
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Graphene oxide reinforced hydrogels for osteogenic differentiation of human adipose-derived stem cells†
Myungkyung Noh‡
a,
Su-Hwan Kim‡ab,
Jiyong Kim‡a,
Ju-Ro Leea,
Gun-Jae Jeonga,
Jeong-Kee Yoona,
Seokyung Kanga,
Suk Ho Bhangc,
Hee Hun Yoona,
Jong-Chan Leea,
Nathaniel S. Hwang *abd and
Byung-Soo Kim*abd
*abd and
Byung-Soo Kim*abd
aSchool of Chemical and Biological Engineering, Seoul National University, Seoul 151-744, Republic of Korea. E-mail: nshwang@snu.ac.kr; byungskim@snu.ac.kr; Fax: +82-2-880-1604; Fax: +82-2-888-1604; Tel: +82-2-880-1635 Tel: +82-2-880-1509
bInterdisciplinary Program for Bioengineering, Seoul National University, Seoul 151-744, Republic of Korea
cSchool of Chemical Engineering, Sungkyunkwan University, Suwon 440-746, Republic of Korea
dN-Bio Institute, Institute of Chemical Processes, Seoul National University, Seoul 151-744, Republic of Korea
First published on 11th April 2017
Abstract
Polyethylene glycol (PEG)-based hydrogels are attractive biomaterials for stem cell culture due to their tunable material properties and mechanical strength. However, the lack of cell adhesion sites has been one of the major obstacles in generating functional tissue constructs using PEG-based hydrogels. To overcome this limitation, we designed graphene oxide (GO)-functionalized polyethylene glycol diacrylate (PEGDA) hydrogels to assign cell adhesion-dependent biofunctionality. The incorporation of GO into three-dimensional PEGDA networks improved cell attachment, engaged focal adhesion, and activated focal adhesion kinase (FAK) signaling of hydrogel-encapsulated human adipose-derived stem cells (hADSCs). Compared to the control PEGDA hydrogel, GO functionalized PEGDA hydrogel (PEGDA-GO) resulted in enhanced cell viability and survival. When subsequently cultured under osteoinductive condition, PEGDA-GO enhanced osteogenic differentiation and stimulated osteogenic phenotypes compared to those in its PEGDA counterpart. Taken together, GO could serve as an effective biofunctionalizing moiety to modulate stem cell adhesion and differentiation.
1. Introduction
Stem cells constantly interact with a myriad of biochemical and biophysical signals from their microenvironment.1 Since this interactive crosstalk between stem cells and their surroundings can affect stem cell behaviors and trigger differentiation,2,3 numerous engineered scaffolds have been extensively investigated to provide biological cues as well as structural support.4 While several types of biomaterials have been investigated for stem cell culture, hydrogels have emerged as attractive candidates for encapsulating stem cells and creating niches that promote tissue regeneration.5,6 Owing to batch-to-batch uniformity and controllability, synthetic polymeric hydrogels have been frequently utilized as scaffolding materials.7 In particular, polyethylene glycol diacrylate (PEGDA) photopolymerizing hydrogels have been intensely investigated for tissue engineering applications because of their adjustable mechanical properties and facile controllable scaffold architecture.7–9 However, PEGDA hydrogels alone do not provide an ideal environment for stem cells, mainly due to their lack of cell adhesion site.10 To incorporate cell adhesive moieties to PEGDA, previous studies have functionalized polyethylene glycol (PEG) networks with integrin adhesive peptides such as arginine-glycine-aspartic acid (RGD)11–13 or co-crosslinked with extracellular matrix (ECM)-derived components.14–16 These studies revealed that the incorporation of cell-adhesive moiety not only promoted cell attachment but also stimulated lineage specific stem cell differentiation. In this regards, proper functionalization of the PEG-based hydrogel can modulate stem cell behaviors and promote fate determination.13,17,18In recent years, graphene oxide (GO) has emerged as an appealing class of biocompatible material for stem cell culture. GO has been reported to promote stem cell growth and differentiation. In particular, human mesenchymal stem cells (hMSCs) seeded on GO-coated surface19 or cultured in GO-conjugated scaffold20,21 promoted cellular growth and osteogenic differentiation. Even though GO may elicit cytotoxicity at high concentration,22,23 recent studies indicate that GO at low concentration is beneficial for cell culture.24,25 In addition, previous in vitro studies have indicated that GO can promote stable stem cell adhesion, growth, and differentiation.26 Stable cellular adhesion can subsequently induce focal adhesion and focal adhesion kinase (FAK)-mediated osteogenic commitment of stem cells.27 Several studies have reported the increase in FAK phosphorylation of hMSCs interacted with GO,20,28,29 so it is rational to think that GO could serve as an effective cellular adhesion platform, which could potentiate further biological signaling for osteogenic commitment of stem cells. Namely, the incorporation of GO would be an effective way to functionalize PEG hydrogel and increase cell adhesion and induce differentiation.
In this work, we designed and created GO-functionalized PEGDA hydrogels (PEGDA-GO) and investigated osteogenic differentiation of human adipose-derived stem cells (hADSCs). As a bio-functionalizing agent, the effect of GO on viability, adhesion, and osteogenic differentiation of hADSCs was investigated and simultaneously compared to that of PEGDA hydrogel.
2. Materials and methods
2.1. Preparation and characterization of PEGA-GO
GO was synthesized from graphite using modified Hummers' method, as previously described.30 Preoxidation of graphite was carried out prior to the oxidation steps. Graphite flakes (1.0 g) were stirred at 80 °C for 6 h in aqueous solution of K2S2O8 (0.5 g) and P2O5 (0.5 g) in concentrated H2SO4, followed by several washing and filtration steps with distilled water using glass frit filter equipped with anodic aluminum oxide (AAO) membrane (0.2 μm of pore size, Whatman, Germany). After dried under vacuum condition at room temperature for 24 h, the preoxidated graphite flakes were vigorously stirred at 0 °C for 40 min in a solution of NaNO3 (0.5 g) in H2SO4. The flask was warmed up to 20 °C and KMnO4 (3.0 g) was slowly added into the flask and stirred for 45 min. After that, the temperature was increased to 35 °C and the mixture was stirred for another 2 h. Brownish mixture was obtained after the temperature was raised up to 98 °C and 2.5 ml of H2O2 (30 wt% aqueous solution) was subsequently added into the flask to complete the oxidation steps, followed by several washing and filtration steps with 250 ml of HCl (10 wt% aqueous solution) using glass frit filter equipped with AAO membrane. To neutralize the final product, GO was thoroughly washed with the large amount of distilled water until the pH value was near 7.0 and then further dried under vacuum condition at room temperature for 48 h. The GO was then PEGylated as previously described by Zhang et al.31 Briefly, 200 mg of GO was added into 10 ml of N,N-dimethylforamide under ultrasonication. In this process, GO was thoroughly exfoliated down to individual sheets and formed stably dispersed GO/DMF solution. The stable GO/DMF suspension was refluxed with 40 ml of SOCl2 at 70 °C for 24 h in CaCl2 tube. Distillation was performed at the end of the reaction to remove excess SOCl2, and 2 g of PEG-1000 and DMSO were added to the mixture. The reaction continued in the mixture at 120 °C for 3 days under magnetic stirring. The resulting suspension was then added to ethanol, filtered through a Buchner funnel under reduced pressure, and dried at 60 °C under vacuum. Final acrylation was performed by reacting as-obtained PEG-GO with acryloyl chloride (Sigma, St. Louis, MO, USA) overnight under N2 gas. For the characterization of acrylated PEG-GO (PEGA-GO), Fourier Transform Infrared Spectroscopy (FT-IR) analyses were carried out on Nicolet 6700 spectrophotometer (Thermo Scientific, USA, spectral range: 3500–500 cm−1, resolution: <0.4 cm−1, detector: high sensitivity DLATGS detector, source: high energy air cooled MIR glower source) using Attenuated Total Reflectance (ATR) equipment (FT-IR/ATR).2.2. Hydrogel preparation and characterization
Acellular hydrogels were prepared in order to study their material properties in terms of swelling ratio and rheological properties. PEGDA (MW 5000; SunBio, Inc., Anyang, Korea) was dissolved at a concentration of 10% (w/v) in phosphate-buffered saline (PBS) containing 0.05% (w/v) Irgacure 2959 (photoinitiator; Sigma). For the preparation of PEGDA-GO hydrogel, PEGA-GO was incorporated into the polymer solution at a concentration of 5 μg ml−1. 40 μl of each polymer solution was pipetted between glass slides separated by 1 mm-thick spacer and photopolymerized under 365 nm UV light (4 mW cm−2) for 7 min. The swelling ratios of hydrogels were calculated as previously described.32 Briefly, the gels at swollen and equilibrated state in PBS were weighted, and subsequently freeze-dried. After lyophilization, the weights of dried constructs were measured. The swelling ratios of hydrogels were calculated according to the following equation.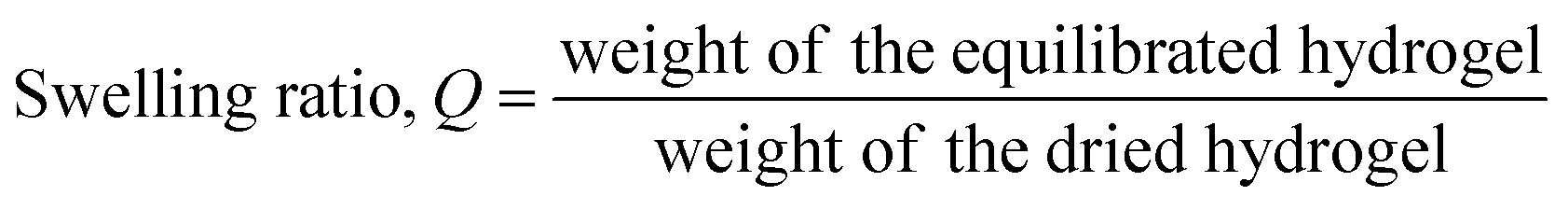
Meanwhile, viscoelastic properties of hydrogels were investigated by performing dynamic strain frequency sweep test. Measurements were carried out on ARES-G2 rheometer (TA Instruments, New Castle, DE, USA) at constant strain (1.0%) and temperature (25 °C). Each construct was placed between 8 mm parallel plates with a gap of 1.3 mm, and the dynamic modulus was recorded from at least three samples of each group. To confirm incorporated GO in PEGDA, Raman spectrometer (T 64000, HORIBA, FR) was used. Ar laser (514 nm) was irradiated to PEGA-GO, PEGDA, and PEGDA-GO and detected Raman signal. The focal length is 640 mm, and aperture is f/7.5.
2.3. Isolation and culture of hADSCs
hADSCs from lipo-aspirates of well-informed and consenting patients (between age 65 and 75) were isolated and cultured as previously described.33 hADSCs isolation procedure was approved by the Borame Hospital Institutional Review Board and Seoul National University (IRB No. 20160113/16-2016-03-021). hADSCs were maintained in growth medium (GM) consisting of Dulbecco's Modified Eagle's Medium (DMEM; Gibco BRL, Gaithersburg, MD, USA) supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco BRL) and 1% (v/v) penicillin/streptomycin (PS; Gibco BRL). Cells were grown under standard culture condition (37 °C, 5% CO2), and the medium was changed every 2 days.2.4. Photoencapsulation and in vitro osteogenic differentiation of hADSCs
hADSCs were encapsulated in PEGDA or PEGDA-GO hydrogel, and the procedure was similar to that of acellular hydrogel formation. Each precursor solution was constituted in sterile PBS containing 1% penicillin/streptomycin (PS; Gibco BRL). Passage 5 hADSCs were gently mixed and suspended in each precursor solution at a concentration of 1 × 107 cells per ml, and 40 μl of cell–polymer suspensions were placed between glass slides of 1 mm spacing. After exposing them to 365 nm UV light (4 mW cm−2) for 7 min, obtained cell-laden constructs were transferred into 24-well culture plates (SPL Life Sciences, Pocheon, Korea). For the osteogenic differentiation of encapsulated cells, hydrogels were cultured up to 3 weeks in osteogenic differentiation medium (ODM) consisting of high-glucose of DMEM (Gibco BRL) supplemented with 50 μg ml−1 ascorbic acid (Sigma), 10 mM β-glycerophosphate (Sigma), 100 nM dexamethasone (Sigma), 10% (v/v) FBS (Gibco BRL), and 1% (v/v) penicillin/streptomycin (PS; Gibco BRL), and the medium was changed every 2 days.2.5. Cellular morphology, viability, and apoptotic activity analyses
The morphological characteristics of the cells either encapsulated within or seeded onto the hydrogels were monitored by F-actin staining. Both cell-laden and cell-seeded constructs were fixed with 4% formaldehyde (PFA; Sigma) for 30 min at room temperature, followed by permeabilization with 0.1% Triton X-100 (Sigma) in PBS for 5 min. To stain F-actin, Alexa Fluor® 488 Phalloidin (Invitrogen, Carlsbad, CA) in 1% (w/v) bovine serum albumin (Sigma) was treated for 20 min at room temperature, and the cell nuclei was simultaneously labeled with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA, USA). The development of actin cytoskeleton and cytoplasmic projections was visualized and imaged either by confocal laser scanning microscope (Eclipse 90i, Nikon, Japan) or by fluorescence microscope (IX71 inverted microscope, Olympus, Tokyo, Japan).To assess the population of encapsulated cells, the amount of cellular deoxyribose nucleic acid (DNA) within hydrogels was quantified at desired time points. Each lyophilized construct was mechanically crushed, and incubated with 1 ml of papain solution containing 125 μg ml−1 papainase type III (Worthington Biomedical, Lakewood, N.J., USA) in 100 mM phosphate buffer (10 mM cysteine, 10 mM EDTA, pH 6.3) at 60 °C for 16 h. DNA contents of digested samples were measured using Quant-iT™ Pico-Green® dsDNA assay kit (Invitrogen) according to the manufacturer's instruction.
Along with the DNA contents measurement, cellular viability and apoptotic activity were also examined by neutral red assay and quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR), which were performed on the cell-laden constructs incubated up to 3 days in GM. For the neutral red assay, the cultured gels were treated with the medium containing 50 μg ml−1 neutral red (Sigma). After 3 hours, the spent medium was removed, and 0.2 ml of acetic acid (1%, v/v) and ethanol (50%, v/v) solution was added to each construct. Within 5 min, red colored-dye was nicely developed in the structures and subsequently extracted. The absorbance at 540 nm, which is proportional to the number of viable cells, was measured using microplate reader (PowerWave X340, Bio-Tek Instruments, VT, USA). In the meantime, qRT-PCR analysis of caspase-3, the pro-apoptotic marker, was carried out on day 3 samples.
2.6. Evaluation of in vitro osteogenic differentiation of encapsulated hADSCs
Osteogenic phenotypes developed by encapsulated hADSCs in each matrix were analyzed through the assays involved in indentifying bone-related biomarkers. For the evaluation of alkaline phosphatase (ALP) activity of hydrogels, p-nitrophenol phosphate (Anaspec®, San Jose, CA, USA) was used as the substrate. The constructs cultured in ODM for 1 week was rinsed twice with PBS and lysed in alkaline lysis buffer. The lysates were centrifuged for 10 min at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g, 4 °C, and the supernatants were collected and subsequently incubated in the glycine buffer containing 2 mg ml−1 of p-nitrophenol phosphate. After 30 min, 3 N sodium hydroxide solution was added to stop the reaction. The absorbance at 405 nm was measured using microplate reader (PowerWave X340, Bio-Tek Instruments).
000 g, 4 °C, and the supernatants were collected and subsequently incubated in the glycine buffer containing 2 mg ml−1 of p-nitrophenol phosphate. After 30 min, 3 N sodium hydroxide solution was added to stop the reaction. The absorbance at 405 nm was measured using microplate reader (PowerWave X340, Bio-Tek Instruments).
To evaluate the extent of calcium deposition within hydrogel networks, cell-laden hydrogels cultured for 3 weeks in ODM were collected either for Alizarin Red S staining or for calcium quantification. For preparing specimens for Alizarin Red S staining, hydrogels were fixed in 4% PFA overnight at 4 °C, dehydrated, and embedded in paraffin using standard techniques. Histological sections obtained from samples (5–7 μm in thickness) were stained with Alizarin Red S solution (pH 4.2; Sigma) to visualize calcium deposition throughout the structures. The amount of calcium contents were also quantified. After rinsed twice with deionized water, the constructs were incubated in 6 N HCl solution. The calcium contents in the lysates were spectrophotometrically quantified with cresolphthalein complexone (Sigma). Three minutes after the addition of reagents, the absorbance of the samples was read at 575 nm using microplate reader (PowerWave X340, Bio-Tek Instruments).
The expression of osteogenic differentiation markers, which is not only transcriptional but also translational, were investigated. qRT-PCR analysis was performed on the hydrogels cultured in ODM for 1 week, 2 weeks, and 3 weeks to examine the relative gene expression of osteogenic markers, including runt-related transcription factor 2 (RUNX2), bone morphogenic protein-2 (BMP-2), and osteopontin (OP). Additionally, immunofluorescence staining of osteocalcin (OC; Abcam, UK) was performed after 3 weeks of culture.
2.7. qRT-PCR
Total ribonucleic acid (RNA) from each hydrogel was extracted with TRIzol reagent (Invitrogen). Reverse transcription reaction was performed to obtain complementary deoxyribonucleic acid (cDNA) from the RNA samples, using AccuPower® RT PreMix (Bioneer Co., Daejeon, Korea). Gene expression profiles were examined via StepOnePlus real-time PCR system (Applied Biosystems, Foster City, CA, USA) with TOPreal™ qPCR 2X PreMIX (SYBR green; Enzynomics, Daejeon, Korea). 55 amplification cycles were performed and each cycle consisted of the following steps; 95 °C for 10 s, 55 °C for 15 s, and 72 °C for 30 s. Obtained data were analyzed through 2−ΔΔCt method.2.8. Immunofluorescence staining
The hydrogel constructs were sliced and subsequently immunostained with antibodies against OC (Abcam). Immunoreactivity was visualized through rhodamine isothiocyanate-conjugated secondary antibodies (Jackson Immuno Research Laboratories Inc., West Grove, PA, USA). The slices were then mounted, while the nuclei of cells was counterstained with DAPI (Vector Laboratories). The images were obtained using confocal microscope (SP8 X STED, Leica, Mannheim, Germany).2.9. Western blot
Western blot was performed to evaluate the molecular signaling pathway of FAK and extracellular signal-regulated kinase (ERK). The cell-laden constructs were cultured for 3 weeks in ODM. Then, the samples were mechanically crushed and lysed in cold lysis buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 1% Nonidet P-40) containing protease inhibitor cocktail for 40 min. After centrifugation at 4 °C (14000 rpm, 20 min), the concentration of protein in collected supernatant was determined by bicinchoninic acid protein assay (Pierce Biotechnology, Rockford, IL, USA). Equal amount of proteins were loaded and separated by 10% (w/v) sodium dodecyl sulfate-polyacrylamde gel (SDS-PAGE). The proteins were then transferred to an Immobilon-P membrane (Milipore Corp., USA) and probed with antibodies (Abcam) against FAK, phosphorylated FAK (pFAK), ERK, and phosphorylated ERK (pERK). The labeled proteins were then incubated with a horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology, USA) for 1 h at room temperature. The blots were developed using an enhanced chemiluminescence detection system (Amersham Bioscience, USA). β-Actin served as the internal control.
2.10. Statistical analysis
Quantitative data from at least triplicate samples were presented as mean value ± standard deviation. Statistical analysis was conducted via one-way single factor analysis of variance (ANOVA) with the Tukey significant difference post hoc test using SPSS software (SPSS Inc., USA). p < 0.05 was considered statistically significant.3. Results
3.1. Synthesis and characterization of PEGA-GO
For the functionalization of PEGDA hydrogel with GO, PEGylation process was performed on GO (Fig. 1). Following the PEGylation process, acrylate group was introduced to the PEG chain. The size of GO used for the study was approximately 10–20 μm, and PEGylated GO (PEG-GO) and PEG-acrylated GO (PEGA-GO) retained were well within the initial GO size distribution (data not shown). The structural features of GO, PEG-GO, and PEGA-GO were examined by FT-IR spectroscopy (Fig. 2A). FT-IR spectrum of GO showed a strong and broad absorption at 3392 cm−1 originating from O–H stretching vibrations. The characteristic stretching absorption bands around 1500 cm−1 correspond to the carbonyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in the COOH units situated at the edges of the GO sheets, whereas the bands of CC bond in graphitic sheet appear around 1300 cm−1. After PEGylation, the ether groups (C–O–C) in PEG imprints absorption bands around 1000 cm−1 in the spectrum. Further acrylation process shifts the C–O–C bands to the right, and the intensity of the bands around 1500 cm−1 rises in the spectrum of PEGA-GO, because of the increased carbonyl groups. From these results, we confirmed the obtained GO derivate as PEGA-GO, and performed subsequent UV-mediated polymerization with PEGDA.
O) in the COOH units situated at the edges of the GO sheets, whereas the bands of CC bond in graphitic sheet appear around 1300 cm−1. After PEGylation, the ether groups (C–O–C) in PEG imprints absorption bands around 1000 cm−1 in the spectrum. Further acrylation process shifts the C–O–C bands to the right, and the intensity of the bands around 1500 cm−1 rises in the spectrum of PEGA-GO, because of the increased carbonyl groups. From these results, we confirmed the obtained GO derivate as PEGA-GO, and performed subsequent UV-mediated polymerization with PEGDA.
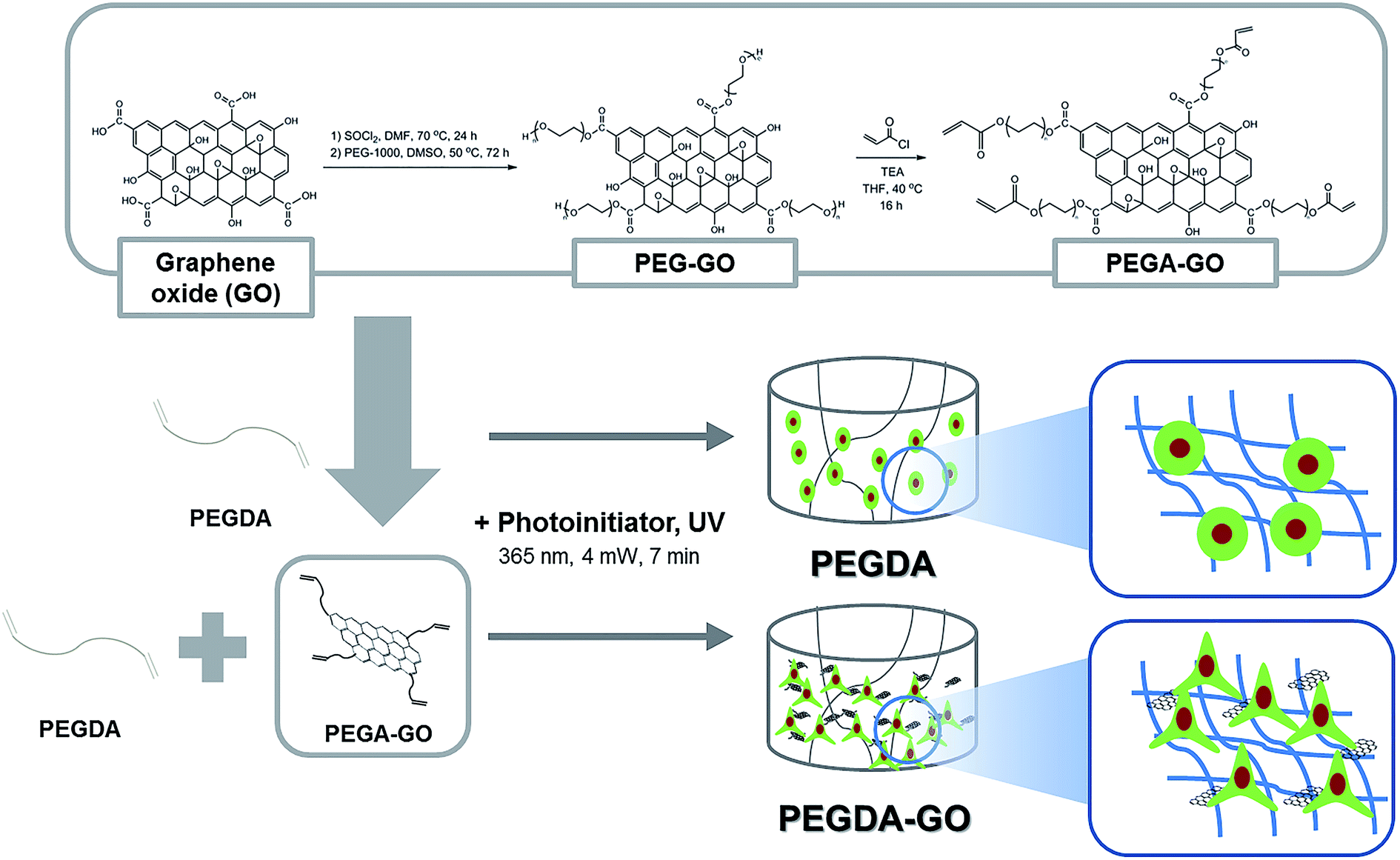 | ||
| Fig. 1 Schematic of PEGDA and PEGDA-GO hydrogel fabrication and chemical modification procedure for PEGA-GO. | ||
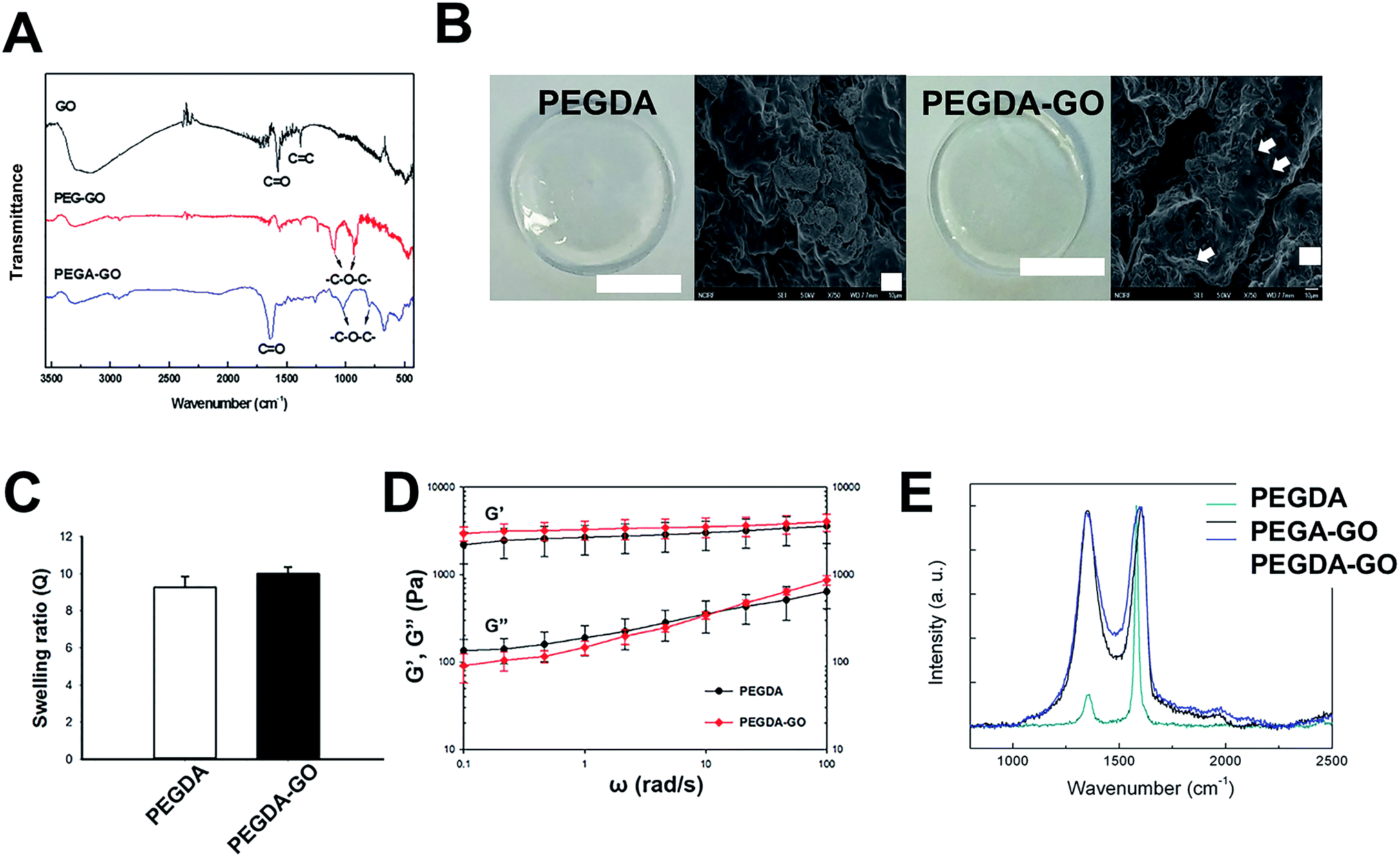 | ||
| Fig. 2 Preparation and characterization of PEGDA and PEGDA-GO hydrogels. (A) Characterization of GO, PEG-GO and PEGA-GO via FT-IR spectroscopy (B) SEM images of photopolymerized PEGDA and PEGDA-GO (scale bar = 10 μm, white arrows indicate GO in PEGDA). (C) Swelling ratio of the hydrogels. No statistical difference was observed between PEGDA and PEGDA-GO. (D) Dynamic modulus measurement in frequency sweep mode. Both shear storage (G′) and shear loss (G′′) modulus were not statistically different between PEGDA and PEGDA-GO. (E) Raman spectrometer analysis of PEGDA, PEGA-GO, and PEGDA-GO. | ||
3.2. Preparation and characterization of hydrogels
Fig. 2B represents the photopolymerized PEGDA and PEGDA-GO hydrogels. With or without functionalizing components, two types of PEGDA-based hydrogels were prepared by the exposure to UV light. It should be noted that the concentration of PEGA-GO was determined depending on its cytotoxicity. According to a preceding research, PEGylated GO derivatives did not cause any cytotoxic events to the cells under the concentration of 6.25 μg ml−1.34 Thus, we employed 5 μg ml−1 of PEGA-GO for the generation of PEGDA hydrogel to prevent any potential cytotoxic event that can be caused by PEGA-GO. As we incorporated PEGA-GO into PEGDA solution at low concentration, there was no significant color change between PEGDA and PEGDA-GO, and PEGDA-GO would sustain its microstructures (Fig. 2B). Up to 150 μg ml−1 of GO concentration, PEGDA-GO preserved its transparent properties (Fig. S2†).To demonstrate the GO effect to cells, we optimized GO concentration to maintain mechanical properties similar to PEGDA hydrogel. To optimize and evaluate the influence of incorporated GO into PEGDA hydrogel, we conducted subsequent characterization in terms of swelling ratio and dynamic modulus. Despite the difference in precursor composition, the swelling ratios of hydrogels did not make any clear distinction in statistical terms (Fig. 2C). Furthermore, the results from dynamic modulus measurement revealed that there was no statistical difference in both shear storage (G′) and shear loss modulus (G′′), which are the indicators of viscoelastic behavior (Fig. 2D). Although GO-functionalized hydrogels did not represent any observable differences in swelling and mechanical behaviors, Raman analysis demonstrated that the GO was incorporated within PEGDA structure (Fig. 2E). Thus, we concluded that 5 μg ml−1 of PEGA-GO conditions could functionalize hydrogel constructs without causing any changes in basic material properties of PEGDA hydrogel.
3.3. Morphological analysis and focal adhesion-related signaling of hADSCs in PEGDA and PEGDA-GO
The morphological changes of hADSCs occurred on cell–hydrogel interface were evaluated by F-actin staining. Cells were uniformly encapsulated in both hydrogels, and they displayed rounded morphologies (Fig. 3A; left panel). Our previous study reported micro-sized protrusions when cells were encapsulated in PEG-based hydrogels containing RGD peptide.11,12,35 Interestingly, similar to the RGD-functionalized hydrogel, distinct cellular protrusions were evident in PEGDA-GO hydrogel. Based on the assumption that the extension of actin fibers was induced by the adhesive interaction of cells with GO, we observed direct morphological responses of the seeded cells to the hydrogel surfaces (Fig. 3A; right panel). As expected, hADSCs developed distinctive protrusions within PEGDA-GO hydrogel due to increased cell adhesion moieties. Furthermore, unlike the cells on PEGDA, cellular spreading also significantly increased via GO-functionalization of hydrogel. From the results, we confirmed that GO-functionalization of PEGDA hydrogel (PEGDA-GO) could induce the modulation of cellular morphology and cell adhesion.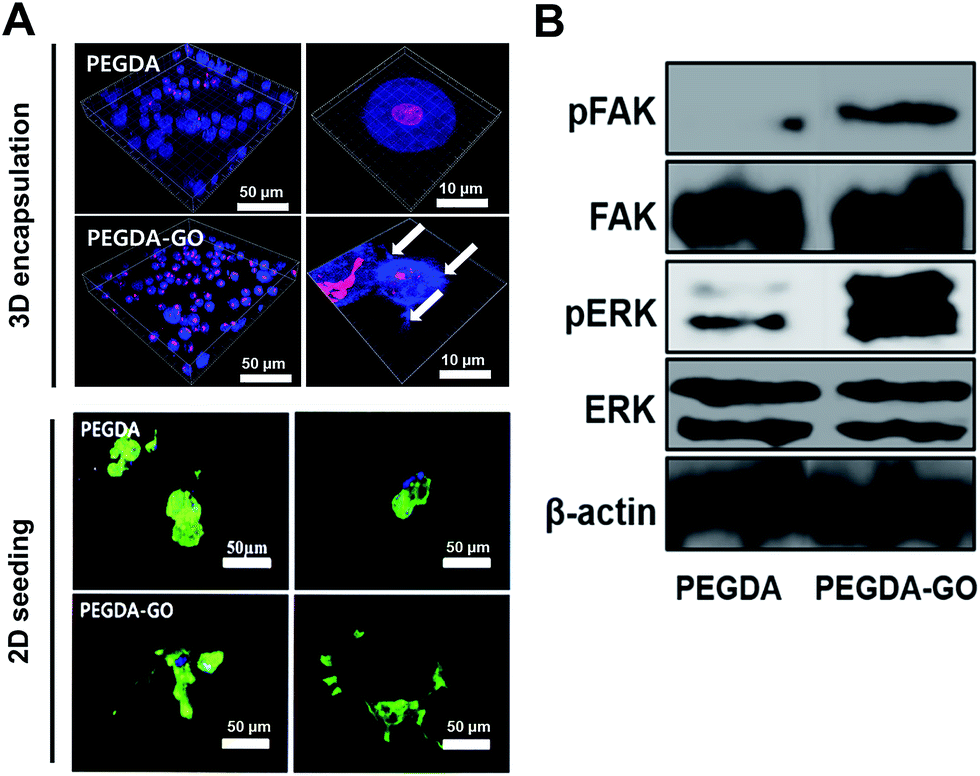 | ||
| Fig. 3 The positive influence of incorporated GO on cell adhesion, FAK phosphorylation, and downstream ERK activation. (A) F-actin staining of hADSCs either encapsulated in three-dimensional networks (3D encapsulation, blue, white arrow indicate the protrusions of hADSC) or seeded on two-dimensional hydrogel surfaces (2D seeding, green). Cell nuclei were counterstained with DAPI (pink in 3D encapsulation, scale bar, left panel = 50 μm, right panel = 10 μm and blue in 2D seeding image, scale bar = 50 μm). (B) Western blot analysis of pFAK, FAK, pERK, and ERK expression. | ||
On the basis of F-actin analysis, further investigation was performed on cell adhesion-related signaling molecules and their downstream pathways. The cell surface receptors such as integrins interact with extracellular cell-adhesive functional groups and clustered to establish focal adhesion. Western blotting was therefore performed to evaluate the expression and phosphorylation level of FAK (Fig. 3B). FAK phosphorylation remarkably increased in PEGDA-GO, which corresponds to the F-actin analyses. Moreover, we investigated the FAK downstream signaling, such as ERK. The phosphorylation level of ERK was also up-regulated in PEGDA-GO, and we deduced that GO induces focal adhesion formation and triggers downstream intracellular signaling via ERK. Thus, in subsequent experiments, we investigated the potential role of GO on directing further intracellular events in stem cells, beyond focal adhesion.
3.4. Cell viability, anti-apoptotic activity, and long-term maintenance of hADSCs in PEGDA-GO
Next, we examined the viability of hADSCs encapsulated in PEGDA and PEGDA-GO (Fig. 4A). The colorimetric evaluation of the cell viability via neutral red assay revealed that there was no statistically difference at day 1. However, at day 3, hADSCs in PEGDA-GO showed improved cell viability compared to those in PEGDA. UV source and photo-initiator also may cause cellular cytotoxicity during photopolymerization however, we incorporated 365 nm UV light (4 mW cm−2 for 7 min) and 0.05% (w/v, Irgacure 2959) photo-initiator, which has been shown to be biocompatible.36 We further investigated the cell proliferation rate through Alamar blue assay, and low concentration of GO in PEGDA had a little effect to cell proliferation (Fig. 4B). The effect of GO on cell proliferation rate in 2D condition correlated to the 3D culture condition (Fig. S3A†).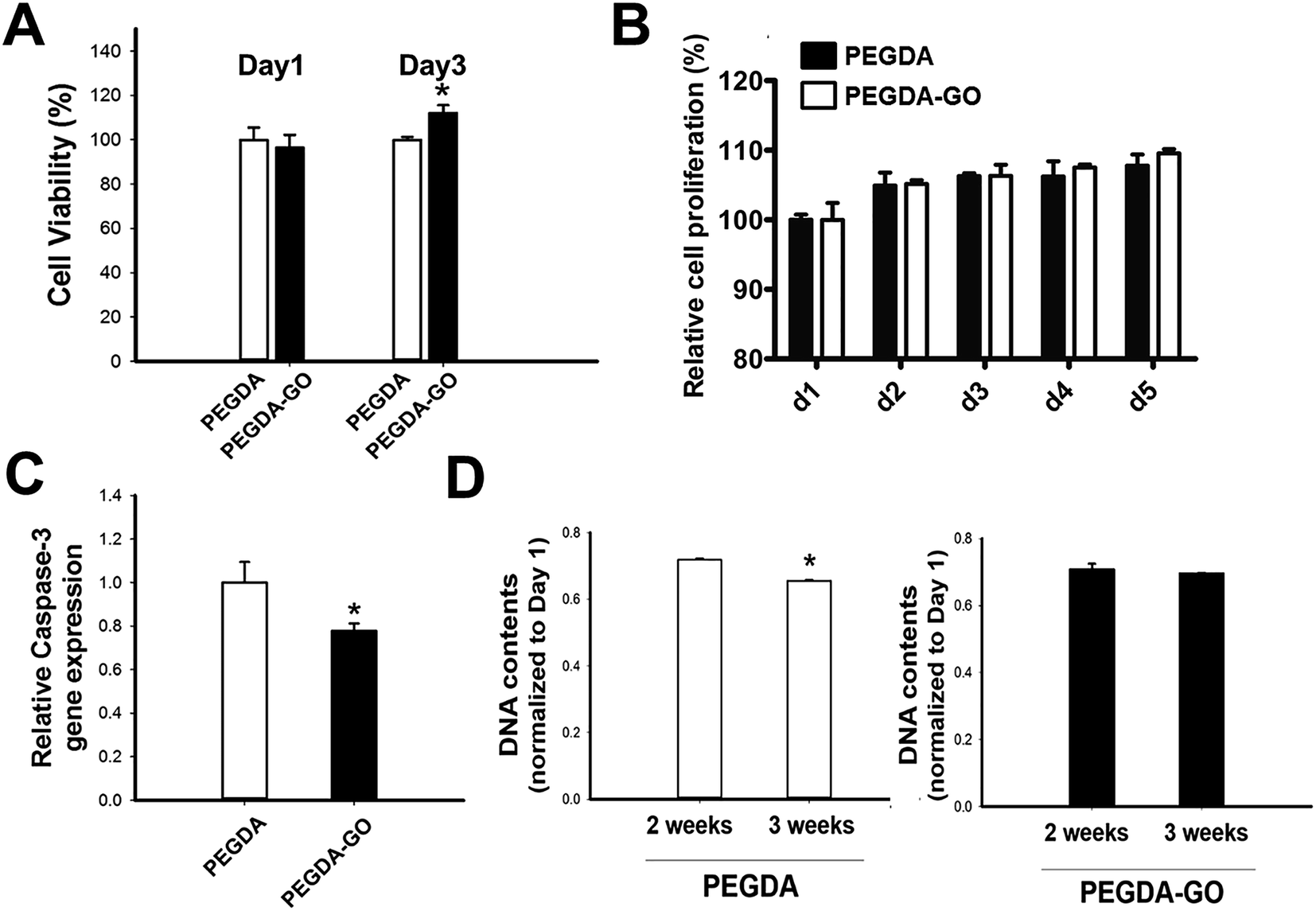 | ||
| Fig. 4 The effect of GO-mediated functionalization on viability, proliferation, anti-apoptotic behavior, and long-term survival of hADSCs. (A) Cell viability evaluated by neutral red assay, at day 1 and day 3 (n = 3, *p < 0.05 versus PEGDA at day 3). (B) Cell proliferation rate analyzed by Alamar blue assay. (C) The apoptotic activity of encapsulated cells investigated through relative mRNA expression of pro-apoptotic caspase-3 at day 3 (n = 3, *p < 0.05). (D) Quantification of DNA contents in PEGDA and PEGDA-GO cultured for 2 weeks and 3 weeks in ODM. The values are normalized by day 1 (n = 3, *p < 0.05). No significant difference occurred between 2 weeks and 3 weeks in PEGDA-GO group. | ||
To further investigate cellular behavior within hydrogels, the messenger RNA (mRNA) expression of pro-apoptotic marker caspase-3 was evaluated. Corresponding to the neutral red assay, the relative expression of caspase-3 was fairly down-regulated in PEGDA-GO group (Fig. 4C). In addition to these results, which were occurred in early culture period, the survival of encapsulated cells in prolonged time points was monitored by DNA quantification (Fig. 4C). DNA contents of PEGDA and PEGDA-GO were similar after 2 weeks, but as additional one week went by, the amount of cellular DNA in PEGDA group decreased at 3 weeks. On the other hand, DNA contents in PEGDA-GO were well maintained for 3 weeks, which was higher than those in PEGDA. Thus, GO-functionalization of hydrogel reduced pro-apoptotic marker expression and enhanced cell viability. The advantageous influence of GO was also found in extended culture period, in respect of improved cell retention within PEGDA-GO hydrogel.
3.5. Enhanced in vitro osteogenic commitment of hADSCs in PEGDA-GO
Based on the bioactive role of GO in terms of cell viability, adhesion, and FAK-ERK activation, we assumed that the osteogenic commitment of hADSCs would be improved in PEGDA-GO. To evaluate the hypothesis, we first examined the relative mRNA expression of osteogenic markers (RUNX2, BMP-2, and OP) in hydrogels during the culture period (Fig. 5A). At 1 week, the mRNA expression of RUNX2, the most specific osteoblast-associated transcription factor,37 was highest in PEGDA-GO. OP, the osteoblast-specific marker for matrix mineralization,38 was the most significantly up-regulated osteogenic gene of hADSCs in PEGDA-GO group. At 2 weeks, the mRNA expression of OP was 2 fold higher for PEGDA-GO compared with PEGDA, and its expression level has further increased more than 3 fold in 3 weeks. BMP-2 is another osteogenic indicator, whose autocrine loop is activated in the cells undergoing osteogenic differentiation.39 After 3 weeks, the differences in BMP-2 gene expression between two groups became distinctive and the values from the cells in PEGDA-GO were about three-fold higher than those from PEGDA hydrogel. | ||
| Fig. 5 Enhanced osteoinductivity achieved by the incorporation of GO. (A) Relative osteogenic marker expression (RUNX2, BMP-2, and OP) at 1 week, 2 weeks and 3 weeks (n = 3, *p < 0.05). (B) Relative ALP activity of hADSCs at 1 week (n = 3, *p < 0.05). (C) Alizarin Red S staining for deposited calcium in PEGDA and PEGDA-GO for 3 weeks, and its quantitative analysis (scale bar = 50 μm, n = 3, *p < 0.05). (D) Immunofluorescence staining of OC performed on hydrogels cultured for 3 weeks in ODM. Cytoplasmic osteocalcin expression was visualized by red fluorescence and cell nuclei were counterstained with DAPI (blue) (scale bar = 75 μm). | ||
Additionally, we examined the relation between the concentration of incorporated GO and OP expression, which is the most upregulated osteogenic gene, to investigate the concentration dependence of GO-mediated osteogenic differentiation (Fig. S1†). Although we decided the GO amount in response to the cellular viability, hADSCs encapsulated with 5 μg ml−1 of GO showed the highest expression of OP. Therefore, we confirmed that this concentration is the optimal condition for osteogenic differentiation of hADSCs in PEGDA-GO hydrogel.
Several osteogenic phenotypes were also examined, such as ALP activity, calcium deposition, and osteocalcin protein expression. ALP is the essential osteogenic marker, whose activity is upregulated during the early phase of osteogenesis.40 At 1 week, ALP activity was elevated in the cells cultured in PEGDA-GO compared to its counterpart (Fig. 5B). Inorganic calcium deposition is one of the general features of bone formation.41 After 3 weeks, the calcium contents were quantified, and hADSCs in PEGDA-GO deposited higher amount of calcium than those in PEGDA (Fig. 5C). The degree of calcium deposition was also qualitatively confirmed by histological analysis with Alizarin Red S staining after 3 weeks (Fig. 5C). In accordance with the calcium quantification result, PEGDA-GO developed intense red color, compared to PEGDA. A trend in 2D culture also showed similar to 3D encapsulation (Fig. S3B and C†). Furthermore, immunofluorescence staining of OC, another specific marker protein residing in cytoplasmic compartment of differentiating osteoblasts,42 was performed in order to confirm the osteogenic marker expression at protein level. After 3 weeks, hADSCs cultured in PEGDA-GO demonstrated the most elevated expression of osteocalcin (Fig. 5D).
4. Discussion
In this work, we modified PEGDA hydrogel with GO to assign biofunctionality. Previous studies showed that GO can adsorb considerable amount of ECM proteins, resulting in enhanced cell to matrix attachment.28,29 GO has carboxyl, carboxylate, carbonyl and hydroxyl functional groups, and GO can interact with a range of biomolecules including serum proteins under typical cell culture condition via electrostatic, covalent, and hydrogen bond.19,43–45 In addition, GO is composed of aromatic rings with reasonable oxygen contents, which leads to hydrophobic property of the material that allows increased adsorption of proteins. Because ECM globular proteins such as fibronectin can directly mediate cell adhesion, increase in ECM protein adsorption onto GO can facilitate initial attachment of stem cells onto the culture substrate and form the foundation of basic cellular behaviors. In the present study, GO functioned as cell-adhesive moiety, and the incorporation of GO in PEGDA hydrogel induced stark improvement in cell adhesion (Fig. 3A). These adhesive behaviors might be mediated by ECM proteins adsorbed onto GO, and the interaction between integrins and ECM proteins could further activate the downstream effectors FAK, which play a critical role in initiating adhesion-dependent cellular responses to matrix environment.46 As shown in Fig. 3B, in accordance with F-actin formation, hADSCs successfully developed FAK autophosphorylation. Considering that the enhancement in cell survival and osteogenic commitment of hADSCs in PEGDA-GO coincided with the development of focal adhesion and enhanced FAK activation in our work, we deduced that the central working mechanism of GO might originate from stable cell adhesion via strong adsorption between GO and ECM proteins.The incorporation of GO into PEGDA hydrogels had positive influences on cell viability and long-term survival. It is widely accepted that FAK plays a central role in transmitting adhesion-dependent survival signals,47,48 and stimulation of FAK signaling cascades followed by cell attachment to the ECM suppresses the activity of apoptotic factors.48 Notably, at day 3, the viability of hADSCs in PEGDA-GO was higher than that of PEGDA group (Fig. 4A), and it coincided with the decreased expression of pro-apoptotic caspase-3, a cysteine protease activated during apoptosis, in PEGDA-GO (Fig. 4C). However, there was no significant difference in proliferation rate between PEGDA and PEGDA-GO up to day 5 (Fig. 4B). Based on our observation of cell adhesion and subsequent FAK activation in PEGDA-GO group, we concluded that GO is capable of supporting cell homeostasis and survival, with respect to the successful retention of cells in PEGDA-GO even in the longer time period (Fig. 4D).
Previous studies have reported the role of GO on directing osteogenic differentiation of stem cells.19–21 We also demonstrated that hADSCs encapsulated in PEGDA-GO hydrogel stimulated the osteogenic phenotypes compared to the cells in PEGDA. RUNX2 gene transcription and ALP activity, which are the harbingers of osteogenesis, were elevated in PEGDA-GO group at 1 week (Fig. 5A and B). Together with enhanced BMP-2 and OP gene expression (Fig. 5A), characteristic osteogenic phenotypes including calcium deposition and osteocalcin expression were increasingly observed in hADSCs from PEGDA-GO hydrogel after 3 weeks (Fig. 5C and D). A preceding research reported that FAK activation and subsequent downstream ERK phosphorylation promote osteogenic differentiation of stem cells.27 As shown in Fig. 3B, hADSCs cultured in PEGDA-GO hydrogel established FAK phosphorylation, and this activation was then intracellularly transmitted to activate ERK. Namely, the enhanced osteogenic commitment of hADSCs in PEGDA-GO could be attributed by the incorporated GO, which promotes cell adhesion and following FAK-ERK pathway. In addition, there might be a connection between activated ERK and the upregulated RUNX2 gene expression occurred at 1 week, based on the critical role of ERK in osteogenesis. According to a study by Ge and colleagues, the activated ERK signaling pathway functions as an integral conduit for the development of osteogenic phenotype development and results in elevated RUNX2 phosphorylation and transcriptional activity.46 Likewise, in our study, FAK-ERK signaling pathway, at least in part, might bridge the signals from GO-adhered ECM proteins to the nucleus and could make impacts on RUNX2 gene activation.
Other potential factors that could accelerate GO-mediated osteogenic differentiation can be found among soluble factors. Dexamethasone is a chemical inducer that directs stem cells into osteogenic lineage,49 and the previous study demonstrated that GO could concentrate dexamethasone on its graphitic plane via π–π interactions.19 Because we cultured the hydrogel constructs under the presence of dexamethasone, the enhanced osteogenic differentiation might be partially explained by localization of dexamethasone within GO moiety. Moreover, we observed that the mRNA expression of BMP-2 was significantly upregulated in PEGDA-GO group compared to PEGDA group (Fig. 5A). Based on the fact that BMP-2 autocrine signaling loop provides positive feedback to differentiating stem cells,39,50 a preceding research employed BMP-2 autocrine signaling as a central mechanism for scaffold-mediated osteogenic differentiation.51 By introducing BMP-2-binding moiety within PEGDA hydrogel, the researchers could attach secreted BMP-2 proteins from differentiating cells in engineered scaffold, and they argued that the sequestered BMP-2 continuously promoted osteogenic differentiation via autocrine pathway. Considering that GO flakes can adsorb BMP-2 protein onto its surface,52,53 GO could sequester the BMP-2 from the encapsulated hADSCs in our PEGDA-GO hydrogel system. Therefore, PEGDA-GO might facilitate the osteogenic differentiation of hADSCs by sequestering de novo synthesized BMP-2 from the cells, which could provide a partial contribution in an autocrine manner during the osteogenesis of hADSCs in PEGDA-GO.
5. Conclusion
In conclusion, we demonstrated that the incorporation of GO could successfully assign biofunctionality to PEGDA hydrogel by enhancing cell attachment and subsequent osteogenic differentiation. hADSCs encapsulated in PEGDA-GO hydrogels resulted in an elevated level of focal adhesion and activation of related kinases (FAK and ERK). Furthermore, hADSCs cultured in PEGDA-GO successfully developed osteogenic phenotypes, and its outcome was more prominent than that of PEGDA. From this result, incorporation of GO into hydrogel can provide new design strategy of hydrogel-based substitutes for tissue regeneration.Acknowledgements
This study was supported by the Korea Health Industry Development Institute (KHIDI), Ministry of Health and Welfare (HI15C0498, HI15C3029 and HI14C3270), Republic of Korea. This study was also supported by National Research Foundation of Korea (Grant number NRF-2016R1E1A1A01943393 and NRF-2012M3A9C6050096).References
- F. Gattazzo, A. Urciuolo and P. Bonaldo, Biochim. Biophys. Acta, 2014, 1840, 2506–2519 CrossRef CAS PubMed.
- F. M. Watt and W. T. Huck, Nat. Rev. Mol. Cell Biol., 2013, 14, 467–473 CrossRef CAS PubMed.
- F. Guilak, D. M. Cohen, B. T. Estes, J. M. Gimble, W. Liedtke and C. S. Chen, Cell Stem Cell, 2009, 5, 17–26 CrossRef CAS PubMed.
- E. Carletti, A. Motta and C. Migliaresi, Methods Mol. Biol., 2011, 695, 17–39 CAS.
- I. M. El-Sherbiny and M. H. Yacoub, Glob. Cardiol. Sci. Pract., 2013, 2013, 316–342 Search PubMed.
- S. H. Kim, H. R. Lee, S. J. Yu, M. E. Han, D. Y. Lee, S. Y. Kim, H. J. Ahn, M. J. Han, T. I. Lee, T. S. Kim, S. K. Kwon, S. G. Im and N. S. Hwang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15426–15431 CrossRef CAS PubMed.
- S. Varghese and J. H. Elisseeff, Adv. Polym. Sci., 2006, 95–144, DOI:10.1007/12_072.
- S. J. Bryant and K. S. Anseth, Biomaterials, 2001, 22, 619–626 CrossRef CAS PubMed.
- C. G. Williams, T. K. Kim, A. Taboas, A. Malik, P. Manson and J. Elisseeff, Tissue Eng., 2003, 9, 679–688 CrossRef CAS PubMed.
- C. R. Nuttelman, M. C. Tripodi and K. S. Anseth, J. Biomed. Mater. Res., Part A, 2004, 68, 773–782 CrossRef PubMed.
- C. R. Nuttelman, M. C. Tripodi and K. S. Anseth, Matrix Biol., 2005, 24, 208–218 CrossRef CAS PubMed.
- N. Huebsch, P. R. Arany, A. S. Mao, D. Shvartsman, O. A. Ali, S. A. Bencherif, J. Rivera-Feliciano and D. J. Mooney, Nat. Mater., 2010, 9, 518–526 CrossRef CAS PubMed.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- S. Varghese, N. S. Hwang, A. C. Canver, P. Theprungsirikul, D. W. Lin and J. Elisseeff, Matrix Biol., 2008, 27, 12–21 CrossRef CAS PubMed.
- N. S. Hwang, S. Varghese, H. J. Lee, P. Theprungsirikul, A. Canver, B. Sharma and J. Elisseeff, FEBS Lett., 2007, 581, 4172–4178 CrossRef CAS PubMed.
- N. S. Hwang, S. Varghese, H. Li and J. Elisseeff, Cell Tissue Res., 2011, 344, 499–509 CrossRef CAS PubMed.
- M. M. Stevens and J. H. George, Science, 2005, 310, 1135–1138 CrossRef CAS PubMed.
- G. Chan and D. J. Mooney, Trends Biotechnol., 2008, 26, 382–392 CrossRef CAS PubMed.
- W. C. Lee, C. H. Lim, H. Shi, L. A. Tang, Y. Wang, C. T. Lim and K. P. Loh, ACS Nano, 2011, 5, 7334–7341 CrossRef CAS PubMed.
- S. Kang, J. B. Park, T. J. Lee, S. Ryu, S. H. Bhang, W. G. La, M. K. Noh, B. H. Hong and B. S. Kim, Carbon, 2015, 83, 162–172 CrossRef CAS.
- Y. Luo, H. Shen, Y. X. Fang, Y. H. Cao, J. Huang, M. X. Zhang, J. W. Dai, X. Y. Shi and Z. J. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 6331–6339 CAS.
- K. H. Liao, Y. S. Lin, C. W. Macosko and C. L. Haynes, ACS Appl. Mater. Interfaces, 2011, 3, 2607–2615 CAS.
- Y. Zhang, S. F. Ali, E. Dervishi, Y. Xu, Z. Li, D. Casciano and A. S. Biris, ACS Nano, 2010, 4, 3181–3186 CrossRef CAS PubMed.
- K. P. Loh, Q. L. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS PubMed.
- O. N. Ruiz, K. A. Fernando, B. Wang, N. A. Brown, P. G. Luo, N. D. McNamara, M. Vangsness, Y. P. Sun and C. E. Bunker, ACS Nano, 2011, 5, 8100–8107 CrossRef CAS PubMed.
- J. Park, B. Kim, J. Han, J. Oh, S. Park, S. Ryu, S. Jung, J. Y. Shin, B. S. Lee, B. H. Hong, D. Choi and B. S. Kim, ACS Nano, 2015, 9, 4987–4999 CrossRef CAS PubMed.
- R. M. Salasznyk, R. F. Klees, W. A. Williams, A. Boskey and G. E. Plopper, Exp. Cell Res., 2007, 313, 22–37 CrossRef CAS PubMed.
- H. H. Yoon, S. H. Bhang, T. Kim, T. Yu, T. Hyeon and B. S. Kim, Adv. Funct. Mater., 2014, 24, 6455–6464 CrossRef CAS.
- J. Park, B. Kim, J. Han, J. Oh, S. Park, S. Ryu, S. Jung, J.-Y. Shin, B. S. Lee and B. H. Hong, ACS Nano, 2015, 9, 4987–4999 CrossRef CAS PubMed.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- S. Zhang, P. Xiong, X. Yang and X. Wang, Nanoscale, 2011, 3, 2169–2174 RSC.
- S. J. Bryant and K. S. Anseth, J. Biomed. Mater. Res., 2002, 59, 63–72 CrossRef CAS PubMed.
- P. A. Zuk, M. Zhu, P. Ashjian, D. A. De Ugarte, J. I. Huang, H. Mizuno, Z. C. Alfonso, J. K. Fraser, P. Benhaim and M. H. Hedrick, Mol. Biol. Cell, 2002, 13, 4279–4295 CrossRef CAS PubMed.
- M. Wojtoniszak, X. Chen, R. J. Kalenczuk, A. Wajda, J. Lapczuk, M. Kurzewski, M. Drozdzik, P. K. Chu and E. Borowiak-Palen, Colloids Surf., B, 2012, 89, 79–85 CrossRef CAS PubMed.
- H. D. Kim, J. Heo, Y. Hwang, S. Y. Kwak, O. K. Park, H. Kim, S. Varghese and N. S. Hwang, Tissue Eng., Part A, 2015, 21, 757–766 CrossRef CAS PubMed.
- C. G. Williams, A. N. Malik, T. K. Kim, P. N. Manson and J. H. Elisseeff, Biomaterials, 2005, 26, 1211–1218 CrossRef CAS PubMed.
- T. Komori, J. Cell. Biochem., 2006, 99, 1233–1239 CrossRef CAS PubMed.
- R. Zohar, S. Cheifetz, C. A. G. McCulloch and J. Sodek, Eur. J. Oral Sci., 1998, 106, 401–407 CrossRef CAS PubMed.
- E. H. Groeneveld and E. H. Burger, Eur. J. Endocrinol., 2000, 142, 9–21 CrossRef CAS PubMed.
- M. F. Pittenger, A. M. Mackay, S. C. Beck, R. K. Jaiswal, R. Douglas, J. D. Mosca, M. A. Moorman, D. W. Simonetti, S. Craig and D. R. Marshak, Science, 1999, 284, 143–147 CrossRef CAS PubMed.
- A. K. Gaharwar, S. M. Mihaila, A. Swami, A. Patel, S. Sant, R. L. Reis, A. P. Marques, M. E. Gomes and A. Khademhosseini, Adv. Mater., 2013, 25, 3329–3336 CrossRef CAS PubMed.
- N. O. Kanbur, O. Derman, T. A. Sen and E. Kinik, Int. J. Adolesc. Med. Health, 2002, 14, 235–244 CrossRef PubMed.
- K. Andre Mkhoyan, A. W. Contryman, J. Silcox, D. A. Stewart, G. Eda, C. Mattevi, S. Miller and M. Chhowalla, Nano Lett., 2009, 9, 1058–1063 CrossRef CAS PubMed.
- C. Gomez-Navarro, J. C. Meyer, R. S. Sundaram, A. Chuvilin, S. Kurasch, M. Burghard, K. Kern and U. Kaiser, Nano Lett., 2010, 10, 1144–1148 CrossRef CAS PubMed.
- J. T. Jeong, M. K. Choi, Y. Sim, J. T. Lim, G. S. Kim, M. J. Seong, J. H. Hyung, K. S. Kim, A. Umar and S. K. Lee, Sci. Rep., 2016, 6, 33835 CrossRef CAS PubMed.
- C. Ge, G. Xiao, D. Jiang and R. T. Franceschi, J. Cell Biol., 2007, 176, 709–718 CrossRef CAS PubMed.
- P. Chiarugi and E. Giannoni, Biochem. Pharmacol., 2008, 76, 1352–1364 CrossRef CAS PubMed.
- P. J. Reddig and R. L. Juliano, Cancer Metastasis Rev., 2005, 24, 425–439 CrossRef PubMed.
- N. Z. Mostafa, R. Fitzsimmons, P. W. Major, A. Adesida, N. Jomha, H. Jiang and H. Uludag, Connect. Tissue Res., 2012, 53, 117–131 CrossRef CAS PubMed.
- E. Schonherr and H. J. Hausser, Dev. Immunol., 2000, 7, 89–101 CrossRef CAS PubMed.
- D. S. W. Benoit, S. D. Collins and K. S. Anseth, Adv. Funct. Mater., 2007, 17, 2085–2093 CrossRef CAS PubMed.
- W. G. La, M. J. Jung, J. K. Yoon, S. H. Bhang, H. K. Jang, T. J. Lee, H. H. Yoon, J. Y. Shin and B. S. Kim, Carbon, 2014, 78, 428–438 CrossRef CAS.
- W. G. La, S. Park, H. H. Yoon, G. J. Jeong, T. J. Lee, S. H. Bhang, J. Y. Han, K. Char and B. S. Kim, Small, 2013, 9, 4015–4060 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02410j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |