DOI:
10.1039/C7RA02547E
(Paper)
RSC Adv., 2017,
7, 23970-23976
New diterpene alkaloids from the marine sponge Agelas mauritiana†
Received
1st March 2017
, Accepted 21st April 2017
First published on 3rd May 2017
Abstract
Chemical investigation of an ethanol extract of the sponge Agelas mauritiana led to the isolation and characterization of five new diterpene alkaloids, namely, (−)-8′-oxo-agelasine B (1), (+)-agelasine B (2), (+)-8′-oxo-agelasine C (3), agelasine V (4), and (+)-8′-oxo-agelasine E (5), along with two known compounds, (−)-8′-oxo-agelasine D (6), and agelasine D (7). The structures of these compounds were determined by interpretation of spectroscopic data and comparison with literature properties. Compounds 1 and 3–5 are the second example of 8′-oxo-agelasine analogs. Compounds 2 and 7 not only exhibited moderate cytotoxicity toward the cancer cell lines PC9, A549, HepG2, MCF-7, and U937 with IC50 values of 4.49–14.41 μM, but also showed potent antibacterial activities against a panel of methicillin-resistant Staphylococcus aureus (MRSA) clinical isolates with MIC90 values of 1–8 μg mL−1.
Introduction
Marine sponges are distinguished as excellent sources for marine natural products, with 283 new compounds reported in 2014 alone.1,2 The Agelas genus is a prolific producer of secondary metabolites containing bromopyrrole derivatives,3,4 sesqui- and diterpenoid alkaloids,5 glycosphingolipids,6 carotenoids,7 steroids,8 and fatty acids.9 Particularly, terpenoid alkaloids from this genus are attractive compounds, which are characterized by a sesqui- or diterpene unit attached to different polar functional groups including 6-amino-5-(formylamino)-4-(methylamino)-1,3-diazine, guanidine, sulfone, and methyladeninum. Since the first sesquiterpenoid alkaloid, agelasidine A, was isolated from the Sponge Agelas sp. in 1983, an array of terpenoid alkaloids have been reported to date.10 These terpenoid derivatives are listed as agelasidines,10–15 agelines,16 agelasines,5,15,17–24 agelasimines,25 gelasines,18 and axistatins,26 which show interesting biological effects, such as cyctotoxic,14,18,25,26 anti-malarial,23 anti-microbial,11–14,16,22,24,26 anti-fouling,19 antifungal,15 as well as inhibitory effects on Na+/K+-ATPase.11,17,20
Our previous chemical investigation of the sponge Agelas mauritiana led to the isolation of four new antimicrobial alkaloids, in which (−)-8′-oxo-agelasine D was the first and the only 8′-oxo-agelasine reported to date.12 As our continued exploration of this sponge in a search for structurally new products with promising bioactivities, we found that the CH2Cl2-soluble portion of an EtOH extract of the title sponge collected in March 2013, was cytotoxic against PC9, A549, and U937 cell lines in a Cell Counting Kit-8 (CCK-8) bioassay (8.5–10.5 μg mL−1) and showed moderate antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA 2010-210) with MIC90 value of 1.0 μg mL−1. Further bioactivity-guided fractionation of this CH2Cl2-soluble portion led to the isolation of five new compounds (1–5), as well as two known compounds (6 and 7). Compounds 1 and 3–5 represent the second example of 8′-oxo-agelasine analogs. All isolated compounds (1–7) were evaluated for their cytotoxic and antibacterial activities. Herein, we describe the isolation, structure elucidation, and bioactivities of 1–7.
Results and discussion
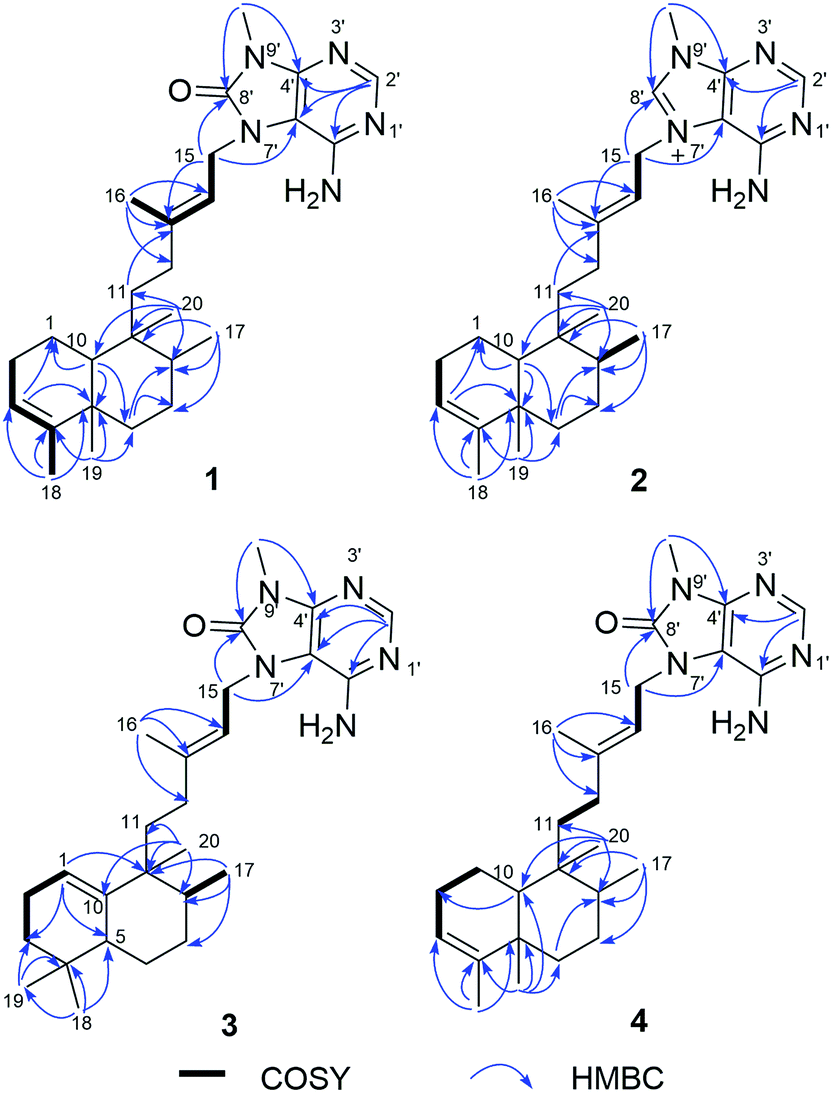
Compound 1 was isolated as a pale yellow amorphous powder, and its molecular formula was deduced to be C26H39N5O from the HRESIMS ion at m/z 438.3232 [M + H]+. The IR bands at 3325, 1718 cm−1 implied the presence of amino and carbonyl functionalities, respectively. UV absorption at 275 nm was in agreement with literature value23 for purine moiety. The 1H NMR spectrum of 1 displayed one deshielded aromatic siglet (δH 8.17), two exchangeable siglets (δH 7.36), one olefin siglet (δH 5.13), one olefin triplet (δH 5.28), four methyl siglets (δH 1.79, 0.95, 0.68, and 3.46), and two methyl doublets (δH 0.75 and 1.53). The 13C NMR and HSQC spectra of 1 revealed 26 carbon signals corresponding to six methyls (including one nitrogen-bearing carbon at δC 26.8), seven methylenes, five methines, and eight quaternary carbons (including one carbonyl carbon at δC 152.6). The above-mentioned NMR data exhibited close resemblance to those of agelasine B,17 except for the purine unit. The corresponding sp2 carbon in purine moiety at δC 141.7 in agelasine B17 was replaced by a carbonyl carbon at δC 152.6 in 1, which was established by the HMBC cross-peaks of the nitrogen methyl protons (δH 3.46) and C-4′ (δC 148.3) and C-8′ (δC 152.6), and of the aromatic proton (δH 8.17) and C-2′ (δC 146.3), C-4′ (δC 148.3), and C-5′ (δC 105.4) (Fig. 2). Moreover, a 1H–15N HMBC experiment was conducted to confirm this 8-oxo-9-N-methyladenine moiety. The observed 1H–13N HMBC correlations of H–NCH3/N-9′ (δN 127.8), H2-15 (δH 4.63)/N-7′ (δN 118.2), H-2′ (δH 8.17)/N-1′ (δN 193.3), and N-3′(δN 224.3) further prove this point. The rest C20H33 alkyl skeleton was assigned to have a clerodane skeleton by analysis of 2D NMR data (Fig. 2). In addition, HMBC correlations from two methylene protons (δH 4.63) to C-13 (δC 144.0), C-5′ (δC 105.4), and C-8′ (δC 152.6) suggested that the 9 N-methyladenine was attached to C-15 via N-7′.
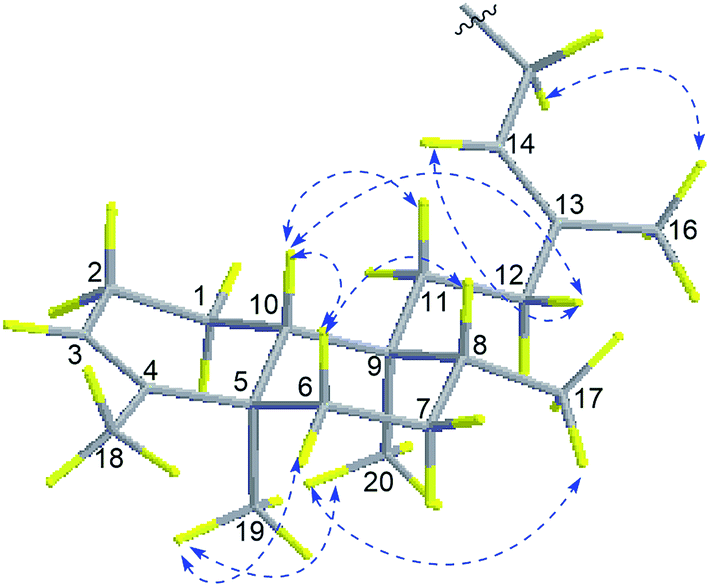
The relative configuration of 1 was deduced from NOESY spectroscopic data (Fig. 3). The NOESY correlations of H2-15 (δH 4.63)/H3-16 (δH 1.79) and the chemical shift of C-16 (δC 16.9) indicated the 13E-configuration of the double bond Δ.13,14 Cross-peaks of H3-19 (δH 0.95)/H3-20 (δH 0.68) and H-6a (δH 1.66), H3-17 (δH 0.75)/H3-20, H-10 (δH 1.27)/H-11a (δH 1.48), H-12b (δH 1.84), and H-6b (δH 1.12), and H-6b/H-8 (δH 1.40) indicated that H3-17, H3-19, H-6a, and H3-20 were α-oriented while H-8, H-6b, H-10, and H2-11 were β-oriented. Further comparison of the [α]25D values of 1 (−33.9, MeOH) with that of agelasine B (−21.5, MeOH17 and −27.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27) suggested the absolute configuration of 1 was probably identical to agelasine B since they had the same relative stereochemistry and the same sign of specific rotation. Thus, the structure of (−)-8′-oxo-agelasine B was concluded to be shown in Fig. 1.
27) suggested the absolute configuration of 1 was probably identical to agelasine B since they had the same relative stereochemistry and the same sign of specific rotation. Thus, the structure of (−)-8′-oxo-agelasine B was concluded to be shown in Fig. 1.
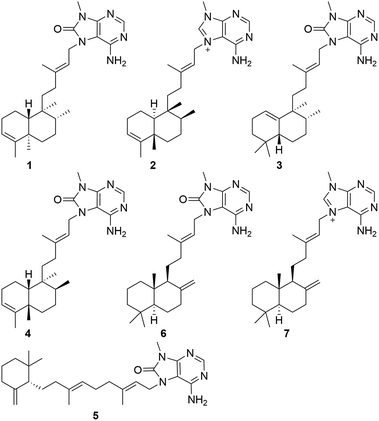 |
| | Fig. 1 The structures of compounds 1–7 from Agelas mauritiana. | |
Compound 2, a white amorphous powder, possessed a molecular formula of C26H39N5, according to its 13C NMR and HRESIMS (m/z 422.3280 [M + H]+, calcd for C26H40N5, 422.3284) data. Detailed analysis of the 1D and 2D NMR spectral data revealed that the planar structure of 2 was the same as agelasine B (Fig. 2).17 The double bond between C-13 (δC 146.2) and C-14 (δC 114.9) possessed the E-geometry, which was established by the NOESY correlations of H2-15 (δH 5.16)/H3-16 (δH 1.79) and H-14 (δH 5.45)/H2-12 (δH 1.97, δH 1.87) and the chemical shift of C-16 (δC 16.7). Moreover, the α-configurations of H-6b (δH 1.10) and H-10 (δH 1.29) were derived from NOESY correlations for H-6b/H-10, while the β-configurations of H-6a (δH 1.65), H3-17 (δH 0.78), H3-19 (δH 0.95), and H3-20 (δH 0.70) were implied from NOESY cross-peaks of H3-19/H-6a and H3-20 and H3-17/H3-20 (Fig. 4). The aforementioned data suggested 2 have identical relative configuration with agelasine B. However, the opposite optical rotation data for them [2 ([α]30D +22.7, MeOH), agelasine B ([α]25D −21.5, MeOH17 and [α]25D −27.227)] suggest 2 differed in absolute configuration at chiral centers with agelasine B. Thus, the structure of 2 was established as a stereoisomer of agelasine B and named (+)-agelasine B.
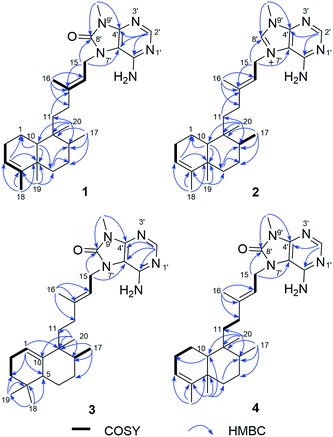 |
| | Fig. 2 Key 1H–1H COSY and HMBC correlations of compounds 1–4. | |
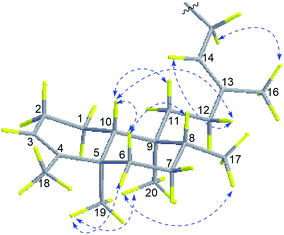 |
| | Fig. 3 Key NOESY correlations of compound 1. | |
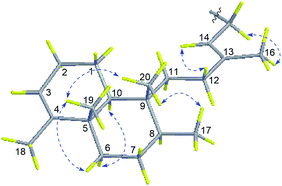 |
| | Fig. 4 Key NOESY correlations of compound 2. | |
Compound 3 showed a [M + H]+ ion peak at m/z 438.3231 in the HRESIMS, corresponding to a molecular formula of C26H39N5O. Comparison of the 1H and 13C NMR spectroscopic data (Tables 1 and 2) of compound 2 suggested a rearranged labdane skeleton for 3, which was further supported by the HMBC correlations from H3-17 (δH 0.83) to C-7 (δC 31.2), C-8 (δC 44.5), and C-9 (δC 42.5), from H3-20 (δH 1.00) to C-8, C-9, C-10 (δC 145.4), and C-11 (δC 29.6), from H3-18 (δH 0.84) to C-3 (δC 31.3), C-4 (δC 31.2), C-5 (δC 43.8), and C-19 (δC 27.8), from H3-19 (δH 0.84) to C-3, C-4, C-5, and C-18 (δC 27.8), from H-1 (δH 5.34) to C-3, C-5, and C-9, and from H-5 (δH 1.49) to C-1 (δC 117.6), C-3, C-7, C-10 (δC 145.4), C-18, and C-19 (Fig. 2). The 13E-configuration of the double bond Δ13,14 was inferred from the NOESY correlations of H2-15 (δH 4.65)/H3-16 (δH 1.79) and H-14 (δH 5.27)/H2-12 (δH 1.81, δH 1.72) and the chemical shift of C-16 (δC 17.0). The NOESY correlations of H3-17/H3-20 and H-6a (δH 1.78) indicated the β-orientation of H3-17, H-6a, and H3-20, while the cross-peaks of H-5/H-6b (δH 1.73) and H-8 (δH 1.28)suggested these protons are α-oriented, establishing the relative configurations of 3 (Fig. 5), in consonance with those of the synthesis product (+)-agelasine C.28 The absolute configurations of 3 was determined by comparison of its optical rotation [α]25D +29 (MeOH) and the synthesis product (+)-agelasine C [α]22D +25 (MeOH). Therefore, the structure of (+)-8′-oxo-agelasine C (3) was defined as shown in Fig. 1.
Table 1 1H NMR spectroscopic data for compounds 1–5 (δ in ppm, J in Hz)
| Position |
1a,c |
2a,d |
3a,c |
4b,c |
5b,c |
| Measured at 600 MHz. Measured at 400 MHz. Measured in CDCl3. Measured in DMSO-d6. |
| 1a |
1.38, m |
1.54, m |
5.34, m |
1.83, m |
2.00, m |
| 1b |
|
|
|
1.59, m |
|
| 2a |
1.99, m |
1.98, m |
2.01, m |
1.97, m |
1.53, m |
| 2b |
1.92, m |
|
|
|
|
| 3a |
5.13, s |
5.13, s |
1.38, m |
5.34, m |
1.46, m |
| 3b |
|
|
1.06, m |
|
1.21, m |
| 5 |
|
|
1.49, dd (12.6, 3.0) |
|
1.67, m |
| 6a |
1.66, d (12.6) |
1.65, dt (13.2, 3.0) |
1.78, m |
1.62, m |
1.38, m |
| 6b |
1.12, m |
1.10, m |
1.73, m |
1.48, m |
|
| 7a |
1.92, m |
1.46, m |
1.41, dd (13.2, 3.6) |
1.43, m |
1.74, m |
| 7b |
1.37, m |
1.38, m |
1.09, m |
1.34, m |
|
| 8 |
1.40, m |
1.42, m |
1.28, m |
1.55, m |
|
| 9 |
|
|
|
|
5.03, s |
| 10 |
1.27, m |
1.29, d (12.6) |
|
1.41, m |
|
| 11a |
1.48, m |
1.47, m |
1.25, s |
1.83, m |
2.11, m |
| 11b |
1.29, m |
1.35, m |
|
1.24, m |
|
| 12a |
1.92, m |
1.97, m |
1.81, m |
2.12, td (13.2, 4.0) |
2.11, m |
| 12b |
1.84, td (12.6, 4.8) |
1.87, td (12.6, 4.8) |
1.72, m |
2.00, m |
|
| 14 |
5.28, t (6.0) |
5.45, t (6.0) |
5.27, t (6.0) |
5.35, m |
5.34, t (5.6) |
| 15 |
4.63, d (6.0) |
5.16, d (6.0) |
4.65, d (6.0) |
4.68, d (5.6) |
4.67, d (5.6) |
| 16 |
1.79, s |
1.79, s |
1.79, d (1.2) |
1.83, s |
1.82, s |
| 17 |
0.75, d (5.4) |
0.78, d (6.6) |
0.83, s |
0.85, d (7.2) |
1.58, s |
| 18 |
1.53, d (1.8) |
1.54, d (1.2) |
0.84, s |
1.62, s |
0.90, s |
| 19 |
0.95, s |
0.95, s |
0.84, s |
1.12, s |
0.82, s |
| 20a |
0.68, s |
0.70, s |
1.00, s |
1.01, s |
4.74, s |
| 20b |
|
|
|
|
4.52, d (2.0) |
| 2′ |
8.17, s |
8.46, s |
8.18, s |
8.19 |
8.18, s |
| 8′ |
|
9.54, s |
|
|
|
| 2′-NH2 |
7.36, br s |
7.90, br s |
7.62, br s |
7.69, br s |
7.66, br s |
| 9′-CH3 |
3.46, s |
3.89, s |
3.50, s |
3.51, s |
3.50, s |
Table 2 13C NMR spectroscopic data for compounds 1–5 (δ in ppm)
| Position |
1a,c |
2a,d |
3a,c |
4b,c |
5b,c |
| Measured at 150 MHz. Measured at 100 MHz. Measured in CDCl3. Measured in DMSO-d6. |
| 1 |
18.3, CH2 |
17.8, CH2 |
117.6, CH |
19.9, CH2 |
32.5, CH2 |
| 2 |
26.8, CH2 |
26.3, CH2 |
23.2, CH2 |
25.7, CH2 |
23.7, CH2 |
| 3 |
120.3, CH |
120.2, CH |
31.3, CH2 |
122.5, CH |
36.3, CH2 |
| 4 |
144.3, C |
143.6, C |
31.2, C |
141.8, C |
34.9, C |
| 5 |
38.1, C |
37.6, C |
43.8, CH |
38.7, C |
53.6, CH |
| 6 |
36.7, CH2 |
36.2, CH2 |
30.1, CH2 |
32.1, CH2 |
24.8, CH2 |
| 7 |
27.4, CH2 |
27.0, CH2 |
31.2, CH2 |
27.2, CH2 |
38.2, CH2 |
| 8 |
36.2, CH |
35.7, CH |
44.5, CH |
37.4, CH |
137.0, C |
| 9 |
38.6, C |
38.2, C |
42.5, C |
38.7, C |
122.5, CH |
| 10 |
46.4, CH |
45.9, CH |
145.4, C |
44.6, CH |
149.3, C |
| 11 |
36.4, CH2 |
35.8, CH2 |
29.6, CH2 |
36.0, CH2 |
26.1, CH2 |
| 12 |
33.0, CH2 |
32.5, CH2 |
33.9, CH2 |
33.7, CH2 |
39.5, CH2 |
| 13 |
144.0, C |
146.2, C |
144.7, C |
144.7, C |
143.3, C |
| 14 |
119.5, CH |
114.9, CH |
119.3, CH |
119.5, CH |
119.8, CH |
| 15 |
40.5, CH2 |
47.0, CH2 |
40.6, CH2 |
40.6, CH2 |
40.6, CH2 |
| 16 |
16.9, CH3 |
16.7, CH3 |
17.0, CH3 |
17.1, CH3 |
16.9, CH3 |
| 17 |
15.9, CH3 |
15.8, CH3 |
16.3, CH3 |
15.3, CH3 |
16.1, CH3 |
| 18 |
17.9, CH3 |
17.7, CH3 |
27.8, CH3 |
19.3, CH3 |
28.4, CH3 |
| 19 |
19.8, CH3 |
19.6, CH3 |
27.8, CH3 |
28.0, CH3 |
26.2, CH3 |
| 20 |
18.2, CH3 |
18.1, CH3 |
23.0, CH3 |
26.2, CH3 |
108.8, CH2 |
| 2′ |
146.3, CH |
155.4, CH |
145.8, CH |
145.6, CH |
145.6, CH |
| 4′ |
148.3, C |
148.9, C |
148.3, C |
148.3, C |
148.3, C |
| 5′ |
105.4, C |
109.2, C |
105.4, C |
105.4, C |
105.4, C |
| 6′ |
143.6, C |
152.3, C |
143.5, C |
143.4, C |
143.3, C |
| 8′ |
152.6, C |
140.9, CH |
152.6, C |
152.6, C |
152.6, C |
| 9′-CH3 |
26.8, CH3 |
31.4, CH3 |
26.9, CH3 |
26.9, CH3 |
26.9, CH3 |
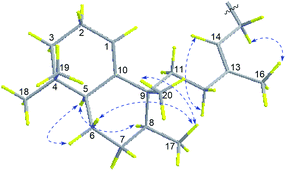 |
| | Fig. 5 Key NOESY correlations of compound 3. | |
Compound 4 had a molecular formula of C26H39N5O based on the [M + H]+ ion at m/z 438.3230 (calcd for C26H40N5O, 438.3233) in the HRESIMS. Extensive analysis of HMBC and COSY correlations revealed that compound 4 shared the same planar structure as 1 (Fig. 2). The NOESY correlation of H2-15 (δH 4.68)/H3-16 (δH 1.83) and H-14 (δH 5.35)/H2-12 (δH 2.12 and 2.00) and the chemical shift of C-16 (δC 17.1) suggested the E-geometry of the Δ13,14 double bond. The NOESY cross-peaks of H3-17 (δH 0.85)/H-11b (δH 1.24) and H-10 (δH 1.41), H-10/H-12a (δH 2.12), H2-11 (δH 1.83 and 1.24), H3-19 (δH 1.12) and H-6a (δH 1.62), and H3-19/H-6a located H3-17, H-10, H-6a, and H3-19 on the same face, while NOESY correlation of H3-20 (δH 1.01)/H-8 (δH 1.55) positioned H3-20 and H-8 on the opposite face (Fig. 6). Thus, the structure of 4 was assigned as a stereoisomer of agelasine B and named agelasine V.
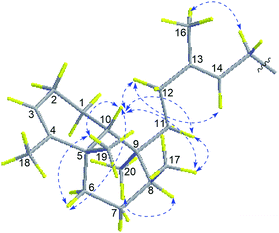 |
| | Fig. 6 Key NOESY correlations of compound 4. | |
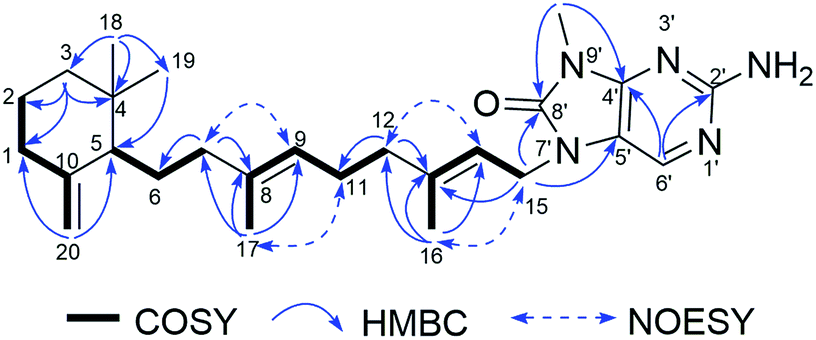
Compound 5, a pale yellow amorphous powder, had a molecular formula of C26H39N5O deduced from the 13C NMR and HRESIMS (438.3229 [M + H]+, calcd for C26H40N5O, 438.3233) data. Compound 5 possessed the same 8-oxo-9-N-methyladenine moiety according to the comparison of its 1D NMR data (Tables 1 and 2) with those of compound 1. The rest 9, 10-seco-labdane skeleton was determined by analysis of 2D NMR data (Fig. 7). The configurations of the double bonds (Δ9,10 and Δ13,14) were established as E based on the NOESY correlations of H3-17 (δH 1.58)/H2-11 (δH 2.11), H-9 (δH 5.03)/H2-7 (δH 1.74), H3-16 (δH 1.82)/H2-15 (δH 4.67), and H2-12 (δH 2.11)/H-14 (δH 5.34) and the chemical shift of C-17 (δC 16.1) and C-16 (δC 16.9). The absolute configuration of 5 was assumed to be the same as (+)-trixagol, whose enantiomer was equate to the terpenoid side chain of (−)-agelasine E in that they both exhibit positive optical rotations [5 ([α]30D +30.6, MeOH), (+)-trixagol ([α]D+14, CHCl3)29 and agelasine E ([α]25D −17.1, MeOH)20]. Accordingly, The structure of (+)-8′-oxo-agelasine E (5) was proposed as shown Fig. 1.
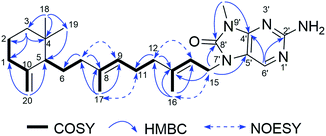 |
| | Fig. 7 Key 1H–1H COSY, HMBC and NOESY correlations of compound 5. | |
All isolated compounds were assessed for their antibacterial activity against a methicillin-susceptible S. aureus (MSSA) strain H608 and four methicillin-resistant S. aureus (MRSA) strains 2010-260, 2010-210, 2010-292, and 2010-300. As shown in Table 3, compounds 2 and 7 exhibited potent activities against MRSA with MIC90 values of 1–8 μg mL−1 while other compounds showed no activity (MIC90 > 64 μg mL−1). The cytotoxic activities of individual compounds were evaluated against the PC9, A549, HepG2, MCF-7, and U937 cell lines using Cell Counting Kit-8 (CCK-8) bioassay (Table 3). Compounds 2 and 7 showed moderate activities against the five cancer cell lines with IC50 values of 4.49–14.41 μM. Other compounds showed no activity (IC50 > 20 μM) except compound 6 show weak cytotoxicity against U937 cell line with IC50 value of 16.89 μM.
Table 3 Antibacterial and cytotoxic activities of compounds 1–7
| Compoundsa |
Antibacterial activity against clinical MRSAd and MSSAe strains (MIC90, μg mL−1) |
Cytotoxic activity (IC50, μM) |
| 2010-260 |
2010-210 |
2010-292 |
2010-300 |
H608 |
PC9 |
A549 |
HepG2 |
MCF-7 |
U937 |
| Compounds 1 and 3–5 were inactive in all assays. Positive control for antibacterial assay. Positive control for cytotoxic assay. Clinical MRSA strains 2010-260, 2010-210, 2010-292, 2010-300. Clinical MSSA strain H608. |
| 2 |
2 |
1 |
2 |
1 |
2 |
5.08 |
14.07 |
9.76 |
7.64 |
4.49 |
| 6 |
>64 |
>64 |
>64 |
>64 |
>64 |
>50 |
>50 |
>50 |
>50 |
16.89 |
| 7 |
4 |
4 |
8 |
8 |
1 |
4.49 |
14.41 |
10.07 |
5.47 |
6.86 |
| Vancomycinb |
1 |
1 |
1 |
0.5 |
2 |
|
|
|
|
|
| Doxorubicinc |
|
|
|
|
|
0.22 |
0.49 |
0.20 |
0.38 |
0.05 |
Conclusions
In conclusion, five new diterpene alkaloids, illustrated by (−)-8′-oxo-agelasine B (1), (+)-agelasine B (2), (+)-8′-oxo-agelasine C (3), agelasine V (4), (+)-8′-oxo-agelasine E (5), along with two known related metabolites, (−)-8′-oxo-agelasine D (6), agelasine D (7), were isolated from the marine sponge Agelas mauritiana. The structures of them were established by interpretation of spectroscopic data and comparison with literature properties. Isolation of compounds 1 and 3–5 provided new examples of 8′-oxo-agelasine analogs. Compounds 2 and 7 showed potent antibacterial and moderate cytotoxic activities. Analysis of the structures of the diterpene alkaloids (1–7) and their antibacterial and cytotoxic activities led to a preliminary summary of the structure–activity relationship (SAR): C-8′-carbonylated compounds (1 and 3–6) can provide lower antibacterial and cytotoxic activities than other analogs (2 and 7). Moreover, the interesting antibacterial activity of compounds 2 and 7 indicated that they could be possible lead candidates as promising anti-MRSA agents.
Experimental section
General experimental procedures
Optical rotation data were measured in MeOH on an Autopol I polarimeter (no. 30575, Rudolph Research Analytical) with a 10 cm length cell. UV were recorded on a Hitachi U-3010 spectrophotometer. IR (KBr) spectra were obtained on Jasco FTIR-400 spectrometer. 1D and 2D NMR spectra were recorded in DMSO-d6 or CDCl3 on a Bruker DRX-600 or on a Bruker DRX-400 MHz NMR spectrometers. HRESIMS data was recorded on a Q-TOF micro YA019 mass spectrometer. Column chromatography (CC) was carried out on Sephadex LH-20 (Amersham Pharmacia Biotech AB), silica gel (200-300 mesh, Qingdao Marine Chemical Inc. China), and reverse phase C18 silica gel (15 μm, Santai Technologies, Inc.). Analytical thin-layer chromatography was carried out using HSGF 254 plates and visualized by spraying with anisaldehyde-H2SO4 reagent. Reversed-phase high-performance liquid chromatography (HPLC) was performed on Waters SunFire™ Prep C18 column (5 μm, 19 × 150 mm) with a Waters 1525 separation module equipped with a Waters 2998 photodiode array detector, and all solvents used for HPLC were of HPLC grade.
Animal material
Samples of Agelas mauritiana were collected along the coast of Yongxing Island in the South China Sea on March 19, 2013. A voucher specimen (no. 13-7) has been deposited at Research Center for Marine Drugs, State Key Laboratory of Oncogenes and Related Genes, Shanghai Jiao Tong University.
Extraction and isolation
The sponge (1.2 kg, wet weight) was cut and percolated with 95% EtOH at room temperature to afford the crude extract (15.2 g), which was suspended in H2O and extracted with EtOAc. The EtOAc-soluble extract (9.3 g) was concentrated under reduced pressure. Subsequently, the EtOAc-soluble extract was partitioned between 90% aqueous MeOH and petroleum ether to give 5.5 g petroleum ether-soluble fraction. The 90% aqueous MeOH fraction was diluted to 60% aqueous MeOH with H2O and extracted with CH2Cl2 to afford a 4.5 g CH2Cl2-soluble fraction. This CH2Cl2-soluble extract was chromatographed on silica gel column eluting with a step gradient of CH2Cl2–MeOH (100:1 to 0:1) to give nine fractions (DA–DI). Fraction DH (1.1 g) was subjected to VLC over silica gel eluting with a CH2Cl2–EtOAc–MeOH–H2O system (10:5:1.5:0.2 and 0:0:1:0) to afford four subfractions (DH1–DH4). Subfraction DH3 (200.1 mg) was directly separated by reversed-phase HPLC (Waters SunFire™ Prep C18, 5 μm, 19 × 150 mm; 10.0 mL min−1; 210, 270 nm) eluting with a CH3CN–H2O–TFA system (50:50:0.1) to give (+)-agelasine B (2, 40.0 mg, tR 8.5 min) and agelasine D (7, 35.2 mg, tR 10.0 min). DH1 (163.6 mg) was subjected to silica gel column chromatography, using a gradient of CH2Cl2–EtOAc–MeOH–H2O solvent system (50:5:1:0.1, 45:5:1:0.1, 40:5:1:0.1, 25:5:1:0.1, 20:5:1:0.1, 20:5:1:0.1, 10:5:1.25:0.2, and 0:0:1:0) to give 5 subfractions (DH1A–DH1E). Subfraction DH1B (60.5 mg) was further purified by reversed-phase HPLC (Waters SunFire™ Prep C18, 5 μm, 19 × 150 mm; 9.0 mL min−1; 210, 270 nm) eluting with a CH3CN–H2O–TFA (55:45:0.1) system to give (−)-8′-oxo-agelasine B (1, 15.0 mg, tR 49.5 min), (−)-8′-oxo-agelasine D (6, 13.1 mg, tR 55.0 min), (+)-8′-oxo-agelasine C (3, 5.7 mg, tR 63.9 min), agelasine V (4, 2.8 mg, tR 52.0 min), (+)-8′-oxo-agelasine E (5, 2.9 mg, tR 70.0 min).
(−)-8′-Oxo-agelasine B (1). Pale yellow amorphous powder; [α]25D −33.9 (c 0.11, MeOH); UV (MeOH) λmax (logε) 216 (4.07), 275 (3.64) nm; IR (KBr) νmax 3325, 2927, 2861, 1718, 1639, 1460, 1376, 1198, 1141 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 438.3232 [M + H]+ (calcd for C26H40N5O, 438.3233).
(+)-Agelasine B (2). Pale yellow amorphous powder; [α]30D +22.7 (c 0.10, MeOH); UV (MeOH) λmax (logε) 214 (4.16), 262 (3.63) nm; IR (KBr) νmax 3320, 2927, 2858, 1683, 1649, 1460, 1379, 1202, 1133, 798, 720 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 422.3280 [M + H]+ (calcd for C26H40N5, 422.3284).
(+)-8′-Oxo-agelasine C (3). Pale yellow amorphous powder; [α]25D +29.0 (c 0.08, MeOH); UV (MeOH) λmax (logε) 214 (4.00), 274 (3.50) nm; IR (KBr) νmax 3325, 2925, 2856, 1721, 1640, 1459, 1373, 1197, 1142 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 438.3231 [M + H]+ (calcd for C26H40N5O, 438.3233).
Agelasine V (4). Pale yellow amorphous powder; [α]30D +36.5 (c 0.08, MeOH); UV (MeOH) λmax (logε) 214 (3.62), 275 (3.12) nm; IR (KBr) νmax 3330, 2926, 2856, 1721, 1641, 1461, 1375, 1198, 1092, 802 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 438.3230 [M + H]+ (calcd for C26H40N5O, 438.3233).
(+)-8′-Oxo-agelasine E (5). Pale yellow amorphous powder; [α]30D +30.6 (c 0.11, MeOH); UV (MeOH) λmax (logε) 203 (3.58), 275 (3.03) nm; IR (KBr) νmax 3325, 2925, 2856, 1721, 1640, 1459, 1373, 1197, 1142 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 438.3229 [M + H]+ (calcd for C26H40N5O, 438.3233).
Antibacterial assay
The in vitro antibacterial assay was carried out as reported before.30 Vancomycin was used as positive control and displayed MIC90 values of 2, 1, 1, 1, and 0.5 μM against methicillin-susceptible S. aureus strain H608 and methicillin-resistant S. aureus strains 2010-260, 2010-210, 2010-292, and 2010-300, respectively.
Cytotoxicity assay
The effects of 1–7 on cell viability was determined using the Cell Counting Kit-8 (CCK-8). The human PC9, A549, HepG2, MCF-7, and U937 cell lines were obtained from the Institute of Biochemistry Cell Biology (Shanghai, China). Cells were seeded in 96 well plates (5 × 103 cells per well). After 24 h incubation, the cells were treated with various concentrations of 1–7 for 72 h. Then the CCK8 solution (10 μL) was added for additional 1 h incubation at 37 °C. The absorbance at 450 nm was measured in a microplate reader (spectra MAX190, Molecular Devices, USA). Independent experiments were performed in triplicate.
Acknowledgements
This research was supported by the National Natural Science Fund for Distinguished Young Scholars of China (81225023), the National Natural Science Fund of China (No. U1605221, 41428602, 41576130, 81502936, 41476121 and 81402844), and the Fund of the Science and Technology Commission of Shanghai Municipality (No. 14431901300 and 15431900900).
Notes and references
- M. Mehbub, J. Lei, C. Franco and W. Zhang, Mar. Drugs, 2014, 12, 4539–4577 CrossRef PubMed.
- J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2016, 33, 382–431 RSC.
- Y. Zhu, Y. Wang, B.-B. Gu, F. Yang, W.-H. Jiao, G.-H. Hu, H.-B. Yu, B.-N. Han, W. Zhang, Y. Shen and H.-W. Lin, Tetrahedron, 2016, 72, 2964–2971 CrossRef CAS.
- T. Yasuda, A. Araki, T. Kubota, J. Ito, Y. Mikami, J. Fromont and J. i. Kobayashi, J. Nat. Prod., 2009, 72, 488–491 CrossRef CAS PubMed.
- D. B. Abdjul, H. Yamazaki, S.-i. Kanno, O. Takahashi, R. Kirikoshi, K. Ukai and M. Namikoshi, J. Nat. Prod., 2015, 78, 1428–1433 CrossRef CAS PubMed.
- J. J. La Clair and A. D. Rodríguez, Bioorg. Med. Chem., 2011, 19, 6645–6653 CrossRef CAS PubMed.
- Y. Tanaka and T. Katayama, Nippon Suisan Gakkaishi, 1982, 48, 531–533 CrossRef CAS.
- J.-F. Hu, M. Kelly and M. T. Hamann, Steroids, 2002, 67, 743–747 CrossRef CAS PubMed.
- D. Tasdemir, B. Topaloglu, R. Perozzo, R. Brun, R. O'Neill, N. M. Carballeira, X. Zhang, P. J. Tonge, A. Linden and P. Rüedi, Bioorg. Med. Chem., 2007, 15, 6834–6845 CrossRef CAS PubMed.
- H. Nakamura, H. Wu, J. i. Kobayashi, Y. Ohizumi, Y. Hirata, T. Higashijima and T. Miyazawa, Tetrahedron Lett., 1983, 24, 4105–4108 CrossRef CAS.
- H. Nakamura, H. Wu, J. Kobayashi, M. Kobayashi, Y. Ohizumi and Y. Hirata, J. Org. Chem., 1985, 50, 2494–2497 CrossRef CAS.
- F. Yang, M. T. Hamann, Y. Zou, M.-Y. Zhang, X.-B. Gong, J.-R. Xiao, W.-S. Chen and H.-W. Lin, J. Nat. Prod., 2012, 75, 774–778 CrossRef CAS PubMed.
- A. Medeiros Maria, A. Lourenço, R. Tavares Maria, M. Curto Maria João, S. Feio Sónia and C. Roseiro José, Zeitschrift für Naturforschung C, 2006, 61, 472–476 Search PubMed.
- J. J. Morales and A. D. Rodríguez, J. Nat. Prod., 1992, 55, 389–394 CrossRef CAS.
- E. P. Stout, L. C. Yu and T. F. Molinski, Eur. J. Org. Chem., 2012, 2012, 5131–5135 CrossRef CAS PubMed.
- R. J. Capon and D. J. Faulkner, J. Am. Chem. Soc., 1984, 106, 1819–1822 CrossRef CAS.
- H. Nakamura, H. Wu, Y. Ohizumi and Y. Hirata, Tetrahedron Lett., 1984, 25, 2989–2992 CrossRef CAS.
- L. Calcul, K. Tenney, J. Ratnam, J. H. McKerrow and P. Crews, Aust. J. Chem., 2010, 63, 915–921 CrossRef CAS.
- T. Hattori, K. Adachi and Y. Shizuri, J. Nat. Prod., 1997, 60, 411–413 CrossRef CAS.
- H. Wu, H. Nakamura, J. i. Kobayashi, Y. Ohizumi and Y. Hirata, Tetrahedron Lett., 1984, 25, 3719–3722 CrossRef CAS.
- K. Ishida, M. Ishibashi, H. Shigemori, T. Sasaki and J. Kobayashi, Chem. Pharm. Bull., 1992, 40, 766–767 CrossRef CAS PubMed.
- X. Fu, F. J. Schmitz, R. S. Tanner and M. Kelly-Borges, J. Nat. Prod., 1998, 61, 548–550 CrossRef CAS PubMed.
- J. Appenzeller, G. Mihci, M.-T. Martin, J.-F. Gallard, J.-L. Menou, N. Boury-Esnault, J. Hooper, S. Petek, S. Chevalley, A. Valentin, A. Zaparucha, A. Al-Mourabit and C. Debitus, J. Nat. Prod., 2008, 71, 1451–1454 CrossRef CAS PubMed.
- T. Kubota, T. Iwai, A. Takahashi-Nakaguchi, J. Fromont, T. Gonoi and J. i. Kobayashi, Tetrahedron, 2012, 68, 9738–9744 CrossRef CAS.
- R. Fathi-Afshar and T. M. Allen, Can. J. Chem., 1988, 66, 45–50 CrossRef CAS.
- G. R. Pettit, Y. Tang, Q. Zhang, G. T. Bourne, C. A. Arm, J. E. Leet, J. C. Knight, R. K. Pettit, J.-C. Chapuis, D. L. Doubek, F. J. Ward, C. Weber and J. N. A. Hooper, J. Nat. Prod., 2013, 76, 420–424 CrossRef CAS PubMed.
- E. Piers and J. Y. Roberge, Tetrahedron Lett., 1992, 33, 6923–6926 CrossRef CAS.
- I. S. Marcos, N. García, M. J. Sexmero, P. Basabe, D. Díez and J. G. Urones, Tetrahedron, 2005, 61, 11672–11678 CrossRef CAS.
- A. K. Bakkestuen and L.-L. Gundersen, Tetrahedron, 2003, 59, 115–121 CrossRef CAS.
- W.-H. Jiao, J. Li, Q. Liu, T.-T. Xu, G.-H. Shi, H.-B. Yu, F. Yang, B.-N. Han, M. Li and H.-W. Lin, Molecules, 2014, 19, 18025–18032 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: HRESIMS, 1D NMR, 2D NMR, IR, UV, and CD spectra. See DOI: 10.1039/c7ra02547e |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a