DOI:
10.1039/C7RA02126G
(Paper)
RSC Adv., 2017,
7, 23293-23300
Preparation of ionic liquid-modified SiO2@Fe3O4 nanocomposite as a magnetic sorbent for use in solid-phase extraction of zinc(II) ions from milk and water samples†
Received
20th February 2017
, Accepted 6th April 2017
First published on 27th April 2017
Abstract
In this study, silica-coated magnetite nanoparticles modified with triethoxysilylpropylpyridinium hexafluorophosphate ionic liquid were synthesized and used as a sorbent for the solid-phase extraction and preconcentration of zinc ions from aqueous solutions. The structure and morphology of the synthesized nanosorbent were characterized by X-ray diffraction, scanning electron microscopy, and Fourier transform infrared spectroscopy. After desorption with acidic ethanol, zinc was quantified by flame atomic absorption spectrometry. The experimental factors affecting the extraction/preconcentration of the analyte were investigated and optimized. Under the optimized experimental conditions, the calibration graph was linear in the concentration range of 0.5–15 ng mL−1 (with an R2 value of 0.998). The limit of detection and the preconcentration factor were 0.17 ng mL−1 and 100, respectively. The relative standard deviation for six replicate determinations of 10 ng mL−1 Zn was 2.3%. The method was validated by the successful analysis of a standard reference material (NIST SRM 1549; non-fat milk powder) and employed for the determination of zinc in several samples of water and milk, with recoveries in the range of 97.0–102.5% for the spiked samples.
Introduction
Zinc (Zn) is an essential trace element in the nutrition of all animals and humans and plays an important role in many bodily functions. It acts as a cofactor in numerous enzymes and performs many biochemical processes.1 A deficiency of zinc is a worldwide nutritional problem that leads to several disorders such as growth retardation, diarrhea, weakening of immunological defenses, eye and skin lesions, impairment of wound healing and other skin diseases.2 However, an excess amount of zinc is harmful and can be toxic when exposure exceeds physiological needs, which can cause several damaging effects in the human body, including alterations in immune response, miscarriage in pregnancy, weight loss, epigastric pain and retardation of growth.3 In uncontaminated waters, the zinc concentration is usually very low and can span a wide range from 10−10 to 10−6 mol L−1.4 Concentrations of zinc of greater than 5.0 mg L−1 affect the potable status of water in alkaline conditions.5 Therefore, it is essential to establish simple, rapid, sensitive and environment-friendly methods for the monitoring of zinc at trace levels in environmental and food samples.
Different analytical techniques have been employed for the determination of trace levels of zinc in various matrices. Commonly used techniques include flame atomic absorption spectrometry (FAAS), electrothermal atomic absorption spectrometry, inductively coupled plasma optical emission spectrometry (ICP-OES), inductively coupled plasma mass spectrometry, X-ray spectrometry, spectrofluorometry, spectrophotometry and electroanalytical techniques. However, the direct determination of zinc is often difficult because of the insufficient sensitivity of these techniques at low concentrations and/or the interference of complex matrices present in real samples. For this reason, a preliminary separation and preconcentration step is often necessary.
Magnetic solid-phase extraction (MSPE) with magnetic nanoparticles (MNPs) has excited great interest in the analytical community in recent years. Magnetic materials can be used as sorbents in SPE. In this procedure, MNPs are added to the sample solution and the target analyte is adsorbed on the surface of the magnetic beads, which are separated from the aqueous solution by means of an external magnetic force. Then, the target analyte is desorbed by an eluent for further determination.6 The SPE agent is used without packing of cartridges, and phase separation can be achieved easily by applying an external magnetic field. However, pure MNPs suffer from an inherent limitation as they tend to agglomerate, which alters their magnetic properties in complex matrices.7–9 Therefore, the modification of these MNPs is essential to overcome such limitations.
Ionic liquids (ILs), as useful environment-friendly green solvents, have recently emerged as promising extraction media owing to their unique physicochemical properties, such as low volatility, high thermal stability, high conductivity and tunable miscibility.10,11 Because ILs can dissolve many kinds of organic, organometallic and inorganic compounds,12 in recent years ILs have attracted great interest in separation science and analytical chemistry.13–15 Ionic liquid-based extraction has some disadvantages, such as lower rates of mass transfer, longer equilibrium times, and difficulty in phase separation, which results in loss of the IL to the aqueous phase.16 These limitations can be overcome by immobilizing ILs on solid substrates, which generates new materials with interesting properties.
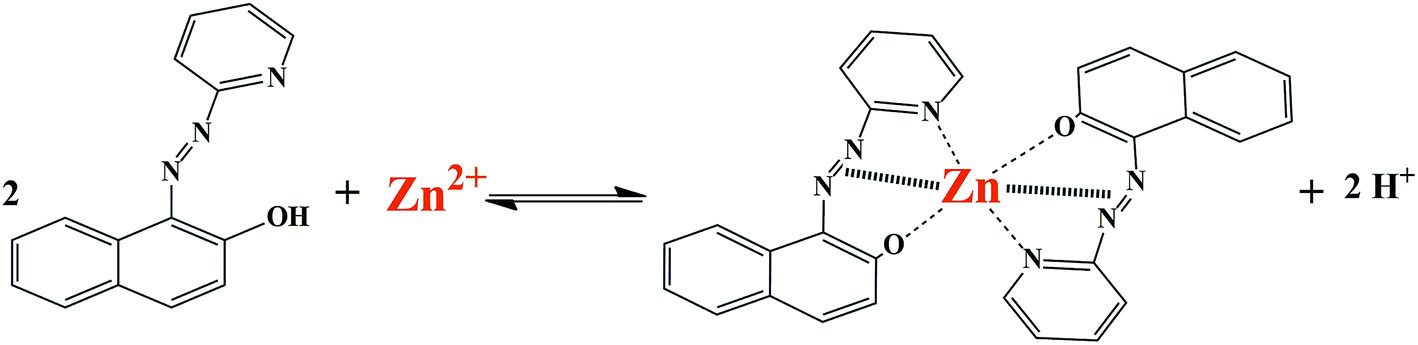
To the best of our knowledge, there has been no report on the use of silica-coated magnetite nanoparticles coated with triethoxysilylpropylpyridinium hexafluorophosphate ionic liquid for the extraction and determination of metal ions. Therefore, in this work ionic liquid-modified silica-coated magnetite NPs were employed as a novel nanosorbent for the extraction and preconcentration of zinc ions. Zn(II) ions were adsorbed by the synthesized nanosorbent in the form of a 1-(2-pyridylazo)-2-naphthol (PAN) complex (Scheme 1). After desorption with acidic ethanol, zinc was quantified by FAAS. The effect of experimental parameters that affected the extraction efficiency was studied and optimized. To evaluate the applicability of the method, it was employed for the determination of Zn(II) ions in different samples of water and milk.
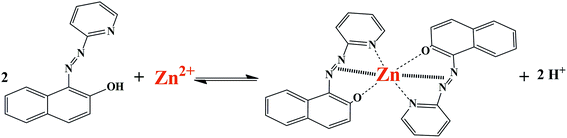 |
| | Scheme 1 Schematic illustration of formation of complex between Zn2+ ion and PAN ligand. | |
Experimental
Apparatus
A Varian model SpectrAA 220 (Mulgrave, Victoria, Australia) flame atomic absorption spectrometer was used for the determination of zinc. It was equipped with a hollow cathode lamp and an air/acetylene burner. The instrumental parameters were as follows: wavelength, 213.9 nm; lamp current, 5 mA; and band pass, 1 nm. The flame composition was controlled at an acetylene flow rate of 1.5 L min−1 and an air flow rate of 3.5 L min−1. To identify the crystal structure of the nanosorbent, powder X-ray diffraction (XRD) measurements were performed at room temperature employing a D8 Advance (Bruker, Germany) instrument with Cu Kα radiation (1.54 Å), an accelerating voltage of 40 kV, and a current of 35 mA. In addition, a Vector 22 (Bruker, Germany) Fourier transform infrared (FT-IR) spectrometer was employed to determine the functional groups present in the sorbent. FT-IR spectra were recorded in the range of 4000–400 cm−1 at a resolution of 4 cm−1 using the conventional KBr pellet technique with a ratio of sample/KBr of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 by mass. An LEO1430vp model scanning electron microscope (Carl Zeiss, Germany) was used to examine the morphological characteristics of the sorbent.
:100 by mass. An LEO1430vp model scanning electron microscope (Carl Zeiss, Germany) was used to examine the morphological characteristics of the sorbent.
Values of pH were measured with an 827 model pH meter (Metrohm Ltd., Switzerland) equipped with a glass combined electrode. A GS-6 model centrifuge (Beckman, USA) and pure N2 gas (99.9995%, Azaroxide, Iran) were used in the preparation process of the ionic liquid. An AEX 1200-4L model electrical furnace (Exciton, Iran) was employed to control the temperature in the synthesis process of Fe3O4 nanoparticles. An ultrasonic bath (SONICA, Italy) was used to disperse the nanoparticles in the solution. An Nd–Fe–B (10000 G) magnet (6.0 mm × 6.0 mm × 3.0 mm) was used for magnetic separation. A PB303 model electronic analytical balance (Mettler Toledo, Switzerland) was used for weighing the solid materials.
Standard solutions and chemicals
All chemicals used were of analytical-reagent grade, and all solutions were prepared with high-purity deionized water (Ghazi Serum Co., Tabriz, Iran). All the plastic and glassware used for the trace analysis were kept in 15% (v/v) nitric acid at least overnight and were rinsed three times with deionized water prior to use. Stock solutions of Zn(II) ions and those used for the interference study (1000 mg L−1) were prepared by dissolving the appropriate amounts of the respective pure nitrate salts (Merck, Darmstadt, Germany) in deionized water. Working standard solutions were obtained daily by suitable stepwise dilution of the stock solutions with deionized water and were shaken just before use. The chelating agent 1-(2-pyridylazo)-2-naphthol (PAN), ethanol, n-butanol, methanol, dichloromethane, tetraethyl orthosilicate (TEOS, 98%), triethoxychloropropylsilane, ammonia solution (25%), NaOH solution (32%) and all salts used were purchased from Merck (Darmstadt, Germany). Toluene, acetonitrile and H2O2 (35%) were purchased from Acros Organics (New Jersey, USA).
Preparation of the nanosorbent
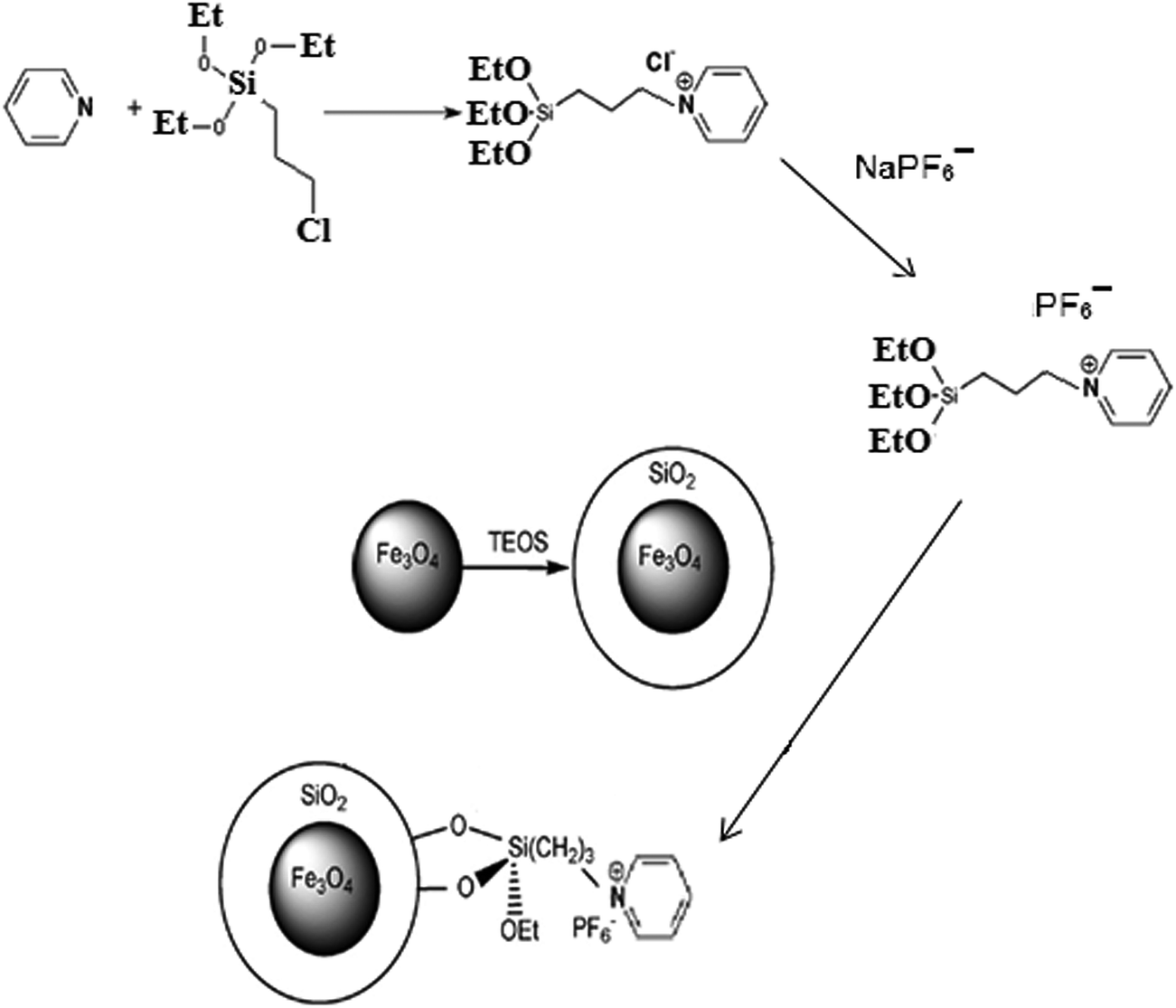
The synthesis of the nanosorbent was carried out via four defined steps. An illustration of the procedure is shown in Fig. 1.
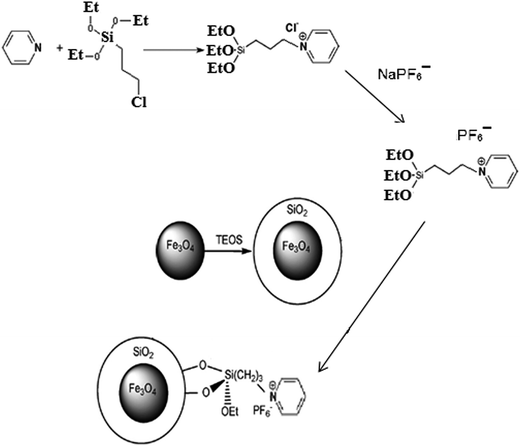 |
| | Fig. 1 Schematic diagram of synthesis of ionic liquid-functionalized silica-coated magnetite nanoparticles. | |
Synthesis of Fe3O4 nanoparticles
A chemical co-precipitation method was used for the preparation of Fe3O4 NPs, as described elsewhere17 with some modifications. In brief, FeCl3·6H2O (11.68 g) and FeCl2·4H2O (4.30 g) were dissolved in 200 mL deionized water under a nitrogen atmosphere with vigorous stirring at 70 °C. Then, 20 mL of 25% aqueous ammonia was injected into the mixture rapidly, and the color of the bulk solution changed from orange to black immediately. After being magnetically separated via an external magnetic field, the MNPs were washed three times with deionized water and were finally dried in an oven at 70 °C.
Synthesis of silica-coated Fe3O4 nanoparticles (Fe3O4–SiO2)
Silica-coated Fe3O4 NPs were prepared according to a procedure described in the literature.18 For this purpose, 50 mg of the synthesized Fe3O4 NPs were dispersed in a mixture of ethanol (160 mL), deionized water (40 mL) and ammonium hydroxide solution (3 mL, 25%). Then, 0.2 mL TEOS was added and the mixture was subsequently stirred at 25 °C for 12 h. Finally, the obtained magnetic nanoparticles (Fe3O4–SiO2) were separated by a magnet from the solution, washed with deionized water to eliminate excess reactants, and dried at 60 °C.
Synthesis of the ionic liquid
Triethoxysilylpropylpyridinium hexafluorophosphate ionic liquid was synthesized using a previously reported method19 with some modifications. In brief, 4 mL pyridine (50 mmol) was mixed with 12 mL 3-chloropropyltriethoxysilane (50 mmol) and then refluxed for 120 h at 90 °C. After cooling down, the resulting mixture was washed with diethyl ether and dried under vacuum at room temperature. Then, 3 mmol sodium hexafluorophosphate solution was added dropwise to 3 mmol of the obtained triethoxysilylpropylpyridinium chloride solution, and the mixture was stirred for 12 h. Finally, the resulting mixture was filtered and the obtained precipitate was washed three times with dichloromethane and dried under vacuum at room temperature.
Synthesis of ionic liquid-functionalized silica-coated Fe3O4 NPs (Fe3O4–SiO2–IL)
In order to prepare ionic liquid-functionalized silica-coated Fe3O4 nanoparticles, 1 g of the as-prepared ionic liquid was dispersed in 30 mL toluene by ultrasonication for 10 min. Afterward, 0.5 g Fe3O4–SiO2 was added and the mixture was refluxed at 80 °C for 24 h. Finally, the obtained product (Fe3O4–SiO2–IL) was separated by a magnet, washed several times with acetonitrile and ethanol, and dried at 60 °C.20
Sample preparation
Water samples. Water samples, including tap water, spring water and mineral water, were collected from local sources. After sampling, they were filtered through Rund filter paper (blue band, no. 300210) to remove suspended particles. An aliquot of 150.0 mL from each sample solution was analyzed by following the procedure described below in the General procedure section.
Milk powder samples. A sample of 50 mg powdered milk (Humana) or NIST SRM 1549 (non-fat milk powder) was heated on a hot plate at a fairly low temperature in a glass beaker containing a mixture of concentrated sulfuric acid (10 mL) and nitric acid (4 mL) to dryness. After that, the sample was cooled to room temperature, and the residue was dissolved in 1.0 mL of 0.1 mol L−1 HNO3.21 After dilution with deionized water, the pH was adjusted to nearly 7 with 0.1 mol L−1 NaOH, and the final volume of the solution was adjusted to 150.0 mL using deionized water. Finally, the concentration of zinc was determined by the procedure described as follows.
General procedure
A sample of 50 mg of the nanosorbent was placed in a 250 mL glassware beaker. Then, a 150 mL portion of a standard or sample solution containing zinc ions in the range of 0.5–15 μg L−1, 4 μmol L−1 PAN and 0.05 mol L−1 borate buffer (pH = 9) was transferred into a beaker. To disperse the nanosorbent throughout the whole solution, the beaker was placed in an ultrasonic bath for 1 min. The adsorption of Zn(II) ions on the nanosorbent was performed under continuous mechanical stirring of the mixture by a shaker for 10 min at room temperature. Finally, the sorbent was gathered to one side of the beaker under an external magnetic field, and the clear supernatant was directly decanted. The isolated sorbent was eluted with 1.5 mL of 0.5 mol L−1 HNO3 in 5% (v/v) ethanol to desorb the analyte. The elution was accelerated by stirring on a shaker for 5 min. This step was also completed with the help of a magnet. The clear solution of the eluent containing Zn(II) ions was transferred into a test tube for subsequent analysis by FAAS.
Results and discussion
Characterization of the nanosorbent
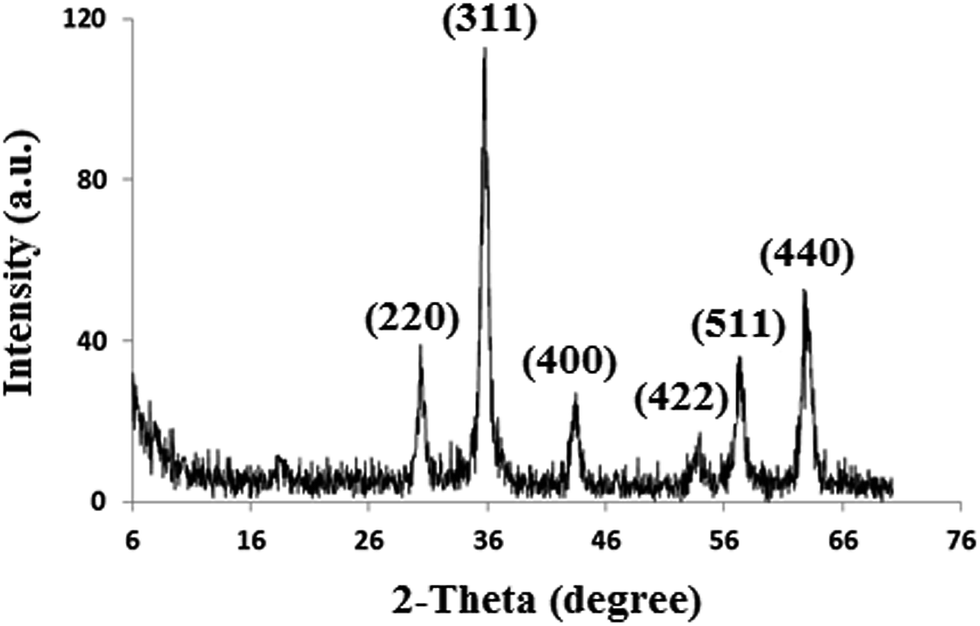
Characterization of the synthesized Fe3O4 NPs, Fe3O4@SiO2 and Fe3O4@SiO2@IL was performed using Fourier transform infrared (FT-IR) spectroscopy, powder X-ray diffraction (XRD) and scanning electron microscopy (SEM). Fig. 2 shows the XRD pattern of Fe3O4 NPs. The characteristic reflections of the (220), (311), (422), (400), (511) and (440) planes of crystalline Fe3O4 can be observed, which is in agreement with the results reported in the literature.22
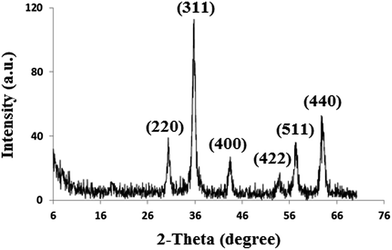 |
| | Fig. 2 XRD pattern of Fe3O4 nanoparticles. | |
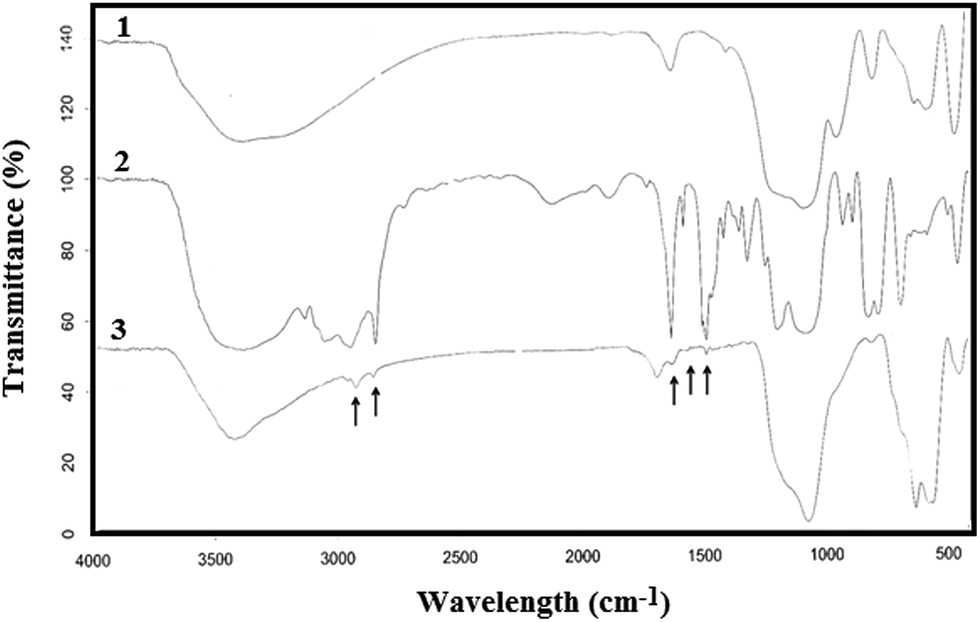
To characterize the surface nature of the nanoparticles, infrared absorption spectroscopy was used. Fig. 3 shows the FT-IR spectra of (1) silica-coated Fe3O4 NPs (Fe3O4@SiO2), (2) the ionic liquid (IL) and (3) ionic liquid-functionalized silica-coated Fe3O4 NPs (Fe3O4@SiO2@IL). For the SiO2–Fe3O4 NPs, the strong band at 565 cm−1 corresponds to Fe–O vibrations of the magnetite core, and the weak band at 800 cm−1 is characteristic of Si–O–Fe linkages, which proves that SiO2 was chemically bonded to Fe3O4. The band at 1100 cm−1 represents Si–O bonds, whereas those at 3420 cm−1 and 1640 cm−1 correspond to the stretching and bending vibrations, respectively, of Si–OH groups. In the FT-IR spectrum of ionic liquid-functionalized silica-coated Fe3O4 NPs, the relatively weak peaks at 2947 cm−1 and 2843 cm−1 represent the stretching vibrations of CH3 and CH2 groups, respectively, present in the ionic liquid, whereas those at 1463 cm−1 and 1381 cm−1 correspond to their respective bending vibrations. The FT-IR spectra prove that the silica-coated Fe3O4 NPs were bonded to the pyridinium-based ionic liquid.18
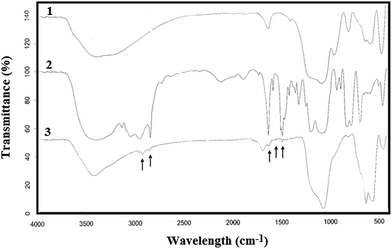 |
| | Fig. 3 FT-IR spectra of (1) silica-coated Fe3O4 nanoparticles (Fe3O4@SiO2); (2) ionic liquid (IL); and (3) ionic liquid-modified silica-coated Fe3O4 nanoparticles (Fe3O4@SiO2@IL). | |
The size and morphology of the as-prepared nanoparticles were studied by scanning electron microscopy (SEM). Fig. 4a shows an SEM image of bare Fe3O4 NPs. The image shows severe aggregation of particles, whereas silica-coated Fe3O4 NPs were well dispersed (Fig. 4b). The SiO2 coating layer could prevent the partial exposure of naked magnetite and thus prevent the aggregation of particles. An SEM image of silica-coated Fe3O4 NPs modified with the ionic liquid (Fe3O4@SiO2@IL) is shown in Fig. 4c. The particles modified with the ionic liquid had an average diameter of about 60 nm and exhibited little aggregation in comparison with the Fe3O4@SiO2 NPs. This type of coating can provide abundant reaction sites for grafting.
 |
| | Fig. 4 SEM images of (a) Fe3O4 nanoparticles; (b) Fe3O4@SiO2 nanoparticles; and (c) ionic liquid-modified silica-coated Fe3O4 nanoparticles (Fe3O4@SiO2@IL). | |
Optimization of conditions for magnetic solid-phase extraction
To determine the most effective and suitable conditions for the separation and preconcentration of zinc from different complex matrices, several experimental parameters were optimized. Optimization of the method was carried out by altering one variable at a time. A 5 ng mL−1 solution of Zn(II) was used for all the measurements, and three replicates were tested in all cases. The recovery percentage, which was calculated from the amount of zinc ions in the starting sample and the amount of zinc ions eluted from the nanosorbent, was used as an analytical criterion to evaluate the influence of the parameters under investigation on the extraction and determination of zinc ions using the present method.
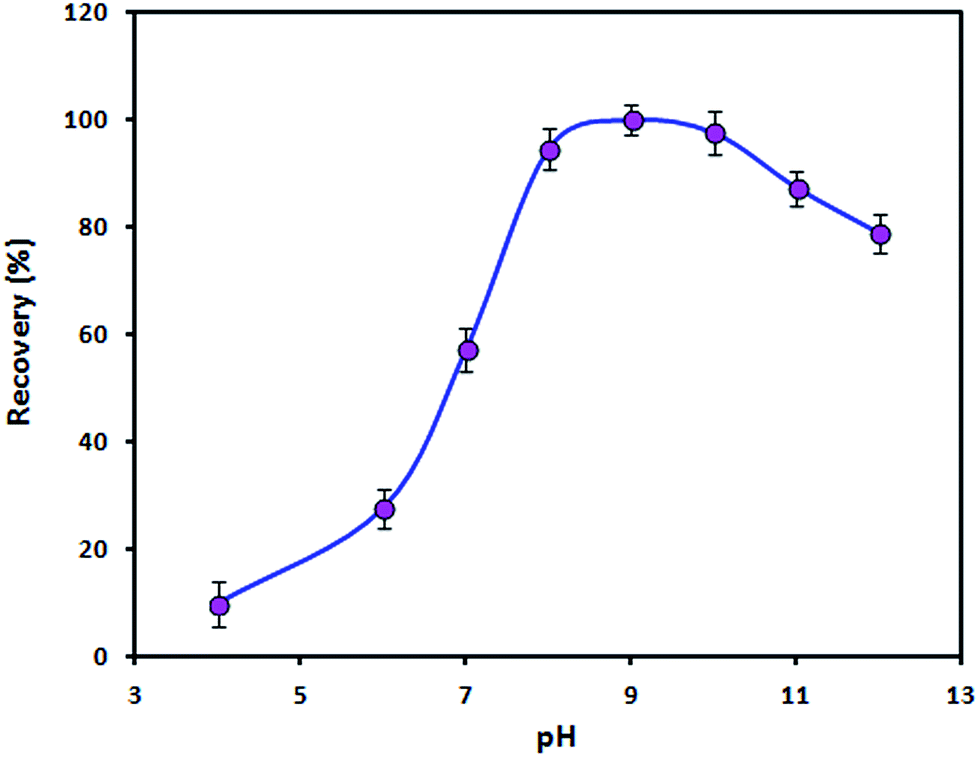
Effect of pH
The influence of the pH value on the recovery of Zn(II) ions was studied by adjusting the pH value of the sample solution in the range of 4–12 with minimal volumes of 0.01 mol L−1 HNO3 and/or NaOH. In this study, the pH value not only affects the surface charge of the adsorbent but also plays an important role in the complexation of Zn(II) ions with PAN. The surface charge of bare Fe3O4 NPs is zero at a pH of ≈7.0 (pHpzc). However, in the presence of the ionic liquid the pHpzc is lower than 7.0. It is obvious that the adsorbent surface is negatively charged when the pH is above the pHpzc. Therefore, electrostatic attraction may occur between positively charged Zn(II) ions and adsorbent particles. On the other hand, very stable complexes between heavy metal ions and PAN form in the pH range of 8–10.23 As shown in Fig. 5, the recovery of the analyte increased with an increase in the pH from 4 to 8 and remained constant between a pH of 8 and a pH of 10 before decreasing at pH values of >10. An increase in the concentration of OH− anions and the precipitation of Zn(II) ions in the form of a hydroxide at a pH of >10 might be ascribed to the observed decrease in the recovery. Therefore, a value of 9 was selected as the working pH. The pH was adjusted using a boric acid/sodium hydroxide buffer solution.
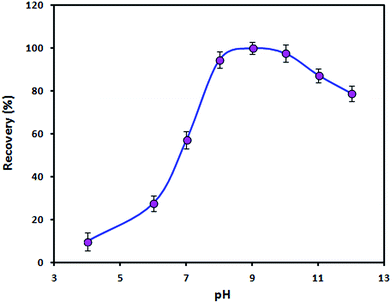 |
| | Fig. 5 Effect of pH on the adsorption of Zn(II) ions by the Fe3O4@SiO2@IL nanosorbent. Utilized conditions: sample volume: 150.0 mL, Zn(II) ion concentration: 5 ng mL−1, PAN concentration: 4.0 μmol L−1, amount of nanosorbent: 50.0 mg, extraction time: 10 min, elution conditions: 1.5 mL of 0.5 mol L−1 HNO3 in ethanol, desorption time: 4 min. | |
Although the mechanism of adsorption of zinc by the ionic liquid is not clear, a complexation reaction occurs between zinc ions and the pyridinium ring of the ionic liquid. According to the hard–soft acid–base theory, the N atom in the pyridinium ring of [propylpyridinium]+ [hexafluorophosphate]− ionic liquid is regarded as a soft base and has a high affinity for soft acids. Hence, zinc ions, as a soft acid, could be extracted by the ionic liquid. It seems that an anionic complex is initially formed between Zn(II), PAN and [PF6]− in the ionic liquid structure at a pH of 9 (borate buffer solution). Then, the anionic complex combines with [propylpyridinium]+ to form a neutral compound, which is extracted by the ionic liquid.24,25
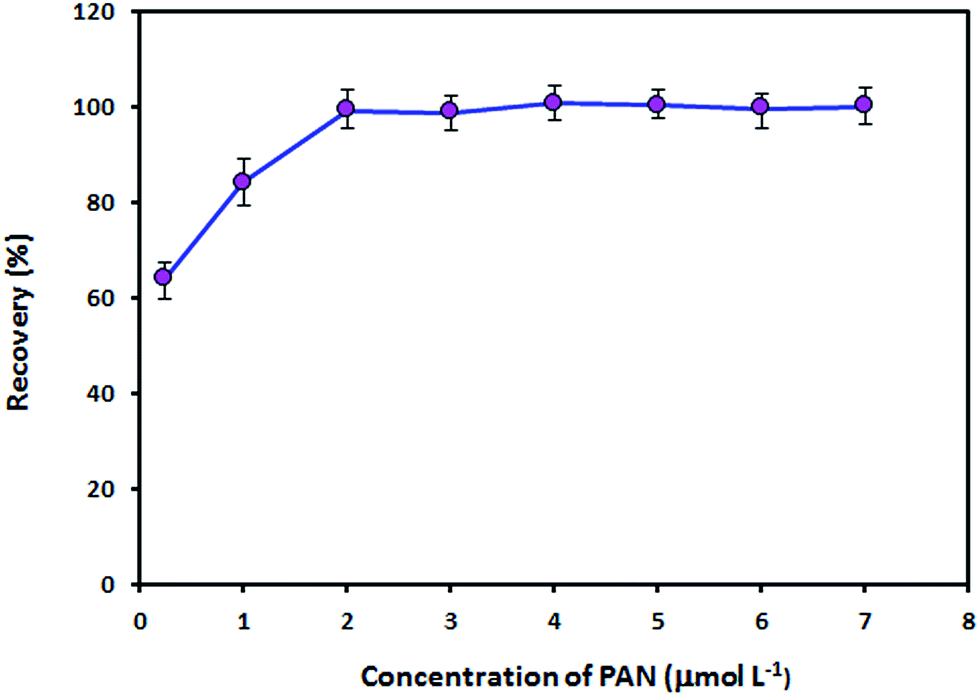
Effect of the concentration of PAN
PAN acts as a tridentate ligand and can form very stable complexes with metal ions via the hydroxyl oxygen atom, the nitrogen atom of the pyridine ring and one of the nitrogen atoms in the azo group.23 Metal ion complexes with PAN are sparingly soluble in water and can be extracted into organic solvents. The effect of the concentration of PAN on the recovery of Zn(II) ions was studied in the concentration range of 0.25–7.0 μmol L−1 PAN. As can be seen from Fig. 6, 2.0 μmol L−1 is the minimum concentration of PAN necessary to achieve maximum recovery. The recovery remained constant from a PAN concentration of 2.0 μmol L−1 to at least 7.0 μmol L−1. Therefore, a PAN concentration of 4.0 μmol L−1 was employed for the subsequent experiments.
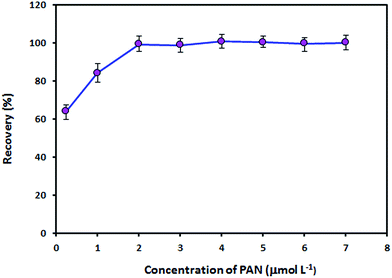 |
| | Fig. 6 Effect of the concentration of PAN on the adsorption of Zn(II) ions by the Fe3O4@SiO2@IL nanosorbent. Utilized conditions: sample volume: 150.0 mL, Zn(II) ion concentration: 5 ng mL−1, pH: 9, amount of nanosorbent: 50.0 mg, extraction time: 10 min, elution conditions: 1.5 mL of 0.5 mol L−1 HNO3 in ethanol, desorption time: 4 min. | |
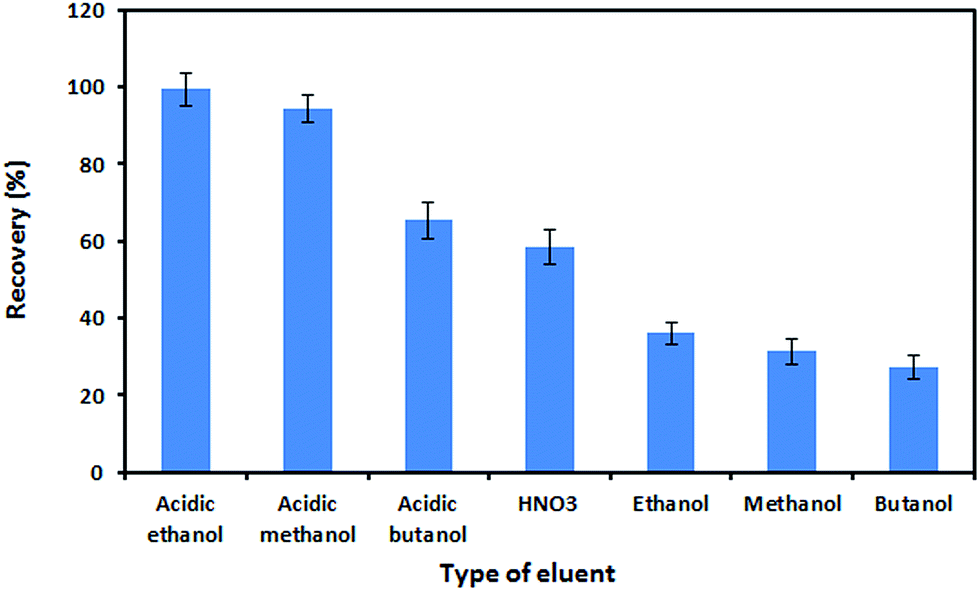
Optimization of desorption conditions
In this work, the desorption of the extracted analytes from the nanosorbent was examined using various reagents such as HNO3, ethanol, methanol, butanol, and their acidic solutions (with HNO3 in each solvent). As shown in Fig. 7, the best recovery was achieved when acidic ethanol was used as an eluent. The concentration of HNO3 in ethanol was also optimized. For this purpose, various concentrations of HNO3 (0.1–1.5 mol L−1) in ethanol were tested for their elution performance. On the basis of the obtained results, 0.5 mol L−1 HNO3 in ethanol was sufficient for the complete elution of the adsorbed analytes. By keeping the eluent concentration at 0.5 mol L−1 HNO3, the effect of the eluent volume on recovery was investigated within the range of 0.5–4.0 mL. The recovery of Zn(II) ions increased as the volume of eluent increased to 1.5 mL and remained constant afterward. Therefore, the optimum volume of eluent was chosen to be 1.5 mL. The effect of the desorption time on recovery was studied in the range of 1–10 min. It was observed that 5 min was sufficient for the complete desorption of the analyte from the sorbent.
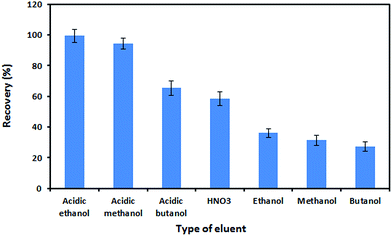 |
| | Fig. 7 Effect of the eluent type on the desorption of the analyte from the Fe3O4@SiO2@IL nanosorbent. Utilized conditions: sample volume: 150.0 mL, Zn(II) ion concentration: 5 ng mL−1, pH: 9, PAN concentration: 4.0 μmol L−1, amount of nanosorbent: 50.0 mg, extraction time: 10 min, elution conditions: 1.5 mL of 0.5 mol L−1 HNO3 in ethanol, desorption time: 4 min. | |
Reusability of the nanosorbent
The nanosorbent could be reused after being regenerated with 1.5 mL of a 0.5 mol L−1 solution of HNO3 in ethanol and 10 mL deionized water. The potential reusability and stability of the nanosorbent were studied by monitoring the change in the recovery of the analyte through several adsorption–elution cycles. The results showed that the Fe3O4@SiO2@IL nanosorbent could be recycled and reused 150 times without a significant reduction in its extraction efficiency. On the basis of the obtained results, the RSD of the absolute recovery values after 150 adsorption/desorption cycles (n = 10, at a Zn(II) ion concentration of 5 ng mL−1) was 3.2% with an average recovery of 98.4%, which suggests the high reusability of the sorbent.
Study of interferences
In order to assess the potential for analytical applications of the recommended procedure, the effect of some potentially interfering ions that may interfere with the extraction and determination of Zn(II) ions in real samples was studied. The results are summarized in Table 1. An ion was considered to interfere when its presence at a certain concentration produced a variation of more than ±5% in the analytical signal of the sample. It can be seen that the presence of major cations and anions had no obvious influence on the determination of Zn(II) ions. This result shows that this procedure could be successfully applied to the determination of Zn(II) ions in natural water and food samples without interference effects.
Table 1 Tolerance limits of interfering ions in the determination of 5 ng mL−1 Zn(II) ions
| Interfering ions |
Interferent to analyte ratioa |
| The ratios in this table are the ratios at which the mentioned ions would cause a variation of ±5% in the signal of the analyte. |
| Na+, K+, Ca2+, Mg2+, Ba2+, PO43−, SO42−, HPO42−, H2PO4−, CH3COO−, NO3−, BO3−, F−, Cl−, Br−, I− |
5000:1 |
| Bi(III), Cu(II), Mn(II), Pb(II), Ag(I), Ni(II), Co(II), Al(III), Fe(III) |
1000:1 |
| Cd(II), Cr(III) |
500:1 |
Analytical figures of merit
Under the optimized experimental conditions, analytical features of the present method such as the linear range of the calibration graph, limit of detection, accuracy and precision were examined. The calibration graph was linear between 0.5 and 15 ng mL−1 with a correlation coefficient of 0.9989 when the preconcentration procedure was employed. The calibration equation was A = 0.05CZn(II) + 0.04, where CZn(II) is the concentration of Zn(II) ions in ng mL−1. The limit of detection (LOD) and limit of quantification (LOQ) of this method, which were defined as 3Sb/m and 10Sb/m (where Sb is the standard deviation of six replicate measurements of the blank and m is the slope of the calibration curve) were 0.17 and 0.5 ng mL−1, respectively. In order to study the precision of the method, a series of six solutions containing 5 ng mL−1 Zn(II) ions were measured on the same day. On the basis of the obtained results, the relative standard deviation (RSD) was 2.3%, which showed the high precision of the method.
Application to real samples
To test the reliability of the present procedure, the method was employed to determine trace amounts of Zn(II) ions in different water samples, i.e., tap water, spring water, mineral water and milk powder. In order to verify the accuracy of the established procedure, recovery experiments were carried out by spiking the samples with different amounts of Zn(II) ions before any pretreatment. Table 2 shows the obtained results. As can be seen, recoveries of between 97.0% and 102.5% were obtained, which confirms the accuracy of the present method. In addition, the reliability of the present procedure and its accuracy were evaluated by employing the procedure for the determination of Zn(II) ions in a more complex matrix. For this purpose, a biological standard reference material, namely, NIST SRM 1549 non-fat milk powder, was analyzed. The assayed value (45.36 ± 1.73 μg g−1) was in good agreement with the certified value (46.1 ± 2.2 μg g−1). It can be concluded that the present method is accurate and free from systematic errors.
Table 2 Determination of zinc ions in water and milk samples (results for recoveries of spiked samples and analysis of a certified reference material)
| Samples |
Zn added (ng mL−1) |
Zn founda (ng mL−1) |
Recovery (%) |
| Mean ± standard deviation of three experiments. Obtained from drinking water system of Azarshahr, Iran. Obtained from Vata Natural Mineral Water Co., Ardabil, Iran. Obtained from spring water from Pirchupan village, Azarshahr, Iran. Obtained from the local pharmacy (Humana milk powder). |
| Tap waterb |
— |
6.42 ± 3.53 |
— |
| 4.0 |
10.52 ± 1.79 |
102.5 |
| 8.0 |
14.33 ± 1.28 |
98.9 |
| Mineral waterc |
— |
Not detected |
— |
| 4.0 |
3.95 ± 3.23 |
98.7 |
| 8.0 |
8.16 ± 2.10 |
102.0 |
| Spring waterd |
— |
3.51 ± 2.77 |
— |
| 4.0 |
7.46 ± 1.02 |
98.7 |
| 8.0 |
11.55 ± 1.34 |
100.5 |
| Samples |
Zn added (μg g−1) |
Zn found (μg g−1) |
Recovery (%) |
| Milk powdere |
— |
28.78 ± 0.78 |
— |
| 4.0 |
32.75 ± 2.50 |
99.2 |
| 8.0 |
36.54 ± 1.71 |
97.0 |
| |
Assayed value (μg g−1) |
Certified value (μg g−1) |
Relative error (%) |
| NIST SRM 1549 |
45.36 ± 1.73 |
46.10 ± 2.2 |
−1.6 |
Comparison with other methods
The limit of detection, enrichment factor and relative standard deviation achieved with the present technique were compared with those of other published preconcentration methods used for the determination of Zn(II) ions in Table 3. The present method has a relatively low LOD and a high enrichment factor comparable with those of other methods. The simple operating procedure makes sample preparation very easy and rapid, and only a few minutes are needed before instrumental analysis can be carried out. The method is presented as a suitable alternative to more expensive instruments for the determination of Zn at trace levels. This methodology is a reproducible, simple and low-cost technique, does not require further instrumentation, and can be used with regular FAAS equipment.
Table 3 Comparison of the developed method with other reported methods used for determination of Znd
| Extraction system |
Determination technique |
EFa |
LODb (ng mL−1) |
RSDc (%) |
Ref. |
| Enhancement or enrichment factor. Limit of detection. Relative standard deviation. SPE: solid-phase extraction, DLLME: dispersive liquid–liquid microextraction, MNPs: magnetic nanoparticles, MSPE: magnetic solid-phase extraction. |
| SPE |
FAAS |
200 |
0.05 |
1–3.1 |
6 |
| Nanoparticle-based SPE |
ICP-OES |
150 |
0.2 |
2.5 |
8 |
| Ionic liquid-based LLME |
FAAS |
60 |
0.22 |
1.92 |
21 |
| Carbon nanotube-based SPE |
FAAS |
25 |
20 |
<10 |
26 |
| Modified MNPs |
ICP-OES |
182 |
0.11 |
4.2 |
27 |
| SPE |
ICP-OES |
125 |
6.5 |
5.0 |
28 |
| Fe3O4@SiO2@IL-based MSPE |
FAAS |
100 |
0.17 |
2.3 |
This work |
Conclusions
In this work, triethoxysilylpropylpyridinium hexafluorophosphate ionic liquid was covalently immobilized on the surface of silica-coated magnetite nanoparticles and employed as an adsorbent for the magnetic solid-phase extraction and preconcentration of trace levels of Zn(II) ions before their determination by FAAS. Covalent immobilization endowed the ionic liquid coating with high stability, which prevented its loss during the extraction and elution processes. This aspect is especially interesting, because it enabled the reuse of the nanosorbent up to 150 times after cleaning with 1.5 mL of 0.5 mol L−1 HNO3 in ethanol and conditioning with 10 mL deionized water. This method represents a sensitive, reproducible, simple, low-cost and environment-friendly technique with analytical characteristics better than or comparable to those of most other methods that can be used for the preconcentration of Zn(II) ions in routine analytical laboratories. The developed method was successfully employed to measure low concentrations of zinc in water and milk samples with high accuracy and precision.
Acknowledgements
The financial support from the Research Council of Azarbaijan Shahid Madani University (Grant no. ASMU/95372-16) is gratefully acknowledged.
References
- D. R. William, Computer Models of Metal Bio-chemistry and Metabolism in Chemical Toxicology and Clinical Chemistry of Metals, Academic Press, New York, 1983 Search PubMed.
- S. M. Z. Al-Kindy, S. T. Al-Bulushi and F. E. O. Suliman, Spectrochim. Acta, Part A, 2008, 71, 676–681 CrossRef PubMed.
- P. Pohl, I. Sergiel and B. Prusisz, Food Chem., 2011, 125, 1504–1509 CrossRef CAS.
- J. De Zuane, Handbook of Drinking Water Quality Standards and Controls, Van Nostrand Rein-hold, New York, 1990 Search PubMed.
- W. W. Borland, B. F. Allan, J. Mce Neilson and J. M. Sommervile, Clin. Chem., 1982, 28, 2450–2451 Search PubMed.
- G. Giakisikli and A. N. Anthemidis, Anal. Chim. Acta, 2013, 789, 1–16 CrossRef CAS PubMed.
- X. Zhao, Y. Shi, T. Wang, Y. Cai and G. Jiang, J. Chromatogr. A, 2008, 1188, 140–147 CrossRef CAS PubMed.
- M. Faraji, Y. Yamini and M. Rezaee, J. Iran. Chem. Soc., 2010, 7, 1–37 CrossRef CAS.
- S. Sadeghi and E. Aboobakri, Microchim. Acta, 2012, 178, 89–97 CrossRef CAS.
- S. Ayata, S. S. Bozkurt and K. Ocakoglu, Talanta, 2011, 84, 212–215 CrossRef CAS PubMed.
- J. Li, Y. Cai, Y. Shi, S. Mou and G. Jiang, Talanta, 2008, 74, 498–504 CrossRef CAS PubMed.
- C. He, S. Li, H. Liu, K. Li and F. Liu, J. Chromatogr. A, 2005, 1082, 143–149 CrossRef CAS PubMed.
- C. F. Poole and S. K. Poole, J. Chromatogr. A, 2010, 1217, 2268–2286 CrossRef CAS PubMed.
- J. Fan, Y. C. Fan, Y. C. Pei, K. Wu and J. J. Wang, Sep. Purif. Technol., 2008, 61, 324–331 CrossRef CAS.
- C. F. Ding, F. Zhao, R. Ren and J. M. Lin, Talanta, 2009, 78, 1148–1154 CrossRef CAS PubMed.
- P. Liang and L. Peng, Talanta, 2010, 81, 673–677 CrossRef CAS PubMed.
- A. Maquieira, H. A. M. Elmahadi and R. Puchades, Anal. Chem., 1994, 66, 3632–3638 CrossRef CAS.
- Y. Jiang, C. Guo, H. Xia, I. Mahmood, C. Liu and H. Liu, J. Mol. Catal. B: Enzym., 2009, 58, 103–109 CrossRef CAS.
- M. Bouri, M. Gurau, R. Salghi, I. Cretescu, M. Zougagh and A. Rios, Anal. Bioanal. Chem., 2012, 404, 1529–1538 CrossRef CAS PubMed.
- Z. Huacong, L. Wei, S. Qinghui, G. Hongshuai, X. Peng, D. Fuli and L. Huizhou, Chin. J. Chem. Eng., 2012, 20, 146–151 CrossRef.
- H. Abdolmohammad-Zadeh and G. H. Sadeghi, Anal. Chim. Acta, 2009, 649, 211–217 CrossRef CAS PubMed.
- G. Cheng, M. He, H. Peng and B. Hu, Talanta, 2012, 88, 507–515 CrossRef CAS PubMed.
- K. L. Cheng and R. H. Bray, Anal. Chem., 1955, 27, 782–785 CrossRef CAS.
- S. T. Fujiwara, R. V. S. Alfaya and Y. Gushikem, Colloids Surf., A, 2001, 178, 135–141 CrossRef CAS.
- M. S. Iamamoto and Y. Gushikem, Analyst, 1989, 114, 983–985 RSC.
- S. Vellaichamy and K. Palanivelu, J. Hazard. Mater., 2011, 185, 1131–1139 CrossRef CAS PubMed.
- M. H. Mashhadizadeh and Z. Karami, J. Hazard. Mater., 2011, 190, 1023–1029 CrossRef CAS PubMed.
- Y. Ciu, X. Chang, X. Zhu, H. Luo and Z. Hu, Microchem. J., 2007, 87, 20–26 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02126g |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Sima Hassanlouei
*,
Sima Hassanlouei