
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Template-directed synthesis of a cubic cyclodextrin polymer with aligned channels and enhanced drug payload†
Vikramjeet Singhac,
Tao Guoa,
Li Wua,
Jianghui Xua,
Botao Liua,
Ruxandra Gref*b and
Jiwen Zhang *a
*a
aCenter for Drug Delivery System, Shanghai Institute of Materia Medica, 201210, Shanghai, China. E-mail: jwzhang@simm.ac.cn
bInstitut de Sciences Moléculaires d'Orsay, UMR CNRS 8214, Université Paris-Sud, Université Paris-Saclay, 91400 Orsay, France
cSchool of Pharmaceutical Sciences, Sun Yat-Sen University, Guangzhou, China
First published on 11th April 2017
Abstract
Despite its 3D porous structure, the pharmaceutical applications of cyclodextrin based cross-linked polymers are limited due to their structural irregularities. To address this issue, a template-directed strategy is used to obtain cubic micro and nano cyclodextrin cross-linked polymer (CD-cubes) from cyclodextrin metal organic frameworks in this study. The well-organized γ-CDs in MOFs were crosslinked by diphenyl carbonate by a facile single step chemical reaction. Scanning electron microscopy and X-ray diffraction analysis revealed the almost perfect cubic shapes of the particles with a disordered internal structure. Contrarily to the non-crosslinked materials which immediately dissolved in water, the CD-cubes were remarkably stable after extensive washing with water. The CD-cubes possessed a mesoporous structure with pore size in the range of 2–4 nm and showed much higher BET surface and 8 times higher adsorption capacity for doxorubicin as compared to conventional cyclodextrin-sponges.
Introduction
In recent years, tremendous efforts have been dedicated to the development of cyclodextrin (CD)-based drug carriers. By improving the physico-chemical properties of the drugs, such as stability, solubility and bioavailability, CDs clearly appear to be not just simple additives, but rather “smart” parts of the drug delivery devices.1,2 Taking advantage of the peculiar structure of the CDs and their unique properties, particles made of crosslinked CDs became popular in recent years.3 In particular, cyclodextrin cross-linked polymers or so called “nanosponges” (CD-sponges) have been proposed as a new nanosized drug delivery system but display inhomogeneous morphologies and size distributions in the micron range. They are advanced cross-linked cyclodextrin polymers nanostructured within a three-dimensional network. Because of their nano-porous structure, CD-sponges can advantageously carry water insoluble (Biopharmaceutical Classification System class-II drugs) and soluble drugs for safe and targeted delivery.4,5 These complexes can be used to increase the dissolution rate, stability and mask unpleasant taste of drugs. Subsequently, the CD-sponges were reported to be synthesized and evaluated for number of applications including loading of drugs, i.e., doxorubicin, resveratrol, camptothecin, paclitaxel, itraconazole and more in drug delivery application.6–10 There are number of different techniques and cross-linkers reported for the synthesis of CLPs according to their applications. The intensive research on structural parameters of CD-sponges including vibrational dynamics, sol–gel transitions, swelling phenomena and biodegradability is now well documented.11–14 In the case of polymer particles in the range of nanometer to millimeter, the control of size and shape still remains very challenging.15–18 To the best of our knowledge, there is no report describing the controlled synthesis of CD-sponges, mainly in reason of experimental difficulties related to the cross-linking reactions.19 Therefore, a precise control of the size and shape, and a good stability of the particles in aqueous media are needed for the development of medical applications of CD-based drug carriers. The template-directed strategy has been proved a powerful method for control synthesis of nanoparticles with desired shapes in recent times.20 The cyclodextrin metal organic frameworks (γ-CD-MOFs) were used as a template in this study for the control synthesis of CD-CLPs for drug delivery applications. γ-CD-MOFs are a class of almost perfectly shaped, crystalline and porous materials build by coordination of γ-CDs and potassium (K) ions.21,22 The γ-CD-MOFs have become very popular in reason of their biocompatibility and potential applications in gas storage and biomedicine.23,24 However, the applications of γ-CD-MOFs in the pharmaceutical field are hampered by the poor water stability of these particles which rapidly disintegrate when exposed to humid conditions. To address the challenge of increasing the stability of γ-CD-MOFs in water, only two strategies have been reported so far. Furukawa et al. used ethylene glycol diglycidyl ether to crosslink γ-CD-MOFs to produce γ-CD-MOF hydrogels.25 However, the reaction was time-consuming, taking more than three days at 65 °C and several steps were required to remove the unreacted cross-linker. Very recently, Li et al. incorporated fullerene (C60) into the hydrophobic cavities of γ-CDs in an attempt to increase the aqueous stability of γ-CD-MOF.26 However, the supramolecular assemblies did not maintain their structure over prolonged times and eventually degraded in water after 24 h incubation due to the weak ligand–CD interaction. Moreover, the occupancy of γ-CD cavity by C60 might also reduce the capacity of γ-CD-MOFs to load drugs. In conclusion, the production of stable porous γ-CD-MOFs capable of efficiently loading drugs remains a major unmet challenge as well.In this paper, we successfully cross-linked the constitutive γ-CDs in γ-CD-MOFs through carbonate linkages using diphenyl carbonate as cross-linker. A facile synthesis route was developed using triethylamine (TEA) as a catalyst, capable to crosslink the γ-CD-MOF in a minimal time of four hours. Particles of well-defined shapes in the nanometer (200–500 nm) and micrometer (2–5 μm) ranges were obtained and the compared with conventional CD-sponges to evaluate their performance.
Experimental
Materials and reagents
The γ-cyclodextrin was obtained from MaxDragon Biochem Ltd. Potassium hydroxide (KOH), cetyl trimethyl ammonium bromide (CTAB), isopropanol, ethanol (EtOH), acetone, dichloromethane (DCM) and N,N-dimethylformamide (DMF) were of analytical grade and purchased from Sinopharm Chemical Reagent Co. Ltd (Beijing, China). Doxorubicin (>99.5% purity) were purchased from Dalian Meilun Biotech Co., Ltd. (Dialian, China). Diphenyl carbonate and triethylamine were ordered from Aladdin (China). All other chemicals were of analytical grade.Synthesis of nano and micrometers sized γ-CD-MOFs
The γ-CD (162 mg, 0.125 mmol) was mixed with 8 equivalent moles of KOH (56 mg, 1 mmol) in 5 mL aqueous solution. The solution was filtered through a 0.45 μm filter membrane into a glass tube, and 0.5 mL of MeOH was added, followed by vapor diffusion of MeOH into the solution at 50 °C. After 6 h, the supernatant was transferred into another glass tube with addition of CTAB (8 mg mL−1) and the solution was incubated at room temperature overnight. The synthesis procedure of nanometer sized CD-MOFs (CD-MOF-nano) were same as that for CD-MOF-micro, except that MeOH with the same volume of supernatant was added before CTAB dissolution. The precipitated washed with isopropanol and dried overnight at 37 °C.Synthesis of cyclodextrin micro and nano cubes
The cross-linked micro and nano cubes of cyclodextrins were directly synthesized using CD-MOFs. The harvested crystals of CD-MOFs (60 mM) were incubated in DMF with diphenyl carbonate (360 mM) at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 molar ratio (CD:DPC). After addition of triethylamine (450 μL), the particles were stirred at 80 °C for different time points (4, 8, 12, 24, 36 and 48 h). After completion of the condensation polymerization reaction, the reaction mixture was cooled to room temperature and 2 volumes of ethanol were added to quench the reaction. Triethylamine was used as a catalyst to enhance the reaction speed. The CD-cubes precipitated were then thoroughly washed with water acetone and ethanol (Soxhlet extraction, 6 h) and finally dried at 60 °C overnight.
:6 molar ratio (CD:DPC). After addition of triethylamine (450 μL), the particles were stirred at 80 °C for different time points (4, 8, 12, 24, 36 and 48 h). After completion of the condensation polymerization reaction, the reaction mixture was cooled to room temperature and 2 volumes of ethanol were added to quench the reaction. Triethylamine was used as a catalyst to enhance the reaction speed. The CD-cubes precipitated were then thoroughly washed with water acetone and ethanol (Soxhlet extraction, 6 h) and finally dried at 60 °C overnight.
Synthesis of γ-CD sponges
The γ-CD sponges were synthesized by the same procedure as described above for the synthesis of CD cubes and harvested at 4 h. Briefly, a 60 mM solution of γ-CD (0.778 g) in 10 mL DMF was prepared in clean and dry round bottom flask and heated up to 80 °C to obtain a clear solution. Then, 360 mM of DPC (0.771 g) was added and the reaction mixture was stirred for 10 min. Finally, 450 μL of triethylamine (C6H15N) was added, followed by gentle stirring for 4 h. After completion of the condensation polymerization reaction, the reaction mixture was cooled to room temperature and 6 volumes of distilled water were added to precipitate the CD-sponges. CD-sponges were then thoroughly washed in ethanol (Soxhlet extraction, 6 h) and distilled water to remove unreacted reagents and byproducts, especially phenol. They were subsequently lyophilized for 12 h using a freeze dryer (SIM international group Co., Ltd., USA) to keep their spongy structure intact. The traces of water were completely removed by oven drying at 50 °C for 2 h under vacuum.Characterization
The FTIR spectra of samples were recorded by using a Thermo-scientific system (Nicolet™ iS™5 FT-IR Spectrophotometer) in the region of 600 to 4000 cm−1. The IR spectrum of diphenyl carbonate and γ-CD were also recorded as controls.Morphological characterizations of all samples were conducted by scanning electron microscopy (SEM, S-3400N, Hitachi). The specimens were immobilized on a metal stub using double-sided adhesive tape and coated with a thin gold film, then observed under definite magnification. Considering the high regularity of γ-CD-MOF crystals and the value of length:width:height ≈ 1, the size of γ-CD-MOFs was measured with the Image Pro Plus 6.0 software on the basis of the obtained SEM images. Transmission electron microscopy (TEM) images were taken with a JEOL 2011 microscope (Japan) operating at 75 kV. For the TEM measurements, the samples were dispersed in ethanol and then dried on a holey carbon film Cu grid.
The crystallinity of the samples was characterized by X-ray powder diffraction (XRPD) analysis. Diffraction patterns of the prepared γ-CD-MOF crystals were obtained using a Bruker D8 Advance diffractometer (Bruker, Germany) at ambient temperature, with tube voltage of 40 kV, tube current of 40 mA in a stepwise scan mode (8° min−1). Diffraction data for the thermal and chemical stability samples were collected on an X/Pert Pro 3040/60 diffractometer (PANalytical, Holland). All the samples were irradiated with monochromatized CuKα radiation and analyzed over a 2θ angle range of 3–40°.
Nitrogen adsorption–desorption isotherms were measured with a liquid nitrogen bath (−196 °C) using a porosimeter (TriStar 3000 V6.05 A, USA). In order to remove the interstitial solvents, the samples were activated by immersing in DCM for three days and dried under vacuum at 50 °C for 12 h. Known amounts of samples (e.g. 150–200 mg) were loaded into the BET sample tubes and degassed under vacuum (10–5 Torr) at 80 °C for 5 h. BET model was applied to measure the specific surface areas of the prepared samples.
Thermogravimetric analysis (TGA) of γ-CD-MOF crystals was performed using a thermal analysis system (NETZSCH 209F3 240-20-382-L, USA) at a heating rate of 10 °C min−1 under nitrogen. Samples were weighed (approximately 5 mg) in a hanging aluminum pan and the weight loss percentage of the samples was monitored from 30 to 400 °C.
Drug adsorption study
Drug adsorption studies were performed by dispersing micro- and nanocubes and CD-sponges (0.1 g) in an aqueous solution of doxorubicin hydrochloride with a concentration of 500 mg L−1 (pH 7.0, 20 mL). The suspension was kept under stirring at 100 rpm at 25 °C. Next, 50 μL of the solution was withdrawn at a time interval of 5, 10, 20, 30, 40, 50 and 60 min and submitted to centrifugation to separate the drug-loaded particles from the supernatants containing the non-entrapped drug.The concentration of doxorubicin in the supernatants was determined by high performance liquid chromatography (HPLC) at the wavelength of 554 nm. Analysis was performed using an LunaTM analytical column Phenomenex C18 (150 × 4.6 mm, 5 μm) at room temperature. The mobile phase was a mixture of methanol and 0.1% trifluoroacetic acid at a ratio of 50:50, and a gradient elution was utilized, i.e., 1 mL min−1 until 14 min, linear increase to 1.5 mL min−1 till 20 min, then maintained for 9 min (20–29 min) before returning back to 1 mL min−1 over 1 min. Detection was performed at λex/em 233 nm and injected volumes were 20 μL. Calibration equations were obtained using least squares regression method on the nominal concentration versus the peak height ratio of doxorubicin to the internal standard.
Results and discussion
Synthesis of CD-cubes
The central aim of this research was to address both the challenges related to the nanosponges' size irregularities and to the CD-MOF instability, in an effort to develop original drug carriers. Keeping in mind the irregular structure of γ-CD cross-linked polymers (sponges) (Fig. S-1†), mesoporous morphologically-controlled cubic micro- and nanoparticles were developed. The cubic γ-CD-MOF crystals with regular nano and micrometer sizes were synthesized according to previous reports.27,28 The SEM images of γ-CD-MOFs (Fig. S-2†) revealed their cubic-shaped morphology and uniform sizes. The γ-CD in CD-MOFs crystals were further crosslinked in same manner as CD-sponges with diphenyl carbonate (DPC) in DMF at 80 °C. The reaction was carried out up to 24 hours, followed by quenching with ethanol and washing in water, acetone and ethanol (Soxhlet extraction, 6 h) to remove unreacted cross-linker and the byproduct phenol. The structure of the resulting CD-cubes (Fig. 1) was investigated by molecular simulations and displayed a high stability and minor CDs displacement as expected. At the same time, the simulation study was also suggested that the nano-channels of MOFs were remained significantly unaffected by cross-linking of CDs. These advantageous features were corroborated by specific surface measurements and the newly developed particles could load significantly higher amounts of drugs as compared to conventional CD-sponges or CD-MOFs. | ||
| Fig. 1 Schematic presentation of the molecular structure and synthesis of CD-cubes starting from CD-MOFs. | ||
The stability of CD-cubes in water was found to be strongly dependent on the crosslinking reaction time. Indeed, particles crosslinked for less than four hours were highly soluble in water, as their parent CD-MOFs. Remarkably, γ-CD-MOFs reacted for more than four hours with DPC were stable in water. Therefore, the cross-linking time was further optimized to achieve the desired stable structure. The stable micro- and nano cubes synthesized at different crosslinking times (4, 8, 12 and 24 h) are denoted herein as micro-4, 8, 12, 24 and nano-4, 8, 12 and 24.
Structural and morphological characterization
The successful cross-linking of CDs by DPC in γ-CD-MOFs was confirmed by FTIR investigations (Fig. S-3†). A characteristic band at 1754 cm−1 corresponding to the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) groups in the crosslinking material appeared in the particles crosslinked for more than four hours. A broad peak at ∼1260 cm−1 represented the O–C–O stretching in the structures. Similar features were present in CDs directly crosslinked using DPC, i.e. CD-sponges (Fig. S-3†). No significant structural differences were observed by FTIR between micro- and nano cubes obtained at different crosslinking intervals (Fig. S-4†). The specific peak at ∼1403 cm−1 attributed to the primary O–H functional groups of CDs seemed more prominent in micro and nano-4 samples, which is another indication of the incomplete cross-linking reaction at four hours. However, this peak is absent in the spectra of the γ-CD nanosponges, possibly because of an almost total disappearance of the O–H groups, efficiently reacted with the DPC crosslinking agent. It can be hypothesized that because of steric hindrance, not all the O–H groups of the CDs in the well-organized CD-MOFs are accessible to the crosslinking agent, contrarily to the direct crosslinking of the CDs in aqueous solution leading to the formation of sponges.
O) groups in the crosslinking material appeared in the particles crosslinked for more than four hours. A broad peak at ∼1260 cm−1 represented the O–C–O stretching in the structures. Similar features were present in CDs directly crosslinked using DPC, i.e. CD-sponges (Fig. S-3†). No significant structural differences were observed by FTIR between micro- and nano cubes obtained at different crosslinking intervals (Fig. S-4†). The specific peak at ∼1403 cm−1 attributed to the primary O–H functional groups of CDs seemed more prominent in micro and nano-4 samples, which is another indication of the incomplete cross-linking reaction at four hours. However, this peak is absent in the spectra of the γ-CD nanosponges, possibly because of an almost total disappearance of the O–H groups, efficiently reacted with the DPC crosslinking agent. It can be hypothesized that because of steric hindrance, not all the O–H groups of the CDs in the well-organized CD-MOFs are accessible to the crosslinking agent, contrarily to the direct crosslinking of the CDs in aqueous solution leading to the formation of sponges.
The morphologies of the micro-24 and nano-24 were observed by scanning and transmission electron microscopies (SEM, TEM) and are presented in Fig. 2. Of note, the shapes of micro-4 and nano-4 cubes were not intact and the defects might be caused by the incomplete cross-linking of γ-CDs and the washing steps (Fig. S-5†). In contrast, regular almost perfect cubic shapes were obtained for micro and nano-8 and 12 (Fig. S-5†). It was found that all the cubes prepared at 8, 12 and 24 hours crosslinking times maintained their cubic structures upon contact with water, contrarily to the previously reported crosslinked MOFs which were rather hydroscopic and formed hydrogels in water and retained this gel state even after drying under vacuum.26
 | ||
| Fig. 2 The graph showing transformation of CD-sponges into perfect cubic forms using CD-MOFs as template through SEM and TEM characterization. Morphological characterization of (A, C) micro cubes-24 (B, D) nano cubes-24 by SEM and TEM, respectively. | ||
PXRD evaluation of CD-cubes
The crystallinity of the CD-based particles was investigated by powder X-ray diffraction (PXRD) (Fig. 3). Parent CD-MOFs displayed a crystalline structure in agreement with previously published data.29,30 Of interest, all samples were crystalline after crosslinking and before washing with water to remove unreacted DPC. However, a gradual loss of crystallinity was noticed when increasing the duration of the cross-linking reaction (Fig. 3) from 4 h to further 8, 12 and 24 h subsequently. After extensive washing in water, the micro- and nano cubes became amorphous (Fig. 3) like CD-sponges (Fig. S-6†). Possibly, washing with water progressively displaced potassium ions which served as connecting metal when building the MOF structures. The coordination network was gradually destroyed by washing, but the γ-CD-MOFs maintained their integrity due to the establishment of a new covalent network built by crosslinking with DPC. It is reasonable to think that the covalent network (Fig. 1) cannot be as organized as the coordinative one consisting of identical molecular arrangements.31,32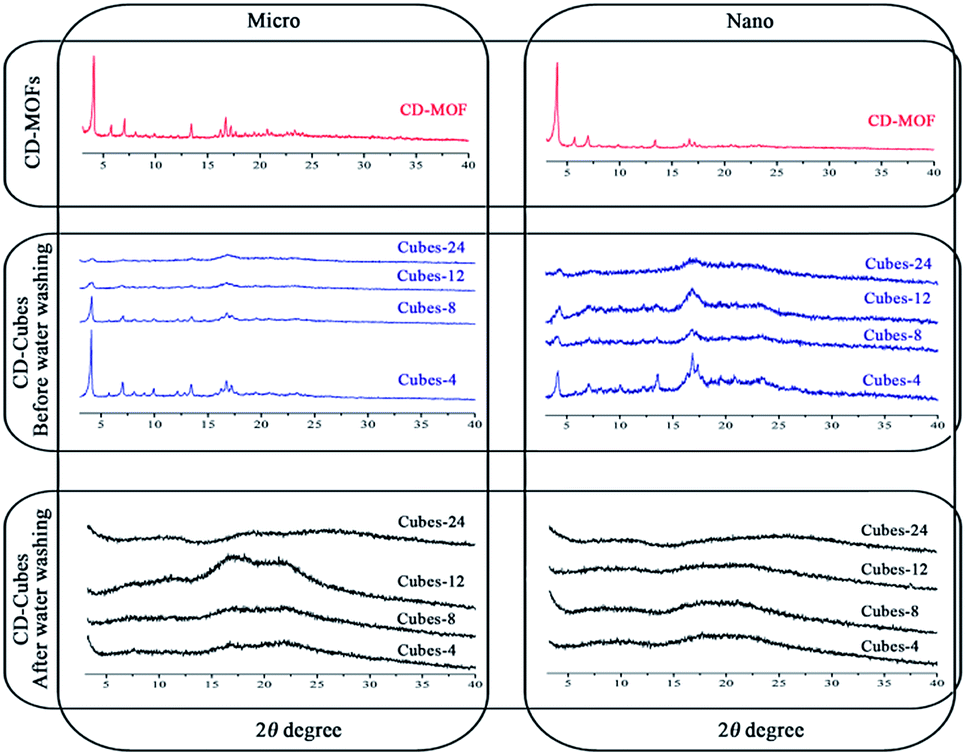 | ||
| Fig. 3 PXRD of cyclodextrins micro and nano cubes synthesized at different time points (before and after water washing) in comparison with parental CD-MOFs structure. | ||
BET measurements and thermal stability
In general, the CD-sponges showed very poor N2 absorption and hence the specific surface area (<1 m2 g−1) might be due to their open structure as reported previously.33 The micro and nano cubes were subjected to BET measurements for specific surface area and pore size evaluations. Typical N2 adsorption isotherms obtained with the micro-24 and nano-24 samples exhibit a type II behavior with relatively large pore volumes and specific surfaces areas. The BET surface areas and pore volumes were 140 and 315 m2 g−1 and 0.0627 cm3 g−1 and 0.145 cm3 g−1 for micro- and nano cubes, respectively. The large BET surface area in comparison to CD-sponges could be attributed to the aligned channels and intact cubic structure of CD-cubes with no doubt. These cubes possess mesopores within the range of 2–4 nm (Fig. 4A). Similar patterns were observed with parent CD-MOFs as shown in Fig. S-7.† However, the capacity to adsorb N2 was decreased by around more than 20% after crosslinking, possibly due to changes in chemical structure (Fig. 4B). Contrarily to γ-CD-MOFs, the CD-sponges exhibited poor both BET surface areas (<1 m2 g−1) and pore volumes (<0.005 cm3 g−1) (Fig. S-7†) which is in agreement with previous reports for CD cross-linked polymers.33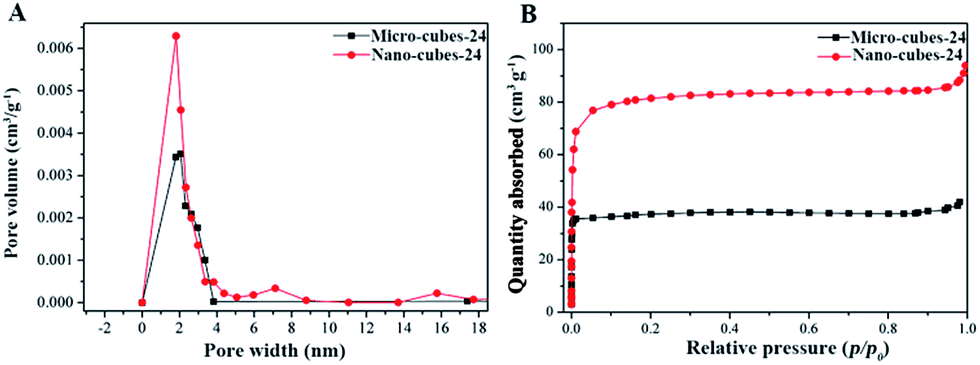 | ||
| Fig. 4 (A) Pore size distributions and (B) N2 absorption patterns for CD micro- and nanocubes. | ||
As previously hypothesized, the formation of numerous crosslinks between the CDs in the nanosponges could create a constraint microenvironment with a reduced available surface area as compared to the organized structure in CD-MOFs (Fig. 1). As shown in Fig. 5A, the thermal stability of γ-CD-MOFs was significantly improved by cross-linking and was similar to the one of CD-sponges. No notable difference in stability was found between micro- and nano cubes synthesized at different time intervals (Fig. S-8†).
 | ||
| Fig. 5 (A) TGA thermograms showing the comparison of CD-cubes with CD-MOFs and CD-sponges. (B) Time dependent doxorubicin adsorption in micro- and nano cubes in comparison with CD-nanosponges. Inset in (B) shows a linear fit of data obtained with cubes using a pseudo-second order kinetic model. | ||
Drug loading study of CD-cubes
The CD-sponges continued to be glorified for the drug delivery applications by several researchers.6–10 Mesoporous stable γ-CD-MOFs were studied for their ability to load drugs of interest. Doxorubicin, a well-known and widely used anticancer drug was loaded in γ-CD sponges and CD-cubes. Non crosslinked γ-CD-MOFs could not be used in reason of their poor stability during the loading process. The adsorption of doxorubicin within micro- and nano cubes was found to be significantly higher (60–80 mg g−1) than within CD-sponges (4–5 mg g−1) (Fig. 5B). In addition, the adsorption capacity was found to increase with the cross-linking reaction time. The highest and lowest absorbed amounts of doxorubicin were found with micro/nano-24 and micro/nano-4, respectively (Fig. S-9†). Indeed, during the loading process, the particles crosslinked during 24 h lost only 5.64% (w/w), whereas the ones crosslinked at 4, 8 and 12 hours lost 49.27 and 17.38% and 11.88%, respectively.The small loss of 24 h samples can attributed to the loss during washing and drying process. It was concluded that sufficient crosslinking was achieved at 24 hours, leading to a better stability upon drug loading. Under these conditions, the adsorption data could be very well fitted using a pseudo-second order kinetic model. The obtained parameters from kinetic model for all curves were presented in Table S-1.†
Conclusions
In a nutshell, stable micro- and nanosized cubic cyclodextrin polymer were obtained by linking the CDs in the regular cubic CD-MOFs through highly stable carbonate linkages. The crosslinking process did not affect the regular shape of the particles, which became amorphous but were highly stable in aqueous media and upon drug loading, contrarily to the non-crosslinked ones. These newly synthesized cubes have pore size in the range of 2–4 nm and specific surface areas of 315 m2 g−1 (nano cubes) and 140 m2 g−1 (micro cubes) allowing efficient drug adsorption. Obtained by a simple method, the γ-CD-MOF cubes combine both the stability of the crosslinked CDs sponges and the regular sizes and shapes of CD-MOFs. Moreover, the available hydroxyl groups at the surface of the γ-CD-MOFs cubes could be further reacted with specific ligands for targeted drug delivery purposes. The scalable and convenient synthesis route opens new applications for CD-MOFs in a wide range of fields including pharmaceutics and material sciences.Acknowledgements
This work was supported by the National Natural Science Foundation of China (81430087). RG acknowledges support from the ANR-14-CE08-0017 grant.Notes and references
- R. Gref and D. Duchene, J. Drug Delivery Sci. Technol., 2012, 2, 223–233 CrossRef.
- B. Gidwani and A. Vyas, BioMed Res. Int., 2015, 15 Search PubMed.
- S. Subramanian, A. Singireddy, K. Krishnamoorthy and M. Rajappan, J. Pharm. Pharm. Sci., 2012, 15, 103–111 CAS.
- F. Trotta, M. Zanetti and R. Cavalli, Beilstein J. Org. Chem., 2012, 8, 2091–2099 CrossRef CAS PubMed.
- G. Tejashri, B. Amrita and J. Darshana, Acta Pharm., 2013, 63, 335–358 CrossRef CAS PubMed.
- R. Cavalli, F. Trotta and W. J. Tumiatti, J. Inclusion Phenom. Macrocyclic Chem., 2006, 56, 209–213 CrossRef CAS.
- S. J. Torne, K. A. Ansari, P. R. Vavia, F. Trotta and R. Cavalli, Drug Delivery, 2010, 17, 419–425 CrossRef CAS PubMed.
- S. Swaminathan, L. Pastero, L. Serpe, F. Trotta, P. Vavia, D. Aquilano, M. Trotta, G. Zara and R. Cavalli, Eur. J. Pharm. Biopharm., 2010, 74, 193–201 CrossRef CAS PubMed.
- K. A. Ansari, P. R. Vavia, F. Trotta and R. Cavalli, AAPS PharmSciTech, 2011, 12, 279–286 CrossRef CAS PubMed.
- S. Swaminathan, P. R. Vavia, F. Trotta and S. Torne, J. Inclusion Phenom. Macrocyclic Chem., 2007, 57, 89–94 CrossRef CAS.
- F. Castiglione, V. Crupi, D. Majolino, A. Mele, B. Rossi, F. Trotta and V. Venuti, J. Raman Spectrosc., 2013, 44, 1463–1469 CrossRef CAS.
- V. Crupi, D. Majolino, A. Mele, L. Melone, C. Punta, B. Rossi and F. Toraldo, Soft Matter, 2014, 10, 2320–2326 RSC.
- F. Castiglione, V. Crupi, D. Majolino, A. Mele, W. Panzeri, B. Rossi, F. Trotta and V. Venuti, J. Inclusion Phenom. Macrocyclic Chem., 2013, 75, 247–254 CrossRef CAS.
- P. Shende, Y. A. Kulkarni, R. S. Gaud, K. Deshmukh, R. Cavalli, F. Trotta and F. Caldera, J. Pharm. Sci., 2015, 104, 1856–1863 CrossRef CAS PubMed.
- A. Abedini, A. A. Bakar, F. Larki, P. S. Menon, M. S. Islam and S. Shaari, Nanoscale Res. Lett., 2016, 11, 287 CrossRef PubMed.
- Y. Politi, T. Arad, E. Klein, S. Weiner and L. Addadi, Science, 2004, 306, 1161–1164 CrossRef CAS PubMed.
- H. Colfen and M. Antonietti, Angew. Chem., 2005, 117, 5714 (Angew. Chem., Int. Ed., 2005, 44, 5576–5591) CrossRef.
- X. Wang, Y. Zhang, W. Luo, A. A. Elzatahry, X. Cheng, A. Alghamdi, A. M. Abdullah, Y. Deng and D. Zhao, Chem. Mater., 2016, 28, 2356–2362 CrossRef CAS.
- F. Trotta, M. Zanetti and R. Cavalli, Beilstein J. Org. Chem., 2012, 8, 2091–2099 CrossRef CAS PubMed.
- B. Zhu, H. Chen, W. Lin, Y. Ye, J. Wu and S. Li, J. Am. Chem. Soc., 2014, 136, 15126–15129 CrossRef CAS PubMed.
- R. A. Smaldone, R. S. Forgan, F. Hiroyasu, J. J. Gassensmith, A. M. Slawin, O. M. Yaghi and J. F. Stoddart, Angew. Chem., Int. Ed., 2010, 49, 8630–8634 CrossRef CAS PubMed.
- R. S. Forgan, R. A. Smaldone, J. J. Gassensmith, F. Hiroyasu, D. B. Cordes, L. Qiaowei, C. E. Wilmer, Y. Y. Botros, R. Q. Snurr and A. M. Slawin, J. Am. Chem. Soc., 2012, 134, 406–417 CrossRef CAS PubMed.
- J. J. Gassensmith, F. Hiroyasu, R. A. Smaldone, R. S. Forgan, Y. Y. Botros, O. M. Yaghi and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 15312–15315 CrossRef CAS PubMed.
- J. M. Holcroft, K. J. Hartlieb, P. Z. Moghadam, J. G. Bell, G. Barin, D. P. Ferris, E. D. Bloch, M. M. Algaradah, M. S. Nassar, Y. Y. Botros, K. M. Thomas, J. R. Long, R. Q. Snurr and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 5706–5719 CrossRef CAS PubMed.
- Y. Furukawa, T. Ishiwata, K. Sugikawa, K. Kokado and K. Sada, Angew. Chem., Int. Ed., 2012, 51, 10566–10569 CrossRef CAS PubMed.
- H. Li, M. R. Hill, R. Huang, C. Doblin, S. Lim, A. J. Hill, R. Babarao and P. Falcaro, Chem. Commun., 2016, 21, 5973–5976 RSC.
- N. Lv, T. Guo, B. Liu, C. Wang, V. Singh, X. Xu, X. Li, D. Chen, R. Gref and J. Zhang, Pharm. Res., 2017, 34, 269–278 CrossRef CAS PubMed.
- K. J. Hartlieb, J. M. Holcroft, P. Z. Moghadam, N. A. Vermeluen, M. M. Algaradah, M. S. Nassar, Y. Y. Botros, R. Q. Snurr and J. F. Stoddart, J. Am. Chem. Soc., 2016, 138, 2292–2301 CrossRef CAS PubMed.
- Y. Marui, T. Kida and M. Akashi, Chem. Mater., 2010, 22, 282–284 CrossRef CAS.
- Z. Moussa, M. Hmadeh, M. G. Abiad, O. H. Dib and D. Patra, Food Chem., 2016, 212, 485–494 CrossRef CAS PubMed.
- A. P. Cote, H. M. El-Kaderi, H. Furukawa, J. R. Hunt and O. M. Yaghi, J. Am. Chem. Soc., 2007, 129, 12914–12915 CrossRef CAS PubMed.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortes, A. P. Cote, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–287 CrossRef CAS PubMed.
- P. L. Meo, G. Lazzara, L. Liotta, S. Riela and R. Noto, Polym. Chem., 2014, 5, 4499–4510 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, SEM images of CD-MOFs and sponges, BET characterizations of CD-MOFs and sponges, FTIR spectra and doxorubicin adsorptions curves. See DOI: 10.1039/c7ra02306e |
| This journal is © The Royal Society of Chemistry 2017 |