
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzymatic synthesis of an electrospinnable poly(butylene succinate-co-dilinoleic succinate) thermoplastic elastomer†
Agueda Sonseca and
Miroslawa El Fray *
*
Division of Biomaterials and Microbiological Technologies, Polymer Institute, Faculty of Chemical Technology and Engineering, West Pomeranian University of Technology, 45 Piastów Ave, 70-311, Szczecin, Poland. E-mail: miroslawa.elfray@zut.edu.pl
First published on 18th April 2017
Abstract
Candida antarctica lipase B was successfully employed for the first time as a biocatalyst to obtain high molecular weight poly(butylene succinate-co-dilinoleic succinate) (PBS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DLS) copolyester via a two-stage method in a diphenyl ether solution from diethyl succinate, 1,4-butanediol, and dimer linoleic diol. The final hard to soft segments were close to the initial feed ratio as confirmed by 1H NMR and FTIR measurements. Thermal analysis revealed the presence of two transitions typical for thermoplastic elastomers: a low temperature glass transition and high temperature melting point. In addition, the optimized synthetic protocol ensured high reaction yield (>74%) and molecular weight (Mw > 34 × 103 g mol−1) high enough to process the copolymer into electrospun mats without defects. The enzymatically synthesized PBS:DLS was able to be processed at 20–25 w/v% concentrations into fibres and depending on the type of solvent, the fibre diameter was controlled in the range of 0.3 to 0.6 μm.
:DLS) copolyester via a two-stage method in a diphenyl ether solution from diethyl succinate, 1,4-butanediol, and dimer linoleic diol. The final hard to soft segments were close to the initial feed ratio as confirmed by 1H NMR and FTIR measurements. Thermal analysis revealed the presence of two transitions typical for thermoplastic elastomers: a low temperature glass transition and high temperature melting point. In addition, the optimized synthetic protocol ensured high reaction yield (>74%) and molecular weight (Mw > 34 × 103 g mol−1) high enough to process the copolymer into electrospun mats without defects. The enzymatically synthesized PBS:DLS was able to be processed at 20–25 w/v% concentrations into fibres and depending on the type of solvent, the fibre diameter was controlled in the range of 0.3 to 0.6 μm.
Introduction
There is an increasing demand for degradable polymers for tissue engineering applications that can be processed into three-dimensional synthetic porous scaffolds. This has led to a great deal of research focused on the synthesis of new aliphatic polyesters and copolyesters with different properties and structures for this target application.1,2 Fibrous scaffolds are favoured for tissue engineering, because of their ability to mimic the structure and properties of the extracellular matrix (ECM), such as anisotropic mechanical properties, interconnected porosity, and high surface area to volume ratio. This approach maximizes the space for cell attachment and tissue in-growth, decreasing the amount of polymer implanted and, at the same time, encouraging effective diffusion and distribution of the cells.3 Towards this aim, electrospinning is a relatively simple technique that provides a straightforward way to produce nano- and microfiber mats for the fabrication of tissue engineered scaffolds.4One of the main applications of these scaffolds is for soft tissue replacements such as myocardium or blood vessels. Ideally, the scaffold should mimic the mechanical properties of native tissues5 thus, elastomeric materials are preferred as they will possess more suitable properties to work in a cyclic loading environment matching the behaviour of native tissues.
In this regard, biodegradable elastomers offer an attractive alternative to non-elastomeric biodegradable polymers currently used for many soft-tissue engineering applications. Several types of biodegradable elastomeric materials have been reported in the literature,6,7 but their main disadvantage is the need to use reinforcing/crosslinking agents, stabilizers or cure systems along with compounding.8,9 Therefore, just a few reports present the fabrication of elastomeric scaffolds obtained by electrospinning, with the need of be processed by blends or sacrificial polymers.10,11
To overcome these drawbacks, a big deal of research has been focused in the development of new thermoplastic elastomers (TPE) as they are the most versatile family of elastomeric materials, thanks to having many of the physical properties of crosslinked elastomers (rubbers) and being processed as thermoplastics. TPEs are (multi)block copolymers composed of thermodynamically incompatible segments built up from rigid (hard) thermoplastic components and soft elastomeric components. This results in microphase separation and formation of a discrete micro- (or nano-) domain structure that stabilizes the system—no need for curing or stabilizing agents.
Poly(butylene succinate) (PBS) is one of the most significant members of the aliphatic polyester family. As its properties can be easily tailored by copolymerization,12,13 it has promising applications as a hard segment building block in bioabsorbable/biocompatible materials for medical purposes. Recently, PBS was used for the synthesis of TPEs, constituting the hard segments in multiblock copolymers, while dimer linoleic acid (DLA) or dimer linoleic diol (DLA–OH) units were used as the soft segments component. Our group has reported on the controlled biodegradability and promising biocompatibility of these materials.14,15 By varying the PBS to DLA (hard to soft segment) ratio, poly(butylene succinate-co-butylene dilinoleate) (PBS:DLA) copolymers covering a wide range of mechanical properties and degradation rates can be synthesized. Importantly, by using dimer linoleic diol (DLA–OH) as soft segment building block instead of dimer linoleic acid (DLA), poly(butylene succinate-co-dilinoleic succinate) multiblock copolymers with interesting elastomeric behaviour at body temperature can also be obtained.10 Therefore, this approach holds the promise for biodegradable and biocompatible medical materials able to mimic the elasticity of human tissues with excellent compliance.
Additionally, electrospinning of elastomeric neat poly(butylene succinate-co-butylene dilinoleate) (PBS:DLA) and poly(butylene succinate-co-dilinoleic succinate) (PBS:DLS), as well as their blends with poly(glycerol sebacate), has been reported to produce scaffolds with properties well-suited for cardiac tissue engineering applications.10,15
While PBS:DLA and PBS:DLS copolymers represent good candidates materials for tailorable scaffolds that mimic the elastic properties of soft tissues ECM, most of the approaches to the synthesis of PBS and its copolymers involve the use of organometallic catalysts at high reaction temperatures (≥200 °C).16 As a result, undesirable degradation and side-reactions can occur due to the high temperature, especially when using thermally or chemically unstable monomers (i.e. siloxane, epoxy and vinyl moieties).17 Further, difficulty to remove residual metals from the catalyst, that strongly interacts with the ester groups, limits their applications as medical materials.18 Finally, environmental concerns are driving demand not only for materials from renewable resources instead of fossil fuels, but also for the use of non-toxic catalysts in the polymer synthesis.
The “green chemistry” approach,19,20 is an emerging field including the use of enzymes as catalysts for polymerization reactions of new polymers, as well as modification/functionalization of existing polymers. Enzymes as polymerization catalysts have been extensively used to produce a variety of useful materials such as polysaccharides, polyesters, polycarbonates and polyamides.21–23 The main advantages of enzymes are related to their versatility as a catalysts: they provide higher enantio-, regio-, and chemoselectivity, are able to lower the activation energy of chemical reactions to operate at mild temperature conditions, and are derived from renewable resources. As a result, enzymatic catalysis provides an effective platform for conducting green polymer chemistry to obtain environment-friendly synthetic process for polymeric materials. Among all the enzymes as a catalyst, Candida antarctica lipase B (CAL-B) is the most widely employed in transesterification and esterification reactions because of its reactivity and high stability in many organic solvents even at elevated temperatures.24 This approach has been extensively used in transesterification reactions to obtain polyesters and copolyesters from a broad range of substrates.25–28 However, although the enzyme-catalyzed materials are extraordinarily advantageous from a toxicity and environmental point of view, the formation of relatively low molecular weight polyesters represents a major drawback especially in the case of the formation of copolyesters. It limits the processability of the obtained polymeric materials into any useful physical form, such as films or electrospun nano/microfiber porous scaffolds for tissue engineering.29
With this background and based on our experience with organometallic synthesis of PBS:DLS copolymers, the objective of this work was to utilize Candida antarctica lipase B (CAL-B) as a catalyst to synthesize poly(butylene succinate-co-dilinoleic succinate) (PBS:DLS) copolymer based on dimer linoleic diol (DLA–OH), with a 70:30 wt% ratio of hard to soft segments. Importantly, the polymerization conditions were optimized in order to achieve a copolymer with sufficiently high molecular weight to fabricate porous scaffolds by electrospinning.
Experimental
Materials
Dichloromethane (DCM: ≥99.5%), diethyl succinate (DS: ≥99%), 1,4-butanediol (BD: 99%) and diphenyl ether (≥99%) were purchased from Sigma Aldrich (Poznan, Poland). Dimer linoleic diol (DLA–OH) Pripol™ 2033 (dimer alcohol ≥ 96.5%), was purchased from Croda Coatings & Polymers, Gouda (The Netherlands). Chloroform (CHCl3: ≥98.5%) was purchased from POCh (Gliwice, Poland), Tetrahydrofuran (THF: ≥99.5%), N,N-dimethylformamide (DMF: ≥99.8%) and methanol (MeOH: ≥99.8%), were purchased from POCh (Gliwice, Poland). Lipase B from Candida antarctica (CAL-B), covalently immobilized on polyacrylate beads (300–500 μm > 95%, Fermase CALB™10000), with a nominal activity of 10000 PLU per g (propyl laurate Units per gram dry weight), was purchased from Fermenta Biotech Ltd. Mumbai and kindly provided from Enzyme Catalyzed Polymers LLC, Akron, OH. Diphenyl ether was stored over 4 Å molecular sieves and CAL-B was dried at 40 °C under vacuum for 18 h prior to use. The other chemicals were used as received.
General procedure for CAL-B-catalyzed synthesis of PBS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :DLS copolymer via two-stage method in diphenyl ether
:DLS copolymer via two-stage method in diphenyl ether
CAL-B (10 wt% of total monomers) was added to a magnetically stirred solution containing 1,4-butanediol (BD, 1.56 g, 17.31 mmol), diethyl succinate (DS. 3.36 g, 19.30 mmol), and dimer linoleic diol (DLA–OH, 1.08 g, 1.99 mmol) in diphenyl ether (200 wt% of total monomers), in a three-necked round-bottom flask, placed in an oil heating bath equipped with a condenser. The amount of the monomers was calculated in order to obtain a multiblock copolymer with poly(butylene succinate) sequences constituting the hard segments and succinate–dimer linoleic diol sequences constituting the soft segments, in approximately 70:30 wt% segment ratio composition. The reaction protocol was adapted and modified from ref. 30 and the two following transesterification stages were performed (Scheme 1): (1) the first step was carried out at the initial temperature of 80 °C under N2 flow at atmospheric pressure. After 1 hour, when the reaction mixture was homogeneous and the CAL-B was well dispersed in the media, the temperature was slowly increased to 95 °C. The collection of ethanol was monitored until half of the theoretical amount of this by-product was collected (2–3 h) and further oligomerization was carried out under 600 torr for 18 h; (2) in the second step, the pressure was reduced to 2 torr while maintaining the reaction at 95 °C for 84 h. Upon completion, the formed product was dissolved in chloroform, the catalyst was removed via filtration and the chloroform solution was concentrated in a rotary evaporator and purified by dropwise precipitation into cold methanol under continuous stirring. The precipitated polymer was filtered, thoroughly washed with cold methanol, collected, and dried under vacuum (225 torr) at 40 °C. No residual solvent from the synthesis stage was detected in the 1H NMR and FTIR spectra. The reaction yield was >74% as calculated from the initial weight of the monomers and the weight of the obtained product after precipitation and drying, taking into account the amount of ethanol collected during the synthesis (step 1).
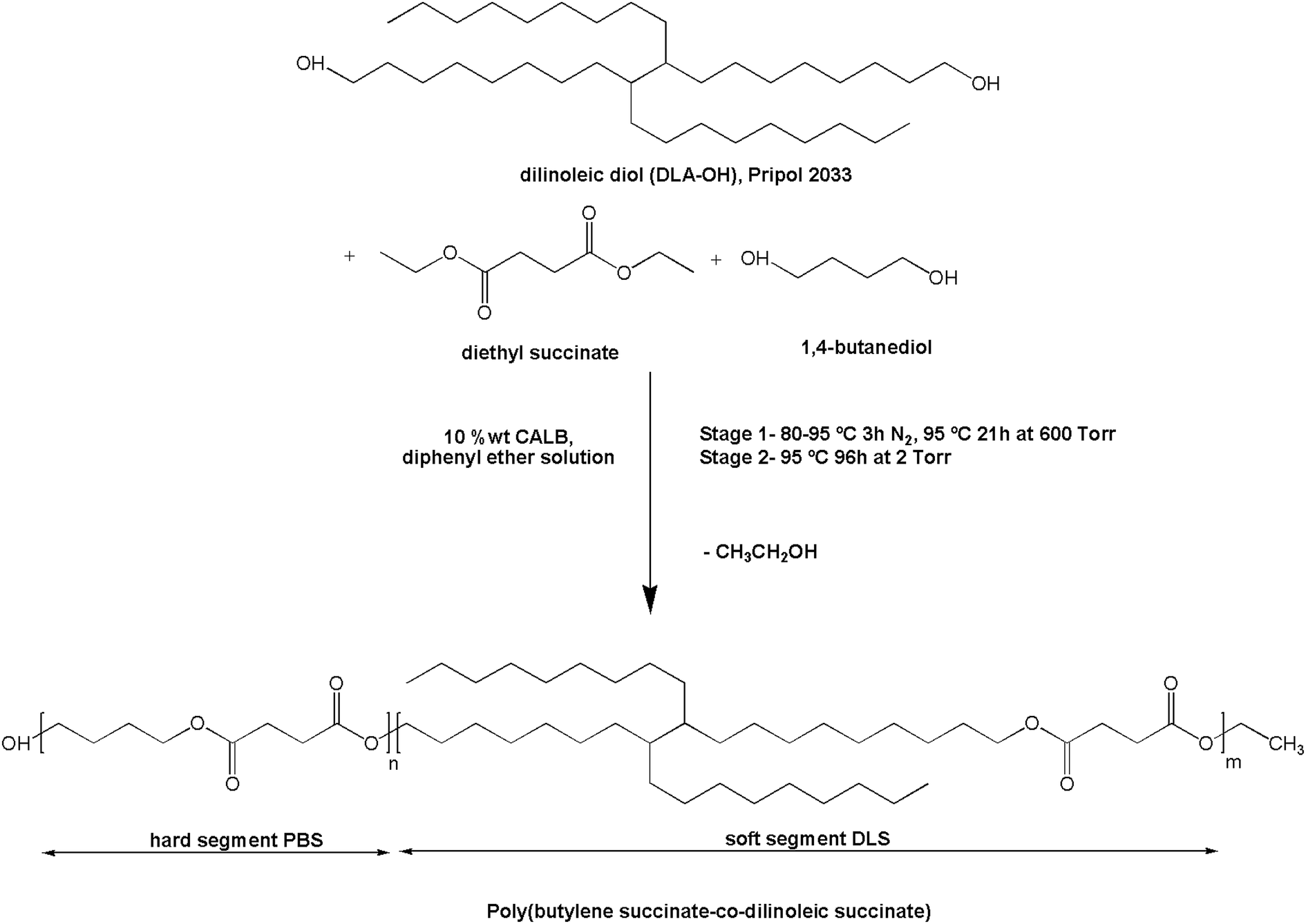 | ||
| Scheme 1 Synthesis conditions for the transesterification of dilinoleic diol, diethyl succinate and 1,4-butanediol catalyzed by CALB to form poly(butylene succinate-co-dilinoleic succinate) copolymer. | ||
Characterization of PBS:DLS copolymer
The chemical structure of the obtained copolymer was verified by nuclear magnetic resonance (1H NMR) and infrared spectroscopy (ATR-FTIR). The 1H NMR (128 scans, 1 second relaxation delay) and 13C NMR (5120 scans, 1 second relaxation delay) spectra were recorded in CDCl3 on a TM Bruker DPX 400 (400 MHz) spectrometer using tetramethylsilane (TMS) as internal reference. The composition was determined by 1H NMR from the ratio of the integrals of the signals associated with the butylene units arising from PBS hard segments at 1.76 ppm (4H) and dimer linoleic diol of the DLS soft segments at 4.12 ppm (6H). FTIR transmission spectra was recorded using a Bruker ALPHA spectrometer with an Attenuated Total Reflectance (ATR) cell between 400 and 4000 cm−1, with a 2 cm−1 resolution. Samples were vacuum-dried prior to measurement, and 32 scans were averaged across the spectral range. The intrinsic viscosity ([η]) was determined in chloroform at 25 °C in an Ubbelhode viscometer (K = 0.00342), and the results were calculated using the Solomon–Ciuta equation.31 The number-average molecular weight (Mn), weight-average molecular weight (Mw), and polydispersity index (PDI) of the PBS:DLS copolymer were determined by gel permeation chromatography (GPC). The experiment was conducted on a Tosoh EcoSEC, equipped with 2 fused silica separation columns and four detection systems: Dual Absorbance UV (UV, Waters 2487, wavelength λ = 254 nm), multi-angle laser light scattering (MALLS, Wyatt mini-DAWN Treos), refractive index (RI, Wyatt OPTILAB DSP), and viscometer (VIS, Wyatt ViscoStar-II). The results were analyzed using ASTRA 6 software (Wyatt Technology). The sample concentration was 2–3 mg mL−1 and CHCl3 (HPLC grade, Alfa Aesar), continuously distilled from CaH2, was used as eluent at a flow rate of 0.7 mL min−1; the injected volume was 15 μL. The run time was 60 min, in order to avoid the aftereffect of the viscometer in the differential refractive index (dRI) signal. The incremental refractive index (dn/dc) was calculated from the concentration and calculated mass, assuming 100% mass recovery from the columns. The thermal behaviour of PBS:DLS was studied in a TA Instruments DSC Q100 differential scanning calorimeter. Measurements were carried out in a heating–cooling–heating cycle over the temperature range from −90 to 200 °C, at a heating/cooling rate of 10 °C min−1, under a N2 atmosphere. The glass transition temperature (Tg) was calculated as the midpoint of the transition in the second heating run. The crystallinity content (Xc%) was calculated as crystallization degree of hard phase evaluated according to the following equation:| Xc% = (0.7 × ΔHm/ΔH0m) × 100 |
Electrospinning process
In order to investigate the ability of the enzymatically synthesized PBS:DLS 70:30 copolymer to be processed by electrospinning, different solution concentrations and solvents were tested to optimize the process. Solvents tested included DCM, CHCl3, DMF, THF, MeOH and their mixtures. All solutions were electrospun using a standard horizontal electrospinning setup. First, all air bubbles were purged and then a syringe pump was used to fee the solutions into the tip of a needle (21 gauge) directly connected to a high voltage power supply. The solutions were electrospun over a grounded flat stainless steel collector covered with aluminium foil. The optimized electrospinning parameters, solution concentration and solvent system were used to produce a thicker uniform nanofiber mat (60 ± 6.1 μm) without structural defects.
Characterization of PBS:DLS scaffolds
Morphological analysis of the electrospun fibers was carried out by means of scanning electron microscopy (SEM, JEOL JSM 6100). Samples were sputtered with gold and observed at 10 kV with a working distance of 14.4–15.7 mm. Fibre diameters were calculated as an average of 30 random measurements over at least two SEM micrographs at 2500× magnification using the ImageJ software (National Institute of Health, USA). A thicker mat was electrospun with experimentally found and optimized parameters and solvent system, and its thermal and mechanical properties were evaluated. The thermal behaviour was studied by using a differential scanning calorimeter (DSC, TA Instruments Q100) under the same conditions as previously described for the bulk material. In addition, the elongation at break, Young's modulus and maximum tensile strength were measured at room temperature by static tensile tests on a universal testing machine (Instron 3366, Instron Co., Ltd., USA) equipped with a 500 N load cell. The measurements were carried out at the crosshead speed of 1 mm min−1 from 0 to 0.5% elongation and of 10 mm min−1 from 0.5% elongation. Sample dimensions were typically 5 mm wide, 50 μm thick and 4 cm long while the length between clamps was 2 cm. Thickness was measured by using a coating thickness gauge (Elcometer Instruments GmbH, Germany) with Elcometer thickness standards, the mechanical data is an average of the six specimens.
Results and discussion
Characterization of PBS:DLS 70:30 copolymer
The synthesis of PBS:DLS 70:30 copolymer was achieved via two-step transesterification process using CAL-B as a catalyst and performed in diphenyl ether solution. The first step of the reaction under N2 and subsequently low vacuum is thought to prevent monomer loss due to evaporation to allow their conversion into non-volatile oligomers. The stepwise decreasing of the vacuum to avoid premature loss of monomers and oligomers at the early stage of the synthesis, together with the high vacuum during the second step that facilitates the removal of the ethanol by-product, as well as the high boiling point of diphenyl ether (allowing for high vacuum without loses) and its high octanol/water partition coefficient (logP = 4.21) (lipase activity is reported to be improved at higher logP than 1.9)32,33 resulted in the convenient promotion of the copolymer chain growth.30 In addition, other commercial forms of the CAL-B immobilized on macroporous acrylic resin particles have been proved to retain good activity at high temperatures in such a solvents as diphenyl ether (>80% of normalized activity at 100 °C after 24 h in diphenyl ether).33 Therefore, one must consider that all the aforementioned conditions led to the formation of a high molecular weight copolyester, helping to the reaction system to remain soluble with well dispersed catalyst and without a significant drop in its activity along the reaction time. Scheme 1 summarizes the general synthesis conditions for the transesterification of DS with BD and DLA–OH, to form poly(butylene succinate-co-dilinoleic succinate) (PBS:DLS) copolymer. Fig. S1–S3 (ESI†) show some possible structures as well as 1H NMR and 13C NMR spectra of the commercial PRIPOL 2033 DLA–OH employed, and are explained there in detail. The structure of the enzymatically synthesized PBS:DLS copolymer is depicted in Fig. 1 and its characteristics are given in Table 1. The composition was determined from 1H NMR confirming a final wt% and molar% composition of hard to soft segments close to the initial feed ratio, confirming that sufficient losses of monomers were avoided with the stepwise decrease of the pressure during the reaction. In 1H NMR, signals appearing at δ1H = 4.09–4.11 ppm (a) are due to the methylene protons of BD segment attached to the oxygen, confirming the reaction between the BD and DS. Methylene protons of DS and the rest from BD appear at δ1H = 2.61 ppm (c), and δ1H = 1.65–1.73 ppm (b), respectively. The signals at δ1H = 4.04–4.08 ppm (d) are due to the methylene protons of DLS segment adjacent to the ester bond confirming the transesterification reaction between the DLA–OH and the DS. The tertiary carbon protons appear at δ1H = 1.57–1.63 ppm (f), methylene groups of DLA appear around δ1H = 1.25 ppm (e) and methyl end groups appear at δ1H = 0.87 ppm (g). The low intensity resonances at δ1H = 3.64–3.68 ppm (a′) are due to the proton signals from the BD methylene units at the end of PBS–OH hard segment chain end groups. Note that no residual signal of unreacted hydroxyl groups from DLA–OH were observed in the 1H NMR spectra (δ1H = 3.61–3.64 ppm). The protons from the DS methylene units at the end of the soft segment chain are not easily visible as they appear in the region near to δ1H = 4.11 ppm where the methylene protons of butanediol are present. Fig. S4 and S5 (ESI†) shows the 13C NMR spectra of the PBS:DLS 70:30 copolymer. The two different methylene carbons next to the oxygen in the ester bonds appear at δ13C = 64.94 ppm (h), due to the reaction between DLA–OH and DS, and at δ13C = 64.17 ppm (a), due the reaction between BD and DS. These facts together with the disappearance of the signal at δ13C = 63.02 ppm, related to the carbon attached to the –OH ending of DLA–OH (Fig. S3, ESI†), as well as the shifting up-field of the (g) signal (–CH2CH2OH, from 32.81 ppm to –CH2CH2O– at 28.58), indicate the formation of the desired structure of the copolymer as well as the incorporation of the DLA–OH between DS units. The signal at δ13C = 172.29 ppm (d) is due to the –CO– from DS attached to DLA–OH, and signals at δ13C = 29.02 ppm (b), and δ13C = 25.21 ppm (c) are due to the –CH2– from DS and BD units, respectively. Low intensity signals due to end-groups were detected at δ13C = 62.2 ppm, δ13C = 29.09 ppm, δ13C = 25.06 ppm and δ13C = 64.57 ppm from OHCH2CH2CH2CH2O– of BD and at δ13C = 172.37 ppm (d′) from –COCH2CH3 and at δ13C = 14.19 ppm (o) from –COCH2CH3 of DS. The signal at δ13C = 14.4 ppm is related to the –CH3 from DLA–OH and the rest of the signals at δ13C = 22.69–31.92 ppm are related to the aliphatic chain from DLA–OH.
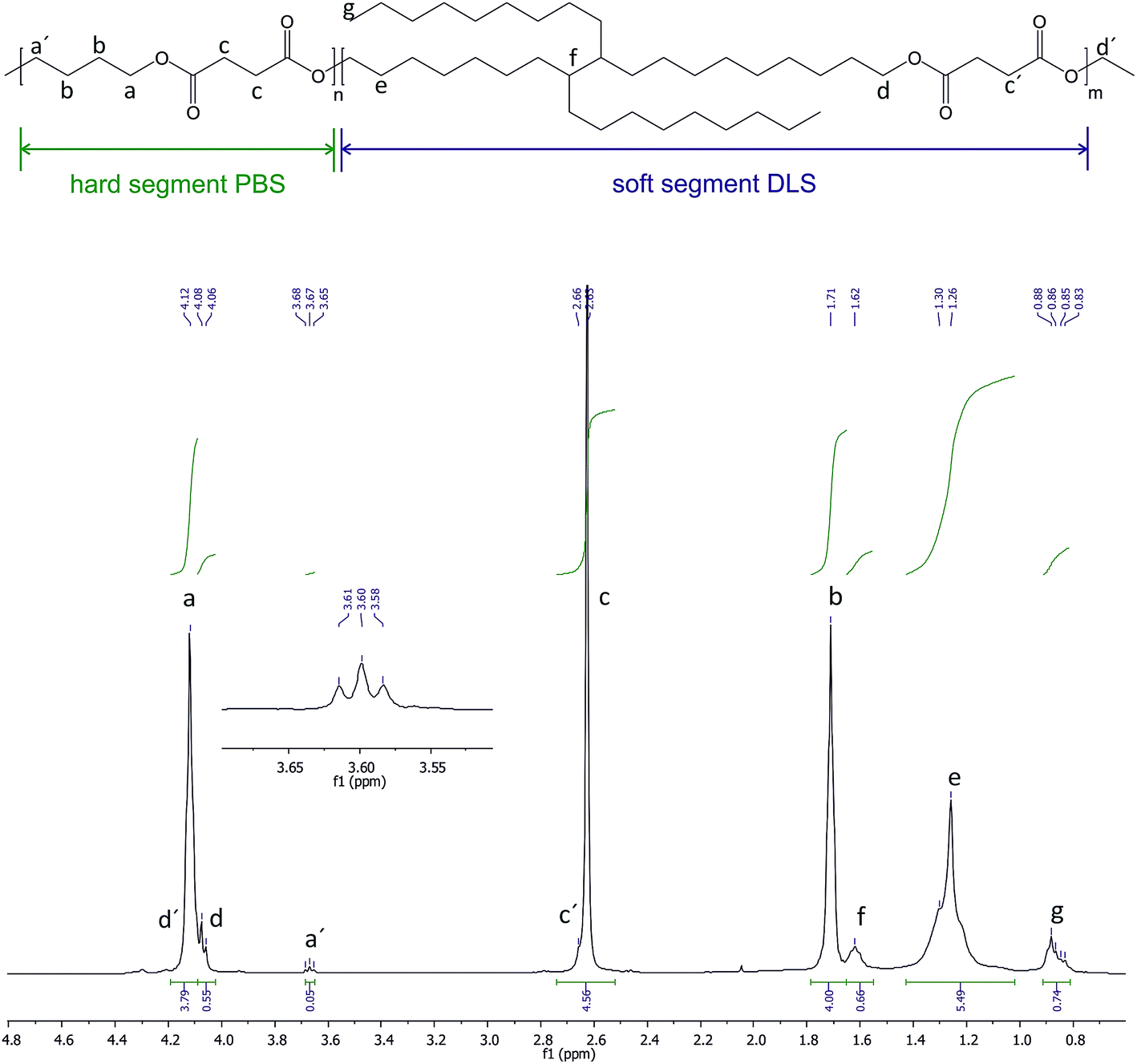 | ||
| Fig. 1 1H NMR of the poly(butylene succinate-co-dilinoleic succinate) 70:30 copolymer. | ||
:DLS copolymer
| Sample | Experimental conditions | 1H NMR | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Hs/Ss (mol%) | Hs/Ss (wt%) | Hs/Ssa (mol%) | Hs/Ssa (wt%) | DPa [Hs] | Mwb (103 g mol−1) | Mnb (103 g mol−1) | Mw/Mnb | [η] [dl g−1] | |
| a Calculated from 1H NMR.b Calculated from GPC/SEC, DP [Hs]-number of hard segment repeating units; Mw – weight average molar mass, Mn – number average molar mass; [η] – intrinsic viscosity. | |||||||||
| Bulk PBS:DLS 70:30 |
89.7/10.3 | 70/30 | 89/11 | 68.5/31.5 | 40 | 34.7 | 12.8 | 2.7 | 0.56 |
Fig. 2 shows the ATR-FTIR spectra of BD and DLA–OH (Fig. 2a and b) monomers and the obtained copolymer processed into electrospun mat (Fig. 2c and d). As can be seen from Fig. 2c (PBS:DLS 70:30 bulk material), the ester formation and the presence of polymerized DLA–OH units is confirmed and is in agreement with 1H NMR results. The characteristic absorptions bands for carbonyl-stretching vibrations (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, 1713 cm−1) and C–O–C stretching (1153 cm−1) of the ester groups in the polymer backbone confirm the successful formation of a polyester. Comparing the spectra for DLA–OH (Fig. 2b) and PBS:DLS copolymer (Fig. 2c), it can be seen that the predominant bands at 2923 cm−1 and 2854 cm−1 correspond to the methylene groups (–CH2–) of the aliphatic long chain soft segments due to the polymerized DLA–OH that at the same time overlap the bands from aliphatic chain (–CH–) from hard segments. The lack of an absorbance band between 3400 and 3500 cm−1 in the copolymer spectra confirms the reaction of the hydroxyl groups from DLA–OH (Fig. 2b) and BD (Fig. 2a)—no OH functionality remains.
O, 1713 cm−1) and C–O–C stretching (1153 cm−1) of the ester groups in the polymer backbone confirm the successful formation of a polyester. Comparing the spectra for DLA–OH (Fig. 2b) and PBS:DLS copolymer (Fig. 2c), it can be seen that the predominant bands at 2923 cm−1 and 2854 cm−1 correspond to the methylene groups (–CH2–) of the aliphatic long chain soft segments due to the polymerized DLA–OH that at the same time overlap the bands from aliphatic chain (–CH–) from hard segments. The lack of an absorbance band between 3400 and 3500 cm−1 in the copolymer spectra confirms the reaction of the hydroxyl groups from DLA–OH (Fig. 2b) and BD (Fig. 2a)—no OH functionality remains.
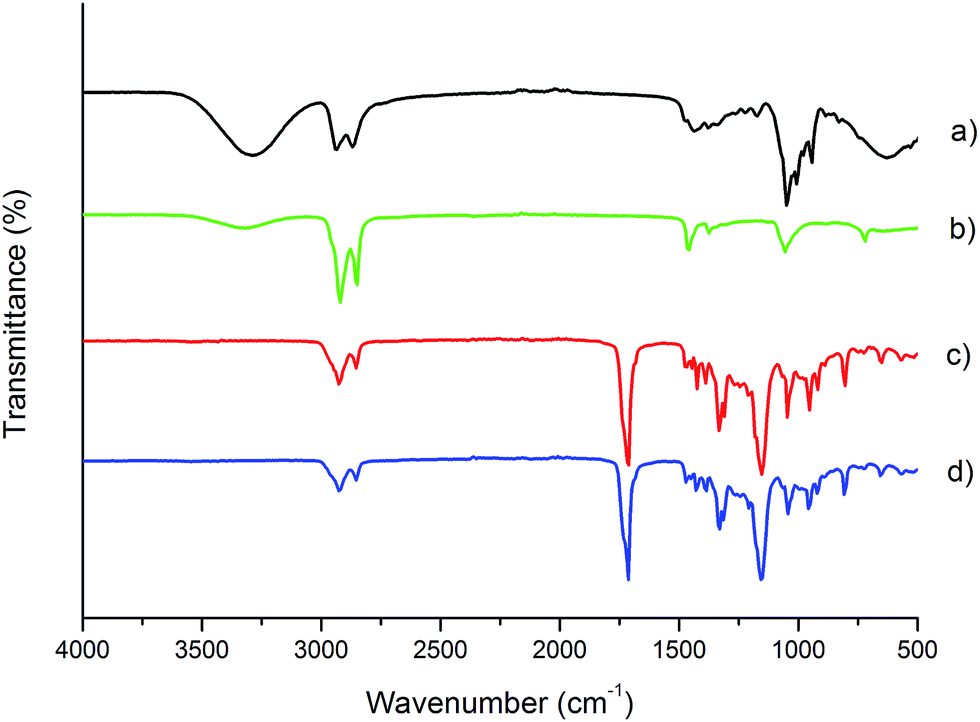 | ||
| Fig. 2 FT-IR spectrum of: 1,4 butanediol (a), Pripol 2033 (b), PBS:DLS 70:30 polymer after precipitation and drying (c) and PBS:DLS 70:30 after electrospinning from a 25 w/v% in a CHCl3:MeOH solution (d). | ||
The number-average molar mass (Mn) and number-average molecular weight (Mw) of the copolyester were estimated by GPC as 12.8 × 103 g mol−1 and 34.7 × 103 g mol−1 respectively, holding a polydispersity index (PDI, Mw/Mn) of 2.7 (Table 1). Although high molecular weight poly(butylene succinate) homopolymer has been achieved under similar conditions,30 just a few reports attempted to enzymatically synthesize copolymers under similar conditions and based on poly(butylene succinate), either with poly((R)-3-hydroxybutyrate)34 or dimethyl itaconate.35 Moreover, for the first time, the enzymatic transesterification between poly(butylene succinate) (PBS), and a fatty dimer alcohol is presented and processed in a useful morphology, suitable for potential biomedical applications on account of the sufficient molecular weight obtained.
In general, the electrospinnability of low molecular weight polymers is strongly limited due to the low amount of molecular entanglements between polymeric chains in solution.36,37 Usually, high molecular weight or high concentrations improve the electrospinning of the polymers favouring the formation of fibres instead of drops.38 Thus, the obtained molecular weight is expected to ensure the electrospinnability of the system using typical solution concentrations for similar copolymers previously synthesized with organometallic catalysts.10
Finally, the thermal properties for the bulk-synthesized copolymer are in agreement with the previous results, confirming that the desired material was obtained. Importantly, as can be seen from Fig. S6 (ESI†) and Table 2, the low temperature Tg of soft segments and high temperature melting Tm of hard segments indicate the formation of thermoplastic segmented elastomer. Additionally, the characteristic temperature transitions reported here for the enzymatically synthesized bulk material are comparable to those of PBS:DLS 70:30 wt% hard to soft segments ratio synthesized previously in our laboratory using Ti(BuO)4 organometallic catalyst.10
:DLS copolymer (powder) and its electrospun fibers from a 25 w/v% in a CHCl3:MeOH solutiona
| Sample | Tc (°C) | Tg (°C) | Tm (°C) | ΔHc (J g−1) | ΔHm (J g−1) | ΔH0m (J g−1) | Xc (%) |
|---|---|---|---|---|---|---|---|
| a Tc – crystallization temperature, Tg – glass transition of soft segments, Tm – melting point of hard segments, ΔHc – crystallization enthalpy; ΔHm – melting enthalpy; ΔH0m – melting enthalpy of 100% crystalline PBS, Xc – degree of crystallization of hard phase from DSC. | |||||||
| Powder PBS:DLS 70:30 |
51.1 | −44.2 | 100.4 | 54.4 | 51.2 | 110.3 | 32.5 |
| Fiber PBS:DLS 70:30 |
48.5 | −42.7 | 100.3 | 63.0 | 59.5 | 110.3 | 37.7 |
Characterization of electrospun mats
It is well-known that the success of forming polymeric fibres during the electrospinning process depends strongly on the selection of the proper solvent system and polymer concentration.39,40 As a result, in the present work, several solvents and binary mixtures were tested at different concentrations until fibres were obtained instead of drops. Then, the other electrospinning process parameters such as voltage, flow rate, and needle-collector distance, were optimized to fix the best conditions in order to obtain PBS:DLS 70:30 copolymer fibres free of beads and other defects. Mainly CHCl3, MeOH, DCM, DMF, THF and their binary mixtures were tested. We observed that the copolymer had limited solubility in DMF, THF and their mixtures either with MeOH and CHCl3, and as a result, the best combinations were the mixtures of either CHCl3 or MeOH with DCM. Due to the volatility of the DCM, all the tested conditions with the 25 w/v% of polymer concentration in DCM:MeOH mixture clogged the needle. As a result, they were not electrospinnable for sufficient time to achieve a significant amount of fibres. Table 3 summarizes the successful tested electrospun systems using different parameters, concentrations and solvents to study their influence over the final morphology of the obtained fibres and the continuation of the process.
:DLS electrospun fibers from a 25 w/v% in a CHCl3:MeOH solution
| Mechanical properties PBS:DLS 70:30 mat |
|
|---|---|
| Young's modulus (MPa) | 24.4 ± 2.8 |
| Ultimate tensile strength (MPa) | 4.0 ± 0.4 |
| Elongation at break (%) | 63 ± 7.0 |
Fig. 3 and 4 illustrate SEM micrographs and diameter distributions of the fibers obtained using DCM:MeOH 7:3 (v/v) mixture as a solvent system. Fig. 5 presents the SEM micrographs and fibre diameter distributions obtained using CHCl3 instead of DCM in the solution. Slight differences in the fibre diameters were observed depending on the electrospinning parameters. Increasing the flow rate from 0.6 to 2 mL h−1 while retaining the same voltage and distance (15 kV and 15 cm), resulted in 25% greater average fibre diameters (Fig. 4a/5a and 4c/5c), as a larger amount of material flows from the needle tip to the collector. However, increasing the flow rate from 0.6 to 2 mL h−1 for collector-needle distances of 20 cm and voltages of 15 kV, resulted in beaded fibres with larger diameters and less homogeneous and wider distributions (Fig. 4b/5b and 4d/5d). These can be due to higher instabilities resulting from the predominant effect of gravitational force over electrical field. The inhomogeneity and beads progressively disappeared with increasing the voltage to 17 kV (Fig. 4e/5e and 4f/5f), i.e. by adjusting the voltage to the flow rate and distance.
 | ||
| Fig. 3 SEM micrographs of poly(butylene succinate-co-dilinoleic succinate) 70:30 copolymer electrospun samples. The polymer concentration was 20 wt/v% in a binary mixture of DCM:MeOH 7:3 for all the samples. The needle-collector distance, flow rate and voltage conditions were as follows: (a) 15 cm, 0.6 mL h−1, 15 kV; (b) 15 cm, 2 mL h−1, 15 kV; (c) 15 cm, 2 mL h−1, 17 kV; (d) 20 cm, 0.6 mL h−1, 15 kV; (e) 20 cm, 2 mL h−1, 15 kV; (f) 20 cm, 2 mL h−1, 17 kV. | ||
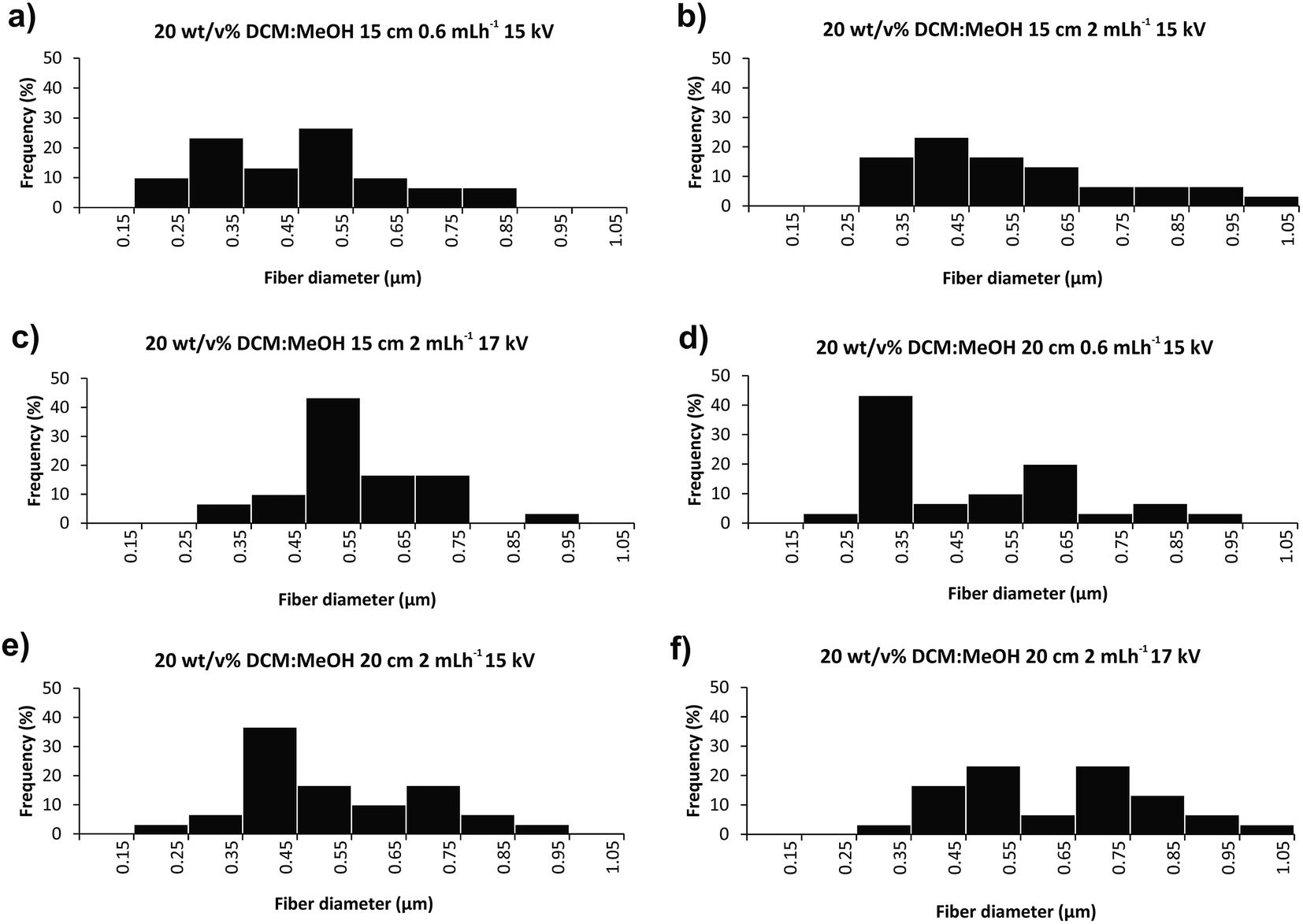 | ||
| Fig. 4 Diameter distribution obtained from SEM micrographs of poly(butylene succinate-co-dilinoleic succinate) 70:30 copolymer electrospun samples. The polymer concentration was 20 wt/v% in a binary mixture of DCM:MeOH 7:3 for all the samples. The needle-collector distance, flow rate and voltage conditions were: (a) 15 cm, 0.6 mL h−1, 15 kV; (b) 15 cm, 0.6 mL h−1, 15 kV; (c) 15 cm, 2 mL h−1, 17 kV; (d) 20 cm, 0.6 mL h−1, 15 kV; (e) 20 cm, 0.6 mL h−1, 15 kV; (f) 20 cm, 2 mL h−1, 17 kV. | ||
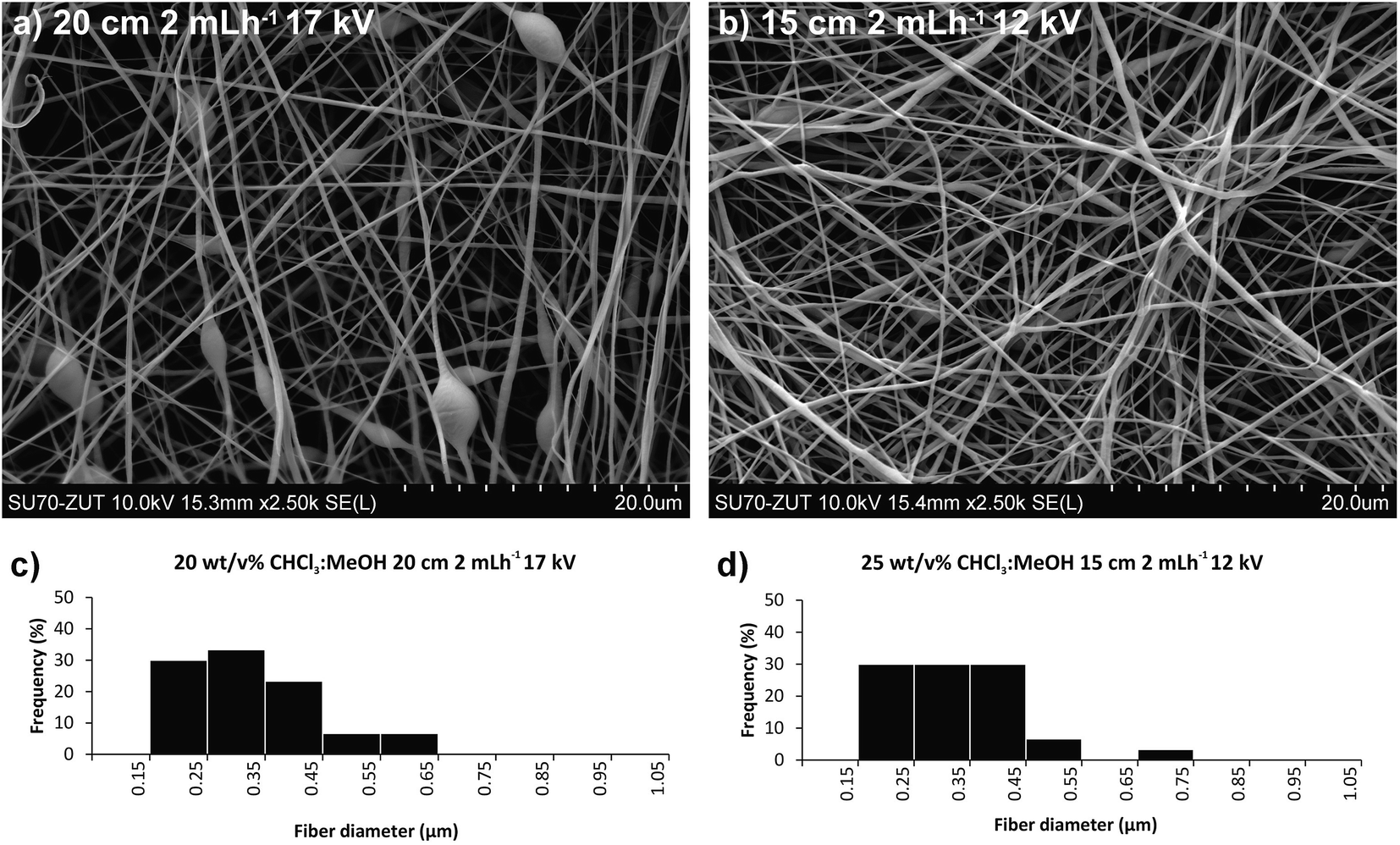 | ||
| Fig. 5 Top; SEM micrographs of poly(butylene succinate-co-dilinoleic succinate) 70:30 copolymer electrospun samples obtained from a binary mixture of CHCl3:MeOH 7:3. (a) The polymer concentration was 20 wt/v%, the needle-collector distance 15 cm, the flow rate 0.6 mL h−1 and the voltage 15 kV; (b) the polymer concentration was 25 wt/v%, the needle-collector distance 15 cm, the flow rate 2 mL h−1 and the voltage 12 kV; Bottom; diameter distribution obtained from SEM micrographs of poly(butylene succinate-co-dilinoleic succinate) 70:30 copolymer electrospun samples from a binary mixture of CHCl3:MeOH 7:3; (c) the polymer concentration was 20 wt/v%, the needle-collector distance 15 cm, the flow rate 0.6 mL h−1 and the voltage 15 kV; (d) the polymer concentration was 25 wt/v%, the needle-collector distance 15 cm, the flow rate 2 mL h−1 and the voltage 12 kV. | ||
In comparison with the DCM:MeOH 7:3 solvent system, by using CHCl3:MeOH 7:3 mixture enabled electrospinning of higher polymer concentrations (25 w/v%) without clogging the needle. Using the same polymer concentration and the same conditions as in the DCM:MeOH 7:3 system, lower average fibre diameters were obtained in case of CHCl3:MeOH. As can be seen in Fig. 5, for the same polymer concentration (20 w/v%) and electrospinning conditions, the solvent system affects strongly the average fibre diameter, reducing it by half, from 0.631 ± 0.2 μm to 0.335 ± 0.11 μm, when DCM:CHCl3 7:3 mixture is used instead of DCM:MeOH 7:3. However, all the conditions tested with the 20 w/v% of polymer in DCM:CHCl3 7:3 resulted in beaded fibers, typically the result of too low concentrations/viscosity.41 However, increasing the polymer concentration to 25 w/v% in CHCl3:MeOH 7:3 (Fig. 5b), resulted in fibres without defects, while retaining smaller diameters (0.334 ± 0.11 μm) and narrower distributions. The optimal conditions to ensure a continuous process and a stable Taylor cone for this higher viscosity solution were found at slightly lower voltage, shorter distance and higher flow rate than for the 20 w/v% DCM:CHCl3 7:3 solution.
The FTIR spectra of a thicker electrospun mat (30 μm) obtained under these optimized conditions (Fig. 2e) showed no marked differences compared to that of the bulk polymer (Fig. 2d), confirming no influence of the electrospinning process or the solvents used on the chemical structure of the copolymer. Likewise, similar thermal properties (Tg, Tc and Tm values) were obtained for the electrospun mat and the bulk synthesized material (Table 2 and Fig. S6†), indicating no influence of the process over the material properties.
Regarding the mechanical properties, the values obtained for the enzymatic PBS:DLS 70:30 fiber mats are comparable to those reported in the literature for related materials obtained by organometallic catalysis.10,15 Due to the higher presence of hard segments −70 wt% in reported material, the fiber mat possesses higher values of Young's modulus (24.2 ± 2.8 MPa) and maximum tensile strength (4.0 ± 0.4 MPa), as well as a decrease in the elongation at break (63 ± 7.0%) in comparison with previously reported values for a mat obtained with a related organometallic PBS:DLS material with lower presence of hard segments (50 wt%). The correlation between the organometallic and enzymatic systems together with our current research work, open the array of possibilities to obtain copolymers containing DLA–OH soft segments able to cover a wide range of properties by adjusting the hard to soft segments ratio.
Conclusions
For the first time, Candida antarctica lipase B enzyme (CAL-B) has been used as a catalyst for co-polymerization of diethyl succinate, dimer linoleic diol and 1,4-butanediol. 1H NMR demonstrated that, a copolyester with dilinoleic molecules as the building block for the soft segments was synthesized via enzyme-catalysed polymerization reaction obtaining a poly(butylene succinate-co-dilinoleic succinate) (PBS:DLS) copolymer with a 70:30 wt% hard to soft segment ratio via an optimized two-stage synthesis method. During the first reaction step, the method allows for the control of the reaction by-product, as well as avoiding losses of monomers during the earlier conversion into oligomers. This method facilitates the future reproducibility of the synthesis, as well as the chain growth, during the second, high vacuum step resulting in high molecular weight and good reaction yields. Thermal and chemical characterization of the material showed that its properties were comparable to those obtained by conventional organometallic synthesis. Ongoing experiments in our laboratory indicate that the described synthetic protocol also works for higher amount of DLS soft segments into the PBS:DLS copolymer, while retaining reaction yields >74%—these will be reported in the future together with kinetics studies. In addition, we found that optimizing the polymerization method yielded a copolyester with sufficiently high molecular weight to be processed into homogeneous electrospun mats without defects. After the optimization of the electrospinning parameters a thicker mat was obtained and its morphological, mechanical and thermal properties were evaluated. Thanks to the positive results described here, further studies will assess the suitability of these materials for biomedical applications, including cardiac tissue engineering.
Acknowledgements
This work is financially supported by National Science Centre, Poland, under the HARMONIA scheme (agreement No. UMO-2014/14/M/ST8/00610). The authors thank Prof. Judit Puskas and Andrew McClain (The University of Akron) for GPC measurements, and Dr Peter Sobolewski for reviewing the English.References
- M. Vert, Biomacromolecules, 2005, 6, 538–546 CrossRef CAS PubMed.
- P. A. Gunatillake and R. Adhikari, Eur. Cells Mater., 2003, 5, 1–16 CrossRef CAS PubMed.
- W. J. Li and J. A. Cooper, in Biomaterials for Tissue Engineering Applications, ed. J. A. Burdick and R. L. Mauck, Springer Vienna, Vienna, 1st edn, 2011, pp. 47–73 Search PubMed.
- B. P. Chan and K. W. Leong, Eur. Spine J., 2008, 17, 467–479 CrossRef PubMed.
- E. Bressan, V. Favero, C. Gardin, L. Ferroni, L. Iacobellis, L. Favero, V. Vindigni, M. Berengo, S. Sivolella and B. Zavan, Polymers, 2011, 3, 509–526 CrossRef CAS.
- Y. D. Wang, G. A. Ameer, B. J. Sheppard and R. Langer, Nat. Biotechnol., 2002, 20, 602–606 CrossRef CAS PubMed.
- C. J. Bettinger, Macromol. Biosci., 2011, 11, 467–482 CrossRef CAS PubMed.
- C. L. E. Nijst, J. P. Bruggeman, J. M. Karp, L. Ferreira, A. Zumbuehl, C. J. Bettinger and R. Langer, Biomacromolecules, 2007, 8, 3067–3073 CrossRef CAS PubMed.
- J. Skrobot, L. Zair, M. Ostrowski and M. El Fray, Biomaterials, 2016, 75, 182–192 CrossRef CAS PubMed.
- L. Liverani, A. Piegat, A. Niemczyk, M. El and A. R. Boccaccini, Eur. Polym. J., 2016, 81, 295–306 CrossRef CAS.
- R. Khosravi, C. A. Best, R. A. Allen, C. E. T. Stowell, E. Onwuka, J. J. Zhuang, Y. U. Lee, T. Yi, M. R. Bersi, T. Shinoka, J. D. Humphrey, Y. Wang and C. K. Breuer, Ann. Biomed. Eng., 2016, 44, 2402–2416 CrossRef PubMed.
- J. Xu and B. H. Guo, Biotechnol. J., 2010, 5, 1149–1163 CrossRef CAS PubMed.
- L. Wu, R. Mincheva, Y. Xu, J.-M. Raquez and P. Dubois, Biomacromolecules, 2012, 13, 2973–2981 CrossRef CAS PubMed.
- A. Kozlowska, D. Gromadzki, M. El Fray and P. Štěpánek, Fibres Text. East. Eur., 2008, 16, 85–88 CAS.
- M. Tallawi, D. C. Zebrowski, R. Rai, J. A. Roether, D. W. Schubert, M. El Fray, F. B. Engel, K. E. Aifantis and A. R. Boccaccini, Tissue Eng., Part C, 2015, 21, 585–596 CrossRef CAS PubMed.
- N. Jacquel, F. Freyermouth, F. Fenouillot, A. Rousseau, J. P. Pascault, P. Fuertes and R. Saint-Loup, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5301–5312 CrossRef CAS.
- R. A. Gross, M. Ganesh and W. Lu, Trends Biotechnol., 2010, 28, 435–443 CrossRef CAS PubMed.
- J. C. Simitzis, L. T. Zoumpoulakis, S. K. Soulis and L. N. Mendrinos, Microchim. Acta, 2001, 136, 171–174 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green chemistry: theory and practice, Oxford University Press, OUP USA, 2000 Search PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- S. Kobayashi and A. Makino, Chem. Rev., 2009, 109, 5288–5353 CrossRef CAS PubMed.
- S. Kobayashi, H. Uyama and S. Kimura, Chem. Rev., 2001, 101, 3793–3818 CrossRef CAS PubMed.
- I. K. Varma, A. C. Albertsson, R. Rajkhowa and R. K. Srivastava, Prog. Polym. Sci., 2005, 30, 949–981 CrossRef CAS.
- E. M. Anderson, K. M. Larsson and O. Kirk, Biocatal. Biotransform., 1998, 16, 181–204 CrossRef CAS.
- H. Uyama and S. Kobayashi, Adv. Polym. Sci., 2006, 194, 133–158 CrossRef CAS.
- H. Uyama, S. Yaguchi and S. Kobayashi, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2737–2745 CrossRef CAS.
- Y. Y. Linko, M. Lämsä, X. Wu, E. Uosukainen, J. Seppälä and P. Linko, J. Biotechnol., 1998, 66, 41–50 CrossRef CAS PubMed.
- A. Mahapatro, B. Kalra, A. Kumar and R. A. Gross, Biomacromolecules, 2003, 4, 544–551 CrossRef CAS PubMed.
- R. K. Srivastava and A. C. Albertsson, Biomacromolecules, 2006, 7, 2531–2538 CrossRef CAS PubMed.
- H. Azim, A. Dekhterman, Z. Jiang and R. A. Gross, Biomacromolecules, 2006, 7, 3093–3097 CrossRef CAS PubMed.
- O. F. Solomon and I. Z. Ciutǎ, J. Appl. Polym. Sci., 1962, 6, 683–686 CrossRef CAS.
- A. Kumar and R. A. Gross, Biomacromolecules, 2000, 1, 133–138 CrossRef CAS PubMed.
- Y. Poojari and S. J. Clarson, Biocatal. Agric. Biotechnol., 2013, 2, 7–11 Search PubMed.
- T. Debuissy, E. Pollet and L. Avérous, Biomacromolecules, 2016, 17, 4054–4063 CrossRef CAS PubMed.
- Z. Yang, X. Zhang, X. Luo, Q. Jiang, J. Liu and Z. Jiang, Macromolecules, 2013, 46, 1743–1753 CrossRef CAS.
- W. Zhong, in An introduction to Health Care and Medical Textiles, Taylor & Francis Group, Lancaster, Pensylvania U.S.A., 1st edn, 2013, pp. 37–68 Search PubMed.
- S. Ramakrishna, K. Fujihara, W. E. Teo, T. C. Lim and Z. Ma, An Introduction to Electrospinning and Nanofibers, World Scientific, Singapur, 2005 Search PubMed.
- E. Llorens, L. J. del Valle, R. Ferrán, A. Rodríguez-Galán and J. Puiggalí, J. Polym. Res., 2014, 21, 360 CrossRef.
- D. Li and Y. Xia, Adv. Mater., 2004, 16, 1151–1170 CrossRef CAS.
- J. Deitzel, J. Kleinmeyer, D. Harris and N. Beck Tan, Polymer, 2001, 42, 261–272 CrossRef CAS.
- H. Fong, I. Chun and D. Reneker, Polymer, 1999, 40, 4585–4592 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02509b |
| This journal is © The Royal Society of Chemistry 2017 |