
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid detection of mercury contamination in water by surface enhanced Raman spectroscopy†
Daniel K. Sarfo ,
Arumugam Sivanesan,
Emad L. Izake and
Godwin A. Ayoko*
,
Arumugam Sivanesan,
Emad L. Izake and
Godwin A. Ayoko*
Queensland University of Technology (QUT), School of Chemistry, Physics and Mechanical Engineering, Nanotechnology and Molecular Science, 2 George st, QLD 4001, Australia. E-mail: g.ayoko@qut.edu.au
First published on 18th April 2017
Abstract
Mercury (Hg) is a potent neurotoxin in fish, wildlife, and humans. The detection of Hg(II) ions in water therefore requires accurate, ultra-sensitive, rapid and cost effective analytical methods. We present a novel nanosensor for the field detection of Hg(II) in water by surface enhanced Raman spectroscopy (SERS). In the new SERS nanosensor, aminodibenzo-18-crown-6 (ADB18C6) was coupled with mercaptopropionic acid and the resultant crown ether derivative (TCE) was self-assembled as a recognition surface layer for Hg(II) onto the surface of a nanostructured gold substrate. The coordination of Hg(II) to the oxygen atoms of TCE led to the spontaneous binding of the metal ion into the cavity of the crown ether layer. This caused the intensity of the Raman band at 1501 cm−1 for the crown ether to increase with the concentration of Hg(II) in the range of 1 × 10−11 M to 1 × 10−6 M. Complexation between TCE and Hg(II) was further confirmed by UV-visible spectrometry, spectrofluorimetry and electrochemistry. The method was successfully applied to the determination of Hg(II) in tap water using a handheld Raman spectrometer and it demonstrated high selectivity towards Hg(II) in the presence of Pb(II) and Cd(II). This eliminated the need for extensive sample preparation and extraction procedures prior to the analysis. The limit of Hg(II) quantification by the new SERS nanosensor and the limit of detection were 1000 fold below the EPA and WHO defined levels for Hg(II) ions in water.
Introduction
Mercury is a heavy transition-metal that is widely released into the environment from anthropogenic sources (e.g. mining, solid waste incineration etc.) and natural activities (e.g. volcanic and oceanic emissions). Even at very low concentrations, Hg(II) has highly toxic and deleterious effects on both the environment and humans.1 Exposure to Hg(II) can lead to brain damage, heart failure, anaemia, kidney damage and nervous system disorders. Because of its abundance, stability and good water solubility, Hg(II) accumulates in water and, subsequently, in humans as well as other living organisms through the intake of Hg(II) contaminated water.2,3 The maximum allowable concentration of Hg(II) in drinking water has been indicated by the U.S. Environmental Protection Agency (USEPA) as 2 μg L−1 (10 nM). Therefore, the rapid detection and removal of Hg(II) are vital for its monitoring, control and remediation.Consequently, various analytical techniques have evolved over the past decade for the detection and quantification of Hg(II) in water. Among these are inductively coupled plasma mass spectrometry (ICP-MS),4 colorimetry and chemiluminescence,5 atomic absorption spectroscopy and X-ray fluorescence spectrometry,6 high performance liquid chromatography,7 neutron activation analysis (NAA)8 and enzyme linked immune sorbent assay (ELISA).9 However, most of these analytical methods require time-consuming sample preparation procedures, expensive instrumental infrastructure and, are therefore, not suitable for remote or on-site applications.
SERS is an ultra-sensitive analytical method that can be utilised for the rapid field detection of Hg(II) in environmental matrices with minimum or no sample pre-treatment.10,11 Many examples of SERS detection of Hg(II) in water were demonstrated recently in literature.12–14 In these examples various approaches were adopted for the metal ion detection, such as the use of Raman reporters or the utilisation of the thymidine–Hg(II)–thymidine coordination chemistry.10
Generally, metal ions are known to have small Raman scattering cross-section with no vibrational modes making their direct detection by SERS difficult. Therefore, SERS detection of Hg(II) usually adopts an indirect approach. In this approach, a Raman reporter is used to detect the presence of Hg(II) in the sample. The SERS signals of the Raman reporter is monitored and changes in the SERS spectrum of the reporter are attributed to its interaction with the mercuric ions.10 Among the Raman active molecules that can be utilised for the SERS detection of Hg(II) are crown ethers. In addition to their Raman activity, crown ethers are excellent ligands that can form complexes with metal ions. The ability of crown ethers to form coordination complexes with different metal ions is highly influenced by the chelating ring size, macrocycle rigidity, and number and type of donor atoms present.15 In addition, the geometric shape, charge density and ionic radius of the metal ions also affect its selective reactivity towards specific crown ethers.16,17 Careful tuning of these factors can lead to the formation of a highly selective complex between a given crown ether and a specific metal ion.18 Many researchers have utilised crown ether derivatives of different crown sizes to host different guest metal ions.19–21 The selective complexation of Hg(II) over a wide range of environmentally relevant metal ions by dibenzo 18-crown-6 azo dye derivatives using UV-visible and fluorescence spectrometry has been reported in the literature.22–24 In this work, a different mode of detection (i.e. SERS) with high sensitivity and a new aminodibenzo-18-crown-6 (ADB18C6) derivative was used. ADB18C6 was coupled with mercaptopropionic acid (MPA) to form a thiolated crown ether derivative (TCE) that was utilised as a recognition molecule for the selective sensing of Hg(II) in water by SERS. The advantage of TCE is its ability to form a self-assembled monolayer by firmly attaching itself onto a SERS substrate through the strong Au–S bond.
To improve the reproducibility of SERS measurements, the firm attachment of the recognition molecule to the surface of a SERS substrate is important. Several approaches have been demonstrated for the attachment of recognition molecules onto SERS substrates. For instance, alkane thiol compounds can be used as a linker to attach recognition molecules to the nanostructures of a gold SERS substrate.25–27 In this scenario, the thiol group of the linker molecule interacts with the gold surface via stable Au–S bond and the other end of the linker molecule may carry an amine group that forms an amide linkage with an active carboxyl group on the recognition molecule.27,28 In this work, MPA was utilised as a linker between a nanostructured gold substrate and the ADB18C6 recognition molecule. The carboxylic functional group of MPA was attached to the primary amine end of ADB18C6 through EDC coupling reaction.29 The gold nanostructures were electrochemically prepared and deposited onto the surface of the solid gold substrate. This approach for preparing the nanostructured gold substrate is relatively fast, does not involve the use of capping agents (which may cause spectral interferences) and eliminates the problem of uncontrolled nanoparticle aggregation. To demonstrate the potential of the new nanosensor for real time analysis, a handheld Raman spectrometer was utilised for the SERS detection of Hg(II) contamination in water samples at the ultra-trace level down to 0.51 pM.
Experimental
Chemicals and reagents
All reagents and solvents were of analytical grade and used without further purification. Concentrated sulfuric acid (98%), hydrogen peroxide solution (30%), mercaptopropionic acid (MPA), 4-amino-dibenzo-18-crown-6 (ADB18C6), gold(III) chloride (HAuCl4), phosphate buffered saline (PBS), N-hydroxysuccinimide (NHS), N-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC), dimethyl sulphoxide (DMSO), Pb(NO3)2, Cd(NO3)2, and Hg(NO3)2 were all purchased from Sigma-Aldrich (USA). All solutions were prepared using ultra-pure water (18.2 MΩ cm@25 °C, Milli-Q).UV-visible and fluorescence studies
To investigate the complex formation between ADB18C6 and Hg(II), 1 mL of 1 mM ADB18C6 (in DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O, 1:1 v/v) solution was mixed with 150 μL of 10 μM HCl. This was followed by the addition of 1 mL of the Hg(II) aqueous solutions in the concentration range of 20 mM, 40 mM, 60 mM and 80 mM. The mixtures were transferred into cuvettes and their UV absorptions were measured in the wavelength range 250 nm to 500 nm. For the same mixtures, fluorescence emissions between 280 nm and 800 nm were measured at an excitation wavelength of 281 nm. To investigate the effect of interfering ions on the UV and fluorescence measurements of Hg(II), the above procedures were repeated after the addition of 1 mL of 80 mM Cd(II) and 80 mM Pb(II) to the ADB18C6/HCl/Hg(II) mixtures.
:H2O, 1:1 v/v) solution was mixed with 150 μL of 10 μM HCl. This was followed by the addition of 1 mL of the Hg(II) aqueous solutions in the concentration range of 20 mM, 40 mM, 60 mM and 80 mM. The mixtures were transferred into cuvettes and their UV absorptions were measured in the wavelength range 250 nm to 500 nm. For the same mixtures, fluorescence emissions between 280 nm and 800 nm were measured at an excitation wavelength of 281 nm. To investigate the effect of interfering ions on the UV and fluorescence measurements of Hg(II), the above procedures were repeated after the addition of 1 mL of 80 mM Cd(II) and 80 mM Pb(II) to the ADB18C6/HCl/Hg(II) mixtures.
ADB18C6 coupling to MPA
EDC (0.2 M) and NHS (0.1 M) were prepared in PBS (10 mM, pH 7.0) and mixed together (50:50, v/v). One mL of MPA (0.4 mM in PBS, pH 7) was then added to 1 mL of EDC/NHS mixture. The reaction mixture was allowed to stand for 15 minutes to ensure the complete activation of the COOH terminal group of MPA. To the MPA/EDC/NHS mixture, 1 mL of ADB18C6 (1 mM in DMSO) was added to form an amide bond between the amine group of the crown ether and the activated COOH group of the MPA. The MPA-coupled ADB18C6 was labelled “TCE” and used directly as a recognition molecule for the detection of Hg(II). The scheme for the TCE ligand formation is shown in ESI material 1 (ESI 1†).
Electrochemical deposition of gold nanostructures on a gold substrate
The surface of the gold substrates were polished with 0.5, 0.3 and 0.05 μm grain sized alumina slurry respectively. The substrates were then rinsed with water and sonicated in Millipore water for about 15 minutes to remove all the physically adsorbed alumina slurry. The polished substrates were then soaked in piranha solution (3 parts of 98% sulfuric acid and 1 part of 30% hydrogen peroxide solution) for 10 minutes and then rinsed with Millipore water. HAuCl4 solution (4 mM in 0.1 M HClO4) was used for the electrochemical deposition of gold nanostructures onto the surface of the gold substrate using the method described by Sivanesan et al.30 after a minor modification where the deposition time was increased from 400 s to 900 s. This was done to increase the number of gold nanostructures and hence the number of hotspots available for SERS enhancement. Finally, the substrates were treated with oxygen plasma for 15 minutes to remove any organic molecule that may become adsorbed to the nanostructured gold surfaces (ESI 2†). The prepared substrates were characterized and used as a SERS active substrate for Raman measurement of Hg(II).Surface functionalisation of the nanostructured gold substrate with crown ethers
The nanostructured gold substrates were functionalised with crown ethers by their incubation in 500 μL of ADB18C6 and TCE solutions overnight. The substrates were then removed from solution and washed with PBS. It was again washed with ultra-pure water to remove all unbounded crown ether and other unreactive molecules from the surface. To prevent the non-specific binding of foreign molecules to the bare area of the nanostructured gold surface, the functionalised substrates were dipped into butanethiol (10−7 M aqueous solution). This procedure was carried out for 2 hours in order to allow the butanethiol molecules to backfill the bare gold sites on the substrate surface via stable Au–S bonds. The functionalised and backfilled nanostructured gold substrates were then washed several times to remove the excess butanethiol from their surfaces.Cyclic voltammetry measurements of the complex formation between Hg(II) and tADB18C6
To confirm the complex formation between the TCE surface layer of the functionalised nanostructured substrates and Hg(II) ions by electrochemistry, the bare nanostructured gold substrate and the TCE-functionalised substrate were first screened by cyclic voltammetry (CV). The voltammograms were acquired in 100 mM PBS solution as an electrolyte. The CV scans (6 cycles) were carried out between −0.5 to 0.6 V at a scan rate of 0.1 V s−1. Ag/AgCl was used as the reference electrode and a platinum wire as an auxiliary electrode. After the first set of CV measurements, Hg(II) standard solutions at 1 μM concentration were loaded onto TCE-functionalized substrates and allowed to stand for 15 min. The CV measurements were then repeated and the oxidation and reduction peaks at 0.40 V and 0.35 V, respectively, of the Hg(II) ion bound substrate were monitored after washing severally with Milli-Q water.SERS detection of Hg(II) by the functionalised nanostructured gold substrates
The TCE-functionalised and ADB18C6-functionalized gold substrates were used for the detection of Hg(II) (ESI 3†). Prior to this, the background spectra of the bare AuNS coated gold substrate was acquired (ESI 4†). These substrates were screened by Raman spectroscopy to acquire their native SERS spectra. The spectrum for MPA was also recorded (ESI 5†). It is noteworthy that these SERS spectra were acquired using a handheld Raman spectrometer with the “reference spectrum” and “clean peaks” modes enabled. With the “reference spectrum” enabled, ambient light and other environmental background as well as any fixed pattern noise is excluded. The “clean peaks” mode is an in built beta correction algorithm that does background correction and removes the effect of fluorescence (ESI 4†). These spectra were also collected in the raster orbital scanning mode (over a wavelength range of 400 cm−1 to 2000 cm−1), which rapidly scans a tightly focused beam in an orbital pattern, allowing lower average power to produce high-integrity data from a larger area of the sample without sample damage. An excitation wavelength of 785 nm laser line was used for the SERS measurements. The laser power at the sample was 10 mW while the scan time was set at 2 accumulations and 3 seconds integration time. After these sets of SERS measurements, 1 mL aliquots of Hg(II) standard solutions, in the concentration range 1 μM to 0.1 pM, were loaded onto the functionalised gold substrates for 5 min to allow for the complex formation between the crown ether surface layer of the substrate and Hg(II) ions. The SERS spectra of the ADB18C6-functionalised and the TCE-functionalized substrates were then repeated after the complex formation (Scheme 1). A calibration curve was plotted between the SERS signal intensity at 1501 cm−1 and the concentrations of the Hg(II) standard solutions.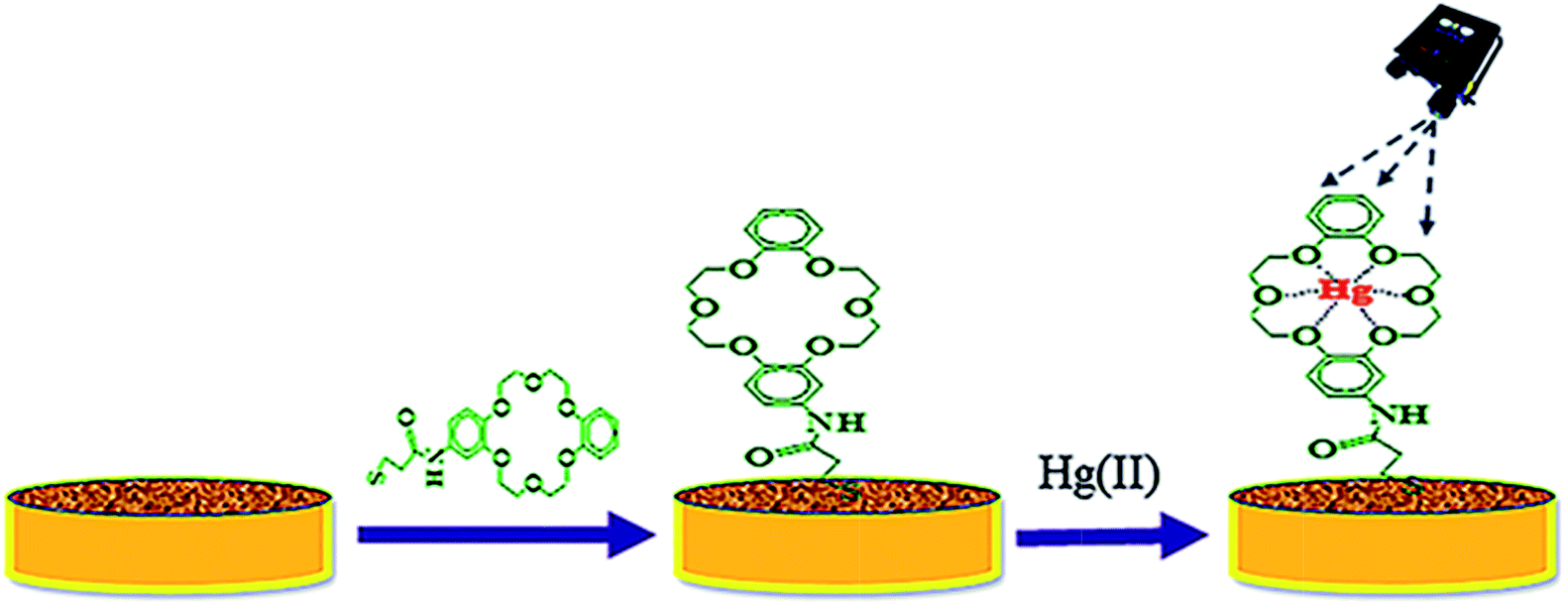 | ||
| Scheme 1 A schematic representation for the modification of nanostructured gold substrate by TCE and subsequent binding of Hg(II). | ||
Determination of detection limit and lower limit of quantification
In order to determine the detection and quantification limits of Hg(II) ions by SERS measurement, seven 0.1 pM Hg(II) bound TCE nanosensors were screened with the handheld Raman spectrometer and the concentration was determined (ESI 6†). The LOD and LOQ were calculated as the standard deviation (σ) × t95% and σ × 10 respectively (where t is the threshold value of student t-distribution with a degree of freedom (n − 1) at 95% confidence interval).31Selectivity of TCE towards Hg(II)
The selectivity of TCE towards Hg(II) was tested using 1 mL aliquots of 1 μM Pb(II), Cd(II) and their mixed ion (Pb(II) and Cd(II)) standard solutions. These solutions were loaded onto individual TCE-functionalised substrates and allowed to stand for 5 min and the SERS spectra were acquired with the handheld Raman spectrometer as previously described. The spectra of the functionalised substrates before and after interaction with the metal ions were compared (ESI 7†).Determination of Hg(II) in tap water by TCE-functionalised gold substrate and handheld Raman spectrometer
To utilise the TCE-functionalised gold substrate as a new nanosensor for Hg(II) contamination studies in the environment, 1 mL of tap water was collected and spiked with 25 μL of 1 μM Hg(II). The spiked water sample was then loaded onto the TCE-functionalised gold substrate and allowed to stand for 15 min. SERS measurements were carried out using the handheld Raman device (n = 3). The concentrations were calculated from the calibration curve and the percentage recovery determined from the relation:where CHg(x), CHg(b) and CHg(s) represent the concentration of: Hg(II) as calculated from the SERS calibration curve, blank sample and the spiked Hg(II) standard respectively.
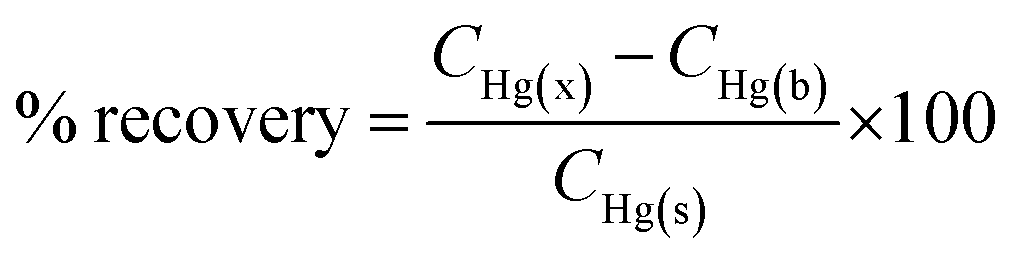
Instrumentation and characterization
The nanostructured gold substrate was characterised by Scanning Electron Microscopy (SEM) to determine the geometry, size and distribution of the developed gold nanostructures. The UV absorption spectra were recorded by Cary 60 UV-visible spectrophotometer (Agilent Technologies, USA). Fluorescence Cary eclipse spectrophotometer was used for fluorescence measurements. SERS measurements were carried out using the handheld ID Raman Mini2 (ocean optics, USA). A μAutolab potentiostat (Metrohm Autolab) with a custom made three-electrode cell setup was also used for all electrochemical measurements.Results and discussion
UV-visible and fluorescence studies on Hg(II)
The absorption spectra of ADB18C6 in the absence and presence of Hg(II) are shown in Fig. 1. ADB18C6 showed a shoulder band at 275 nm and absorption bands at 280 nm and 295 nm respectively (Fig. 1A). The absorption band at 295 nm can be attributed to the n → π* transitions between the oxygen atoms electron lone pairs and the crown ether ring of ADB18C6. The band at 280 nm can be attributed to π → π* transitions within the unsaturated aromatic benzene rings on the crown ether molecule. The shoulder at 275 nm may also be due to n → π transitions between the amino group and the benzene ring of the aniline moiety of the crown ether system.32,33 Upon the addition of different concentrations of Hg(II) ions, the absorption band at 295 nm decreased and was completely quenched at 80 μM of Hg(II) concentration (Fig. 1B spectra a–d). The decrease in the 295 nm band intensity indicated a host–guest interaction between Hg(II) and 18-crown-6 where the Hg(II) metal ions are chelated by the oxygen atoms of the crown ether.29,34 This leads to the formation of a stable complex between ADB18C6 and Hg(II). A closer look at the spectra (a–d in Fig. 1B) reveals an isosbestic point at 287 nm, which is attributed to the presence of both the Hg(II)–ADB18C6 complex and the free ADB18C6 in solution.35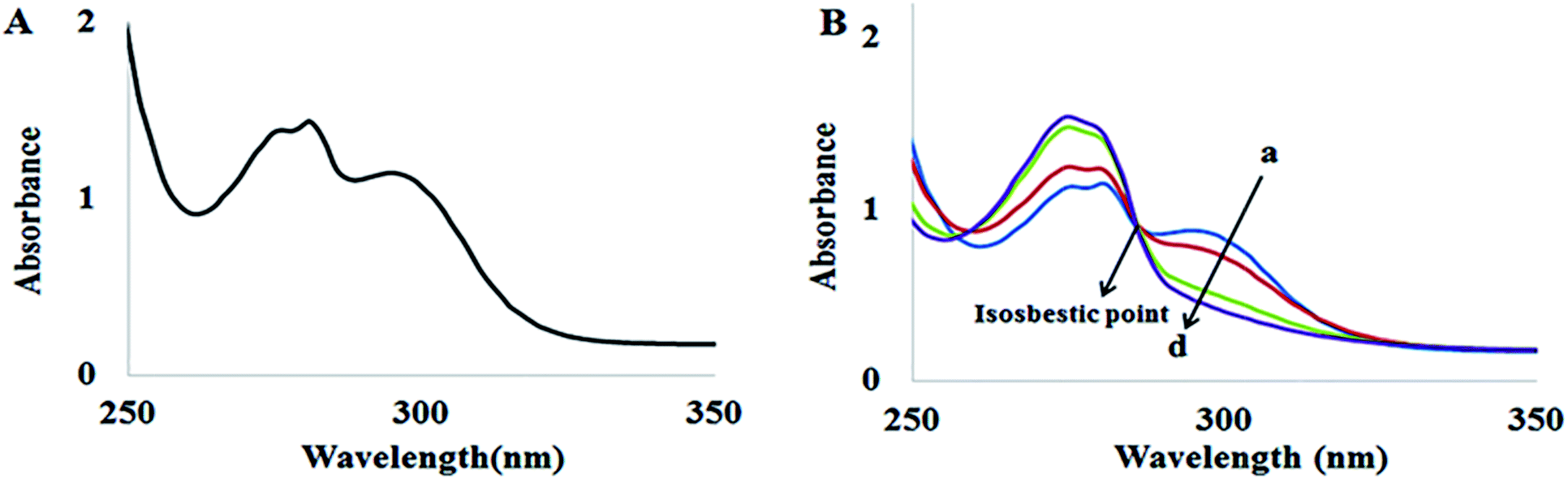 | ||
| Fig. 1 UV-visible spectra of (A) ADB18C6 and (B) ADB18C6 in different concentrations of Hg(II): (a) 20 (b) 40, (c) 60 and (d) 80 μM. | ||
The fluorescence spectra of ADB18C6 in the absence and presence of Hg(II) are likewise shown in Fig. 2. As indicated by the figure, emission bands at 311 nm, 360 nm, 563 and 612 nm were observed in the native spectra of ADB18C6. After the addition of different concentrations of Hg(II), the band at 311 nm increased while the intensity of the band at 360 nm quenched gradually (spectra a–e). Moreover, the addition Hg(II) led to an increase in the intensity of the emission bands at 563 nm and 612 nm (Fig. 2). The quenching of the band at 360 nm and the increased intensity of the 311 nm, 563 nm and 612 nm bands can be attributed to the formation of the Hg(II)–ADB18C6 complex.
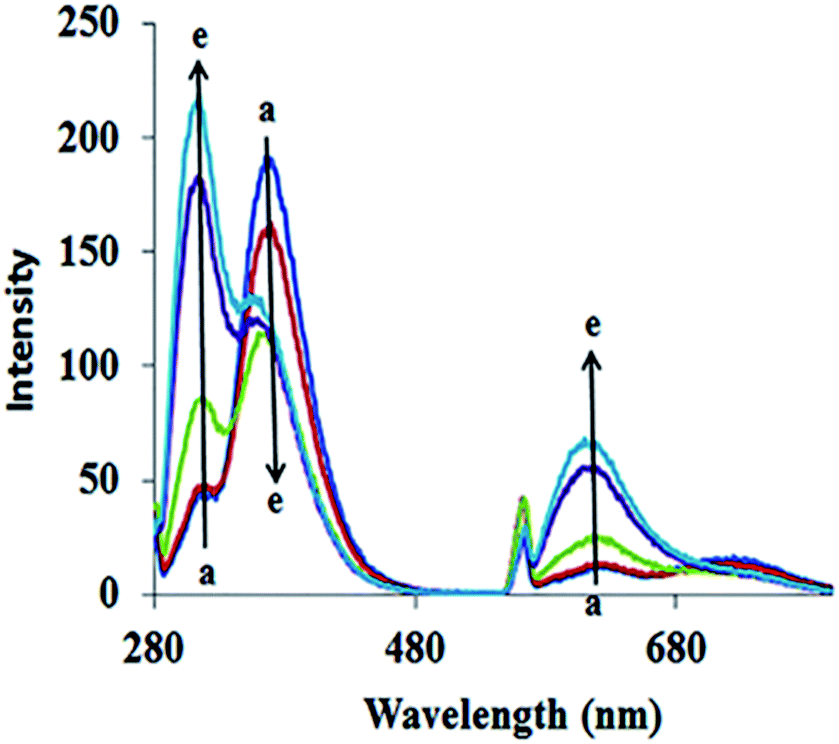 | ||
| Fig. 2 Fluorescence spectra of ADB18C6 in different concentrations of Hg(II): (a) 0, (b) 20, (c) 40, (d) 60 and (e) 80 μM. | ||
Selectivity of ADB18C6 crown ether towards Hg(II)
The selectivity of ADB18C6 towards Hg(II) over 19 other metal ions (i.e. K+, Li+, Na+, Ag+, Ca2+, Mg2+, Ba2+, Sr2+, Ni2+, Co2+, Cd2+, Pb2+, Cu2+, Fe3+, Al3+, La3+, Eu3+ and Dy3+) has been reported in literature.22,23 In this work, cadmium(II) and Pb(II) ions were chosen for the selectivity studies using UV visible and fluorescence spectroscopy because of the similar chemical properties between Cd(II), Pb(II) and Hg(II) and the fact that these toxic heavy metals are among the commonly known environmental pollutants. The UV absorption (Fig. 3A) and fluorescence emission spectroscopy (Fig. 3B) showed no observable changes in the absorption and fluorescence spectra of ADB18C6 after the addition of Cd(II) and Pb(II). These results confirmed the selective complex formation between ADB18C6 and Hg(II).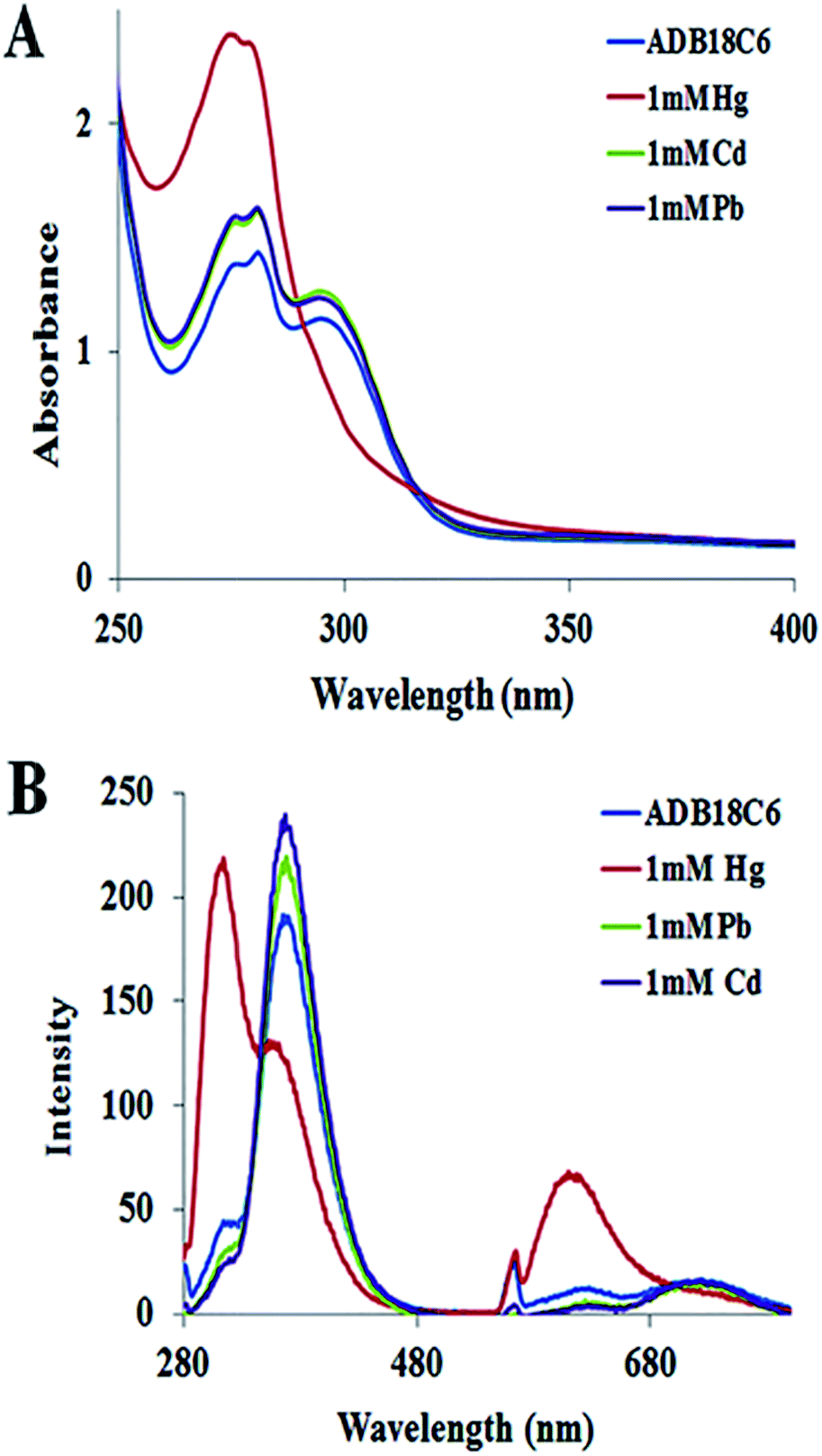 | ||
| Fig. 3 (A) UV absorption spectra and (B) fluorescence emission spectra of ADB18C6 crown ether after reaction with Hg(II), Cd(II) and Pb(II) ions respectively. | ||
Thiolation of ADB18C6 with MPA
To utilise ADB18C6 for the nanosensing of Hg(II) by SERS, it is necessary to immobilise the crown ether onto the surface of a SERS substrate where it acts as a recognition layer for the mercuric ions. For this purpose, the carboxylic acid group of the alkane thiol (i.e. MPA) was coupled to the crown ether using the EDC/NHS coupling reaction to produce the thiolated crown ether “TCE”. Upon interaction with gold surface, the free thiol (SH) terminal group of TCE forms a stable Au–S bond with the gold nanostructures to assemble the crown ether molecules in a highly ordered monolayer with a common orientation. The assembled crown ether was subsequently used to bind Hg(II) ions.Study of complex formation between Hg(II) and TCE by electrochemistry
To confirm the complex formation between the crown ether and Hg(II) ions on the functionalized nanostructured surface, the TCE-functionalized gold substrate was screened by cyclic voltammetry (CV). The cyclic voltammogram of the bare nanostructured gold substrate (bare Au), the TCE-functionalized gold substrate (Au/TCE) and the TCE-functionalised gold substrate after binding with Hg(II) are shown in Fig. 4. As indicated by the figure, there was no observable redox peak(s) in the voltammogram of the bare nanostructured gold substrate. However, two well-defined redox peaks appeared in the voltammogram of TCE-functionalised gold substrate at 0.20 V (oxidation peak) and 0.14 V (reduction peak) respectively. New oxidation and reduction peaks at 0.40 V and 0.35 V, respectively, appeared in the voltammogram of TCE-functionalised gold substrate after its interaction with Hg(II) ions (Au/TCE/Hg). The emergence of these two peaks indicated the binding of Hg(II) to the host crown ether36–39 and they may be attributed to the inclusion of Hg(II) in the cavity of 18-crown-6 moiety of TCE and the accompanied corresponding changes to the redox potential of the substrate.40,41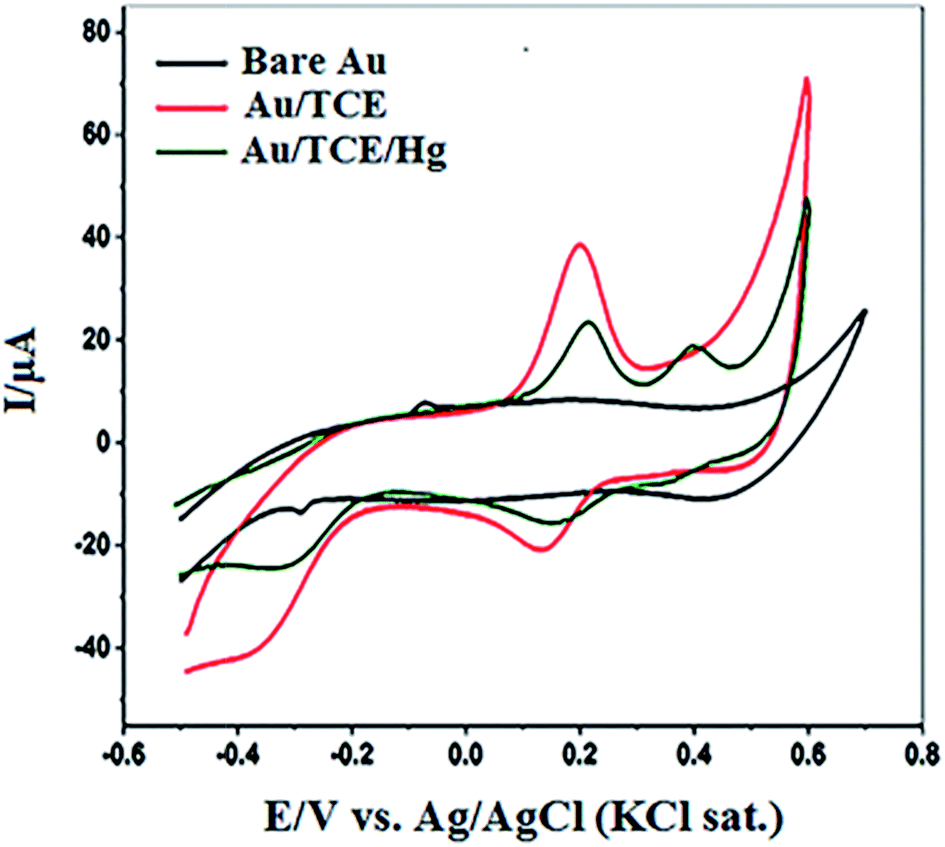 | ||
| Fig. 4 Cyclic voltammogram of bare nanostructured gold substrate (bare Au), TCE-functionalized gold substrate (Au/TCE) and TCE-functionalised gold substrate after complex formation with Hg(II) [(Au/TCE/Hg)]. | ||
SERS detection of Hg(II) by the TCE-functionalized gold substrate
Some research groups11,42–44 have reported SERS studies that utilise colloidal nanoparticles for the detection of mercury contamination in various environmental matrices. While some of these SERS sensors feature high selectivity, the colloidal nanoparticles are quiet unstable since they are prone to uncontrolled aggregation and, in effect, create non-reproducibility challenges in their SERS measurements. Unlike colloidal substrates, solid SERS-active substrates offer the advantages of easy handling, long term stability and adaptability to in-field screening.45,46 We recently developed a solid nanostructured gold substrate for highly reproducible SERS measurements. The substrate was manufactured by electrochemical deposition of gold nanostructures onto a gold surface to deliver high density of hot spots and high SERS enhancement factor.30,45 In the present work, we functionalised this substrate with ADB18C6 and TCE and used it as a nanosensor for the detection of Hg(II). The SERS spectra for both ADB18C6 and TCE after their immobilisation onto the nanostructured gold substrate (Fig. 5) were recorded with the handheld ID Raman mini 2 and the corresponding band assignments given in Table 1. Since the handheld device has the ability to perform fluorescence and background interference corrections, the spectra is reported as raw data. This saves time, giving it advantage over a bench top spectrometer.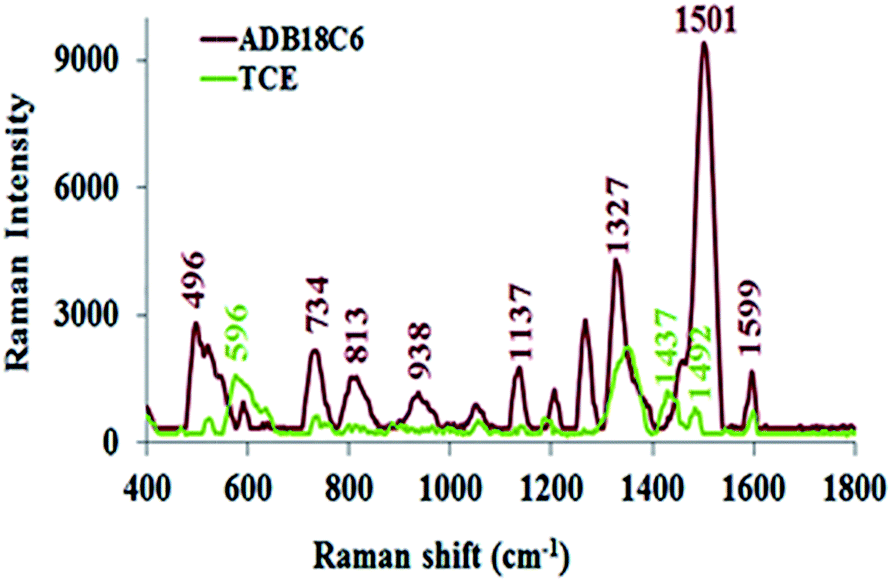 | ||
| Fig. 5 SERS spectra of ADB18C6 and TCE on nanostructured gold substrate. | ||
| Raman shift/cm | Band assignment | References | ||
|---|---|---|---|---|
| ADB18C6 | TCE | ADB18C6 | TCE | |
| 496 | 596 | δ (C–O–C), sym | Au–S | 47 |
| 734 | Cph–H, out of plane bending | 48 | ||
| 813 | ν (C–O–C), sym | 47 | ||
| 938 | Cph–H, out of plane bending | 48 | ||
| 1137 | ν (C–O–C), asym | 47 and 48 | ||
| 1327 | CH2-wag, C–C–H deformation | 47–49 | ||
| 1440 | 1437 | H–C–H deformation | ✓ | 47 |
| 1501 | 1492 | Cph–O str., Cph–H | ✓ | 48 |
| 1599 | 1602 | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C str. (aromatic) C str. (aromatic) |
✓ | 50 |
When ADB18C6 is adsorbed onto the substrate, Au–N and Au–O bonds are formed between the substrate's gold nanostructures and the amine and oxygen groups of ADB18C6 respectively. As a result, we propose that the crown ether moiety assumed a horizontal position on the gold surface. The appearance of the out-of-plane vibrational modes at 734 and 938 cm−1 indicated that, these modes of vibration adopted a perpendicular orientation on the gold surface while the crown ether moiety lies horizontal to this nanostructured gold surface.51,52 In this orientation, since the ADB18C6 molecule lies close to the nanostructured gold substrate's surface, it experiences a strong localised surface plasmon resonance effect (LSPR). The close proximity of the ADB18C6 molecule to the LSPR and the fact that its scattering tensor components for the out-of-plane vibration modes lie perpendicularly to the gold surface, led to a strong Raman intensity enhancement observed in the spectra of ADB18C6.51,52
The Raman spectra of TCE has relatively low intensities compared to the spectra of ADB18C6 (Fig. 5). These changes may be attributed to a non-horizontal orientation of TCE onto the substrate's surface. Thiolated ADB18C6 forms a strong Au–S bond between its MPA moiety and the nanostructures of the gold substrate. Therefore, TCE may assume a more vertical position on the substrate's surface when compared to that of ADB18C6.26,53,54 The vertical orientation of TCE causes the crown ether structure to lie further away from the effective range of the LSPR. Since the LPSR effect decays exponentially with the distance from the gold surface,28 a significant reduction in the Raman signal intensity was observed for TCE. The Raman bands at 1501 cm−1 experienced a strong reduction in its intensity as well as a bathochromic shift to 1492 cm−1.51,55 In addition, the low intensities of the bands at 1492 cm−1 and 1599 cm−1 suggested that the phenyl rings of the crown ether structure are not directly adsorbed onto the substrate surface and, therefore, their interaction with the LSPR is weak.27 The vertical orientation of TCE also causes the out-of-plane vibrational modes at 734 cm−1 and 938 cm−1 to lie parallel to the surface of the substrate.52,54 Hence, these vibrational modes become Raman inactive leading to a quenching of the 734 cm−1 and 938 cm−1 bands in the Raman spectrum of TCE.
The TCE-functionalisation gold substrate was utilised for the SERS detection of Hg(II) ions. Upon complexation of Hg(II) ions with the TCE surface layer of the functionalised substrate, the Raman spectrum of TCE experienced significant changes (Fig. 6). As illustrated by Fig. 6, the bands at 1327 cm−1 and 1501 cm−1 and the H–C–H deformation band at 1454 cm−1 reappeared. These changes may be attributed to the structural deformation of TCE upon its complexation with Hg(II).48,56 In addition, the affinity of Hg(II) to gold may cause the mercuric ions to move close to the substrates surface. This will force the TCE to move to the proximity of the substrates surface in order to chelate the mercuric ions that lie at the gold interface. These combined effects cause the crown ether structure to align itself within the LSPR of the SERS substrate.49 This in turn, led to the strong enhancement of the Raman spectrum of TCE–Hg(II) complex compared to that of TCE alone.
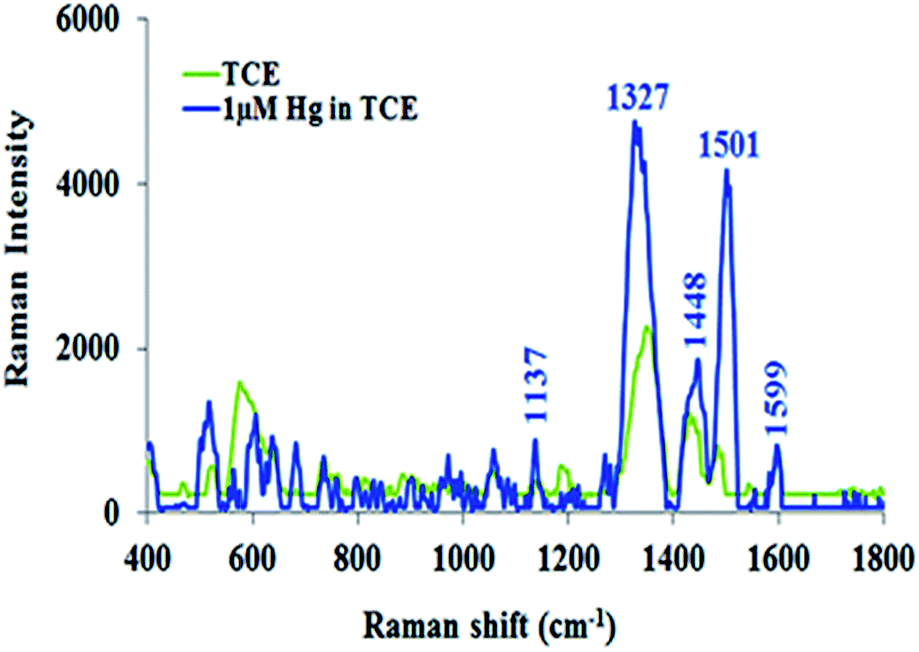 | ||
| Fig. 6 SERS spectra of TCE and TCE–Hg(II) complex. | ||
SERS quantification of Hg(II) in the environment by TCE-functionalised gold nanosensor
The TCE-functionalised gold nanosensor was utilized for SERS quantification of Hg(II) in aqueous solutions. For the quantification of Hg(II) by SERS, the intensity of the Raman band at 1501 cm−1 was recorded at different concentrations of Hg(II). The Raman signal intensity at 1501 cm−1 was found to increase with the concentration of Hg(II) (Fig. 7 inset). A linear relationship was obtained between the SERS signal intensity and the corresponding log concentration of Hg(II) (Fig. 7). Close correlation (R2 = 0.9909) was found over the Hg(II) concentration range of 1 μM to 0.1 pM. The LOD and LOQ of the new nanosensing method were obtained at a standard deviation (σ) of 0.21 pM from seven measurements and estimated to be 5.1 × 10−13 M (0.51 pM) and 2.07 × 10−12 M (2.07 pM) respectively. The LOD and LOQ values of the SERS nanosensor are 1000 fold lower than the recommended EPA and WHO limits of Hg(II) in water (i.e. 10 nM and 30 nM respectively).10,57 Other SERS sensors for Hg(II) have been demonstrated in the literature. For example, Shaban et al.59 developed a SERS sensor for Hg(II) by functionalising porous anodic alumina membrane with CoFe2O4 nanoparticles and growing carbon nanofibers on the inner walls of the functionalised membrane to capture the target analyte. The method development of the sensor was complex and the detection limit for Hg(II) was only 1 μg per L (1 PPb). Ren et al.43 synthesised and functionalised Ag nanoparticles with crystal violet, as a Raman reporter, and utilised them to determine Hg(II) down to 90 pM. The disadvantage of this method is that, the Raman detection of Hg(II) was carried out directly within the Ag colloids. This mode of detection is well known to suffer from non-reproducible SERS measurements due to the inability to control the formation of hot spots.60,61 Wang et al.62 used gold nanoparticles that were functionalised with anti Hg antibody and a Raman reporter to detect Hg(II) by SERS based competitive immunoassay. The method was selective and sensitive down to 0.4 pM. However, this method is disadvantaged by the use of expensive antibody and Raman reporter as well as the limited shelf life of the functionalised nanoparticles due to the potential loss of the antibody activity. By contrast, the developed sensor in this work addresses these challenges. Besides the ease of using this sensor in the field, its LOD is relatively lower making it more sensitive than those for some other methods reported previously (Table 2). In addition, due to the chemical nature of the used crown ether and its immobilisation on the nanostructured substrate via stable Au–S bonds it is more stable than the sensor functionalised by an antibody.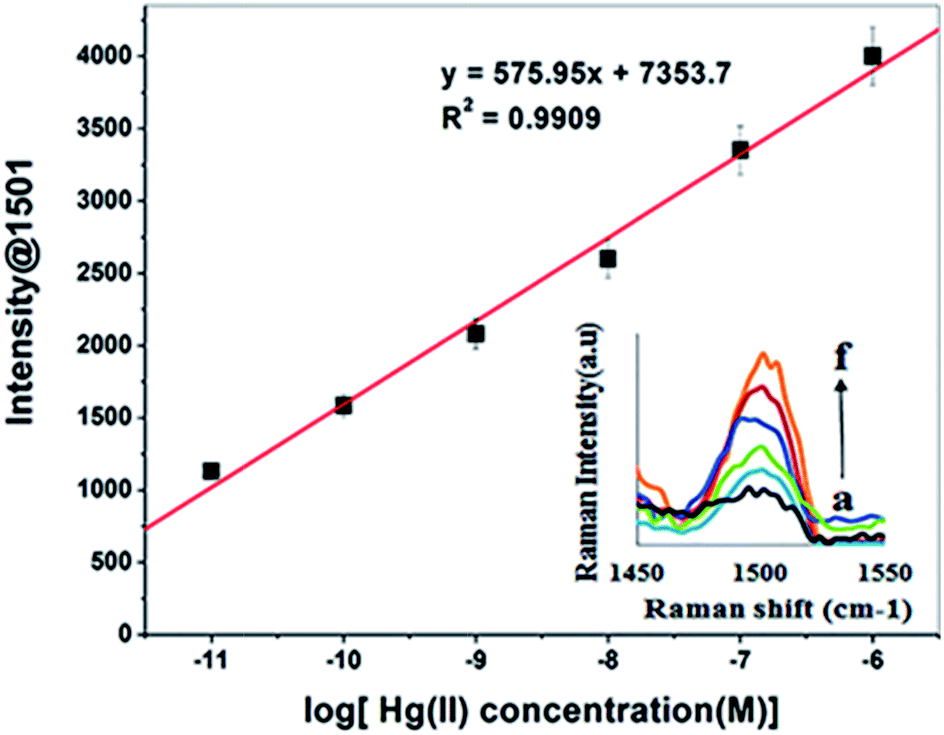 | ||
| Fig. 7 A linear correlation plot of the Raman intensity (at 1501) versus the logarithm of Hg(II) concentration and corresponding SERS spectra (inset) from (a) 1 × 10−11 M to (f) 1 × 10−6 M. | ||
| Sensing material | Analytical technique | Medium | LOD | Reference |
|---|---|---|---|---|
| Dibenzo-18-crown-6 azo dye derivative | Spectrofluorometry | Water | 1.25 × 10−8 M | 22 |
| Dibenzo-18-crown-6 azo dye derivative | UV-visible spectroscopy | Water | 2.90 × 10−8 M | 23 |
| Microporous poly(2-mercaptobenzothiazole) film | Electrochemistry | Water | 1.0 × 10−10 M | 58 |
| Thiolated dibenzo-18-crown-6 derivative | SERS | Water | 5.10 × 10−13 M | This work |
The TCE-functionalised gold nanosensor was utilised for the detection of Hg(II) in tap water. The TAP water was contaminated with Hg(II) (2.5 × 10−8 M), loaded onto the TCE-functionalised gold nanosensor and screened by a handheld Raman spectrometer. This concentration was chosen because it represents a case of Hg(II) contamination in water that can cause toxicity (since it is above the EPA and WHO defined levels for Hg(II) ions in water). The mean prediction concentration of Hg(II) was 1.67 × 10−8 M (n = 3) with a % recovery of 93.33 ± 16.17. The results obtained for the spiked water sample indicated the potential of this nanosensor for the fast, sensitive and selective field analysis of mercury contamination in water.
Conclusion
In this work, a new field-deployable crown ether-based SERS sensor for detecting Hg(II) ions in water has been demonstrated. The new nanosensor showed high sensitivity and selectivity towards Hg(II). The LOD (0.51 pM) and LOQ (2.07 pM) of this new mercury nanosensor were 1000 fold below the EPA and WHO defined levels for Hg(II) ions in water. The TCE-functionalised nanostructured gold substrate provided a convenient platform for the field detection of Hg(II) in water by surface enhanced Raman spectroscopy. In view of these advantages, this nanosensor provides a promising rapid and easy assay that can be used for the field detection of Hg(II) in water. Investigations on the fabrication of more sensitive, rapid, easy-to-use, selective and field deployable nanosensors for other environmental toxicants are in progress in our laboratory.Acknowledgements
We thank the Ghana Atomic Energy Commission (GAEC) for the study leave granted and the Queensland University of Technology (QUT) for their support through the QUT Postgraduate Research Award (QUTPRA) and the QUT International HDR Tuition Fee Sponsorship to DKS. The authors also acknowledge the staff of Central Analytical Research Facility (CARF) of QUT, Natalia Danilova and Llew Rintoul (PhD) for the support. Access to CARF was supported by the generous funding from the Science and Engineering Faculty of QUT.References
- J. Duan, M. Yang, Y. Lai, J. Yuan and J. Zhan, Anal. Chim. Acta, 2012, 723, 88–93 CrossRef CAS PubMed.
- E. Tan, P. Yin, X. Lang, H. Zhang and L. Guo, Spectrochim. Acta, Part A, 2012, 97, 1007–1012 CrossRef CAS PubMed.
- C. Jiang, Z. Guan, S. Y. R. Lim, L. Polavarapu and Q.-H. Xu, Nanoscale, 2011, 3, 3316–3320 RSC.
- M. M. Lynam, B. Klaue, G. J. Keeler and J. D. Blum, J. Anal. At. Spectrom., 2013, 28, 1788–1795 RSC.
- S. Cai, K. Lao, C. Lau and J. Lu, Anal. Chem., 2011, 83, 9702–9708 CrossRef CAS PubMed.
- E. Marguí, P. Kregsamer, M. Hidalgo, J. Tapias, I. Queralt and C. Streli, Talanta, 2010, 82, 821–827 CrossRef PubMed.
- X. Jia, Y. Han, X. Liu, T. Duan and H. Chen, Spectrochim. Acta, Part B, 2011, 66, 88–92 CrossRef.
- T. Osawa, Y. Hatsukawa, P. W. Appel and H. Matsue, Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms, 2011, 269, 717–720 CrossRef CAS.
- Y. Zhang, X. Li, G. Liu, Z. Wang, T. Kong, J. Tang, P. Zhag, W. Yang, D. Li and L. Liu, Biol. Trace Elem. Res., 2011, 144, 854–864 CrossRef CAS PubMed.
- Z. Sun, J. Du and C. Jing, J. Environ. Sci., 2016, 39, 134–143 CrossRef PubMed.
- E. Chung, R. Gao, J. Ko, N. Choi, D. W. Lim, E. K. Lee, S.-I. Chang and J. Choo, Lab Chip, 2013, 13, 260–266 RSC.
- Y. Du, R. Liu, B. Liu, S. Wang, M.-Y. Han and Z. Zhang, Anal. Chem., 2013, 85, 3160–3165 CrossRef CAS PubMed.
- M. Liu, Z. Wang, S. Zong, H. Chen, D. Zhu, L. Wu, G. Hu and Y. Cui, ACS Appl. Mater. Interfaces, 2014, 6, 7371–7379 Search PubMed.
- P. Ma, F. Liang, Q. Diao, D. Wang, Q. Yang, D. Gao, D. Song and X. Wang, RSC Adv., 2015, 5, 32168–32174 RSC.
- J. W. Steed, Coord. Chem. Rev., 2001, 215, 171–221 CrossRef CAS.
- F. Faridbod, M. R. Ganjali, R. Dinarvand, P. Norouzi and S. Riahi, Sensors, 2008, 8, 1645–1703 CrossRef CAS PubMed.
- G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723–2750 CrossRef CAS PubMed.
- A. H. Bond, M. L. Dietz and R. Chiarizia, Ind. Eng. Chem. Res., 2000, 39, 3442–3464 CrossRef CAS.
- A. Alizadeh, M. Khodaei, C. Karami, M. Workentin, M. Shamsipur and M. Sadeghi, Nanotechnology, 2010, 21, 315503 CrossRef CAS PubMed.
- A. R. Fakhari, A. R. Khorrami and M. Shamsipur, Sep. Purif. Technol., 2006, 50, 77–81 CrossRef CAS.
- S. A. Hamidinia, G. E. Steinbaugh, W. L. Erdahl, R. W. Taylor and D. R. Pfeiffer, J. Inorg. Biochem., 2006, 100, 403–412 CrossRef CAS PubMed.
- P. Ncube, R. W. Krause, D. T. Ndinteh and B. B. Mamba, Water SA, 2014, 40, 175–182 CrossRef CAS.
- Z. Yan, H. Lei, N. Li and L. Hong, Spectrochim. Acta, Part A, 2011, 79, 661–665 CrossRef CAS PubMed.
- A. R. Fakhari, M. R. Ganjali and M. Shamsipur, Anal. Chem., 1997, 69, 3693–3696 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS PubMed.
- H. Hakkinen, Nat. Chem., 2012, 4, 443–455 CrossRef PubMed.
- R. Agoston, E. L. Izake, A. Sivanesan, W. B. Lott, M. Sillence and R. Steel, Nanomedicine: Nanotechnology, Biology and Medicine, 2015, 633–641 Search PubMed.
- J. Hughes, E. L. Izake, W. B. Lott, G. A. Ayoko and M. Sillence, Talanta, 2014, 130, 20–25 CrossRef CAS PubMed.
- R. A. Sperling and W. Parak, Philosophical Transactions of the Royal Society of London A: Mathematical, Physical and Engineering Sciences, 2010, 368, 1333–1383 CrossRef CAS PubMed.
- A. Sivanesan, W. Adamkiewicz, G. Kalaivani, A. Kamińska, J. Waluk, R. Hołyst and E. L. Izake, Analyst, 2015, 140, 489–496 RSC.
- G. O. Duodu, A. Goonetilleke and G. A. Ayoko, Talanta, 2016, 150, 278–285 CrossRef CAS PubMed.
- T. Milja, V. Krupa and T. Rao, RSC Adv., 2014, 4, 30718–30724 RSC.
- S. Wiktorowicz, R. Duchêne, H. Tenhu and V. Aseyev, Polym. Chem., 2014, 5, 4693–4700 RSC.
- S. Anandhakumar and J. Mathiyarasu, Microchim. Acta, 2013, 180, 1065–1071 CrossRef CAS.
- N. Vasimalai and S. A. John, J. Lumin., 2011, 131, 2636–2641 CrossRef CAS.
- F. Harnisch and S. Freguia, Chem.–Asian J., 2012, 7, 466–475 CrossRef CAS PubMed.
- J. B. Sperry and D. L. Wright, Chem. Soc. Rev., 2006, 35, 605–621 RSC.
- D. Wei, M. J. Bailey, P. Andrew and T. Ryhänen, Lab Chip, 2009, 9, 2123–2131 RSC.
- C.-W. Hsu and M.-C. Yang, Sens. Actuators, B, 2008, 134, 680–686 CrossRef CAS.
- P. D. Beer, Chem. Soc. Rev., 1989, 18, 409–450 RSC.
- P. D. Beer, Z. Chen and M. I. Ogden, J. Chem. Soc., Faraday Trans., 1995, 91, 295–302 RSC.
- D. Han, S. Y. Lim, B. J. Kim, L. Piao and T. D. Chung, Chem. Commun., 2010, 46, 5587–5589 RSC.
- W. Ren, C. Zhu and E. Wang, Nanoscale, 2012, 4, 5902–5909 RSC.
- L. Guerrini, I. Rodriguez-Loureiro, M. A. Correa-Duarte, Y. H. Lee, X. Y. Ling, F. J. G. de Abajo and R. A. Alvarez-Puebla, Nanoscale, 2014, 6, 8368–8375 RSC.
- H. Wei, S. M. H. Abtahi and P. J. Vikesland, Environ. Sci.: Nano, 2015, 2, 120–135 RSC.
- K. Fukami, M. L. Chourou, R. Miyagawa, Á. Muñoz Noval, T. Sakka, M. Manso-Silván, R. J. Martín-Palma and Y. H. Ogata, Materials, 2011, 4, 791–800 CrossRef CAS PubMed.
- A. Feofanov, A. Ianoul, V. Oleinikov, S. Gromov, O. Fedorova, M. Alfimov and I. Nabiev, J. Phys. Chem., 1996, 100, 2154–2160 CrossRef CAS.
- M. Dułak, R. Bergougnant, K. M. Fromm, H. R. Hagemann, A. Y. Robin and T. A. Wesołowski, Spectrochim. Acta, Part A, 2006, 64, 532–548 CrossRef PubMed.
- J. B. Heyns, L. M. Sears, R. C. Corcoran and K. T. Carron, Anal. Chem., 1994, 66, 1572–1574 CrossRef CAS.
- D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli, The handbook of infrared and Raman characteristic frequencies of organic molecules, Elsevier, 1991 Search PubMed.
- X. Gao, J. P. Davies and M. J. Weaver, J. Phys. Chem., 1990, 94, 6858–6864 CrossRef CAS.
- E. Le Ru, S. Meyer, C. Artur, P. Etchegoin, J. Grand, P. Lang and F. Maurel, Chem. Commun., 2011, 47, 3903–3905 RSC.
- S. Y. Eom, Y. R. Lee, H. L. Kim and C. H. Kwon, Bull. Korean Chem. Soc., 2014, 35, 875 CrossRef CAS.
- A. Sivanesan and S. A. John, Langmuir, 2008, 24, 2186–2190 CrossRef CAS PubMed.
- P. Gao and M. J. Weaver, J. Phys. Chem., 1985, 89, 5040–5046 CrossRef CAS.
- V. Shivaiah and S. K. Das, Inorg. Chem., 2005, 44, 7313–7315 CrossRef CAS PubMed.
- H. Li, J. Zhai, J. Tian, Y. Luo and X. Sun, Biosens. Bioelectron., 2011, 26, 4656–4660 CrossRef CAS PubMed.
- X.-C. Fu, X. Chen, Z. Guo, C.-G. Xie, L.-T. Kong, J.-H. Liu and X.-J. Huang, Anal. Chim. Acta, 2011, 685, 21–28 CrossRef CAS PubMed.
- M. Shaban and A. Galaly, Sci. Rep., 2016, 6, 1–9 CrossRef PubMed.
- R. Tantra, R. J. Brown and M. J. Milton, J. Raman Spectrosc., 2007, 38, 1469–1479 CrossRef CAS.
- M. De Jesus, K. Giesfeldt and M. Sepaniak, J. Raman Spectrosc., 2004, 35, 895–904 CrossRef CAS.
- Y. Wang, S. Chen, C. Wei, M. Xu, J. Yao, Y. Li, A. Deng and R. Gu, Chem. Commun., 2014, 50, 9112–9114 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02209c |
| This journal is © The Royal Society of Chemistry 2017 |