
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reverse thermo-responsive hydrogels prepared from Pluronic F127 and gelatin composite materials†
Mei-Yu Yehab,
Jiong-Yao Zhaoa,
Yi-Ru Hsiehc,
Jhong-Hua Linc,
Fang-Yi Chenc,
Rajan Deepan Chakravarthyc,
Pei-Chun Chungb,
Hsin-Chieh Lin *c and
Shih-Chieh Hung*bd
*c and
Shih-Chieh Hung*bd
aGraduate Institute of Biomedical Sciences, China Medical University, Taichung 40402, Taiwan
bIntegrative Stem Cell Center, China Medical University Hospital, Taichung 40447, Taiwan. E-mail: hung3340@gmail.com
cDepartment of Materials Science and Engineering, National Chiao Tung University, Hsinchu 30010, Taiwan. E-mail: hclin45@nctu.edu.tw
dInstitute of New Drug Development, China Medical University, Taichung 40402, Taiwan
First published on 18th April 2017
Abstract
Gelatin is an important hydrocolloid which has widespread applications in the pharmaceutical industry and biomedical science. However, despite its significance, clinical applications of gelatin are largely limited because of its gel-to-solution transition around human body temperature. In this work, we developed a series of F127–gelatin composite hydrogels with reverse thermo-responsive properties. The stiffness of F127–gelatin hydrogels can be adjusted by varying the types and contents of gelatin (type A, GA and type B, GB). The results of rheological data showed that F127–GA gels have higher gel strengths than those of F127–GB, which might be due to different isoelectric points between GA and GB. In addition, F127–gelatin gels showed significant cell viability and biocompatibility for human mesenchymal stem cells, indicating that these discovered hydrogels can serve as potential biomaterials and implantable scaffolds for applications in tissue engineering and regenerative medicine.
Introduction
Stimuli-responsive hydrogels have great potential for a wide range of applications such as bioactive sensors, enzyme assays, materials for tissue engineering and carriers for drug delivery.1–5 The most common method for preparing such hydrogels is through physical crosslinking between (macro)molecules. Physical crosslinking is the collection of noncovalent interactions between (macro)molecules such as π–π stacking, hydrogen bonding, dipole–dipole interactions, hydrophobic interactions and other van der Waals forces, which are generally responsible for the molecular self-assembly and hydrogelation.6–9 Physical crosslinked hydrogelation has been extensively studied, induced through various stimuli, including temperature,10 pH change,11,12 light,13 electrical,14 enzymes,15,16 ultrasound,12,17 and solvent polarity.18 Among all the physical stimuli, temperature has attracted a great deal of attention due to its ability to change features such as hydrophobic–hydrophilic balance, conformation and solubility, which is potentially useful for a variety of applications.19Gelatin is one of the temperature responsive polymers, which is the denatured product of collagen. Generally, gelatin is divided into two types based on the fabrication process: type A gelatin (GA) is obtained from an acid process while type B gelatin (GB) is produced from an alkaline process. Different processes lead to differences in the isoelectric point (pI) and the pH of gelatin.20 It possesses excellent biocompatibility, biodegradability, non-immunogenicity, and has capacity to assist cell attachment and proliferation.21–23 However, the gelatin hydrogels are in the solution state around human body temperature and form physical thermo-reversible gels on cooling.24 The gelatin hydrogels thus have poor mechanical strength, weak elasticity and low shape stability,25 which significantly limit their biomedical applications at physiological temperatures. Various strategies have been employed to overcome this problem, for example adding chemical crosslinker to gelatin, such as glutaraldehyde,26 carbodiimide27 and epoxy28 compounds; chemical modification of gelatin to photo-crosslinkable materials, such as gelatin methacrylamide.29 Nevertheless, gelatin hydrogels prepared using these chemical methods are cytotoxic and induce chronic inflammation. Therefore, the non-toxic physical crosslinking method has been chosen in this study. Herein, the amphiphilic copolymer, Pluronic F127 (F127) was used, since F127 possess the ability to form a physically crosslinked gel under the appropriate conditions.30,31 Moreover, it is known that mechanical properties of gel scaffolds can affect the behavior of cells.32 In this work, gelatin was incorporated into F127 to alter the gelation and stiffness properties of the composite hydrogels by adjusting the hydrophobic–hydrophilic balance in the system (Scheme 1). A series of F127–gelatin composite hydrogels were developed by using different types and contents of gelatin (GA and GB). The aims of this study are threefold: first, to fabricate gelatin-containing hydrogels which are stable under physiological temperature; second, to construct the composite hydrogels with tunable mechanical strengths; third, to evaluate the biocompatibility of the composite hydrogels with human mesenchymal stem cells (hMSCs). We successfully obtained the thermo-responsive and stiffness-tunable composite hydrogels, making them potential candidates as injectable biomaterials for tissue engineering and drug delivery applications. Furthermore, the thermo-reversible characteristic of the gels can be easily dissolved for cell recovery by cooling the composite hydrogels below their gelation temperature, which is attractive for fundamental study of cell–material interactions in three-dimensional (3D) cultures.
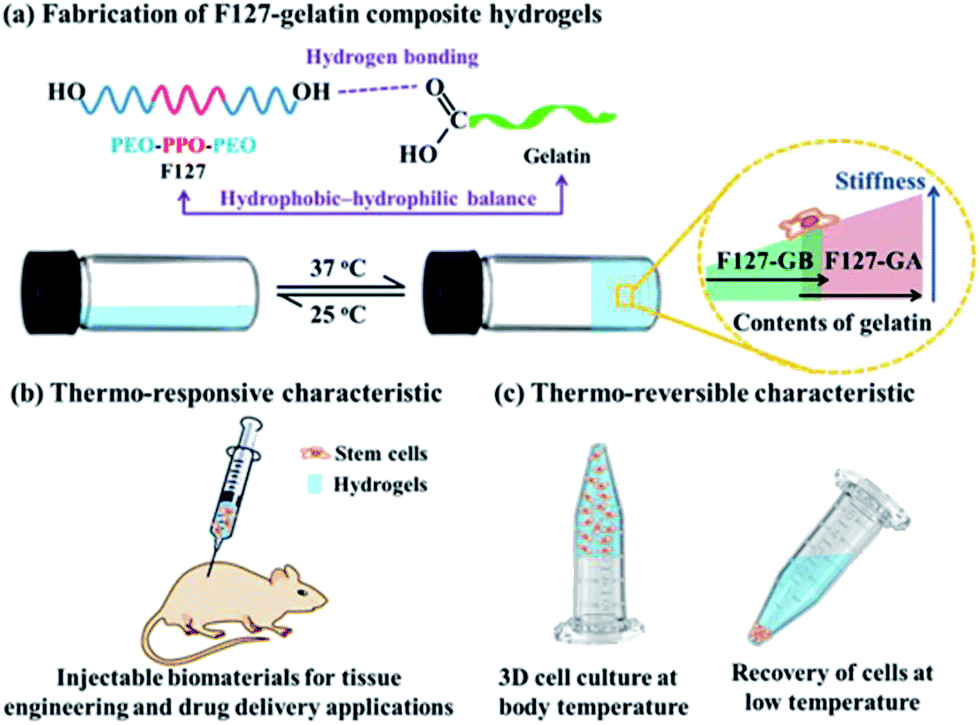 | ||
| Scheme 1 Fabrication of F127–gelatin composite hydrogels and their potential bioapplications (PEO: ethylene oxide; PPO propylene oxide). | ||
Results and discussion
The thermo-responsive composite hydrogels of F127–GA were prepared with various ratios of GA and F127 (16 wt%). Hydrogel samples with GA content of 1, 2, 3, 4 and 5 wt% were labelled as A1–A5 respectively. Initially, we just obtained two phase separated gels and these results may be due to the different hydrophile–lipophile balance between F127 and GA.33,34 To overcome this problem, we introduce a nontoxic and biocompatible surfactant, i.e. sodium carboxymethyl cellulose,35,36 into the system of the F127–GA and furnished the homogeneous gels (Fig. S1†). Similarly, samples with gelatin GB were prepared labeled as B1–B5. The thermo-responsive behaviors of A1–A5 and B1–B5 hydrogels were studied by the inverted tube method and all samples showed temperature dependent sol–gel–sol-transitions (Fig. 1). As depicted in Fig. 1, the A1–A5 and B1–B5 were in solution phases when the temperatures lower than their phase transition temperatures but formed transparent hydrogels with the temperatures higher than the sol–gel transition temperatures (Tsol–gel). The Tsol–gel values are around 29 °C for A1–A3 and 30–34 °C for B1–B3, respectively, indicating that these samples are in solution phase at room temperature (Table S1†). The gel–sol transition temperatures (Tgel–sol) of A1–A5 and B1–B5 are all higher than 48 °C, demonstrating all the materials are stable gels at body relevance temperature (Table S1 and Fig. S2†). In addition, it is interesting to note that the increasing the amount of gelatin would result in an increase in Tgel–sol (from 48 to 77 °C), which might be due to the enhancement of hydrogen bonding interactions between F127 and gelatin.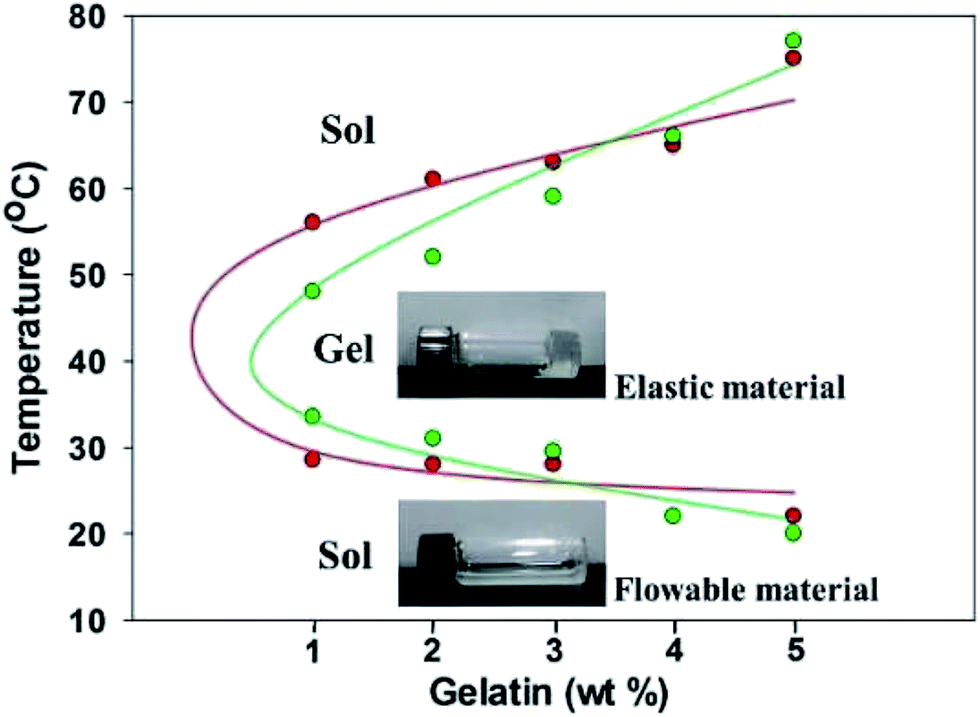 | ||
| Fig. 1 Phase diagrams of the composite hydrogels of A1–A5 and B1–B5 in aqueous solution. Red: A1–A5; green: B1–B5. | ||
It has been intensively investigated that the substrate stiffness could affect the response of the soft tissue cells and it may play an important role in directing cell behavior.32 For example, the substrate stiffness of neurons and epithelial cells are 0.1–1 kPa. Myoblasts require intermediate compliance substrates (around 4 kPa) to maintain function and chondrocytes display increased growth and proliferation markers on hard gels with stiffness of 10 kPa. Dynamic rheology was performed at 37 °C to investigate the mechanical properties of hydrogels A1–A5 and B1–B5. As depicted in Fig. 2, the frequency sweep data of A1–A5 and B1–B5 reveal typical solid-like rheological behavior with storage modulus (G′) dominating the loss modulus (G′′) over the investigated oscillating frequency (0.1–100 rad·s−1). The G′ value of single-component F127 gel (16 wt%) is 5.8 kPa at 37 °C (Fig. S3†). For mixed gels, the G′ values show strong dependence on the types and contents of gelatin materials. Those values are in the range of 3.24–6.68 kPa for A1–A5 and 0.95–3.69 kPa for B1–B5, respectively, see Fig. 2 and Table S1.† Notably, the higher G′ value of single-component F127 is expected compared to those of A1, A2 and B1–B5 because gelatin is in the solution state at 37 °C. In addition, the storage moduli of A1–A5 distinctly higher than those of the corresponding B1–B5, which might be ascribed to the different pI between GA and GB (GA has a pI between 7.0 and 9.0 while GB has a pI between 4.8 and 5.5).20 Furthermore, F127–gelatin hydrogels show enhanced storage moduli with increasing GA contents of the composite hydrogels. The G′ value of A2 and A3 are 1.5 and 2.0 times larger than that of A1, respectively (Fig. 2a). Similarly, in the case of F127–GB hydrogels, the G′ of B2 and B3 are nearly 2.0 and 4.0 times larger than that of B1 (Fig. 2b). Notably, when the contents of gelatin (GA and GB) were higher than 3 wt%, the magnitudes of the storage moduli were found to remain nearly constant. All these results suggest that gelatin-containing hydrogels have tunable mechanical strengths and stability gels at physiological temperature. Furthermore, the gelation kinetic rates of the composite hydrogels were very fast (within 30 s) when the temperature was raised to 37 °C, as shown in Fig. S4,† which is highly desirable for injectable in situ forming hydrogel materials.
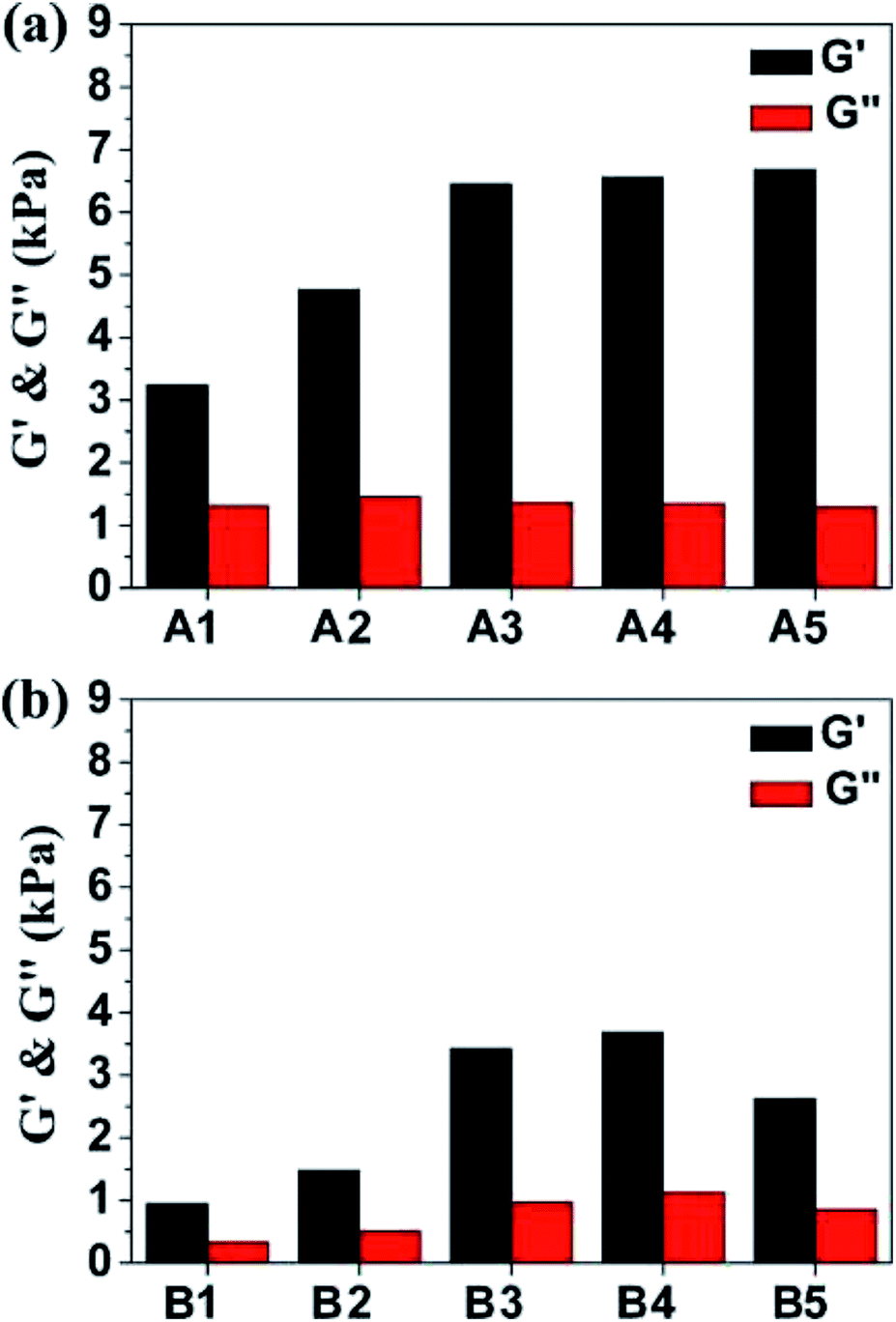 | ||
| Fig. 2 Frequency sweeps of the composite hydrogels of (a) A1–A5 and (b) B1–B5 at 37 °C. Black: G′; red: G′′. | ||
The hMSCs are multipotent stromal cells that can differentiate into a variety of cell types, such as adipocytes, chondrocytes, osteoblasts and so on.37 Next, the biocompatibility of hydrogels of A1–A3 and B1–B3 on hMSCs were evaluated by WST-1 assay (2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium sodium salt).38 Human MSCs were cultured for 2 days with the extraction medium of hydrogels of A1–A3 as well as B1–B3 and subjected for the cell viability experiments. As displayed in Fig. 3, the cell viability assay by WST-1 shows that the experimental hydrogels (A1–A3 and B1–B3) are biocompatible. Notably, the cell viability of A3 was increased significantly compared to that of A1 after 2 days (i.e. 90% for A1 and 110% for A3) and no significant differences in cell viability were found in F127–GB. Moreover, the survival data of the hMSCs soaked in A1–A3 and B1–B3 were evaluated with Live/Dead assay, which label of live cells with calcein AM (green) and of dead cells with EthD-1 (red). The 2 days Live/Dead results (Fig. S5†), are consistent with the WST-1 assay, shows that A3 has better biocompatibility and cell viability in comparison with A1, A2 and B1–B3. These results illustrated that higher concentrations of GA may enhance cell viability and proliferation in hMSCs whereas GB did not have any enhancement effect on concentrations up to 3 wt%. The thermal, mechanical and cell viability properties of A1–A3 and B1–B3 suggested that they are potential biomaterials and could be suitable for tissue engineering applications.
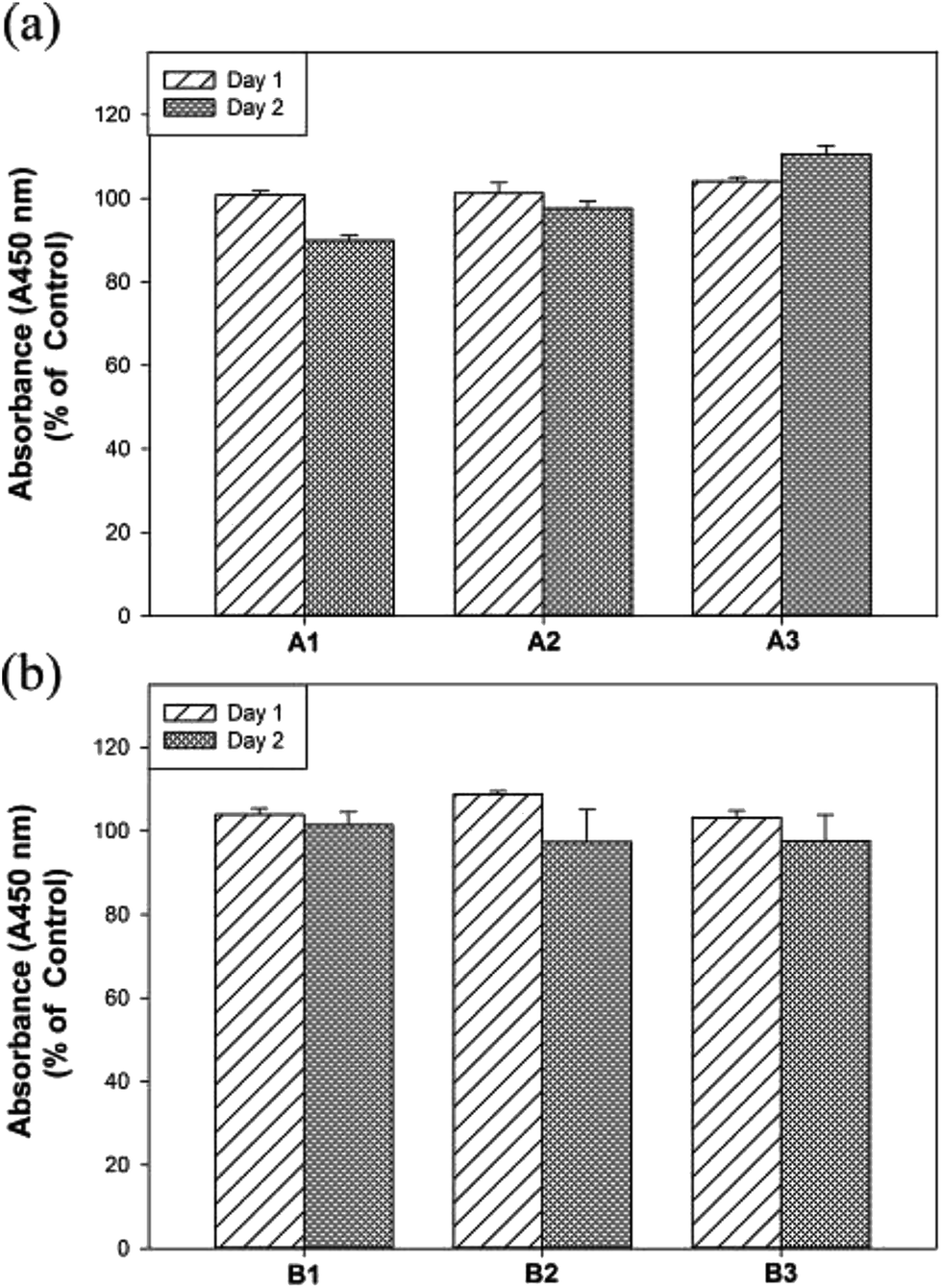 | ||
| Fig. 3 The cell viability assay by WST-1 on hMSCs in (a) A1–A3 and (b) B1–B3 for 2 days. | ||
Further, we attempt to understand microstructure–property relationships of these composite hydrogels at 25 and 37 °C respectively. The scanning electron microscopy (SEM) was used to determine the microstructure of the composite hydrogels. The SEM images revealed the bulk deposition for A1 at 25 °C and the hierarchical architectures of the layered nanocomposite was obtained at 37 °C, indicating that the self-assembly of A1 occurred at body temperature (Fig. 4a and d). At higher concentration of GA, i.e. A2 and A3, the composite hydrogels self-assembled into fiber-like structures which may due to strong non-covalent interactions of entrapped water in the hydrogel at 37 °C (Fig. 4e and f). It is worthy to note that the width of the nanofibers are around 75–300 nm which is falling within the range of natural extracellular matrix.39 The SEM images of B1–B3 were similar to those of A1–A3 (see Fig. S6† for detailed) and these results are consistent with the phase transition diagram data shown in Fig. 1. The X-ray diffraction (XRD) measurement was used to investigate the supramolecular structures formed by A1–A5 and B1–B5 in water. The XRD patterns of the dried samples of A1–A5 and B1–B5 were collected in Fig. S7.† The diffraction peaks were observed at d-spacing values of 6.41 and 3.36 Å, which is corresponding to aromatic–aromatic stacking interactions and hydrogen-bonding interactions for peptides.40,41
 | ||
| Fig. 4 SEM micrographs of vertical cross-sections of the composite hydrogels of A1–A3. (a–c) at 25 °C; (d–f) at 37 °C (scale bar: a–c: 1 μm; d–f: 100 nm). | ||
Temperature-dependent UV-vis absorption and circular dichroic (CD) spectra were used to further investigate the self-assembly of the composite materials from solution to gel state. As shown in Fig. 5a, the UV-vis spectra of A1 in water exhibited the absorption bands which are consistent with the tyrosine and phenylalanine side chains in gelatin.42 The absorption intensities increased with the temperature from 10 to 60 °C, indicating significant intermolecular aromatic–aromatic interactions. In Fig. 5b, the temperature-dependent CD spectra were used to characterize the self-assembly of gelatin in A1. It revealed a negative band at 240 nm when the temperatures are higher than 35 °C, suggesting the typical self-assembly of peptides in the mixture of A1.43 The temperature-dependent UV-vis and CD spectra of B1 were shown in Fig. S8,† and it showed similar results compared with A1. Fig. 5c showed thermo-reversible properties of the hydrogel A1 during heating and cooling treatment. Upon heating the solution to body temperature the semi-transparent solution disappeared completely and a transparent hydrogel was formed. Further cooling the hydrogel to room temperature, the semi-transparent solution reappeared because of the disassembly of the physical crosslinked nanostructures (see Fig. S9† for other reversible hydrogels). We further studied the effect of temperature on rheological properties of the hydrogel A1. The A1 solution was heated to 37 °C and cooled to 25 °C in cycles, and the obvious changes in G′ and G′′ were observed over time (Fig. 5d). The G′ of A1 hydrogels are greater than the corresponding G′′ when increasing temperature from 25 to 37 °C, pointing to sol–gel transition occurred at this stage. The reversible nature of newly discovered A1 was confirmed by carrying out the temperature-step experiment over three cycles. These results suggesting that the in situ formed composite hydrogels may support cell survival and the recovery of the 3D cultured cells can be simply accomplished by cooling the culture system to room temperature.
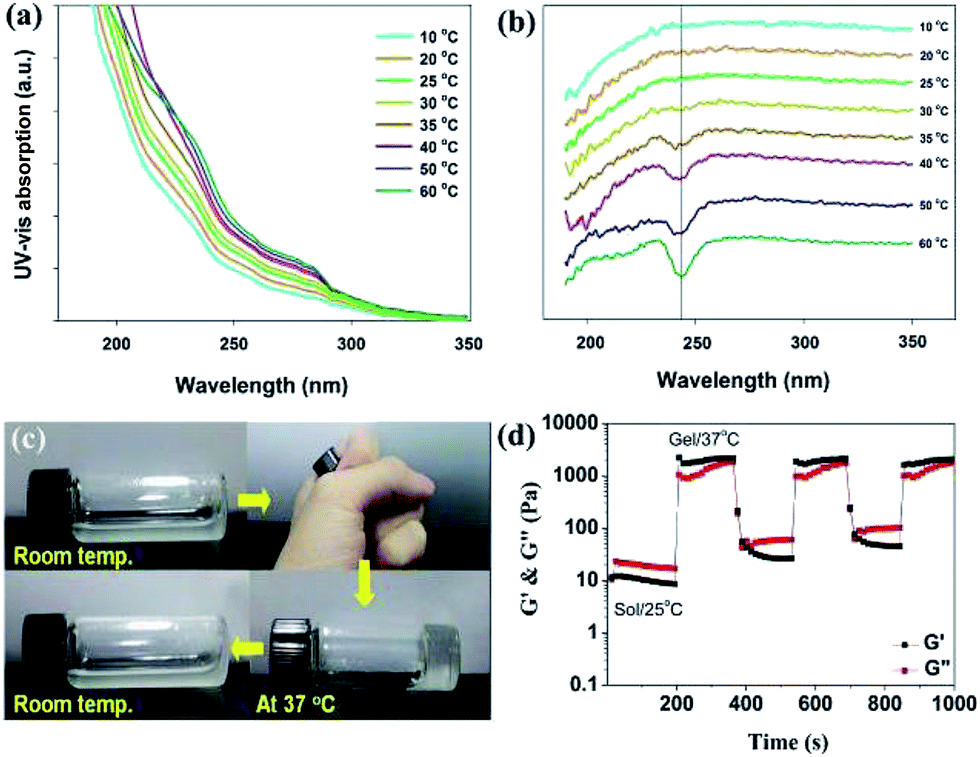 | ||
| Fig. 5 Temperature-dependent (a) UV-vis absorption and (b) CD spectra of A1. (c) Vial inversion tests provide visual evidence that the A1 solution formed a hydrogel when the temperature was higher than the sol–gel transition temperature and formed a solution when the temperature back to the room temperature. (d) Rheology test of A1 solution with thermal cycles of heating (37 °C) and cooling (25 °C). | ||
Conclusions
In conclusion, we have prepared a series of F127–gelatin (F127–GA and F127–GB) composite hydrogels and examined their gelation behavior, physical and biological properties. SEM images, temperature-dependent UV-vis absorption and CD spectra showed that F127–gelatin can self-assemble at physiological temperatures to form fibrous hydrogels. In addition, F127–gelatin hydrogels exhibit tunable stiffness especially F127–GA gels show higher gel strengths compared with those of F127–GB, which might be ascribed to the different isoelectric points between GA and GB. Furthermore, the cell viability of hMSCs were tested and demonstrated that F127–gelatin have good biocompatibility. Vial inversion tests and rheological results (cycled between 25 °C and 37 °C) demonstrated that F127–gelatin composite hydrogels are thermo-reversible. This study and further developments of F127–gelatin hydrogels may open up a new avenue in tissue engineering and regenerative medicine.Experimental section
Materials
Pluronic F127 (F127), gelatins from porcine skin (type A, GA) and bovine skin (type B, GB) were purchased from Sigma. Sodium carboxymethyl cellulose (SCMC) was supplied by Aldrich. All aqueous solutions were prepared with double-distilled deionized water (dd H2O) in whole experiment process.Preparation of composite hydrogels
F127 water solution (16 wt%) was prepared by adding a weighed amount of polymer to ice-cold dd H2O under constant stirring.44 The dispersion was then placed in a refrigerator overnight to obtain a clear solution. 1 wt% GA water solution was prepared by adding 0.01 wt% SCMC and heating the mixture up at 60 °C under constant magnetic stirring until a pale yellow clear solution was obtained. The thermo-reversible composite hydrogel of A1 was prepared by blending F127 with GA solution and the mixture was heated and continuous stirred until a homogeneous solution was achieved. The corresponding hydrogel was obtained by cooling down the solution to body temperature. The thermo-reversible composite hydrogels of A2, A3, A4 and A5 were prepared via a similar route, except that GA contents of 2, 3, 4 and 5 wt%, respectively. For comparison, the hydrogels of B1–B5 were also prepared by blending F127 with GB solution.Inverted tube method
Gelation tests were performed by weighing a compound (2.0 mg) in a screw-capped 2 mL vial (diameter: 10 mm), adding a solvent (0.20 mL), sealing the vial tightly, heating it until the compound had dissolved, and then cooling the vial to room temperature. Gelation was considered to have occurred when a solid-like material was obtained that did not exhibit gravitational flow (inverted test tube method) during a period of 5 min.45Rheological tests
Rheological tests were conducted using a TA rheometer (DHR-1) and a 40 mm parallel plate. The hydrogel sample (400 μL) was placed on the parallel plate for the angular frequency sweep test (test range: 0.1–100 rad s−1; strain, 0.8%; 15 points per decade; sweep mode, “log”; temperature, 25 °C).Scanning electron microscopy
Samples were visualized with a JEOL JSM-6700F scanning electron microscope at an accelerating voltage of 15 kV and a working distance of 6.3 mm. Hydrogels were applied directly onto silicon wafers and the samples were allowed to air dry.Biocompatibility of A1–A3 and B1–B3 hydrogels
The biocompatibility of A1–A3 and B1–B3 hydrogels was evaluated by culturing the human mesenchymal stem cells (hMSCs) with the extraction medium. The extraction medium was prepared by immersing the composite hydrogels of A1–A3 and B1–B3 in Dulbecco's Modified Eagle Medium (DMEM, Gibco) at a volume ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 (hydrogel volume to medium volume) for 2 days at 37 °C. The hMSCs seeded in 96-well plates were cultured with DMEM for 1 day at 37 °C under 5% CO2. Followed by, the DMEM was replaced with extraction medium and the cells were allowed to grow for 2 days. The hMSCs cultured with extraction medium (hydrogels of A1–A3 and B1–B3) and DMEM were subjected to cell viability assay using the Cell Proliferation Reagent WST-1 (Roche) and the optical density of the resulting solution was measured at 450 nm, using Synergy H1 Hybrid Reader (BioTek). Cells that had not been subjected to treatment with the test hydrogels were used as the control.
:10 (hydrogel volume to medium volume) for 2 days at 37 °C. The hMSCs seeded in 96-well plates were cultured with DMEM for 1 day at 37 °C under 5% CO2. Followed by, the DMEM was replaced with extraction medium and the cells were allowed to grow for 2 days. The hMSCs cultured with extraction medium (hydrogels of A1–A3 and B1–B3) and DMEM were subjected to cell viability assay using the Cell Proliferation Reagent WST-1 (Roche) and the optical density of the resulting solution was measured at 450 nm, using Synergy H1 Hybrid Reader (BioTek). Cells that had not been subjected to treatment with the test hydrogels were used as the control.
Acknowledgements
We would like to thank Prof. Hui-Yi Lin and School of Pharmacy, China Medical University for instrument support. This work has been financially supported by the Ministry of Science and Technology of the Republic of China, Taiwan (grant MOST 105-2119-M-009-012-; MOST 105-2113-M-039-008-MY2; MOST 105-2622-E-009-018-CC3); the Novel Bioengineering and Technological Approaches to Solve Two Major Health Problems in Taiwan sponsored by the Taiwan Ministry of Science and Technology Academic Excellence Program under Grant Number: MOST 105-2633-B-009-003.Notes and references
- T. M. Reineke, ACS Macro Lett., 2016, 5, 14–18 CrossRef CAS
.
- N. Sood, A. Bhardwaj, S. Mehta and A. Mehta, Drug Delivery, 2016, 23, 758–780 Search PubMed
- X. Zhang, Y. Guan and Y. Zhang, Biomacromolecules, 2012, 13, 92–97 CrossRef CAS PubMed
- O. D. Krishna and K. L. Kiick, Biopolymers, 2010, 94, 32–48 CrossRef CAS PubMed
- J. D. Ehrick, S. K. Deo, T. W. Browning, L. G. Bachas, M. J. Madou and S. Daunert, Nat. Mater., 2005, 4, 298–302 CrossRef CAS PubMed
- S. Bhattacharjee and S. Bhattacharya, Chem. Commun., 2015, 51, 7019–7022 RSC
- E. A. Appel, M. W. Tibbitt, M. J. Webber, B. A. Mattix, O. Veiseh and R. Langer, Nat. Commun., 2014, 6, 6295 CrossRef PubMed
- K. Zhou, S. Yang, G. Zhao, Y. Ning and C. Xu, RSC Adv., 2015, 5, 61719–61724 RSC
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS
- J. Hu, Z. Ge, Y. Zhou, Y. Zhang and S. Liu, Macromolecules, 2010, 43, 5184–5187 CrossRef CAS
- H. Wu, L. Zhu and V. P. Torchilin, Biomaterials, 2013, 34, 1213–1222 CrossRef CAS PubMed
- W. Chen and J. Du, Sci. Rep., 2013, 3, 2162 Search PubMed
- J. S. Katz and J. A. Burdick, Macromol. Biosci., 2010, 10, 339–348 CrossRef CAS PubMed
- C. Alvarez-Lorenzo, B. Blanco-Fernandez, A. M. Puga and A. Concheiro, Adv. Drug Delivery Rev., 2013, 65, 1148–1171 CrossRef CAS PubMed
- A. J. Harnoy, I. Rosenbaum, E. Tirosh, Y. Ebenstein, R. Shaharabani, R. Beck and R. J. Amir, J. Am. Chem. Soc., 2014, 136, 7531–7534 CrossRef CAS PubMed
- C. D. Ma, L. Adamiak, D. S. Miller, X. Wang, N. C. Gianneschi and N. L. Abbott, Small, 2015, 11, 5747–5751 CrossRef CAS PubMed
- H. Xia, Y. Zhao and R. Tong, Adv. Exp. Med. Biol., 2016, 880, 365–384 CrossRef PubMed
- K. P. Mineart, X. Jiang, H. Jinnai, A. Takahara and R. J. Spontak, Macromol. Rapid Commun., 2015, 36, 432–438 CrossRef CAS PubMed
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS
- R. Schrieber and H. Gareis, Gelatine Handbook: Theory and In-dustrial Practice, 1st edn, Wiley-VCH, 2007 Search PubMed
- S. Huang and X. Fu, J. Controlled Release, 2010, 142, 149–159 CrossRef CAS PubMed
- A. T. Neffe, A. Loebus, A. Zaupa, C. Stoetzel, F. A. Muller and A. Lendlein, Acta Biomater., 2011, 7, 1693–1701 CrossRef CAS PubMed
- Y.-H. Lin, H.-F. Liang, C.-K. Chung, M.-C. Chen and H.-W. Sung, Biomaterials, 2005, 26, 2105–2113 CrossRef CAS PubMed
- Q. Feng, K. Wei, S. Lin, Z. Xu, Y. Sun, P. Shi, G. Li and L. Bian, Biomaterials, 2016, 101, 217–228 CrossRef CAS PubMed
- R. Dash, M. Foston and A. Ragauskas, Carbohydr. Polym., 2013, 91, 638–645 CrossRef CAS PubMed
- A. Bigi, G. Cojazzi, S. Panzavolta, K. Rubini and N. Roveri, Biomaterials, 2001, 22, 763–768 CrossRef CAS PubMed
- H.-C. Liang, W.-H. Chang, L. H.-F. Iang, M.-H. Lee and H.-W. Sung, J. Appl. Polym. Sci., 2004, 91, 4017–4026 CrossRef CAS
- P. B. van Wachem, R. Zeeman, P. J. Dijkstra, J. Feijen, M. Hendriks, P. T. Cahalan and M. J. A. van Luyn, J. Biomed. Mater. Res., 1999, 47, 270–277 CrossRef CAS PubMed
- X. Fang, J. Xie, L. Zhong, J. Li, D. Rong, X. Li and J. Ouyang, J. Mater. Chem. B, 2016, 4, 1070–1080 RSC
- D. Cohn, G. Lando, A. Sosnik, S. Garty and A. Levi, Biomaterials, 2006, 27, 1718–1727 CrossRef CAS PubMed
- W. S. Shim, J. S. Yoo, Y. H. Bae and D. S. Lee, Biomacromolecules, 2005, 6, 2930–2934 CrossRef CAS PubMed
- I. Levental, P. C. Georges and P. A. Janmey, Soft Matter, 2007, 3, 299–306 RSC
- B. Zeeb, Enzymatic Stabilization and Formation of Food Nano- And Microstructures, Logos Verlag Berlin, 2015 Search PubMed
- H. Sun, Q. Meng, S. Tang, J. Su, Q. Yin, L. Chen, W. Gu, H. Yu, Z. Zhang, S. Wang and Y. Li, Mol. Pharmaceutics, 2015, 12, 3323–3331 CrossRef CAS PubMed
- A. A. Oun and J.-W. Rhim, Carbohydr. Polym., 2015, 127, 101–109 CrossRef CAS PubMed
- M. S. A. Rani, S. Rudhziah, A. Ahmad and S. Mohamed, Polymers, 2014, 6, 2371–2385 CrossRef CAS
- Y.-L. Wang, S.-P. Lin, S. R. Nelli, F.-K. Zhan, H. Cheng, T.-S. Lai, M.-Y. Yeh, H.-C. Lin and S.-C. Hung, Macromol. Biosci., 2016 DOI:10.1002/mabi.201600192
- L.-M. Yin, Y. Wei, Y. Wang, Y.-D. Xu and Y.-Q. Yang, Int. J. Med. Sci., 2013, 10, 68–72 CrossRef CAS PubMed
- V. Jayawarna, A. Smith, J. E. Gough and R. V. Ulijn, Biochem. Soc. Trans., 2007, 35, 535–537 CrossRef CAS PubMed
- S. Aravinda, N. Shamala, C. Das, A. Sriranjini, I. L. Karle and P. Balaram, J. Am. Chem. Soc., 2003, 125, 5308–5315 CrossRef CAS PubMed
- A. Asano, T. Taniguchi, M. Sasaki, H. Hasegawa, Y. Katsuya and M. Doi, Acta Crystallogr., 2001, E57, o834–o838 Search PubMed
- S. Barazzouk and C. Daneault, Nanomaterials, 2012, 2, 187–205 CrossRef CAS PubMed
- S. Maity, S. Sarkar, P. Jana, S. K. Maity, S. Bera, V. Mahalingam and D. Haldar, Soft Matter, 2012, 8, 7960–7966 RSC
- I. R. Schmolka, J. Am. Oil Chem. Soc., 1977, 54, 110–116 CrossRef CAS
- X. Z. Wang, X. Q. Li, X. B. Shao, X. Zhao, P. Deng, X. K. Jiang, Z. T. Li and Y. Q. Chen, Chem.–Eur. J., 2003, 9, 2904–2913 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01118k |
| This journal is © The Royal Society of Chemistry 2017 |