
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of α-aminophosphonates using solvate ionic liquids†
Daniel J. Eyckens and
Luke C. Henderson *
*
Institute for Frontier Materials, Deakin University, 75 Pigdons Road, Geelong, Victoria 3216, Australia. E-mail: luke.henderson@deakin.edu.au
First published on 25th May 2017
Abstract
A range of α-aminophosphonates were accessed in high yields and very rapidly, using solvate ionic liquids as the reaction media. Reactions typically required less than 10 minutes to go to completion and precipitation of these products into water excludes the use of traditional work up procedures, giving the products in very high crude purity. Excellent functional group tolerance for both the aldehyde and amine reaction partners was observed, and a range of bis-aminophosphonates derived from aromatic diamines were also accessed in high yield and purity.
Introduction
α-Aminophosphonates are of great interest to organic chemists as they have vast applications in fields such as medicine,1–10 agriculture, and in environmental decontamination.5,11,12 They are considered phosphorus analogues to α-amino acids and the phosphorus' geometry has served as a useful tool in medicinal chemistry.13–16The synthesis of these compounds via the Kabachnik–Fields or Pudovik reaction (Scheme 1) is relatively straight forward, using nucleophilic attack of di-alkyl or -aryl phosphite with an imine.12 The imine species can be generated in situ, as part of a three-component reaction, or preformed and added to the phosphite, both methodologies have been presented in the literature.12 Additionally, several reports employ excess of the amine or dialkyloxy-phosphite (in some cases both) which limits the broad appeal of this reaction.
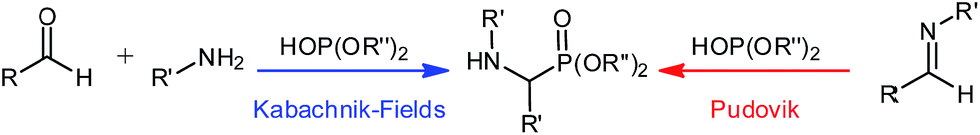 | ||
| Scheme 1 Common approaches to the synthesis of α-aminophosphonates. | ||
A large amount of work has been focused on optimizing the synthesis of these molecules using a variety of Lewis acid catalysts,17,18 such as CeCl3–proline complex,19 GaI3,20 In(OTf)3, YbCl3, SmI2, MoO2Cl2,21 Sc(OTf)3, and Dy(OTf)3 (ref. 12) among others.19,22 These efforts have given a variety of means to access these compounds, though the use of these Lewis acids always pose several problems: disposal of heavy metals and the reaction solvent, the high toxicity of these materials, their sensitivity to atmospheric conditions, and their expensive nature.
Ionic liquids (ILs), in their many forms, have been used in many areas of chemistry from materials science through to solvents to facilitate organic transformations.23–29 The use of ILs as solvents for this reaction have been reported with some excellent results,30–34 particularly for the pseudo-ionic liquid 5.0 M lithium perchlorate in dithethyl ether (5 M LPDE), or the use of LiClO4 as an additive to the reaction mixture.18,35–40 These reactions were able to isolate α-aminophosphonates in very high yield using reaction times under 1 hour.
Despite these benefits, the use of 5.0 M LPDE as the reaction media experiences several limitations, which include: enhanced sensitivity to atmospheric conditions, problematic solvent preparation, inability to be heated (due to potential explosive nature of the ClO4− anion), and reagent restriction as not all reagents will be soluble in the pseudo-ionic liquid. Also, while the use of ionic liquids is an improvement over the use of metal-based catalysts, these procedures still use large amounts of organic solvent in the recovery and purification of the synthesised phosphonate. Accessing these compounds in high yield, short reaction time, and with minimal purification remains a challenge in organic synthesis.
We recently reported the use of solvate ionic liquids (SILs), being equimolar mixtures of LiTFSI in tri- or tetra-glyme (referred to as [G3(Li)]TFSI or [G4(Li)]TFSI, respectively), as suitable reaction media for electrocyclisation reactions and characterised them using Kamlet–Taft parameters (Fig. 1).41,42
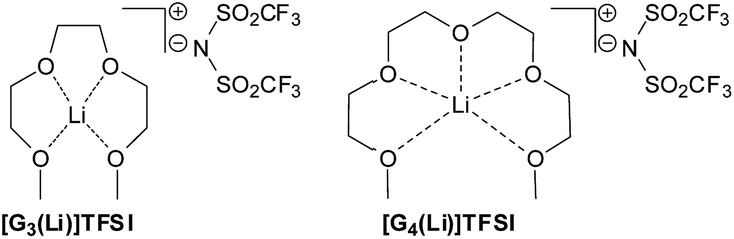 | ||
| Fig. 1 Simplified structures of solvate ionic liquids used in this work. | ||
Indeed these solvate ionic liquids serve as a stable and easily-handled surrogate to 5.0 M LPDE, while often giving superior reaction outcomes. They are able to be heated without concern, present minimal toxicity,43 are simple and cheap to produce, and can be stored over molecular sieves to maintain their anhydrous nature, if required. This work sought to determine if the synthesis of α-aminophosphonates could be successfully carried out in solvate ionic liquids and if so, how does this compare to other ionic liquid and molecular solvents.
Herein we report the application of solvate ionic liquids as a solvent for the rapid and high yielding synthesis of α-aminophosphonates in a three-component reaction. This reaction proceeds at room temperature in 5 minutes, with the majority of crude products being obtained by precipitation and are analytically pure, with minimal workup.
This work began by examining the Kabachnik–Fields reaction (i.e. three-component reaction) to minimize reagent handling and as it represents the more efficient reaction pathway compared to pre-synthesising the imine. As a model system for optimisation, benzaldehyde 1, aniline 2, and diphenyl phosphite were chosen (Table 1).
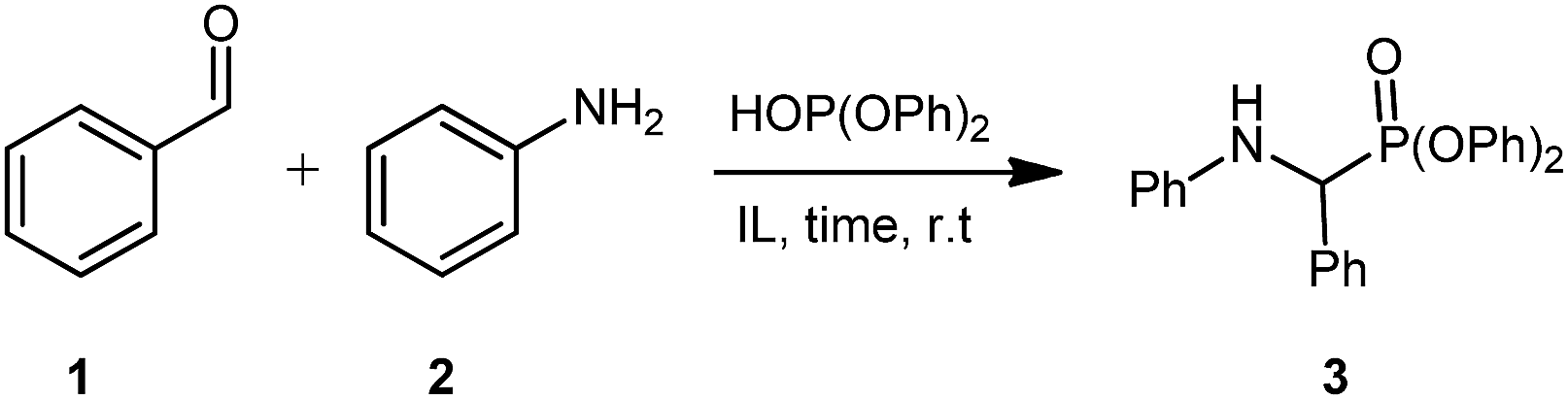
Initially, the three-component reaction was stirred at room temperature for 30 minutes in [G3(Li)]TFSI, accessing pure 3 in a very promising yield of 82% (Table 1, entry 1). In an effort to reduce reaction time, without sacrificing the yield, the reaction time was reduced to 5 minutes (Table 1, entry 2), giving a excellent yield of 87%. Further reduction to 1 minute (Table 1, entry 3) gave the desired product in a slightly depressed yield of 78%. Using this, a 5 minute reaction was considered the optimal due to excellent yield and the reaction occurring on a very practical timescale. Repeating this reaction in [G4(Li)]TFSI, gave the desired product in an excellent yield of 91% (Table 1, entry 4). Note that the isolation of these compounds was achieved via precipitation of the crude products into water, and not via chromatographic methods.
Nevertheless, with these conditions in hand, our attention turned to assessing the functional group tolerance of this reaction in SILs (Table 2). During the reaction scoping process, many aniline derivatives were observed to react extremely quickly, with some forming the solid product almost instantly, preventing further stirring.
|
||||
|---|---|---|---|---|
| Entry | Compound | R | Solvent | Yielda (%) |
| a Isolated yield. | ||||
| 1 | 5a | 4-NO2 | [G3(Li)]TFSI | 64 |
| 2 | [G4(Li)]TFSI | 25 | ||
| 3 | 5b | 4-Cl | [G3(Li)]TFSI | 96 |
| 4 | [G4(Li)]TFSI | 96 | ||
| 5 | 5c | 4-OH | [G3(Li)]TFSI | 82 |
| 6 | [G4(Li)]TFSI | 92 | ||
| 7 | 5d | 4-F | [G3(Li)]TFSI | 84 |
| 8 | [G4(Li)]TFSI | 68 | ||
| 9 | 5e | 3-Cl | [G3(Li)]TFSI | 83 |
| 10 | [G4(Li)]TFSI | 59 | ||
| 11 | 5f | 3-CF3 | [G3(Li)]TFSI | 86 |
| 12 | [G4(Li)]TFSI | 81 | ||
| 13 | 5g | 3,5-CF3 | [G3(Li)]TFSI | 54 |
| 14 | [G4(Li)]TFSI | 60 | ||
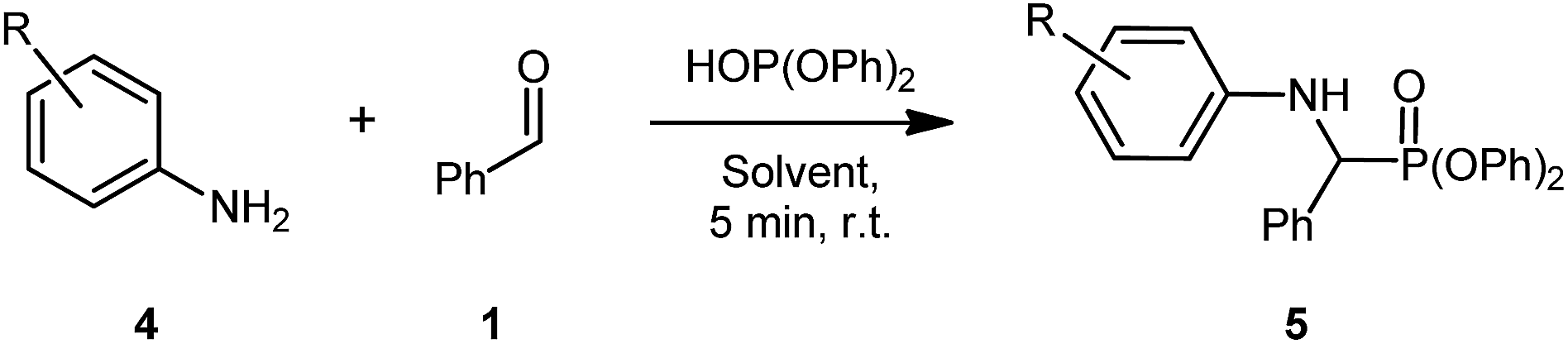
The first reaction using these conditions and 4-nitroaniline proceeded in good yield for [G3(Li)]TFSI (64%) and poor yield in [G4(Li)]TFSI (25%) (Table 2, entries 1 & 2, respectively). The poor yield for the latter SIL was attributed to both the poor nucleophilicity of the aniline and the problematic isolation of the product, due to repeated recrystalisation. Interestingly, the introduction of a halogen-substitution on the aniline gave excellent yields, with both [G3(Li)]TFSI and [G4(Li)]TFSI fetching an exemplary yields of 96% each (Table 2, entries 3 & 4, respectively). A similar yield was obtained from the electron-donating 4-aminophenol in [G3(Li)]TFSI (82%) and [G4(Li)]TFSI (92%) (Table 2, entries 7 & 8, respectively). Incorporating 4-fluoroaniline into the given reaction conditions gave an excellent yield in [G3(Li)]TFSI (84%, Table 2, entry 7). Though when [G4(Li)]TFSI was used, a slightly diminished yield of 68% (Table 2, entry 8) was obtained. Similarly, 3-chloroaniline proceeded well in [G3(Li)]TFSI (83%, Table 2, entry 9), a result not consistent in [G4(Li)]TFSI (59%, Table 2, entry 10). Exploring the effect of 3-(trifluoromethyl)aniline in this reaction revealed a excellent yields of 86% and 81% in [G3(Li)]TFSI and [G4(Li)]TFSI, respectively (Table 2, entries 11 & 12, respectively). Introducing a bis-trifluoromethylaniline derivative demonstrated the robust nature of the reaction conditions, with both [G3(Li)]TFSI and [G4(Li)]TFSI proceeding in good yields of 54% and 60%, respectively (Table 2, entries 13 & 14, respectively). It is worth noting as well that the isolation of some products from [G4(Li)]TFSI was slightly more difficult than the [G3(Li)]TFSI, whereby the isolated material contained traces of the glyme from the IL. The repeated precipitation to remove these traces of glyme may be responsible for the slightly reduced yield in certain instances. With this final example of the reaction's high functional group tolerance, attention was then turned to the effect of aldehyde substitution (Table 3).
|
||||
|---|---|---|---|---|
| Entry | Compound | R | Solvent | Yielda (%) |
| a Isolated yield.b This material possessed some of the α-hydroxyphosphonate (17% by 1H NMR) resulting from direct attack of the phosphite on 4-nitrobenzaldehyde. | ||||
| 1 | 7a | 4-Br | [G3(Li)]TFSI | 90 |
| 2 | [G4(Li)]TFSI | 91 | ||
| 3 | 7b | 4-Me | [G3(Li)]TFSI | 90 |
| 4 | [G4(Li)]TFSI | 86 | ||
| 5 | 7c | 4-NO2 | [G3(Li)]TFSI | 69 |
| 6 | [G4(Li)]TFSI | 59b | ||
| 7 | 7d | 4-F | [G3(Li)]TFSI | 77 |
| 8 | [G4(Li)]TFSI | 78 | ||
| 9 | 7e | 2-OH | [G3(Li)]TFSI | 84 |
| 10 | [G4(Li)]TFSI | 76 | ||
| 11 | 7f | 3,4-Cl | [G3(Li)]TFSI | 74 |
| 12 | [G4(Li)]TFSI | 79 | ||
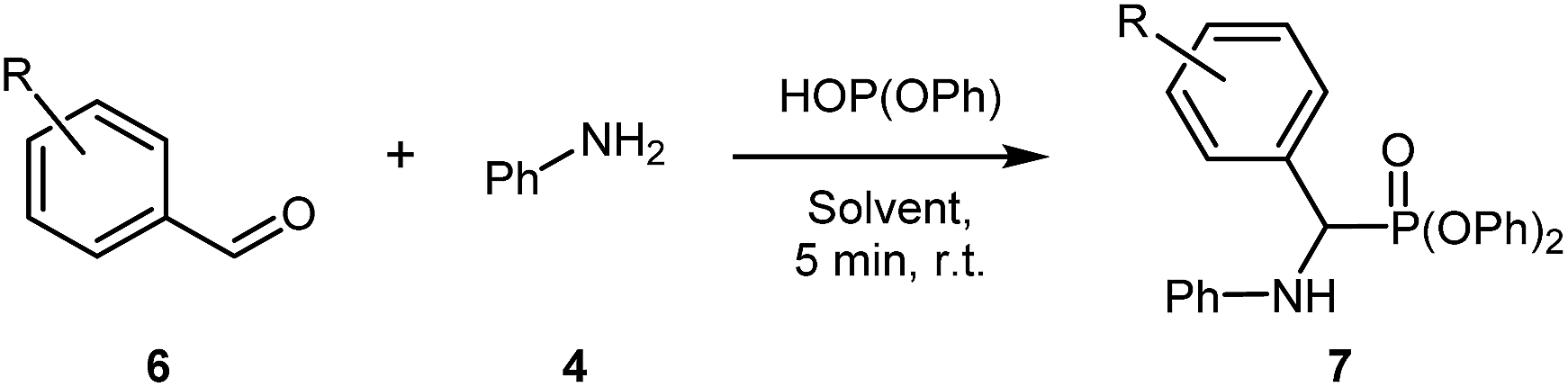
Varying the aldehyde component of the reaction, and maintaining the aniline, scoping began with the use of 4-bromobenzaldehyde, which proceeded in excellent yield of 90% and 91% in both [G3(Li)]TFSI and [G4(Li)]TFSI (Table 3, entries 1 & 2, respectively). Utilizing p-tolualdehyde also gave encouraging yields of 90% in [G3(Li)]TFSI and 86% in [G4(Li)]TFSI (Table 3, entries 3 & 4, respectively). Good yields of 69% and 59% were observed in [G3(Li)]TFSI and [G4(Li)]TFSI, respectively, when using 4-nitrobenzaldehyde as the aldehyde source (Table 3, entries 5 & 6, respectively). The reaction also proceed well with the application of 4-fluorobenzaldehyde, obtaining the final product in good yields of 77% and 78% in [G3(Li)]TFSI and [G4(Li)]TFSI (Table 3, entries 7 & 8, respectively). The use of salicylaldehyde also showed good promise, giving pure product in 84% and 76% in [G3(Li)]TFSI and [G4(Li)]TFSI, respectively (Table 3, entries 9 & 10, respectively) and inclusion of a bis-substituted aldehyde (3,4-dichlorobenzaldehyde) further exemplified the ability of the SILs to assist this reaction, giving the product 7f in good yields of 74% in [G3(Li)]TFSI and 79% in [G4(Li)]TFSI (Table 3, entries 11 & 12, respectively). With this data in hand our attention turned to the examination of carrying out multiple reactions on bis-amines.
The reactions proceeded very smoothly within 5 minutes, giving the desired product for a range of functional groups. The yields were variable depending on the SIL and the electronics of the aldehyde. No discernable trend or difference was noted between either the [G3(Li)]TFSI or [G4(Li)]TFSI, with similar yields given for each example presented (Table 4).

Repeating this process but using the ortho- and meta-substituted diamino anilines (Scheme 2), gave varied results. As may be expected, the formation of 11 proceeded in a good but reduced yield in both [G3(Li)]TFSI or [G4(Li)]TFSI of 25% and 34%, respectively. This was attributed to the increased steric hinderance imposed by the meta-substitution of the parent diamine 10. This was consistent with our observations for the formation of 13, possessing ortho-substitution where only 9% was isolated for [G3(Li)]TFSI and no product was able to be isolated when using [G4(Li)]TFSI.
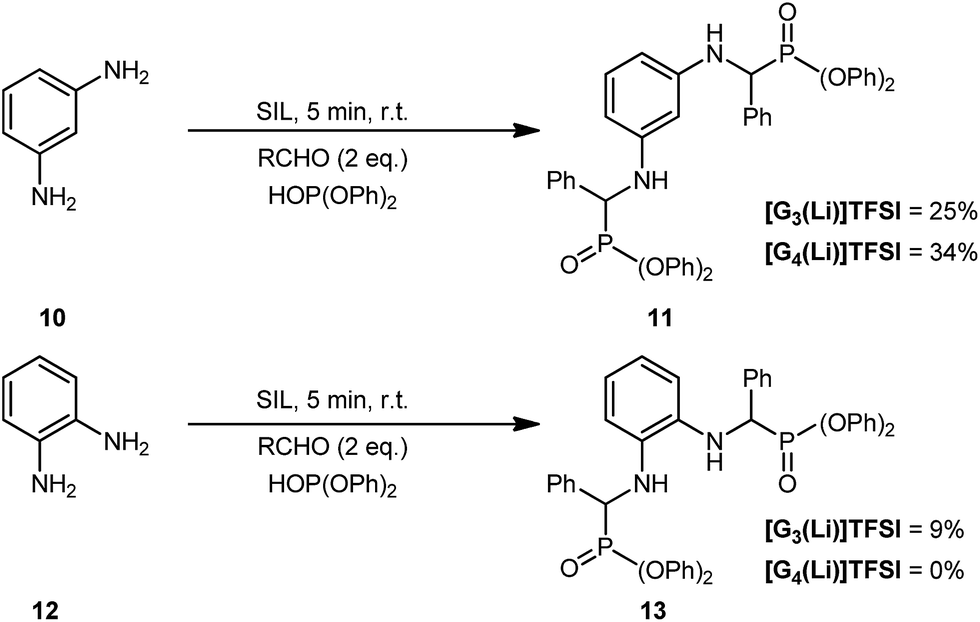 | ||
| Scheme 2 Common approaches to the synthesis of α-aminophosphonates. | ||
A final aspect of this work which was considered challenging and had not been investigated previously is the synthesis of non-symmetric bis-α-aminophosphonates, using two different aldehydes (Scheme 3). There is a considerable challenge in this procedure considering the speed at which the reaction takes place, thus controlling the regiochemistry will be difficult. Experimentally, the use of two aldehydes of similar electronic nature were chosen, reduce any bias in the reacting system, e.g. the use of a reactive aldehyde followed by an unreactive aldehyde would favour the formation of the non-symmetric product.
 | ||
| Scheme 3 Non-symmetric bis-α-aminophosphonate synthesis. | ||
Therefore, in this case the first aldehyde was added to the solution of phosphite and amine in [G3(Li)]TFSI, then after 5 minutes the second aldehyde was added. Using p-tolualdehyde and benzaldehyde, the target non-symmetric-α-aminophosphonate 14 was successfully synthesised in a moderate yield of 43% in [G3(Li)]TFSI, 26% in [G4(Li)]TFSI, and in only 10 minutes (Scheme 3). In each case the standard precipitation described was also successful, giving 14 in analytical purity.
Representative experimental procedure
A round bottom flask was charged with aldehyde (1.00 mmol), which was dissolved in either [G3(Li)]TFSI or [G4(Li)]TFSI (0.5 mL). Aniline (1.00 mmol) was then added, before the addition of diphenyl phosphite (0.230 mL, 1.20 mmol) and stirred at room temperature for the given time period. Diethyl ether (10 mL) was added at the conclusion of the reaction, before the addition of deionised water (10 mL) causing a fine precipitate to form. The removal of diethyl ether under reduced pressure afforded a suspension of precipitate in the aqueous phase, which was then filtered washing with excess water and petroleum spirits (40–60 °C).Conclusions
In conclusion, the use of solvate ionic liquids as excellent reaction media for the Kabachnik–Fields reaction has been shown. A wide range of α-aminophosphonates were able to be synthesised in 5 minutes with simple precipitation giving the desired compound in >95% purity. Extension of this methodology to bis-α-aminophosphonates, using para-diaminobenzene was also successful, in the same 5 minute reaction duration and in high yield. Using ortho- and meta-diaminobenzene gave the desired products but in lower yield (34% and 9%, respectively) presumably due to steric influences. Finally, synthesis of a non-symmetric bis-α-aminophosphonate was achieved, using sequential addition of the aldehydes in excellent yield of 43%. No discernible trend with respect to which SIL was optimal for a given reaction, though it was noted that removal of G4 was more challenging compared to the shorter G3 analogue, this is presumably due to its slightly more ‘organic’ nature.Acknowledgements
The authors would like to thank the Institute of Frontier Materials (IFM) for a postgraduate scholarship to DE.Notes and references
- N. Ali, S. Zakir, M. Patel and M. Farooqui, Eur. J. Med. Chem., 2012, 50, 39–43 CrossRef CAS PubMed.
- R. Damiche and S. Chafaa, J. Mol. Struct., 2017, 1130, 1009–1017 CrossRef CAS.
- L. Jin, B. Song, G. Zhang, R. Xu, S. Zhang, X. Gao, D. Hu and S. Yang, Bioorg. Med. Chem. Lett., 2006, 16, 1537–1543 CrossRef CAS PubMed.
- I. H. Kim, Y. K. Park, H. Nishiwaki, B. D. Hammock and K. Nishi, Bioorg. Med. Chem., 2015, 23, 7199–7210 CrossRef CAS PubMed.
- A. Mucha, P. Kafarski and L. Berlicki, J. Med. Chem., 2011, 54, 5955–5980 CrossRef CAS PubMed.
- Q. Wang, M. Zhu, R. Zhu, L. Lu, C. Yuan, S. Xing, X. Fu, Y. Mei and Q. Hang, Eur. J. Med. Chem., 2012, 49, 354–364 CrossRef CAS PubMed.
- K. P. Boroujeni, E. R. Shirazi and M. M. Doroodmand, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 191, 683–688 CrossRef.
- L. Maier, Phosphorus, Sulfur Silicon Relat. Elem., 1990, 53, 43–67 CrossRef CAS.
- C. Sampath, P. Harika and N. Revaprasadu, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 191, 1081–1085 CrossRef.
- C. Subramanyam, S. Thaslim Basha, G. Madhava, S. Nayab Rasool, S. Adam, S. Durga Srinivasa Murthy and C. Naga Raju, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 267–270 CrossRef CAS.
- E. D. Naydenova, P. T. Todorov and K. D. Troev, Amino Acids, 2010, 38, 23–30 CrossRef CAS PubMed.
- G. Keglevich and E. Balint, Molecules, 2012, 17, 12821–12835 CrossRef CAS PubMed.
- J. Joossens, P. Van der Veken, G. Surpateanu, A.-M. Lambeir, I. El-Sayed, O. M. Ali, K. Augustyns and A. Haemers, J. Med. Chem., 2006, 49, 5785–5793 CrossRef CAS PubMed.
- D. Hocková, D. T. Keough, Z. Janeba, T.-H. Wang, J. de Jersey and L. W. Guddat, J. Med. Chem., 2012, 55, 6209–6223 CrossRef PubMed.
- J. W. De Schutter, J. Park, C. Y. Leung, P. Gormley, Y.-S. Lin, Z. Hu, A. M. Berghuis, J. Poirier and Y. S. Tsantrizos, J. Med. Chem., 2014, 57, 5764–5776 CrossRef CAS PubMed.
- Y. Wu, C. J. Aquino, D. J. Cowan, D. L. Anderson, J. L. Ambroso, M. J. Bishop, E. E. Boros, L. Chen, A. Cunningham, R. L. Dobbins, P. L. Feldman, L. T. Harston, I. W. Kaldor, R. Klein, X. Liang, M. S. McIntyre, C. L. Merrill, K. M. Patterson, J. S. Prescott, J. S. Ray, S. G. Roller, X. Yao, A. Young, J. Yuen and J. L. Collins, J. Med. Chem., 2013, 56, 5094–5114 CrossRef CAS PubMed.
- Y. Cai, Y. Li, M. Zhang, J. Fu and Z. Miao, RSC Adv., 2016, 6, 69352–69356 RSC.
- N. Azizi and M. R. Saidi, Eur. J. Org. Chem., 2003, 2003, 4630–4633 CrossRef.
- C. D. G. da Silva, A. R. Oliveira, M. P. D. Rocha, R. Katla, E. R. Botero, É. C. da Silva and N. L. C. Domingues, RSC Adv., 2016, 6, 27213–27219 RSC.
- P. Sun, Z. Hu and Z. Huang, Synth. Commun., 2004, 34, 4293–4299 CrossRef CAS.
- R. G. de Noronha, C. C. Romão and A. C. Fernandes, Catal. Commun., 2011, 12, 337–340 CrossRef CAS.
- S. Bhagat and A. K. Chakraborti, J. Org. Chem., 2007, 72, 1263–1270 CrossRef CAS PubMed.
- K. M. Beggs, M. D. Perus, L. Servinis, L. A. O'Dell, B. L. Fox, T. R. Gengenbach and L. C. Henderson, RSC Adv., 2016, 6, 32480–32483 RSC.
- L. C. Henderson and N. Byrne, Green Chem., 2011, 13, 813–816 RSC.
- L. C. Henderson, M. T. Thornton, N. Byrne, B. L. Fox, K. D. Waugh, J. S. Squire, L. Servinis, J. P. Delaney, H. L. Brozinski, L. M. Andrighetto and J. M. Altimari, C. R. Chim., 2013, 16, 634–639 CrossRef CAS.
- T. T. Megan, C. H. Luke, B. Nolene and M. P. Frederick, Curr. Org. Chem., 2012, 16, 121–126 CrossRef.
- N. Debeljuh, C. J. Barrow, L. Henderson and N. Byrne, Chem. Commun., 2011, 47, 6371–6373 RSC.
- M. Maghe, C. Creighton, L. C. Henderson, M. G. Huson, S. Nunna, S. Atkiss, N. Byrne and B. L. Fox, J. Mater. Chem. A, 2016, 4, 16619–16626 CAS.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS PubMed.
- T. Olszewski, Synthesis, 2014, 46, 403–429 CrossRef.
- J. S. Yadav, B. V. S. Reddy and P. Sreedhar, Green Chem., 2002, 4, 436–438 RSC.
- K. P. Boroujeni, E. R. Shirazi and M. M. Doroodmand, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 683–688 CrossRef CAS.
- D. Fang, J. Yang and C. Ni, Heteroat. Chem., 2011, 22, 5–10 CrossRef CAS.
- R. Hajinasiri, Z. Hossaini and F. Rostami-Charati, Heteroat. Chem., 2011, 22, 625–629 CrossRef CAS.
- B. S. Babu and K. K. Balasubramanian, Tetrahedron Lett., 1998, 39, 9287–9288 CrossRef CAS.
- P. A. Grieco, J. J. Nunes and M. D. Gaul, J. Am. Chem. Soc., 1990, 112, 4595–4596 CrossRef CAS.
- A. Heydari, Tetrahedron, 2002, 58, 6777–6793 CrossRef CAS.
- Y. Pocker and G. T. Spyridis, J. Am. Chem. Soc., 2002, 124, 7390–7394 CrossRef CAS PubMed.
- S. Sankararaman and J. E. Nesakumar, Eur. J. Org. Chem., 2000, 2003–2011 CrossRef CAS.
- R. Sudha, K. Malola Narasimhan, V. Geetha Saraswathy and S. Sankararaman, J. Org. Chem., 1996, 61, 1877–1879 CrossRef CAS PubMed.
- D. J. Eyckens, M. E. Champion, B. L. Fox, P. Yoganantharajah, Y. Gibert, T. Welton and L. C. Henderson, Eur. J. Org. Chem., 2016, 913–917 CrossRef CAS.
- D. J. Eyckens, B. Demir, T. R. Walsh, T. Welton and L. C. Henderson, Phys. Chem. Chem. Phys., 2016, 18, 13153–13157 RSC.
- P. Yoganantharajah, D. J. Eyckens, J. L. Pedrina, L. C. Henderson and Y. Gibert, New J. Chem., 2016, 40, 6599–6603 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04407k |
| This journal is © The Royal Society of Chemistry 2017 |