
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and electrochemical performance of a polymer-derived silicon oxycarbide/boron nitride nanotube composite
M. A. Abass a,
A. A. Syeda,
C. Gervaisb and
G. Singh*a
a,
A. A. Syeda,
C. Gervaisb and
G. Singh*a
aMechanical and Nuclear Engineering Department, Kansas State University, Manhattan, Kansas 66506, USA. E-mail: gurpreet@ksu.EDU; Fax: +1-785-532-7057; Tel: +1-785-532-7085
bSorbonne Universités, UPMC Univ Paris 06, CNRS, Collège de France, UMR 7574, Chimie de la Matière Condensée de Paris, CC 174, 4 place Jussieu F-75005, Paris, France
First published on 18th April 2017
Abstract
Polymer-derived ceramics (PDCs) such as silicon oxycarbide (SiOC) have shown promise as electrode materials for rechargeable Li-ion batteries (LIBs) owing to the synergy between their disordered carbon phase and hybrid bonds of silicon with oxygen and carbon. This study describes the synthesis and characterization of a new type of PDC composite comprising SiOC embedded with varying wt% of boron nitride nanotubes (BNNTs) and supported on reduced graphene oxide. Electron microscopy and a range of spectroscopic characterization techniques were employed to confirm the polymer-to-ceramic transformation and presence of a disordered carbon phase. The composites were tested as electrode materials for LIB and symmetric supercapacitor applications. As an electrode material for LIB, SiOC–BNNT paper composite containing 0.25 wt% BNNT demonstrated the highest charge capacity of 410 mA h g−1 (at 100 mA g−1) which stabilized at 238 mA h g−1. On the contrary, composite electrode with 0.5 wt% BNNT demonstrated the highest specific capacitance corresponding to 78.93 F g−1 at 1 A g−1 with a cyclic retention of 86% after 185 cycles. This study shows that presence of BNNTs in the composite influenced the free carbon phase in SiOC and its electrochemical performance. As such, the composite may be used as a flexible electrode material for electrochemical energy storage devices.
1. Introduction
Ever since their discovery in the early 1960s, organosilicon-based polymer-derived ceramics (PDCs) have attracted the attention of researchers.1 This class of ceramics is well known for its superior thermal stability, high oxidation resistance and remarkable mechanical properties. Unlike conventional ceramics, the physical properties of PDCs can be tuned by varying the chemical composition of the pre-ceramic polymer and the pyrolysis environment.2 Recent studies have shown that silicon-based PDCs can accommodate large amounts of Li+ (approximately three times that of graphite) without significant volume changes and cyclic stress during lithiation and delithiation processes.3–5 The observed Li storage property of PDCs is attributed to their amorphous nanodomain structure which is composed of graphitic carbon chains wrapped around nano-sized silicon-based domains such as silicon carbide (in silicon oxycarbide, SiOC) or silicon nitride (in silicon carbonitride, SiCN).3–6Among various PDC electrodes reported in the literature, SiOC has emerged as the most promising candidate for electrochemical energy storage applications.3,6,7 Structurally, SiOC is comprised of disordered carbon and Si in an amorphous tetrahedral structure wherein Si atoms share bonds with O and C without any C–O bonds.8,9 However, SiOC suffers from high voltage hysteresis or energy inefficiency and low charge transfer primarily due to its electrically insulating nature.10
Various approaches have been employed to improve the Li+ cycleability of SiOC without compromising its desirable properties.3,11 One of the approaches utilizes a porous form of SiOC (aerogels) as anode materials for Li-ion batteries (LIBs) in order to enhance the surface area of SiOC and diffusion kinetics of Li+ into SiOC.6 Kasper et al. reported Li+ cycling in SiOC correlates with size and amount of the free carbon phase in SiOC, which in turn are influenced by the pyrolysis temperature. Results revealed the Li+ charge capacity of SiOC decreases with pyrolysis temperature beyond 1200 °C.12 Bhandavat et al. demonstrated improvement in Li+ charge capacity and cycleability in SiOC by chemically interfacing with carbon nanotubes (CNTs) as conducting fillers. In this configuration, the fillers can be conveniently dislodged into the open amorphous structure of SiOC. More recently, synthesis of lightweight freestanding SiOC-based composite electrodes has been reported. This approach precludes the need for an organic binder and metallic supports known for contributing to the electrode's dead weight. Following this rationale, David et al. synthesized a SiOC–graphene paper composite that demonstrated a remarkable charge capacity of 702 mA h g−1 at a current density of 100 mA g−1.3 Recently, Kolathodi et al. investigated the performance of a layer-by-layer SiOC graphene composite (LBL–SiOC–rGO) and reported a reversible capacity of 750 mA h g−1 that stabilizes to 400 mA h g−1 at a current density of 50 mA g−1.7 These findings have shown that SiOC can be made into thin films with reduced graphene oxide (rGO) acting as a structural support.7 The presence of a graphene support is expected to reduce internal resistance of the composite by providing a conducting pathway for electroactive species and enhance the surface area of the composite, thus increasing the accessible sites for interaction of the composite with the electrolyte.13
Recent studies have shown promising results for the use of SiOC as electrode material for supercapacitor application. Kim et al. studied the capacitance of SiOC–carbon nanofiber composites and concluded that the observed capacitance is due to the synergistic effect of the carbon nanofiber's double-layer capacitance and the SiOC's pseudocapacitance.14 Tolosa et al. reported synthesis of freestanding electrodes made from electrospun SiOC fiber mat and compared the capacitance with traditional SiOC electrode on copper foil. The freestanding electrode demonstrated a superior capacity due to elimination of binders, which are known to diminish available pore site in SiOC for storing ions.15 Kolathodi et al. investigated the capacitive property of an LBL–SiOC–rGO composite. They concluded that SiOC is capable of being intercalated between the layers of rGO, thus improving the composite's available porous sites for charge storage.7
Structural modification of the properties of PDCs via interfacing with other nanomaterials have reported in the literature. Common examples of interfacing materials that have been reported in the literature includes CNT,16 boron nitride (BN),5 and graphite.17 In previous study, we showed that charge capacity of freestanding SiCN-based LIB electrode can be enhanced by interfacing SiCN with exfoliated BN nano sheets.18 The enhancement in Li+ insertion capacity was attributed to an increase in free carbon phase in SiCN due to interfacing with BN nanosheets.5 Inspired by these findings, we investigate the synthesis and electrochemical performance of SiOC modified with BN in the form of nanotubes (SiOC–BNNT) as potential electrode material for LIBs and supercapacitors. To the best of our knowledge, this is the first studies investigating the feasibility of synthesizing a freestanding paper composite comprising of BNNT-modified SiOC in which the filler (BNNTs in this case) shows no appreciable morphological degradation after polymer-to-ceramic conversion of the composite. The BNNTs are expected to suppress SiOC particles against re-aggregation, enhance the thermal stability and mechanical property of the composites. In addition, rGO would provide conducting path for electroactive species during the electrochemical process and the required mechanical support to synthesize the BNNT-modified SiOC ceramic as a thin film for lightweight electrochemical energy storage applications.13
2. Experimental
2.1 Materials
Sulfuric acid (H2SO4, 96.4%), sodium nitrate (NaNO3, 99.2%), potassium permanganate (KMnO4, 99.4%), hydrogen peroxide (H2O2, 31.3% solution in water), hydrochloric acid (HCl, 30% solution in water) and ethanol (C2H5OH, 99.9%) were purchased from Fischer Scientific. All materials were used as received without further purification.2.2 Preparation of boron nitride nanotube-modified SiOC ceramic
The BNNT-modified SiOC (SiOC–BNNT) ceramic was synthesized via pyrolysis of tetravinyl cyclotetrasiloxane (TTCS, Gelest, PA) using a similar procedure that has been reported in the literature.3 Varying wt% of BNNTs (Sigma-Aldrich) – with respect to the active weight of TTCS – were dissolved in a liquid mix of TTCS and 1 wt% of dicumyl peroxide (cross-linking agent). The liquid solution was cross-linked at 250 °C in argon (Ar) (heating rate of 100 °C h−1) for 3 h, resulting in the formation of an infusible mass. The cross-linked BNNT-modified TTCS was then crushed into fine powders and pyrolyzed at 1000 °C for 10 h in Ar atmosphere resulting in a fine black BNNT–SiOC ceramic powder termed SiOC–BNNT-2 wt%, SiOC–BNNT-0.5 wt% and SiOC–BNNT-0.25 wt% for the 2 wt%, 0.5 wt% and 0.25 wt% BNNT-modified SiOC ceramic, respectively.2.3 Preparation of ceramic paper composite
Graphene oxide (GO) was synthesized via a modified Hummer's method that has been described in the literature.19 A colloidal suspension of GO (20 ml) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) of deionized water and isopropanol (Fisher Scientific) was sonicated for 10 min and then SiOC–BNNT powder was slowly added to the GO suspension. The resulting composite suspension was further sonicated for 1 h and then stirred for another 6 h. The composite suspension was filtered by vacuum filtration through a 10 μm filter membrane (HPLC grade, Millipore). The paper composite was carefully removed from the filter membrane, dried and thermally reduced in an Ar-filled tube furnace at 500 °C for 2 h. The papers were cut and then used as electrodes for electrochemical energy storage.
:1 (v/v) of deionized water and isopropanol (Fisher Scientific) was sonicated for 10 min and then SiOC–BNNT powder was slowly added to the GO suspension. The resulting composite suspension was further sonicated for 1 h and then stirred for another 6 h. The composite suspension was filtered by vacuum filtration through a 10 μm filter membrane (HPLC grade, Millipore). The paper composite was carefully removed from the filter membrane, dried and thermally reduced in an Ar-filled tube furnace at 500 °C for 2 h. The papers were cut and then used as electrodes for electrochemical energy storage.
2.4 Physical characterization
The morphology of the synthesized paper composite electrodes was analyzed using scanning electron microscopy (SEM) on a Carl Zeiss EVO MA10 system with an accelerating voltage of 5 to 30 kV. Transmission electron microscopy (TEM) was used to characterize the structure of the paper composite electrodes using a Phillips CM100 under an accelerating voltage of 100 kV. X-ray photoelectron spectra (XPS) of the SiOC–BNNT ceramic were collected on a PHI Quantera SXM-03 Scanning XPS Microprobe using a monochromatic Al Kα radiation (beam size < 9 μm) at 45° angle of incidence. The chemical state of B in SiOC–BNNT ceramic was studied using 11B magic angle spinning nuclear magnetic resonance spectroscopy (11B MAS NMR). The spectrum was acquired at 7.0 T using a Bruker Avance 300 wide-bore spectrometer operating at a Larmor frequency of 96.35 MHz equipped with a 4 mm Bruker probe with a rotor spinning at 12 kHz. The spectrum was referenced using BF3(OEt)2 (δ = 0 ppm) and acquired using a spin–echo θ–τ–2θ pulse sequence and a recycle delay of 1 s. Raman spectra were collected using a He–Ne laser wavelength of 633 nm (laser power of 17 mW) on a confocal Raman imaging system (Horiba Jobin Yvon LabRam ARAMIS). Phase transformation of the samples was observed using a PANalytical Empyrean X-ray diffractometer operated at room temperature, with Cu Kα radiation (λ = 1.5418 Å) and nickel filter. Fourier transform infrared spectroscopy (FT-IR) spectra were obtained using an Agilent Cary 630 FT-IR spectrometer operated in the attenuated total reflectance (ATR) mode.2.5 Electrode fabrication and electrochemical measurements
The pyrolyzed paper composites were cut into circles using a-14.3 mm-diameter punch (total weight loading varied from 1 to 2 mg cm−2) and used as working electrode in the Li coin cell setup. The electrolyte solution was made by adding 4 to 5 drops of 1 M lithium hexafluorophosphate (LiPF6, Alfa Aesar) to a 1:1 v/v mixture of dimethyl carbonate (Sigma-Aldrich) and ethylene carbonate (Sigma-Aldrich) with an ionic conductivity of 10.7 mS cm−1. A glass separator (diameter of 19 mm and thickness of 25 μm) pre-soaked in the electrolyte solution was placed between the paper electrode (anode) and pure Li metal (cathode, 14.3 mm diameter and 75 μm thick, Alfa Aesar). The entire setup was assembled into a LIR 2032 battery coin cells in an Ar-filled glove box. Electrochemical performance of the coin cell battery was evaluated using a multichannel BT2000 Arbin test unit sweeping between 2.5 V and 10 mV vs. Li/Li+ at a current density of 50 mA g−1 (where 1 mA g−1 ≡ 1.5 mA cm−2). The efficiency of the cell (η) was calculated using the following expression:| η = Crev/Cch × 100 | (1) |
Symmetric two-electrode cell configuration was designed to study the electrochemical properties and performance of the paper electrodes as a supercapacitor. A 2 cm × 2 cm Whatman filter paper pre-soaked in a 6 M potassium hydroxide (KOH, 87.2%, Fisher Scientific) aqueous electrolyte separated the two 0.25 cm2 triangle-shaped paper electrodes. Testing was carried out on a CHI660E (CH Instruments, Inc.) electrochemical workstation. Cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) measurements were performed in a potential window of 0 to 0.9 V at different scan rates and current densities, respectively. The specific capacitance of the tested electrodes was calculated from the CV and GCD using the following expressions:
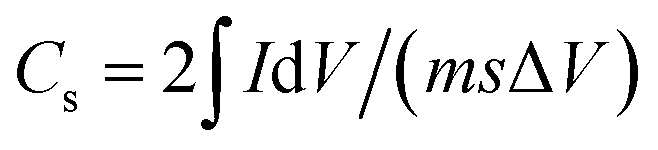 | (2) |
| Cs = 2IΔt/(mΔV) | (3) |
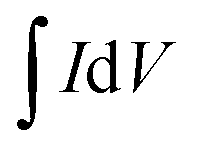 , and s is the specific capacitance (F g−1), response or applied current (A), discharge time (s), average mass of the electrodes (g), potential window (V, excluding IR drop during discharge process in the GCD), integrated area under the CV loop (F), and the scan rate (mV s−1), respectively. An electrochemical impedance spectroscopic (EIS) investigation was carried out in the frequency range of 1 MHz to 10 mHz at open circuit potential with an alternating voltage of 5 mV amplitude.
, and s is the specific capacitance (F g−1), response or applied current (A), discharge time (s), average mass of the electrodes (g), potential window (V, excluding IR drop during discharge process in the GCD), integrated area under the CV loop (F), and the scan rate (mV s−1), respectively. An electrochemical impedance spectroscopic (EIS) investigation was carried out in the frequency range of 1 MHz to 10 mHz at open circuit potential with an alternating voltage of 5 mV amplitude.
3. Results and discussion
3.1 Synthesis of SiOC–BNNT/rGO composite
Modification of SiOC ceramic with BNNT was achieved via vigorous physical mixing of TTCS with BNNTs. The polymer-to-ceramic conversion of BNNTs-containing TTCS was achieved via pyrolysis in an Ar environment. The ceramic powder was crushed into fine powders and then made into a freestanding electrode using vacuum filtration of the fine powders in a dispersion of GO. Heat treatment of the mix resulted in the conversion of GO to rGO. In this context, rGO provides the structural support and pathway for electrons to the SiOC–BNNT particles.3.2 Morphological and structural characterization
SEM images of the synthesized SiOC–BNNT-2 wt% paper are presented in Fig. 1(a)–(c). The resulting composite contains chunks of SiOC with uneven size distribution. Insets b and c in Fig. 1 shows an enlarged image of the selected section in Fig. 1(a). The insets also show that the SiOC chunks were dispersed on rGO sheets and that the fibrous structure of the BNNTs was retained after pyrolysis. Inset c shows that BNNTs were embedded in the SiOC–rGO, potentially reducing the agglomeration of SiOC and, enhancing the porosity of the paper composite. TEM images of the SiOC–BNNT-2 wt% paper are shown in Fig. 1(e)–(g). These images confirm the BNNTs retained their pristine tubular morphology and became embedded in the SiOC–rGO composite (inset f and g). Inset f shows that the average internal diameter of BNNT dispersed in the ceramic matrix is approx. 4 nm – which is same as the internal diameter of the as received BNNTs. The selected area electron diffraction (SAED) pattern obtained from the red dotted spots in the image is shown in the inset of Fig. 1(e). The SAED shows diffused concentric rings typical of an amorphous material, specifically SiOC in this case.20 | ||
| Fig. 1 (a–c) SEM images of SiOC–BNNT-2 wt% paper composite, insets (b and c) shows zoomed image of the regions shown in (a), (d–f) TEM images of SiOC–BNNT-2 wt% paper composite, inset (e) shows a section of the tubular BNNT, inset (f) shows the internal diameter of BNNT shown in inset (e). | ||
The surface elemental composition of the SiOC–BNNT-2 wt% composite was characterized by XPS as shown in Fig. 2(a)–(f). The XPS survey scan (Fig. 2(a)) confirmed the presence of silicon, nitrogen, carbon, boron, and oxygen in the composite. As shown in Fig. 2(b), a lower energy peak at 103.21 eV was fitted to Si 2p, suggesting the presence of Si–O bonds that are detectable at the surface of the composite.21 In Fig. 2(c), a single peak at 398.64 eV is assigned to B–N for the N 1s band.21 Similarly, a single low energy peak at 191.19 eV (Fig. 2(d)) indicating the presence of B–N bonds further confirms that the BNNTs in the composite maintained its chemical state after interfacing with SiOC. A high-resolution XPS of C 1s is displayed in Fig. 2(e). Peaks at 284.70 eV, 284.93 eV, 286.58 eV and 288.86 eV could be assigned to C–C (free carbon), C–Si, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CO(O) bonds, respectively.20 Also, peaks at 532.52 eV and 533.33 eV corresponding to Si–O and SiO2, respectively were observed in the O 1s band in Fig. 2(f).5
O and CO(O) bonds, respectively.20 Also, peaks at 532.52 eV and 533.33 eV corresponding to Si–O and SiO2, respectively were observed in the O 1s band in Fig. 2(f).5
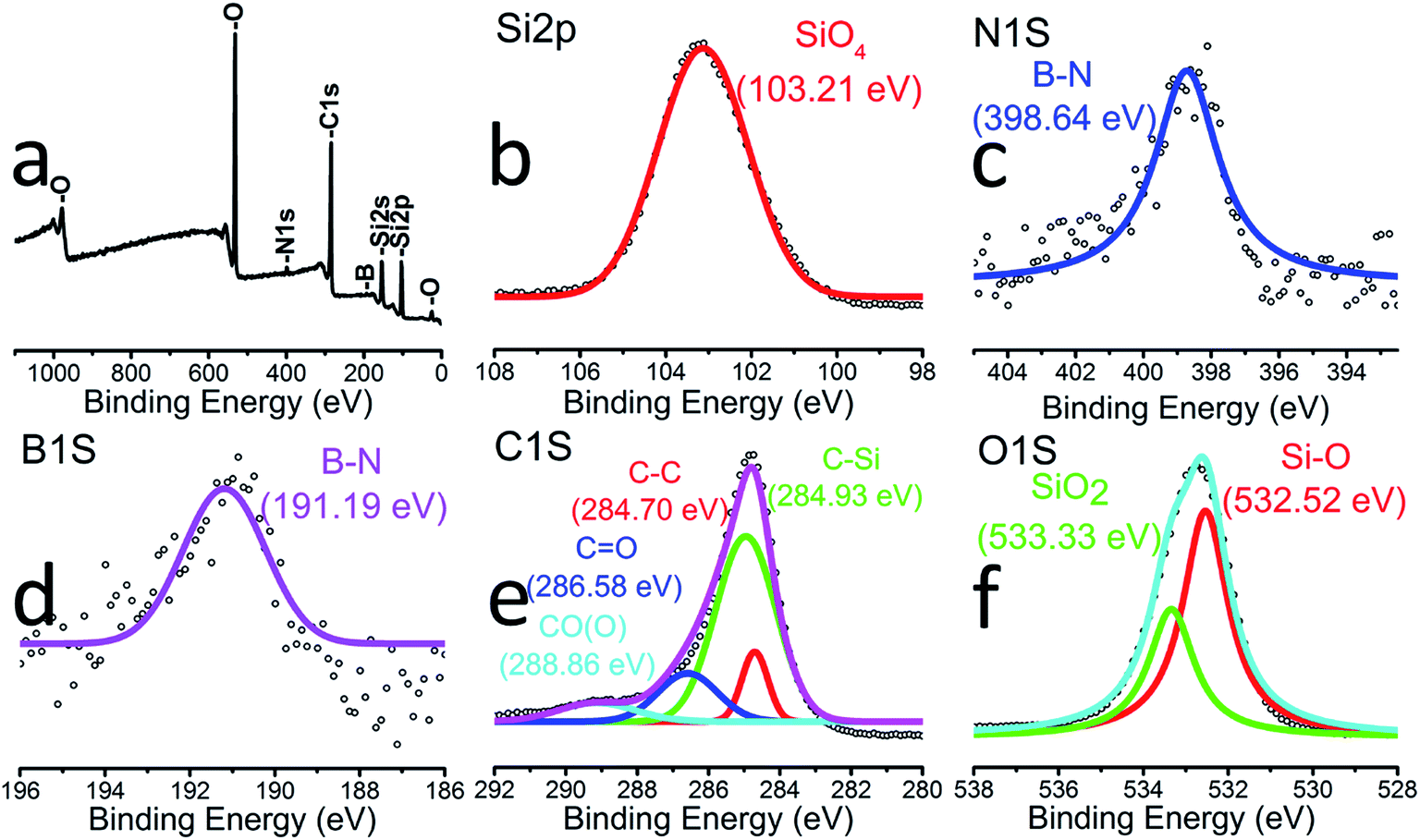 | ||
| Fig. 2 XPS showing (a) survey spectrum SiOC–BNNT-2 wt% (b) Si 2p (c) N 1s (d) B 1s (e) C 1s (f) O 1s. | ||
The type of boron sites in the synthesized ceramic powder containing 2 wt% BNNT was investigated using solid-state 11B MAS NMR (Fig. 3(a)). The spectrum is characteristic of B–N bonds in trigonal BN3 environments (δiso = 28 ppm, CQ = 3.0 MHz, ηQ = 0.2) present in the BNNTs.5,22 This observation affirms the XPS spectrum of B 1s (Fig. 2(d)), thereby confirming the presence of BN and absence of tetragonal BO4 groups in the BNNTs after polymer-to-ceramic conversion of the composite.
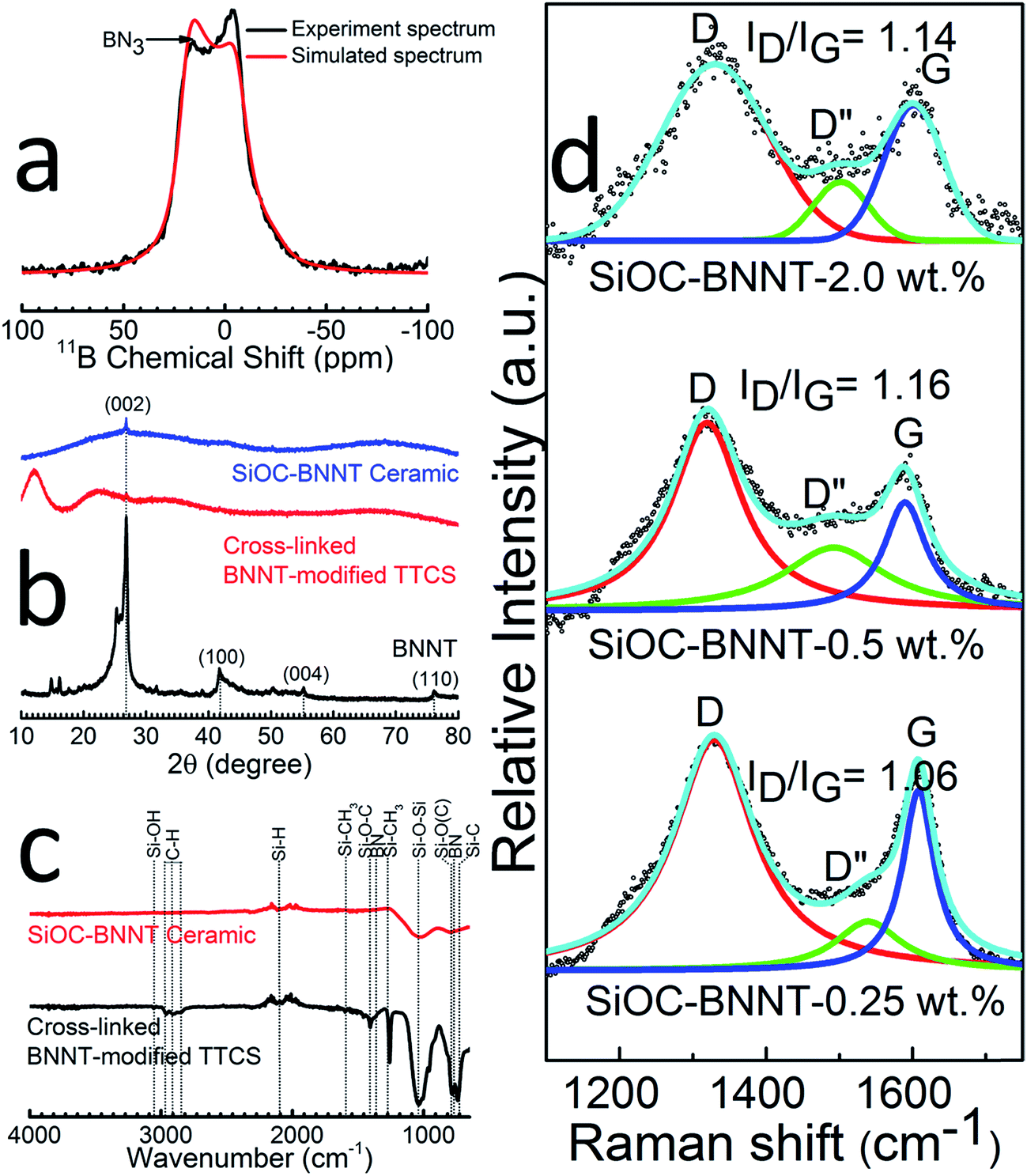 | ||
| Fig. 3 (a) 11B MAS NMR experimental and simulated spectrum of SiOC–BNNT-2 wt% ceramic, (b) XRD peaks of cross-linked TTCS polymer and SiOC–BNNT-2 wt% ceramic in comparison with ‘as-obtained’ BNNTs, (c) FT-IR spectra of BNNT-modified (cross-linked) polymer and ceramic containing 2 wt% BNNT, (d) Raman spectra of SiOC–BNNT ceramic paper with varying wt% of BNNTs. | ||
The comparative crystallinity of the cross-linked 2 wt% BNNT-modified TTCS polymer, SiOC–BNNT-2 wt% ceramic to the as-obtained BNNTs is presented in Fig. 3(b). Both cross-linked and pyrolyzed specimen displayed characteristic 002 peaks of h-BN indicating retention of BNNTs within the ceramic's structure after successive heat treatment. In addition, the ceramic sample displayed no visible peaks except the BN 002 peak, confirming that the SiOC phase in the synthesized composite is amorphous.5 This observation is in agreement with the SAED diffused concentric ring pattern in the inset of Fig. 1(e).
Structural transformation of the cross-linked polymer (white) to the ceramic (black) was also studied using FT-IR spectroscopy (Fig. 3(c)). FT-IR spectra of the cross-linked polymer indicated absorption peaks corresponding to Si–OH (3056 cm−1), C–H (2852–2978 cm−1), Si–CH3 (1276 and 1594 cm−1), Si–O–C (1415 cm−1), Si–O–Si (1037 cm−1), Si–O(C) (791 cm−1) and Si–C (725 cm−1).3,20,23 Characteristic peaks (774 cm−1 and 1367 cm−1) due to BN were also observed in the cross-linked polymer.23,24 Polymer-to-ceramic transformation was confirmed by FT-IR spectra of the ceramic which indicate breaking of bonds corresponding to Si–CH3 and C–H and dampening of prominent Si–O–Si and BN peaks.10 The only visible bands in the SiOC–BNNT ceramic were those of Si–H, Si–O–Si, BN and SiC. This observation corroborates the decrease in the BN 002 peak intensity shown in the XRD spectra (Fig. 3(b)). In addition, the absence of Si–O–B peaks in FT-IR spectra of the ceramic suggests no appreciable chemical reaction had occurred between the TTCS polymer and BNNT during pyrolysis.20
Raman spectroscopic analysis was carried out in order to study the free carbon phase in the composite. This data is presented in Fig. 3(d) which shows two distinct peaks corresponding to the D and G bands typical of incompletely graphitized carbon atoms in SiOC (disordered carbons).11 Emergence of D and G bands is attributed to disorder-induced vibrations of 6-membered aromatic rings and plane bond stretching of a sp2 pair of hybridized carbon atoms, respectively.25 In addition, the Raman spectra revealed a broad D′′ band with low intensity, suggesting the presence of amorphous carbon in the SiOC–BNNT samples. Also, the position of the D and G bands shifted towards lower wavenumbers with an increasing concentration of BNNT in SiOC. More so, the D to G band intensity ratio (ID/IG) was the lowest for SiOC–BNNT–rGO-0.25 wt%, indicating a lower ordering and smaller size growth of graphite crystals.26
3.3 Electrochemical characterization of SiOC–BNNT
The electrochemical performance of the binder-free SiOC–BNNT paper composites (containing 40 wt% of rGO) was tested as electrode material for LIBs using a Li half-cell configuration containing a pure Li metal counter electrode. Specific capacity values of the tested materials were calculated with respect to the total weight of the electrode material including the active material. The 1st and 2nd cycle charge–discharge profiles of the tested ceramic composite at a current density of 100 mA g−1 are presented in Fig. 4(a)–(c). The shape of charge–discharge profile of the tested samples are similar. When comparing the shape of the 1st cycle discharge profiles, an irreversible capacity near 0.11 V, 0.10 V and 0.12 V corresponding to the 2.0 wt%, 0.5 wt% and 0.25 wt% BNNT-modified ceramic paper composite, respectively was observed. This irreversible capacity is attributed to be due to the formation of a solid-electrolyte interphase (SEI) layer.3 The 1st cycle discharge profile of the tested samples also exhibited a long plateau below 0.3 V, indicating an irreversible lithium insertion. Wilamowska et al. suggested that the observed irreversible voltage plateau is due to the presence of oxygen-rich SiO sites that irreversibly react with Li during insertion.27 The modeling and ab initio simulation of Li insertion in SiOC ceramics by Kroll et al. have also shown that the enthalpy of Li insertion into oxygen-rich sites in SiOC is low and promoted by free carbons, thus resulting in the ease of binding Li with these sites.28 However, in same study, Peter et al. reported that free carbons are also capable of enhancing the reversible storage of Li.28 In the 2nd cycle, Li insertion capacity of the 2.0 wt%, 0.5 wt% and 0.25 wt% BNNT-modified ceramic composite dropped to ca. 369 mA h g−1, 301 mA h g−1 and 440 mA h g−1, respectively.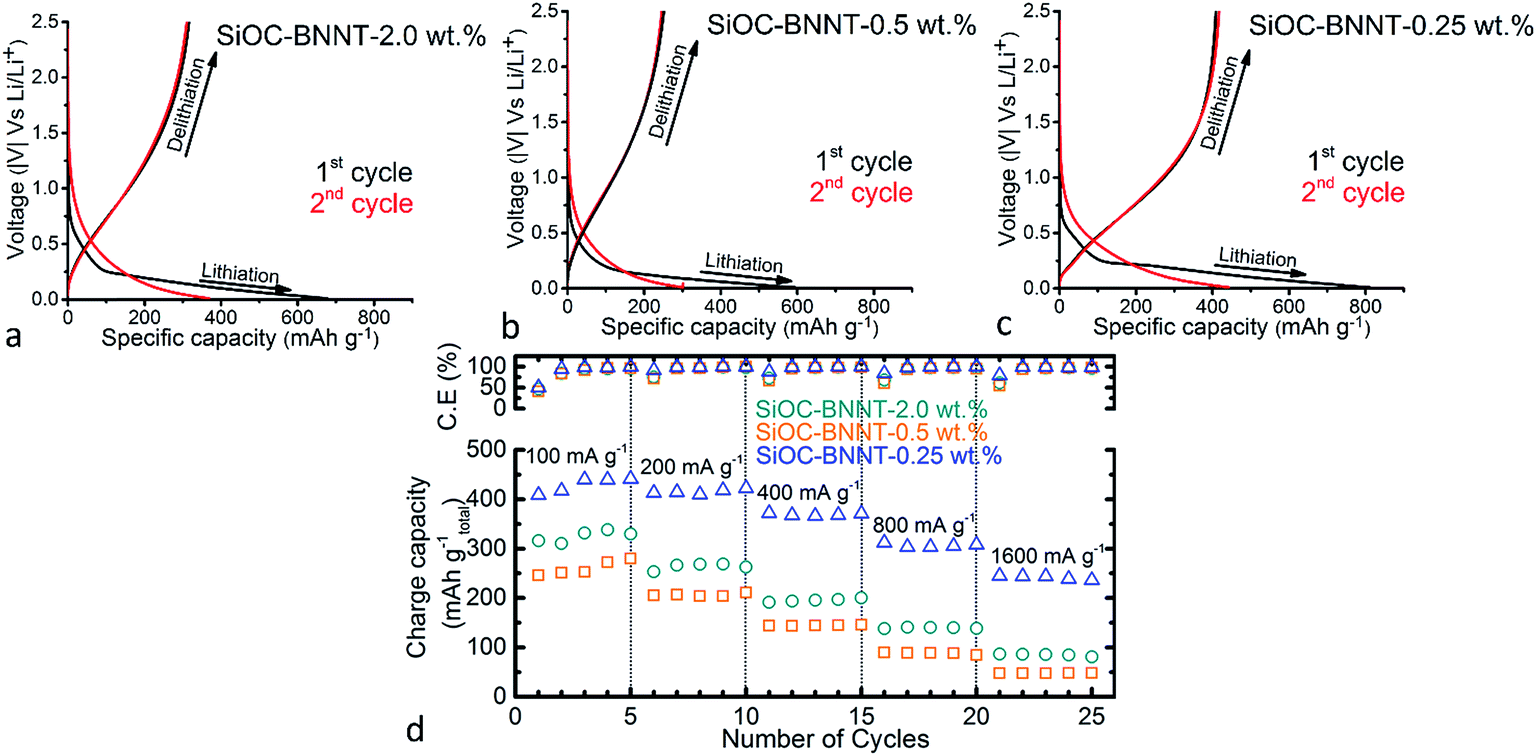 | ||
| Fig. 4 (a–c) 1st and 2nd cycle charge–discharge profile of SiOC–BNNT paper composite, (d) charge capacity of all electrodes asymmetrically cycled at different current densities and corresponding cyclic efficiencies. | ||
Among the tested ceramic paper composites, SiOC–BNNT-0.25 wt% demonstrated the highest 1st cycle Li insertion and extraction capacity of ca. 812 mA h g−1 and 410 mA h g−1, respectively with a 1st cycle loss of 51%. Table 1 summarizes the 1st cycle lithiation capacity (Cch), reversible delithiation capacity (Crev), irreversible capacity (Cirr) and corresponding efficiency (η) of the tested paper composites.
| Material | Cch (mA h g−1) | Crev (mA h g−1) | Cirr (mA h g−1) | η (%) |
|---|---|---|---|---|
| SiOC–BNNT-2.0 wt% | 675 | 317 | 358 | 46.9 |
| SiOC–BNNT-0.5 wt% | 596 | 250 | 346 | 41.9 |
| SiOC–BNNT-0.25 wt% | 812 | 410 | 402 | 50.4 |
The composite ceramics were further cycled asymmetrically by doubling the current density (starting with 100 mA g−1) every five cycles. As shown in Fig. 4(d), the 0.25 wt% demonstrated a high rate performance, which remains stable at 238 mA h g−1 even at 1600 mA g−1 after 25 cycles. On the other hand, the 0.5 wt% and 2.0 wt% BNNT-modified ceramic paper composites demonstrated a Li extraction capacity of 47 mA h g−1, and 82 mA h g−1 (at a current density of 1600 mA g−1), respectively after 25 cycles. More so, the cyclic efficiency plot shown in Fig. 4(d) indicates all tested electrodes demonstrated a stable cyclic efficiency when cycled with increasing current density.
So far, SiOC–BNNT-0.25 wt% remains the best performing electrode material among the BNNT-modified ceramic paper composites. The observed trend suggests a reduction in charge capacity of the BNNT-modified composites due to the presence of BNNTs, a known insulator.29 It is inferred that modification of SiOC ceramic with BNNT at higher wt% could degrade its property due to the insulating property of BN, resulting in lower electrochemical performance as LIB anode29 – as previous studies has shown that neat SiOC supported on rGO demonstrated higher charge capacity when tested as LIB anodes.3 Surprisingly, the SiOC–BNNT-2.0 wt% ceramic paper composite displays a higher charge capacity than the SiOC–BNNT-0.5 wt% sample. This disparity is attributed to the higher ordering of graphitic planes in SiOC–BNNT-0.5 wt%, (as evidenced by the Raman spectra shown in Fig. 3(d)), thus offering a lesser disordered carbon phase for Li insertion.26
The electrochemical performance of the SiOC–BNNT ceramic paper composites at varying wt% of BNNTs was analyzed in a two electrode setup. The testing was carried out in 6 M KOH(aq) at room temperature. The CV, GCD and Nyquist plots of the electrode materials are presented in Fig. 5(a)–(c). Previous research has shown that SiOC supported on graphene sheets is capable of storing charges via faradaic processes due to the presence of oxygen in its structure.7 The CV curve (at 20 mV s−1) of the SiOC–BNNT paper composite at various concentration of BNNT is presented in Fig. 5(a). The electrode materials displayed a noticeable oxidation peak at ca. 0.5 V – with the SiOC–BNNT-0.5 wt% paper composite showing the largest oxidation peak. More so, the SiOC–BNNT-0.5 wt% displayed the largest CV area among the tested electrode materials with a specific capacitance of 66.88 F g−1 (according to (2)). In this configuration, the SiOC–BNNT-0.5 wt% paper composite demonstrated the optimum concentration of BNNT for electrochemical energy storage as a supercapacitor electrode.
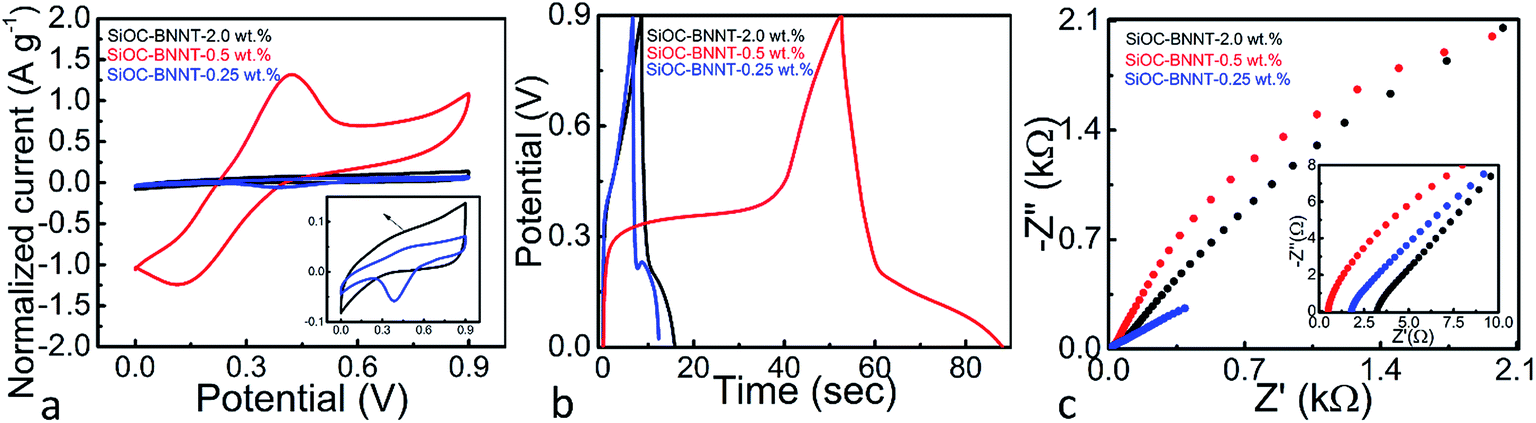 | ||
| Fig. 5 (a) CVs of SiOC–BNNT paper composite at 20 mV s−1 (inset shows expanded CV of the 2.0 wt% and 0.25 wt% paper composite electrode), (b) GCD of SiOC–BNNT paper composite at a current density of 1.0 A g−1, (c) Nyquist plots for SiOC–BNNT paper composite (inset shows expanded plot in the high frequency region). | ||
Further, the GCD analysis of SiOC–BNNT paper composite were carried out at a current density of 1 A g−1 (Fig. 5(b)). The GCD profiles of SiOC–BNNT paper composites were not uniform, suggesting the presence of faradaic charge transfer process in the material.7 In addition, the SiOC–BNNT-0.5 wt% electrode material demonstrated the least steepest discharge slope with a capacitance of 52.96 F g−1 (according to (3)). The superior charge–discharge properties of SiOC–BNNT-0.5 wt% paper composite might be due to its higher surface area in contact with the electrolyte as suggested by the Raman spectra shown in Fig. 3(d) which indicates higher ordering and larger size of its graphitic crystals.26,30 Moreover, the decrease in electrochemical charge storage performance in SiOC–BNNT-2.0 wt% paper composite is attributed to the difficulty in dispersing BNNTs in SiOC at higher concentration.20
Electrochemical resistance of the SiOC–BNNT paper composite was characterized using Nyquist plots obtained from electrochemical impedance spectroscopy. Fig. 5(c) shows the Nyquist plots and magnified high-frequency region in the inset. The intercept of the curve with the X-axis in the high-frequency region indicates the interfacial resistance of the cell setup which is believed to be due to the contributing resistance of the electrode material, electrolyte, and current collector.4 As observed in the magnified high-frequency region, the SiOC–BNNT-0.5 wt% paper composite displays the lowest interfacial resistance with a value corresponding to 0.52 Ω – which is comparable to that of SiOC previously reported in the literature.7 In addition, none of the tested electrode materials displayed a semicircle in the high-frequency region, suggesting a very low interfacial resistance due to the conductive nature of the electrode materials.31 Also, SiOC–BNNT-0.5 wt% paper composite displayed a steeper frequency tail in the low-frequency region which indicates a higher mass transfer of ions between the electrolyte and its pores.32
The SiOC–BNNT-0.5 wt% paper composite was tested using the same setup described earlier – wherein the electrode material was made into a symmetric supercapacitor and tested using a 6 M KOH(aq) electrolyte at room temperature. The CV and GCD plots of SiOC–BNNT-0.5 wt% paper composite are presented in Fig. 6(a) and (b). The CVs of SiOC–BNNT-0.5 wt% paper composite showed quasi-rectangular plots with broad reduction and oxidation peaks around 0.1 V and 0.4 V, respectively. The relative position of the redox peaks was very similar to that of neat SiOC paper previously reported by our group.7 As such, the charge storage mechanism of SiOC–BNNT paper composite is also assumed to be faradaic.33 In addition, the CV profile of SiOC–BNNT-0.5 wt% paper composite remained unchanged up to lower scan rates, indicating enhanced electrode–electrolyte interaction which translates to higher charge storage performance. Corresponding GCD plots presented in Fig. 6(b) show a similar non-linear charge–discharge profile that has been observed in LBL–SiOC–rGO, which is in agreement with the CV plots shown in Fig. 5(a) – suggesting the SiOC–BNNT-0.5 wt% paper composite is pseudocapacitive.7 At a current density of 1.0 A g−1, SiOC–BNNT-0.5 wt% paper composite displays a capacitance of 78.93 F g−1.
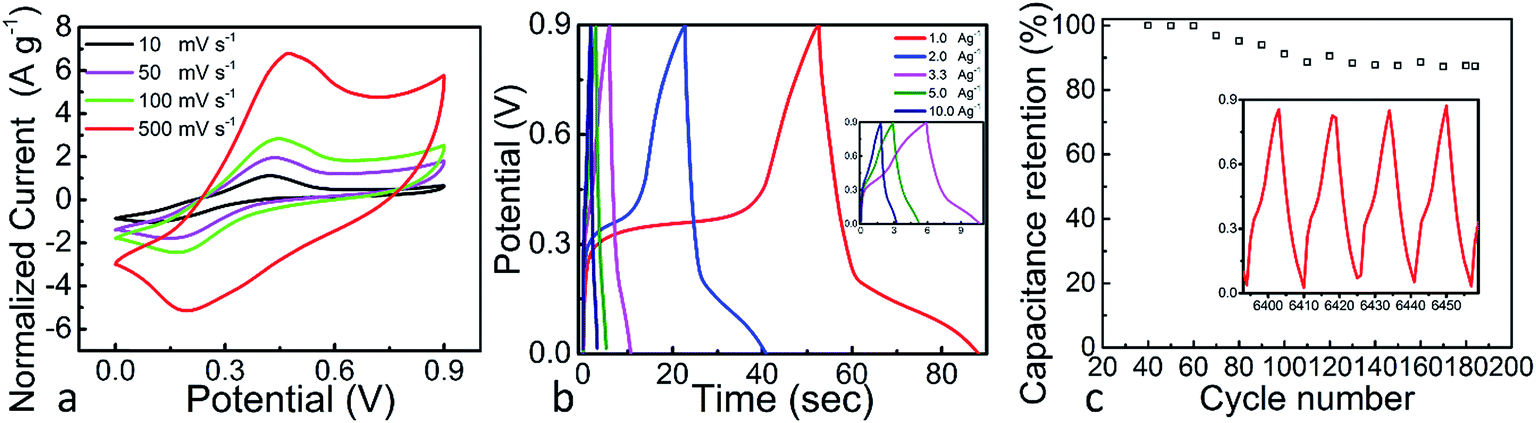 | ||
| Fig. 6 (a) CVs of SiOC–BNNT-0.5 wt% paper composite at different scan rates, (b) GCD curves of SiOC–BNNT-0.5 wt% paper composite at various current densities, (c) capacitance retention plot of SiOC–BNNT-0.5 wt% paper composite as a function of cycle number at a current density of 1 A g−1. | ||
The cyclic stability of SiOC–BNNT-0.5 wt% paper composite was studied. Fig. 6(c) presents the cyclic stability of SiOC–BNNT-0.5 wt% paper composite after 185 cycles at a current density of 1.5 A g−1. The cyclic retention was 86% with a corresponding specific capacitance of 26.67 F g−1 (according to (3)) – which is comparable to that of some carbonaceous materials that have been reported in the literature.34,35 Yet, lower than neat SiOC supported on rGO that has been studied in our group.7
Overall, application of SiOC–BNNT paper composite as LIB anode is more promising than supercapacitor electrode. The presence of BNNT in the SiOC microstructure may also confer additional functional properties that could enable such electrode material to be coupled with devices that can only be powered at high temperature. PDCs are also well known for their chemical stability. Therefore, the synthesized SiOC–BNNT composite may be used as an electrode material for devices that are routinely used in harsh conditions or corrosive environments.
4. Conclusion
This study explored the feasibility of modifying TTCS preceramic polymer with BNNTs, its structural evolution to SiOC–BNNT ceramic upon pyrolysis at high temperature, and potential application as an electrode material for LIB and a symmetric supercapacitor. Structural characterization reveals the absence of significant reaction between the BNNTs and SiOC. As such, the morphology of the BNNTs was preserved after polymer-to-ceramic transformation of the composites. In addition, the presence of disordered carbon known for their electrochemical activity was confirmed in the SiOC–BNNT paper composites. Thus, the synthesized composite is electrochemically active.As an electrode material for LIB, the SiOC–BNNT paper composite containing 0.25 wt% BNNT demonstrated the highest 1st cycle lithiation capacity corresponding to 812 mA h g−1. The charge capacity of the composite decreased with an increase in the BNNT content of the ceramic paper composites. The observed negative trend was attributed to an increasing content of BNNTs in the composites, consequently dampening the electroactive property of the resulting electrode. On the other hand, SiOC interfaced with 0.5 wt% BNNT demonstrated the highest capacitance when tested as an electrode material for symmetric supercapacitor. The superior capacitance of the 0.5 wt% electrode material could possibly be due to the higher ordering of its graphitic planes which result in increased surface area of the paper composite.
Acknowledgements
Financial support from United States National Science Foundation grant number NSF CBET-1335862 and NSF CAREER CMMI-1454151 is gratefully acknowledged. Thanks are due to Mr Gary Zito for access to the transmission electron microscope facilitiy at Colorado School of Mines.References
- P. Colombo, Polymer Derived Ceramics: From Nano-Structure to Applications, DEStech Publications, Lancaster, Pennsylvania, 10th edn, 2010 Search PubMed.
- P. Colombo, G. Mera, R. Riedel and G. D. Soraru, J. Am. Ceram. Soc., 2010, 93, 1805–1837 CAS.
- L. David, R. Bhandavat, U. Barrera and G. Singh, Nat. Commun., 2016, 7, 10998 CrossRef CAS PubMed.
- L. David, K. M. Shareef, M. A. Abass and G. Singh, RSC Adv., 2016, 6, 53894–53902 RSC.
- L. David, S. Bernard, C. Gervais, P. Miele and G. Singh, J. Phys. Chem. C, 2015, 119, 2783–2791 CAS.
- V. S. Pradeep, D. G. Ayana, M. Graczyk-Zajac, G. D. Soraru and R. Riedel, Electrochim. Acta, 2015, 157, 41–45 CrossRef CAS.
- M. S. Kolathodi, L. David, M. A. Abass and G. Singh, RSC Adv., 2016, 6, 74323–74331 RSC.
- C. G. Pantano, A. K. Singh and H. Zhang, J. Sol-Gel Sci. Technol., 1999, 14, 7–25 CrossRef CAS.
- A. Saha, R. Raj and D. L. Williamson, J. Am. Ceram. Soc., 2006, 89, 2188–2195 CAS.
- R. Bhandavat and G. Singh, J. Phys. Chem. C, 2013, 117, 11899–11905 CAS.
- A. Tolosa, B. Krüner, N. Jäckel, M. Aslan, C. Vakifahmetoglu and V. Presser, J. Power Sources, 2016, 313, 178–188 CrossRef CAS.
- J. Kaspar, M. Graczyk-Zajac and R. Riedel, J. Power Sources, 2013, 244, 450–455 CrossRef CAS.
- Z. Fan, J. Yan, L. Zhi, Q. Zhang, T. Wei, J. Feng, M. Zhang, W. Qian and F. Wei, Adv. Mater., 2010, 22, 3723–3728 CrossRef CAS PubMed.
- C. H. Kim and B. H. Kim, Electrochim. Acta, 2014, 117, 26–33 CrossRef CAS.
- A. Tolosa, B. Krüner, N. Jäckel, M. Aslan, C. Vakifahmetoglu and V. Presser, J. Power Sources, 2016, 313, 178–188 CrossRef CAS.
- S. Sarkar, J. Zou, J. Liu, C. Xu, L. An and L. Zhai, ACS Appl. Mater. Interfaces, 2010, 2, 1150–1156 CAS.
- M. Graczyk-Zajac, C. Fasel and R. Riedel, J. Power Sources, 2011, 196, 6412–6418 CrossRef CAS.
- C. Zhi, Y. Bando, C. Tang and D. Golberg, J. Phys. Chem. B, 2006, 110, 8548–8550 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- S. Xie, Y. Wang, Y. Lei, B. Wang, N. Wu, Y. Goua and D. Fang, RSC Adv., 2015, 5, 64911–64917 RSC.
- J. G. Alauzun, S. Ungureanu, N. Brun, S. Bernard, P. Miele, R. Backov and C. Sanchez, J. Mater. Chem., 2011, 21, 14025 RSC.
- J. Li, S. Bernard, V. Salles, C. Gervais and P. Miele, Chem. Mater., 2010, 22, 2010–2019 CrossRef CAS.
- J. Hong, K. Y. Cho, D.-G. Shin, J. I. Kim and D. H. Riu, RSC Adv., 2015, 5, 83847–83856 RSC.
- P. M. Sudeep, S. Vinod, S. Ozden, R. Sruthi, A. Kukovecz, Z. Konya, R. Vajtai, M. R. Anantharaman, P. M. Ajayan and T. N. Narayanan, RSC Adv., 2015, 5, 93964–93968 RSC.
- F. Tuinstra and L. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- V. S. Pradeep, M. Graczyk-Zajac, R. Riedel and G. D. Soraru, Electrochim. Acta, 2014, 119, 78–85 CrossRef CAS.
- M. Wilamowska, V. S. Pradeep, M. Graczyk-Zajac, R. Riedel and G. D. Sorarù, Solid State Ionics, 2014, 260, 94–100 CrossRef CAS.
- P. Kroll, MRS Proc., 2011, 1313, 1–6 Search PubMed.
- Q. Weng, X. Wang, X. Wang, Y. Bando and D. Golberg, Chem. Soc. Rev., 2016, 45, 3989–4012 RSC.
- Q. Ke and J. Wang, J. Mater., 2016, 2, 37–54 Search PubMed.
- Z. Wu, L. Li, Z. Lin, B. Song, Z. Li, K.-S. Moon, C.-P. Wong and S.-L. Bai, Sci. Rep., 2015, 5, 10983 CrossRef PubMed.
- H. Hu, Z. Hu, X. Ren, Y. Yang, R. Qiang, N. An and H. Wu, Chin. J. Chem., 2015, 33, 199–206 CrossRef CAS.
- M. S. Kolathodi, M. Palei and T. S. Natarajan, J. Mater. Chem. A, 2015, 3, 7513–7522 CAS.
- P. Bondavalli, C. Delfaure, P. Legagneux and D. Pribat, J. Electrochem. Soc., 2013, 160, A601–A606 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, Carbon, 2013, 56, 155–166 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |