
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphene entanglement in a mesoporous resorcinol–formaldehyde matrix applied to the nanoconfinement of LiBH4 for hydrogen storage†
A. Gasnier *a and
F. C. Gennariab
*a and
F. C. Gennariab
aConsejo Nacional de Investigaciones Científicas y Técnicas, CONICET, Instituto Balseiro, U.N. Cuyo, Centro Atómico Bariloche (CNEA), S.C. de Bariloche, Río Negro, R8402AGP, Argentina. E-mail: aurelien.gasnier@cab.cnea.gov.ar
bInstituto Balseiro, Universidad Nacional de Cuyo, Argentina
First published on 26th May 2017
Abstract
A new, easy and versatile method for graphene inclusion within resorcinol–formaldehyde is presented and applied to the nanoconfinement of LiBH4. After the initial formation of a graphene hydrogel, the resin precursors were allowed to diffuse through the aqueous phase at room temperature. Depending on the precursor's concentration, after curing and pyrolysis, the materials presented a specific area of about 600 m2 g−1, with a pore size as low as 6.1 nm without macropores, and pore volume as high as 1.53 cm3 g−1. Once filled with LiBH4 by melt-infiltration, the differential scanning calorimetry (DSC) of these materials was typical of nanoconfined hydrides, with broader, flatter and lower transition, melting and decomposition temperatures. Hydrogen release was confirmed for temperatures as low as 253 °C in the presence of graphene, with a total hydrogen release of 13 wt% at 400 °C, close to the expected theoretical value. The absence of diborane formation was confirmed by IR and is a good indication of these materials' reversibility. After rehydrogenation at 400 °C for 5 h under 60 bar H2, the hydrogen release was close to 6 wt%. Microscopic observations and pore-size analysis indicated that the presence of graphene could be beneficial to the pore filling.
1. Introduction
As an energy vector, hydrogen is an efficient and environmentally-friendly alternative to fossil fuels. One of the main constrains for its technological application is to develop a safe, fast and reversible hydrogen storage medium under mild conditions, with high gravimetric and volumetric capacities. Despite the interest of the scientific community, no solution has been found so far that could meet the conditions of commercial application.Solid-state hydrogen storage based on reversible metal hydrides offers several advantages compared to other approaches of storing hydrogen. In particular, lithium borohydride (LiBH4) is a good candidate because of its large weight capacity (18.4% of mass loss in the case of total dehydrogenation), although its application is hindered by slow kinetics and sluggish reversibility. Indeed LiBH4 decomposes around 500 °C with toxic diborane (B2H6) liberation, and it requires temperatures of 600 °C under 350 bar H2 to be rehydrogenated. Also, the formation of closoboranes is a major issue, as they hinder the reverse formation of LiBH4 from dehydrogenated products. Thus many strategies have been proposed to improve the kinetics of LiBH4 decomposition and its reversibility.1–5 Those strategies mostly rely on the incorporation of destabilizing hydrides likes MgH2,1 and catalytic transition metal,2 oxides or chlorides.3
Nanoconfinement is a more recent method, proposed to overcome the inherent drawbacks of hydrides, LiBH4 in particular.6 Several authors exposed that when hydrides are confined in nanosized pores, their decomposition kinetics are greatly augmented.7–11 To obtain matrices sprinkled with mesopores of controlled size, amorphous carbon materials based on resorcinol–formaldehyde and highly ordered nanoporous carbons are usually considered because they offer a great versatility of study.10,12–17 Several other carbon-based materials were also applied to the nanoconfinement of borohydrides, such as: carbon fibers,18,19 carbon nanotubes,20 polymers,21 and graphene.22 It was observed that smaller pores limit the formation of diborane during the dehydrogenation of LiBH4; this prevents the subsequent transformation of diborane toward stable closoborane salts, and improves the reversibility of boron-based hydrogen storage systems.23
The outstanding physical properties of graphene carbon allotrope motivated the emergence of myriad ways to modify it (functionalization, doping, nanoparticle decoration),24–26 applied in some cases to hydrogen storage.27–31 We considered that on one hand the excellent thermal conductivity of graphene could compensate the poor conductivity of amorphous carbon, and on the other hand this material could represent a versatile platform to bring a range of nanosized catalyst to the nanoconfined material.32 Moreover it has been demonstrated that graphene is able to wrap around borohydrides, enhancing their hydrogen release and uptake.22 While the chemical synthesis of graphene is scalable, it is usually fouled with many chemical and structural defects. In particular it is difficult to get rid of all the oxygenated functions, which could be detrimental to the LiBH4 integrity. Yet, structural defects (principally holes in the graphene sheets), typical of chemically synthesized graphene, might be beneficial to the infiltration of LiBH4 within the matrix.33,34 Still, it is difficult to obtain graphene matrices with mesopores of controlled distribution and macropores represent a major fraction of the total pore volume, which won't present a nanoconfinement effect over the hydride.35 Moreover, graphene three dimensional structures are often obtained as hydrogels that shrink by several orders of magnitude during air-drying. To circumvent this, specific amenities (super critical CO2 drying, lyophilisation) are needed whereas the pore structure can still be affected.35
Kim and Worsley described a method to incorporate graphene within a resorcinol matrix where the graphene oxide solution forms the aqueous media in which resorcinol and formaldehyde will be dissolved before curing the resin.36,37 Nevertheless, resorcinol–formaldehyde resins pore size is dependent on the concentration of a basic catalyst in the mixture and graphene oxide presents many carboxylic functions that might interfere with pore-size control by the catalyst concentration. We assumed that our objective to later employ chemically modified and/or decorated graphene would be even more problematic. Nonetheless, it is reasonable to assume that the reduced graphene solid would interact much less than its soluble graphene oxide counterpart with the resorcinol–formaldehyde solution during the polymerisation–reticulation step. As graphene can conveniently be reduced to a hydrogel by ascorbic acid and this being displaced out of the matrix by diffusion with water washing later on,35 resorcinol and formaldehyde could migrate within the graphene matrix prior to the cure step.
We propose here an original method to entangle graphene in a solid matrix: at first a graphene oxide solution is reduced to obtain a hydrogel, which aqueous part is replaced by diffusion of a resorcinol/formaldehyde solution. Afterward the mixture is cured to obtain a solid resin that is subjected to pyrolysis. The influence of the incorporation of graphene on the matrix pore-size is presented.
The pores were filled with LiBH4 by melt-impregnation to determine the effects of graphene on the transition, fusion and dehydrogenation temperatures of LiBH4. Finally we examine the behaviour of these materials for hydrogen release kinetics and their reversibility. Their morphological aspect with and without LiBH4 is discussed, before and after hydrogen release/uptake cycle.
2. Experimental
2.1. Synthesis of materials
All chemicals were employed as received without purification. Graphite flakes (product number 33246-1) was supplied by Aldrich. Ascorbic acid (99.0%), potassium permanganate (99.0%), sulphuric acid (98%), hydrochloric acid (35%), diethyl ether (98.0%), resorcinol (98.5%) were purchased from Cicarelli, hydrogen peroxide (30%), and formaldehyde (40%) from Biopack while phosphoric acid (85%) was provided by Merck.Materials were mechanically milled under argon atmosphere using a P6 Pulverissette planetary device, with an 80 cm3 milling chamber and 5 stainless balls. 1.5 g LiBH4 (Sigma Aldrich, 90%) was milled during 300 min at 400 RPM, with a sequence of 10 min milling and 10 min pause. To reduce morphological impact and Fe contamination the resins were milled for a short time: 5 reverse repetitions of 2 min milling at 200 RPM, 1 min pause.
Every hydride material was handled inside a glove box to avoid contact with air (content of oxygen and water <10 ppm). After their synthesis the resins were dried for 3 h at 300 °C under vacuum and placed inside the glove box for handling.
Graphene oxide was synthesized according to the Tour's method because it affords highly soluble sheets that can be used on a broader range of concentration.38
To obtain the resins modified with graphene (Fig. 1), the dry graphene oxide flakes were dispersed at 3 mg mL−1 by ultrasonication in distilled water. 10 mL of this mixture were placed in plastic bottles with five mass equivalents of ascorbic acid. After resting 80 h at 40 °C a black hydrogel set; next the aqueous phase was washed three times with excess water over 72 h. 46 g of aqueous resorcinol/formaldehyde solutions (named A to D, according to the precursors concentration, see Table 1) were prepared. 10 g of these solutions were isolated as reference material (noted with the “R” prefix in the following sections) and the remnant was employed to fill the graphene hydrogel with the resin precursors (“G” prefix).
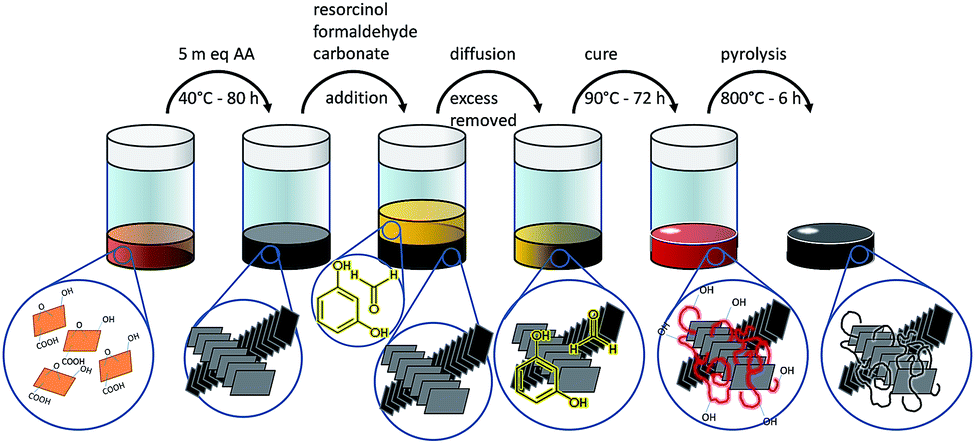 | ||
| Fig. 1 Schematic representation of the incorporation of graphene within a resorcinol–formaldehyde matrix. | ||
| Resin type | A | B | C | D |
| Resorcinol (g) | 11.85 | 14.88 | 11.89 | 14.86 |
| Formaldehyde (g) | 17.92 | 22.27 | 17.55 | 22.22 |
| Water (g) | 16.20 | 8.83 | 16.52 | 8.84 |
| Carbonate (mg) | 24.3 | 28.5 | 37.7 | 81.9 |
| Org. fraction (%) | 40 | 50 | 40 | 50 |
| R/B ratio | 500 | 500 | 310 | 180 |
During the first 24 h at RT, 6 g of these resorcinol–formaldehyde solutions were poured in the flask with the graphene hydrogel every 4 h, gently swirled and allowed to rest; then the solution was replaced with a fresh aliquot (doing so one can expect a concentration close to 94% of the reference solution). Afterwards the mixtures (GA to GD) were placed 24 h at 50 °C next to their reference (RA to RD) and eventually all these were cured for 72 h at 90 °C. Then the resins were allowed to cool to RT and the residual water was displaced with 3 washings of excess acetone over 72 h. The resins were leaved to dry for 3 days in the hood and finally the red glassy monoliths (homogeneously filled with a black core when graphene is present, ESI1†) were broken to small pieces, placed in a quartz tube under nitrogen flux and pyrolized at 800 °C for 6 h to obtain black chunks.
To impregnate the carbon matrix with LiBH4, both materials were separately ball-milled to powders, then they were gently mixed together manually in a glove box with a pillar and mortar for 30 min. In the following sections the filling percentage will be given as a volume per volume, so a 50% filling means that 50% of the considered matrix pore volume (determined by BJH) is expected to be filled with a precise mass of LiBH4, given a density of 0.67 g cm−3 for this hydride. Typically 50.0 mg of LiBH4 mixed with 100 to 200 mg of carbon material (according to their respective textural parameters) were placed in a sampler within an autoclave. The impregnation of the porous carbonaceous matrix was realized by melting LiBH4 at 300 °C under 60 bar H2 for 30 min. To be characterized the obtained samples were transferred back to the glove box, whereas hydrogen release was measured right after cooling the impregnation product without taking the products out of the autoclave.
2.2. Material characterization
Textural parameters of the samples were studied using a Micromeritics ASAP 2020 analyzer. N2 adsorption isotherms were collected at −196 °C on 0.1 g of sample, after surface cleaning in vacuum at 300 °C overnight. Surface area and pore distribution were obtained applying the BET and BJH methods, respectively. Morphological and agglomerate size distribution analyses of the samples were performed by scanning electron microscopy (SEM Nova Nano 230, FEI Company). The powder samples were dispersed on carbon-tape and observed with secondary electrons.Fourier transform infrared spectroscopy of the gas phase was realized with a Perkin Elmer Spectrum 400 with MCT detector. The gases released after heating the sample at different temperatures (100 °C, 200 °C, 300 °C and finally 400 °C) were collected in a degassed quartz optical cell with KBr windows and optical length of 9 cm. The gas phase spectra were taken at room temperature (RT) with a resolution of 0.5 cm−1. The solid samples were grounded with dry KBr, pressed to pellets and placed in a specially designed cell, closed in an argon-filled glove box.
Hydrogen sorption kinetics were obtained using a modified Sieverts-type equipment, coupled with a mass flow controller. The sample was placed in a stainless reactor, inside an autoclave that was connected to the Sieverts device. Before the first dehydrogenation the sample was heated up to the melting temperature of LiBH4 (300 °C) under hydrogen pressure (60 bar) and kept at this temperature for 30 min, then allowed to cool freely to RT. Dehydrogenation curves were obtained by heating at 5 °C min−1 up to 400 °C with a hydrogen back pressure of 0.5 ± 0.1 bar. The rehydrogenation curves were measured at 400 °C for 5 h at an initial hydrogen pressure of 60 bars. The amount of absorbed/desorbed hydrogen was determined with a relative error of ±5%.
Thermal desorption behaviour of the hydride phases was studied by differential scanning calorimetry (DSC, TA 2910 calorimeter), using heating rates of 5 °C min−1 and argon flow rate of 122 mL min−1. The heat flow was normalized with respect to the mass of LiBH4.
3. Results and discussion
3.1. Effect of pore size and filling fraction of the matrix on LiBH4 transition, melting and dehydrogenation temperatures
Fig. 2 presents the textural parameters of resins RA to RD (Fig. 2A) and their graphene-containing counterparts GA to GD (Fig. 2B). As expected, the average pore size is dependent on the organic/water fraction and the resorcinol/base values. Without graphene the pore distribution is very narrow with an average pore diameter comprised between 4.6 and 15.6 nm (Table 2). When graphene is present, small pores can still be obtained according to the same parameters, but the pore size distribution is broader and displaced towards higher diameter (from 6.1 to 26 nm) than their reference counterparts. We believe this is due to the slightly lower concentration and the necessary handling of the samples during the first 24 h when filling the graphene hydrogel with the RF sol by successive washings. Even if graphene affects the pore size, this effect is limited (25 to 125% increase of average pore diameter) and in contrast to graphene aerogels the macropore population is negligible. On the other hand, the specific area is hardly affected by the presence of graphene (from 13% decrease to 6% increase) while the total pore volume increases when graphene is present (from 14% to 54% increase), which is beneficial for our purpose (as higher mass of LiBH4 can fit in a given percentage of volume filling). Thus, the presence of graphene does not impair the possibility of obtaining carbonaceous materials with pores below ten nanometers and to control their size with precursor's concentration.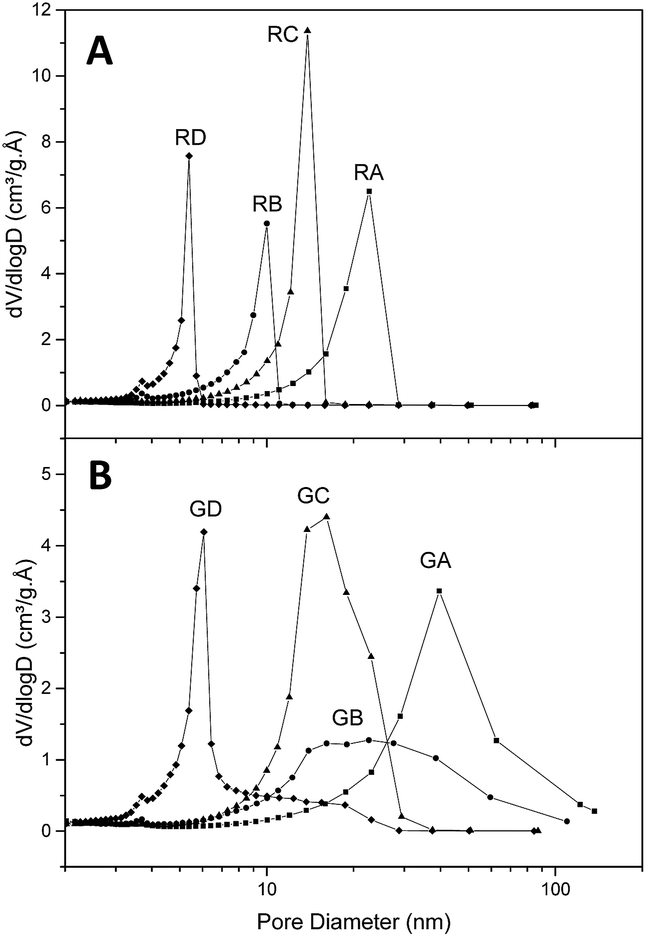 | ||
| Fig. 2 Pore-size distribution of resins without (A) and with graphene (B). | ||
| a BET surface area (m2 g−1).b BJH desorption cumulative volume of pores between 1.7 nm and 300 nm diameter (cm3 g−1).c BJH desorption average pore diameter (4V/A) (nm). | ||||||||
|---|---|---|---|---|---|---|---|---|
| Resin | RA | GA | RB | GB | RC | GC | RD | GD |
| Surfacea | 599 | 603 | 655 | 569 | 759 | 675 | 578 | 611 |
| Volumeb | 1.18 | 1.53 | 0.65 | 1.0 | 1.23 | 1.4 | 0.48 | 0.68 |
| Diameterc | 15.6 | 26 | 7.2 | 16 | 10.8 | 13.6 | 4.6 | 6.1 |
Once the presence of nanosized pores within the matrix was verified, the effect of these pores with respect to LiBH4 decomposition was studied. DSC of bulk and matrix-filled LiBH4 at 30, 50 and 70% are presented in ESI2 and 3–4† respectively, with heat flow normalized with respect to the mass of hydride within the sample.
The DSC plot of bulk LiBH4 up to 400 °C (ESI2†) presents two endothermic peaks: first the hexagonal to orthorhombic transition around 119 °C, followed by the melting at 290 °C. LiBH4 decomposition cannot be seen below 400 °C but a third endothermic peak near 495 °C was already described by Liu et al. and attributed to this phenomenon.23 These peaks are present for nanoconfined samples at different temperatures and in some cases their sharpness decreases.
As nanoconfinement effects are usually more marked for lower pore filling,13,23 we present the DSC curves of nanoconfined LiBH4 at 30% volume filling without (Fig. 3A) and with graphene (Fig. 3B). At first glance, Fig. 3 highlights two trends when pores get smaller: peak temperature decreases and peak definition gets blunt; in certain cases some peaks can even disappear. The effect of nanoconfinement on the definition of the peaks was already discussed in several papers,13,23,32,39,40 and it was attributed to the increasing disorder – the absence of long order crystal – of nanosized LiBH4 particles.32,40,41 The decrease of the peaks temperature is due to the enhanced BH4− mobility when nanoconfined,42,43 and the more pronounced contribution of mobile hydrogen species within smaller pore.17 Both effects can be attributed to the decrease of coherence length, and ultimately can conduce to the disappearance of transition and melting peaks with pore size below 4 nm, once LiBH4 gets amorphous.23 Fig. 3C and D illustrate the impact of LiBH4 filling ratio. It can be seen that the peaks get more defined with higher filling percentage; in particular, the decomposition peak is absent for any resin at 30% filling, while it is present in every case at 70%.
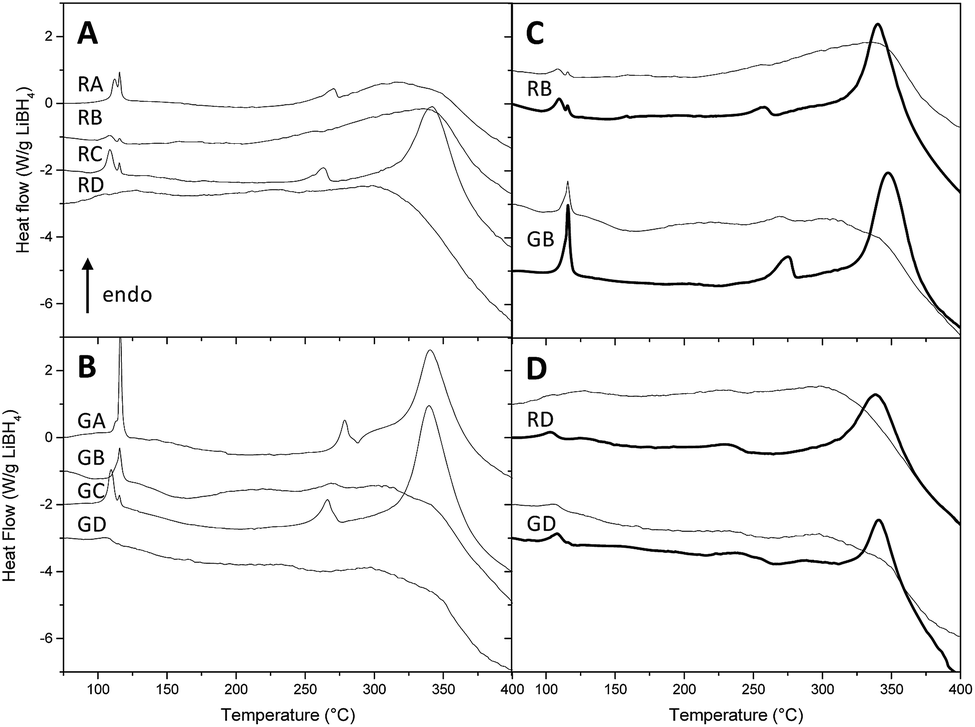 | ||
| Fig. 3 DSC plots of 30% filled resins without (A) and with graphene (B). DSC plots of 30% (fine line) and 70% (bold line) filled resins RB/GB (C) and RD/GD (D) without (upper) and with graphene (lower). | ||
Fig. 4 summarizes for each class of peak the effect of pore size. From 104 °C to 116 °C, the observed peaks are attributed to the hexagonal to orthorhombic transition (Fig. 4A). Without graphene, two populations of transition peaks are distinguished by DSC: from 104 °C to 112 °C the peak temperature (squares) is dependent from pore size, while a second population (stars) stays around higher values of temperature (115 °C to 116 °C). To explain this behaviour two hypotheses may be formulated: (a) the first population of peaks – dependant on pore volume – may be related to nanoconfined LiBH4, while the second population could be related to non-infiltrated LiBH4, i.e. hydride that stays on the outer part of the resin (thus independent on the pore size). Yet, it is intriguing that the resins with the smallest pores (RD), which should be harder to fill completely, do not present any peak around 115 °C (that could be attributed to non-infiltrated LiBH4). (b) Another hypothesis in better agreement with this observation could be that the first population of peak is related to the outer sphere of the nanoconfined hydride (in contact with carbonaceous content and subject to superficial tension) while the second population is related to the inner sphere of the nanoparticle, thus acting like bulk material with higher transition temperature. In this last case, the contribution of the inner sphere should be less noticeable the smaller the particles are. When graphene is present, a similar trend can be presumed; nevertheless it is harder to distinguish two transition peaks on the DSC curves, probably because the pore size distribution is broader in the presence of graphene, promoting an also broader transition peak, harder to resolve from the second one.
 | ||
| Fig. 4 Temperature of transition (A), melting (B), and decomposition (C) according to pore size without (filled symbol) and with graphene (empty symbol). Figures (A to B) present results for 30% pore filling, figure (C) for 50% pore filling. In figure (A) stars represent the second transition peak. Materials submitted latter on to volumetric investigation are indicated by small (RB, GB) and large (RD, GD) grey circle. | ||
The melting temperature (Fig. 4B) appears to be remarkably dependant on the pore size: while in LiBH4 transition temperatures vary by 10 °C, its melting temperatures are comprised within a 50 °C window. With decreasing pore diameter, melting occurs at lower temperature and its peak appears less sharp. Moreover, while transition temperature varies linearly with a broad dispersion, melting temperature varies in a linear and steady fashion down to 7 nm and then abruptly below 6 nm. This is consistent with previous reports, in which below a certain pore size nanoconfined LiBH4 becomes amorphous.23
At 30% filling the LiBH4 decomposition peak is absent in several samples, otherwise it stands between 340 °C and 341 °C (Fig. 3). This can be attributed to the change in the baseline curvature that occurs above 325 °C; nevertheless, the disappearance of the decomposition peak can also be a typical evidence of nanoconfinement. As nanoconfinement gets less noticeable with higher filling, we present the plot of transformation temperature against pore size at 50% filling (Fig. 4C), because at this percentage the peak is observed for any resin. For pore-size comprised between 4 and 14 nm, the temperature of decomposition increases by more than 10 °C; interestingly when graphene is present the 16 nm pore diameter resin (GB) presents a decomposition temperature very close to the temperature observed for resins with 6 nm pore diameter (GD). This fact is consistent with the flattened shape of the peaks displayed by the same resins in Fig. 3B, and indicates that those materials can present lower hydrogen sorption temperatures. Hence, despite the ill-defined distribution of pore size of resin GB (Fig. 2B), the properties of resins B and D, with and without graphene were further studied. In addition, as DSC revealed a remarkable influence of the filling percentage, we decided to check how the hydrogen kinetic and reversibility would change between 30 and 70% filling.
3.2. Hydrogen desorption properties and reversibility
Non-isothermal desorption curves are presented in Fig. 5, with desorbed H2 expressed in percentage of the mass of LiBH4 actually placed in the reactor. The curve associated to bulk LiBH4 decomposition is also presented, for reference (Fig. 5A). During the first desorption (Fig. 5A and B), it appeared that the kinetics of H2 liberation are principally influenced by the filling percentage of the matrix: the higher the LiBH4 filling, the slower the H2 release. To reach 1% hydrogen release, the slower material (resin B with graphene at 70% filling) needs an increase in temperature of 31 °C with respect to the faster one (resin D without graphene at 30% filling) (see ESI7–9† for numerical values). This difference is scarce probably because we selected resins B and D for their better DSC behaviour, but if compared with bulk LiBH4 the difference of temperature at 1% release is comprised between 60 and 90 °C according to the resin and its filling. Graphene hinders hydrogen release kinetics, but less than pore filling. Given the small amount of sample (roughly 300 mg total) and the slow non-isothermal process (5 °C min−1), the enhancement of thermal conduction expected from graphene might not traduce as better kinetics of hydrogen release. Interestingly, graphene is more detrimental at 70% filling (10 °C increase at 1% hydrogen release) than at 30% (3 °C increase at 1% hydrogen release). Finally, the resin type is the most influent factor, lower pore size (RD) presenting lower desorption temperature (249 °C at 1% hydrogen release with 30% filling). More importantly, all materials presented a very similar total mass of released hydrogen (with respect to the total mass of LiBH4), close to the maximum expected value (13.8%) for the usually considered desorption pathway (eqn (1)):| LiBH4(s) → LiH(s) + B(s) + 3/2H2(g) | (1) |
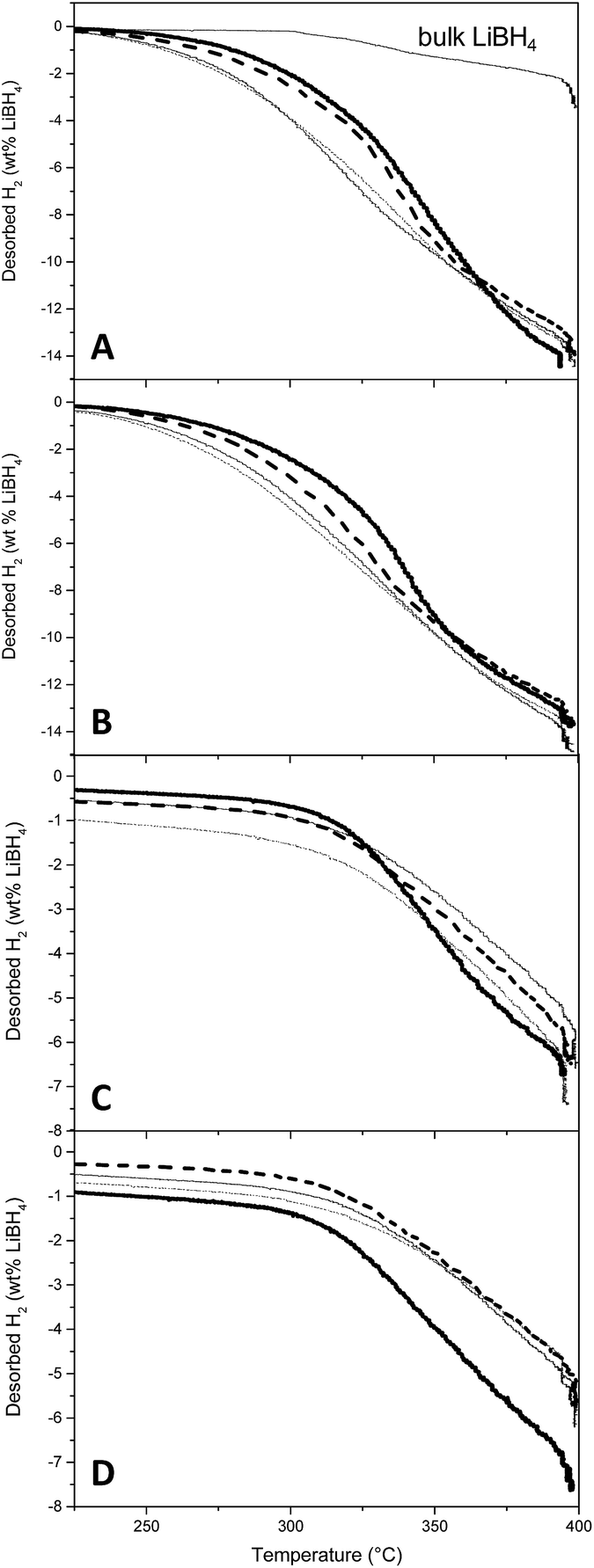 | ||
| Fig. 5 First (A and B) and second (C and D) desorption curves at 5 °C min−1 in the absence (dashed line) and presence (plain line) of graphene. RB/GB (A and C) and RD/GD (B and D) were loaded at 30% (fine line) and 70% (bold line). The desorption curve of bulk LiBH4 under the same conditions is indicated for reference (A). | ||
This last point is a good indication that the presence of graphene (and possible residual oxygenated functions) is not detrimental to the chemical integrity of LiBH4. It also suggests that nanoconfinement prevents the release of diborane and the concomitant formation of closoboranes, in agreement with previous observations.23 To confirm this a sample resin GB filled at 70% was placed in a tube and heated to 100 °C, 200 °C, 300 °C and finally 400 °C. At each step the products of desorption were analyzed by gas-FTIR and diborane was not observed. At the end of the experiment the presence of H2 was confirmed with a gas detector. The absence of closoborane formation should traduce in an increased reversibility of the nanoconfined hydride. The absence of [B12H122−] was also confirmed by solid-state FT-IR after hydrogen cycling. While the freshly impregnated samples presented clear bands attributed to LiBH4, the band at 2480 cm−1, characteristic of closoboranes by-products was absent (ESI6†). Moreover the dehydrogenated samples did not present any band characteristic of LiBH4, confirming the apparent complete dehydrogenation of our nanoconfined samples.
Once the hydrogen flux was negligible, the materials were submitted to rehydrogenation at 400 °C under 60 bar H2 until pressure remained steady (approximately 5 h). Then the samples were submitted to a second dehydrogenation step (Fig. 5C and D). While the rehydrogenation of bulk LiBH4 is ineffective under these conditions (not shown), our samples liberated 6–7% H2. Trends are harder to define for the second hydrogen release, but no detrimental effect of graphene can be highlighted. It seems that resins with bigger pores could present an improved reversibility: possibly smaller pores can be clogged more easily. This will be corroborated by SEM and BET analysis in the corresponding section.
At 30% filling resins RB and GB present a very similar behaviour whether graphene is present or not. This is beneficial because as demonstrated by BJH experiments, the presence of graphene notably improves the impregnation capacity of the resin (1.0 cm3 g−1 for GB vs. 0.65 cm3 g−1 for RB). While resin RD presents better dehydrogenation kinetics, its practical use is hindered by its low volume capacity (0.48 cm3 g−1). Considering these facts, the resin GB with 30% filling is an interesting compromise.
Fig. 6 presents the SEM observations of resins RB (A–C) and GB (D–F). The materials were characterized before loading (A and D), after loading at 70% (B and E) and after a cycle of desorption–absorption–desorption (C and F). Resins B clearly show differences of pore size with and without graphene. The presence of graphene promotes bigger pores but with more fractal structures, which is correlated with the broader pore distribution observed by BJH. This could explain the more efficient filling of the resins when graphene is present. Some planar structures maybe attributed to the presence of the lamellar planes of graphene (Fig. 6D and ESI5,† see arrows). After filling both resins with the hydride at 70%, it is evident that in the absence of graphene many LiBH4 materials remained at the surface of the resin particles (Fig. 6B, see arrow), while in its presence the pores do not seem clogged, and the spill of hydride at the surface is less evident (Fig. 6E). After two dehydrogenation steps RB presents a much clogged surface (Fig. 6C), while with graphene pores are still available, even if extruded structures with brighter contrast might be present as if they were subjected to various melting process (Fig. 6F).
 | ||
| Fig. 6 SEM images of resins RB/GB in the absence (A–C) and presence (D–F) of graphene, without LiBH4 (A and D), filled at 70% with LiBH4 (B and E), and after two desorptions (C and F). | ||
These observations were confirmed by N2 isotherms (Fig. 7): after filling the RB matrix the smaller pores are still present (thus not filled with LiBH4), while when graphene is present the whole pore distribution disappeared. In particular, after a 70% filling, the value of the remaining pore volume (Table 3) is similar in both matrices (0.31), but while in the presence of graphene this remaining volume is very close to the expected value (1.0 − 1.0 × 0.7 = 0.3), when graphene is absent this value is higher than expected (0.65 − 0.65 × 0.7 = 0.24). These observations could indicate that without graphene a portion of LiBH4 remains outside of the smaller pores, while graphene promotes a homogeneous filling. It has been suggested by Mason et al. that the boron atoms released during the melting step get trapped within graphene defects and enhance the plane wettability towards LiBH4.33 Thus, the structural defects of graphene may explain the observed improvement of pore filling when graphene is present in the resin. After two dehydrogenation steps both matrices present an increase in pore volume with respect to the recently filled matrices. As stated by Mason,33 in our samples LiH is probably ejected from the pores, promoting the observed decrease in kinetics and reversibility for each desorption/absorption cycles.
 | ||
| Fig. 7 Pore-size distribution of RB (A) and GB (B), without LiBH4 (black squares), after pore-filling at 70% with LiBH4 (white circles), and after two desorptions (grey stars). | ||
| a BET surface area (m2 g−1).b BJH desorption cumulative volume of pores between 1.7 nm and 300 nm diameter (cm3 g−1).c BJH desorption average pore diameter (4V/A) (nm). | ||||||
|---|---|---|---|---|---|---|
| Resin | RB | GB | ||||
| Life cycle | Empty | Filled | Desorbed | Empty | Filled | Desorbed |
| Surfacea | 655 | 102 | 177 | 569 | 36 | 92 |
| Volumeb | 0.65 | 0.31 | 0.44 | 1.0 | 0.31 | 0.55 |
| Diameterc | 7.2 | 8.3 | 7.8 | 16 | 25 | 19 |
4. Conclusions
This work is a proof of concept for the incorporation of graphene within a resorcinol–formaldehyde matrix by diffusion of the resin precursors through a graphene hydrogel. Depending on the precursor's concentration, the matrix average pore size ranged from 6 to 26 nm consistently with their reference counter-part. When LiBH4 was nanoconfined in the matrix presenting the smallest pores, the temperature of hydrogen liberation was lowered by 85 °C with respect to bulk LiBH4. Neither the kinetics nor the mass percentage of H2 release suffered critically from the presence of graphene (∼14% for the first and ∼6% for the second cycle), and the liberation of diborane appeared to be inexistent. Instead, graphene seemed beneficial to the matrix filling with the hydride: on one hand by increasing pore volume and on the other hand by improving the loading of the pores. More importantly, the faculty to incorporate graphene within a resorcinol–formaldehyde matrix should open avenues to further functionalize or decorate it with a catalyst. Currently our group is using this methodology to study the effect of graphene decorated with transition-metal-nanoparticles over the hydrogen sorption properties of LiBH4 nanoconfined in resorcinol–formaldehyde matrices.Acknowledgements
The authors thank to CONICET (National Council of Scientific and Technological Research), CNEA (National Commission of Atomic Energy) and ANPCyT (PICT 2013 No. 1052 and PICT 2015 No. 2576). We thank the Material Characterization Department for providing SEM service.References
- J. J. Vajo, S. L. Skeith and F. Mertens, J. Phys. Chem. B, 2005, 109, 3719–3722 CrossRef CAS PubMed.
- J. Yang, A. Sudik and C. Wolverton, J. Phys. Chem. C, 2007, 111, 19134–19140 CAS.
- M. Au and A. Jurgensen, J. Phys. Chem. B, 2006, 110, 7062–7067 CrossRef CAS PubMed.
- P. Mauron, F. Buchter, O. Friedrichs, A. Remhof, M. Bielmann, C. N. Zwicky and A. Züttel, J. Phys. Chem. B, 2008, 112, 906–910 CrossRef CAS PubMed.
- K. Miwa, N. Ohba, S. Towata, Y. Nakamori and S. Orimo, J. Alloys Compd., 2005, 404–406, 140–143 CrossRef CAS.
- X. Yu, Z. Tang, D. Sun, L. Ouyang and M. Zhu, Prog. Mater. Sci., 2017, 88, 1–48 CrossRef.
- S. Zhang, A. F. Gross, S. L. Van Atta, M. Lopez, P. Liu, C. C. Ahn, J. J. Vajo and C. M. Jensen, Nanotechnology, 2009, 20, 204027 CrossRef PubMed.
- M. Fichtner, Nanotechnology, 2009, 20, 204009 CrossRef PubMed.
- P. Adelhelm, J. Gao, M. H. W. Verkuijlen, C. Rongeat, M. Herrich, P. J. M. Van Bentum, O. Gutfleisch, A. P. M. Kentgens, K. P. De Jong and P. E. De Jongh, Chem. Mater., 2010, 22, 2233–2238 CrossRef CAS.
- T. K. Nielsen, K. Manickam, M. Hirscher, F. Besenbacher and T. R. Jensen, ACS Nano, 2009, 3, 3521–3528 CrossRef CAS PubMed.
- A. Gutowska, L. Li, Y. Shin, C. M. Wang, X. S. Li, J. C. Linehan, R. S. Smith, B. D. Kay, B. Schmid, W. Shaw, M. Gutowski and T. Autrey, Angew. Chem., Int. Ed., 2005, 44, 3578–3582 CrossRef CAS PubMed.
- W. Li, A. Lu, C. Weidenthaler and F. Schüth, Chem. Mater., 2004, 16, 5676–5681 CrossRef CAS.
- A. F. Gross, J. J. Vajo, S. L. Van Atta and G. L. Olson, J. Phys. Chem. C, 2008, 112, 5651–5657 CAS.
- T. K. Nielsen, U. Bösenberg, R. Gosalawit, M. Dornheim, Y. Cerenius, F. Besenbacher and T. R. Jensen, ACS Nano, 2010, 4, 3903–3908 CrossRef CAS PubMed.
- P. Ngene, P. Adelhelm, A. M. Beale, K. P. De Jong and P. E. De Jongh, J. Phys. Chem. C, 2010, 114, 6163–6168 CAS.
- S. Cahen, J. B. Eymery, R. Janot and J. M. Tarascon, J. Power Sources, 2009, 189, 902–908 CrossRef CAS.
- D. T. Shane, R. L. Corey, C. McIntosh, L. H. Rayhel, R. C. Bowman, J. J. Vajo, A. F. Gross and M. S. Conradi, J. Phys. Chem. C, 2010, 114, 4008–4014 CAS.
- S. Thiangviriya and R. Utke, Int. J. Hydrogen Energy, 2015, 40, 4167–4174 CrossRef CAS.
- P. Plerdsranoy and R. Utke, Int. J. Hydrogen Energy, 2015, 40, 7083–7092 CrossRef CAS.
- F. Agresti, A. Khandelwal, G. Capurso, S. Lo Russo, A. Maddalena and G. Principi, Nanotechnology, 2010, 21, 065707 CrossRef PubMed.
- P. Plerdsranoy, N. Wiset, C. Milanese, D. Laipple, A. Marini, T. Klassen, M. Dornheim and R. Gosalawit-Utke, Int. J. Hydrogen Energy, 2015, 40, 392–402 CrossRef CAS.
- L. Chong, X. Zeng, W. Ding, D. Liu and J. Zou, Adv. Mater., 2015, 27, 5070–5074 CrossRef CAS PubMed.
- X. Liu, D. Peaslee, C. Z. Jost, T. F. Baumann and E. H. Majzoub, Chem. Mater., 2011, 23, 1331–1336 CrossRef CAS.
- H. Bai, C. Li and G. Shi, Adv. Mater., 2011, 23, 1089–1115 CrossRef CAS PubMed.
- K. Bhowmik, A. Mukherjee, M. K. Mishra and G. De, Langmuir, 2014, 30, 3209–3216 CrossRef CAS PubMed.
- W. Feng, Y. Wang, J. Chen, L. Wang, L. Guo, J. Ouyang, D. Jia and Y. Zhou, Carbon, 2016, 108, 52–60 CrossRef CAS.
- J. Xu, Y. Li, J. Cao, R. Meng, W. Wang and Z. Chen, Catal. Sci. Technol., 2015, 5, 1821–1828 CAS.
- L. Wei and Y. Mao, Int. J. Hydrogen Energy, 2016, 41, 11692–11699 CrossRef CAS.
- Y. Zhu, J. Zou and X. Zeng, RSC Adv., 2015, 5, 82916–82923 RSC.
- S. Nachimuthu, P. J. Lai and J. C. Jiang, Carbon, 2014, 73, 132–140 CrossRef CAS.
- M. Lototskyy, J. M. Sibanyoni, R. V. Denys, M. Williams, B. G. Pollet and V. A. Yartys, Carbon, 2013, 57, 146–160 CrossRef CAS.
- J. Shao, X. Xiao, X. Fan, L. Zhang, S. Li, H. Ge, Q. Wang and L. Chen, J. Phys. Chem. C, 2014, 118, 11252–11260 CAS.
- T. Mason and E. H. Majzoub, J. Phys. Chem. C, 2014, 118, 8852–8858 CAS.
- S. D. House, X. Liu, A. A. Rockett, E. H. Majzoub and I. M. Robertson, J. Phys. Chem. C, 2014, 118, 8843–8851 CAS.
- X. Zhang, Z. Sui, B. Xu, S. Yue, Y. Luo, W. Zhan and B. Liu, J. Mater. Chem., 2011, 21, 6494–6497 RSC.
- C. H. J. Kim, D. Zhao, G. Lee and J. Liu, Adv. Funct. Mater., 2016, 26, 4976–4983 CrossRef CAS.
- M. A. Worsley, P. J. Pauzauskie, T. Y. Olson, J. Biener, J. H. Satcher and T. F. Baumann, J. Am. Chem. Soc., 2010, 132, 14067–14069 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- J. Shao, X. Xiao, X. Fan, X. Huang, B. Zhai, S. Li, H. Ge, Q. Wang and L. Chen, Nano Energy, 2015, 15, 244–255 CrossRef CAS.
- X. Liu, D. Peaslee, C. Z. Jost and E. H. Majzoub, J. Phys. Chem. C, 2010, 114, 14036–14041 CAS.
- Z. Z. Fang, P. Wang, T. E. Rufford, X. D. Kang, G. Q. Lu and H. M. Cheng, Acta Mater., 2008, 56, 6257–6263 CrossRef CAS.
- X. Liu, E. H. Majzoub, V. Stavila, R. K. Bhakta, M. D. Allendorf, D. T. Shane, M. S. Conradi, N. Verdal, T. J. Udovic and S.-J. Hwang, J. Mater. Chem. A, 2013, 1, 9935–9941 CAS.
- M. H. W. Verkuijlen, P. Ngene, D. W. De Kort, C. Barré, A. Nale, E. R. H. Van Eck, P. J. M. Van Bentum, P. E. De Jongh and A. P. M. Kentgens, J. Phys. Chem. C, 2012, 116, 22169–22178 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02288c |
| This journal is © The Royal Society of Chemistry 2017 |