P. Berlinguette and S. Trudel, Chem. Commun., 2016, 52, 1513–1516 RSC.
- E. Tsuji, A. Imanishi, K. Fukui and Y. Nakato, Electrochim. Acta, 2011, 56, 2009–2016 CrossRef CAS.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- M. R. Gao, W. C. Sheng, Z. B. Zhuang, Q. R. Fang, S. Gu, J. Jiang and Y. S. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed.
- Y. S. Yang, F. L. Liang, M. R. Li, T. E. Rufford, W. Zhou and Z. H. Zhu, Chemsuschem, 2015, 8, 2193–2197 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- Y. T. Meng, W. Q. Song, H. Huang, Z. Ren, S. Y. Chen and S. L. Suib, J. Am. Chem. Soc., 2014, 136, 11452–11464 CrossRef CAS PubMed.
- F. Song and X. L. Hu, Nat. Commun., 2014, 5, 4477 CAS.
- J. Y. C. Chen, J. T. Miller, J. B. Gerken and S. S. Stahl, Energy Environ. Sci., 2014, 7, 1382–1386 CAS.
- Y. S. Yang, W. Zhou, R. C. Liu, M. R. Li, T. E. Rufford and Z. H. Zhu, ChemElectroChem, 2015, 2, 200–203 CrossRef CAS.
- F. Y. Cheng, J. A. Shen, B. Peng, Y. D. Pan, Z. L. Tao and J. Chen, Nat. Chem., 2011, 3, 79–84 CrossRef CAS PubMed.
- K. J. May, C. E. Carlton, K. A. Stoerzinger, M. Risch, J. Suntivich, Y. L. Lee, A. Grimaud and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 3264–3270 CrossRef CAS.
- R. D. L. Smith, M. S. Prevot, R. D. Fagan, S. Trudel and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 11580–11586 CrossRef CAS PubMed.
- C. J. Zhang, R. D. Fagan, R. D. L. Smith, S. A. Moore, C. P. Berlinguette and S. Trudel, J. Mater. Chem. A, 2015, 3, 756–761 CAS.
- D. G. Tong, W. Chu, P. Wu, G. F. Gu and L. Zhang, J. Mater. Chem. A, 2013, 1, 358–366 CAS.
- S. Gupta, N. Patel, A. Miotello and D. C. Kothari, J. Power Sources, 2015, 279, 620–625 CrossRef CAS.
- D. P. He, L. B. Zhang, D. S. He, G. Zhou, Y. Lin, Z. X. Deng, X. Hong, Y. Wu, C. Chen and Y. D. Li, Nat. Commun., 2016, 7, 12362 CrossRef CAS PubMed.
- M. Gong and H. J. Dai, Nano Res., 2015, 8, 23–39 CrossRef CAS.
- X. Y. Lu and C. A. Zhao, Nat. Commun., 2015, 6, 6616 CrossRef CAS PubMed.
- J. Masa, P. Weide, D. Peeters, I. Sinev, W. Xia, Z. Y. Sun, C. Somsen, M. Muhler and W. Schuhmann, Adv. Energy Mater., 2016, 6, 1502313 CrossRef.
- L. X. Wang, J. C. Li, Y. Q. Wang, L. J. Zhao and Q. Jiang, Chem. Eng. J., 2012, 181, 72–79 CrossRef.
- J. D. Ocon, T. N. Tuan, Y. M. Yi, R. L. de Leon, J. K. Lee and J. Lee, J. Power Sources, 2013, 243, 444–450 CrossRef CAS.
- Y. J. Shih, C. H. Liu, W. C. Lan and Y. H. Huang, Chemosphere, 2014, 111, 232–237 CrossRef CAS PubMed.
- S. L. Shang, Y. Wang and Z. K. Liu, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 024302 CrossRef.
- O. Ozdemir, M. Usta, C. Bindal and A. H. Ucisik, Vacuum, 2006, 80, 1391–1395 CrossRef CAS.
- S. N. Ishmaev, S. L. Isakov, I. P. Sadikov, E. Svab, L. Koszegi, A. Lovas and G. Meszaros, J. Non-Cryst. Solids, 1987, 94, 11–21 CrossRef CAS.
- Y. S. Chou, J. W. Stevenson, T. R. Armstrong and L. R. Pederson, J. Am. Ceram. Soc., 2000, 83, 1457–1464 CrossRef CAS.
- S. Rades, S. Kraemer, R. Seshadri and B. Albert, Chem. Mater., 2014, 26, 1549–1552 CrossRef CAS.
- H. X. Li, X. F. Chen, M. H. Wang and Y. P. Xu, Appl. Catal., A, 2002, 225, 117–130 CrossRef CAS.
- H. X. Li, Y. D. Wu, H. S. Luo, M. G. Wang and Y. P. Xu, J. Catal., 2003, 214, 15–25 CrossRef CAS.
- N. Patel, A. Miotello and V. Bello, Appl. Catal., B, 2011, 103, 31–38 CrossRef CAS.
- N. Patel, R. Fernandes, G. Guella, A. Kale, A. Miotello, B. Patton and C. Zanchetta, J. Phys. Chem. C, 2008, 112, 6968–6976 CAS.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M. J. Cheng, D. Sokaras, T. C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Norskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- J. B. Gerken, S. E. Shaner, R. C. Masse, N. J. Porubsky and S. S. Stahl, Energy Environ. Sci., 2014, 7, 2376–2382 CAS.
- J. A. Haber, D. Guevarra, S. H. Jung, J. Jin and J. M. Gregoire, ChemElectroChem, 2014, 1, 1613–1617 CrossRef CAS.
- J. A. Haber, C. X. Xiang, D. Guevarra, S. H. Jung, J. Jin and J. M. Gregoire, ChemElectroChem, 2014, 1, 524–528 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02558k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |