
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMelatonin enhances the anti-tumor effect of sorafenib via AKT/p27-mediated cell cycle arrest in hepatocarcinoma cell lines†
Fei Long‡
a,
Chengyong Dong‡a,
Keqiu Jianga,
Yakun Xua,
Xinming Chib,
Deguang Suna,
Rui Lianga,
Zhenming Gaoa,
Shujuan Shao*b and
Liming Wang *a
*a
aDivision of Hepatobiliary and Pancreatic Surgery, Department of General Surgery, The Second Affiliated Hospital of Dalian Medical University, Dalian, Liaoning, China. E-mail: wangbcc259@163.com; Fax: +86-0411-84671291; Tel: +86-0411-84671291
bKey Laboratory of Proteomics, Dalian Medical University, Dalian, China. E-mail: shaoshujuan2006@126.com; Fax: +86-0411-86110314; Tel: +86-0411-86110314
First published on 18th April 2017
Abstract
Hepatocellular carcinoma (HCC) is the fifth most common cancer worldwide, often diagnosed in late stages when most therapeutic methods are not very effective. The introduction of the multikinase inhibitor sorafenib as the standard of care has opened a window of hope for patients with advanced HCC, patients with very poor prognosis; however, patients usually develop acquired resistance to sorafenib limiting its therapeutic benefits. Melatonin (MT), an indoleamine compound produced in the pineal gland, has shown a substantial beneficial effect in increasing the efficacy of common anticancer drugs and decreasing their toxic effects. Here we demonstrate that MT potentiated the sorafenib-mediated inhibition of cell viability and colony formation in HCC cell lines. Moreover, combined treatment of MT and sorafenib enhanced the cell cycle arrest of HCC cells at the G0/G1 phase. Co-treatment of sorafenib and MT was found to upregulate p27, an inhibitor of several cyclin-dependent kinases (CDK), and downregulate p-AKT, c-myc, cyclin D1 and CDK4/6 protein expression. Furthermore, overexpression of p-AKT using SC79 reversed the effect of sorafenib and MT combination on cell viability and growth of HCC cells. These results suggest that the AKT pathway might be critical for the enhanced anticancer effect observed after co-treatment with MT and sorafenib. Taken together, our findings demonstrated that AKT/p27-mediated cell growth arrest induced by MT increased the sensitivity of HCC cells to the effect of sorafenib.
1. Introduction
Hepatocellular carcinoma (HCC) is one of the most common malignant tumors worldwide with approximately more than half a million new cases diagnosed annually.1 HCC is highly resistant to chemotherapeutic agents. Indeed, there is a lack of efficient chemotherapy drugs, leaving local ablation, surgical resection, and liver transplantation as the only therapeutic options for patients diagnosed with early-stage HCC. Unfortunately, the disease is often diagnosed in late stages when most patients are not suitable for local ablation or surgery. The introduction of the multikinase inhibitor sorafenib as the standard of care has opened a window of hope for advanced HCC patients with very poor prognosis.2 Sorafenib has been shown to successfully inhibit tumor cell proliferation and vasculogenesis by targeting RAF/MEK/ERK, as well as JAK/STAT pathways.3,4 However, the low rate of tumor response, most likely due to primary and acquired drug resistance, has limited the overall therapeutic effect of sorafenib on HCC patients.5,6 In addition, many patients have multiple undesirable side effects.7 Thus, a synergistic combinatorial approach that includes sorafenib at low doses could be a better therapeutic strategy in HCC to improve the anti-tumor effect of sorafenib and, at the same time, decrease its toxicity.Although synthesis of melatonin (MT) occurs in multiple organs, the major site of MT synthesis is the pineal gland. At physiological concentrations, MT functions as a major regulator of the circadian rhythms and sleep.8,9 Moreover, it has been shown to regulate obesity,10 the immune systems11 and oncostasis.12–14
In addition, previous studies have also shown MT plays a role in tumor growth and gene regulation in cancer cells.15,16 MT inhibited cell proliferation of human B-lymphoma cells,17 human myeloid leukemia cells HL-60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 and human neuroblastoma cancer cells.19 Furthermore, MT exerts a protective function by preventing cell damage,20–22 particularly in neural cells.23,24 A plethora of evidence has unequivocally demonstrated that MT decreases the toxicity associated with chemotherapeutic agents both in animals as well as in patients.25,26
18 and human neuroblastoma cancer cells.19 Furthermore, MT exerts a protective function by preventing cell damage,20–22 particularly in neural cells.23,24 A plethora of evidence has unequivocally demonstrated that MT decreases the toxicity associated with chemotherapeutic agents both in animals as well as in patients.25,26
In this study, we aim to investigate the beneficial impact of combining MT to sorafenib, the standard of care, in HCC and the mechanism by which MT improves the anti-tumor effect of sorafenib.
2. Experimental
2.1 Cell lines and cell culture
Human HCC cell lines Bel7402, SMMC-7721 and the normal hepatocyte cell lines L02 were purchased from KeyGen Biotech Company (Nanjing, China). Cell lines were cultured in RPMI Medium 1640 (Gibco, Gaithersburg, MD, USA), supplemented with 10% fetal bovine serum (FBS) (PAN BIOTECH, Aidenbach, Germany) and 1% penicillin/streptomycin (100 U ml−1, Gibco). Cells were incubated in a humidified atmosphere containing 5% CO2 at 37 °C.2.2 Reagents and antibodies
MT was obtained from Sigma (St. Louis, MO, USA) and was dissolved in an appropriate amount of DMSO. Sorafenib and SC79 were purchased from Selleckchem (Houston, TX, USA) and were also dissolved in an appropriate amount of DMSO. The above three reagents were added to the complete cell culture medium to attain the stipulated concentration. The maximum concentration of DMSO in media was 0.1% and the concentration of DMSO in the control groups was 0.1% constantly. The Cell Counting Kit-8 (CCK8) was purchased from Dojindo (Kumamoto, Japan). Rabbit polyclonal antibodies for total AKT, phospho-AKT (p-AKT) (Ser473), total ERK, phospho-ERK (p-ERK) (Thr202/Tyr204), p21, cyclin D1, phospho-Rb (p-Rb) (Ser807/811) and β-actin were purchased from Cell Signaling (Danvers, MA, USA). Rabbit polyclonal antibodies for p27, CDK4, CDK6, cyclin E1, cyclin A1 and c-myc were obtained from Protein Tech (Chicago, IL, USA). All the secondary antibodies were purchased from Protein Tech (Chicago, IL, USA).2.3 Cell viability assay
Cell viability was tested using the CCK-8 assay in accordance with the manufacturer's instructions. Hepatocellular carcinoma (HCC) cells (2 × 103) were seeded into 96-well plates and cultured in 100 μl of cell culture medium. After 24 hours, cells were treated with MT and/or sorafenib at the indicated concentrations for 48 or 72 hours (in the presence or absence of AKT activator SC79 [8.0 μg ml−1] for 48 hours). Then, the medium was replaced by 100 μl of a mixture of cell culture medium (90 μl) and CCK-8 reagent (10 μl) and cells were incubated at 37 °C or 2 hours. Finally, the optical density was determined at a wavelength of 450 nm by an EnSpireTM 2300 Multilabel Reader (PerkinElmer, Waltham, MA, USA). Three replicates were measured for each test. At least three independent experiments were performed.2.4 Clonogenic survival assay
Cells were seeded into a 6-well culture dish in quadruplicate (800 cells per well). After 48 hours of incubation, cultures were replaced with fresh medium containing MT and/or sorafenib in the indicated concentrations for 2 weeks. During this time the medium was renewed every 4 days. After 2 weeks, cell colonies were washed with phosphate buffered saline (PBS) buffer for three times, fixed in paraformaldehyde (PFA) for 15 minutes, and stained using crystal violet (Sigma-Aldrich, St. Louis, MO, USA) for 15 minutes at room temperature. Clones were visually counted and colony formation efficiency was calculated.2.5 Cell cycle analysis
Cells were seeded into a 6-well culture dish. After 24 hours of incubation, cultures were replaced by fresh medium containing MT and/or sorafenib at the indicated concentrations for 48 hours (in the presence or absence of AKT activator SC79 [8.0 μg ml−1] for 48 hours) and cells were harvested by trypsinization, and, then, fixed overnight at 4 °C with 75% cold ethanol. Fixed cells were treated with 100 μl of RNase A (10 μg ml−1) at 37 °C for 30 minutes and stained with 400 μl of PI (50 μg ml−1) at room temperature in dark conditions for 30 minutes. The cell cycle profiles were obtained using a BD FacsCalibur (BD, Franklin Lanes, NJ, USA) flow cytometer.2.6 Western blot analysis
Cells were cultured in 10 cm-diameter dishes and treated either with DMSO, single agent or combination (in the presence or absence of AKT activator SC79 [8.0 μg ml−1]) for appropriate time-points. Western blot analyses were performed using standard procedures. Actin was used as a loading control. The signal was detected using secondary antibodies coupled with HRP (Jackson laboratory). The luminescent signal was detected using a LAS-3000 Luminescent Image analyzer (Fujifilm, Valhalla, NY).2.7 Animal study
BALB/c nude female mice (4 weeks of age) were used in this study and were purchased from the Dalian Medical University SPF Laboratory Animal Center (Dalian, China). Mice, weighing between 13 and 18 g, were fed in a specific pathogen-free area. To evaluate the therapeutic efficacy of MT and sorafenib in human HCC cell lines SMMC-7721 using a murine model, SMMC-7721 cells (5 × 106 in 100 μl PBS) were injected subcutaneously near the axillary fossa of nude mice. After two weeks, when tumor diameter reached 3 mm × 4 mm, tumor cell-inoculated mice were randomly divided into four treatment groups, each containing four mice. Group A, treated with normal saline (NS); group B treated with 25 mg kg−1 MT (i.p.); group C treated with 10 mg kg−1 sorafenib (orally); group D treated with MT (i.p.) and sorafenib (orally) and each group was administrated every day. Tumors were measured using a caliper every 3 days and the formula V = 1/2 (width2 × length) was used to calculate the tumor volume. The weights of the mice were also recorded every 3 days. On day 32 after cell inoculation, all mice were sacrificed with ether anesthesia and tumor weight in each mouse was measured.All experiments were performed in compliance with the recommendations established by the Animal Care and Ethics Committee of Dalian Medical University. The protocol was approved by the Animal Care and Ethics Committee of Dalian Medical University.
2.8 Statistical analysis
All experiments were repeated at least three times. Means and standard deviations were calculated from at least three different measurements. GraphPad Prism software was used for statistical analysis. Statistical comparisons were implemented using one-way ANOVA test. A p-value <0.05 was considered to be statistically significant. SPSS v13.0 software was applied for all statistical analyses.3. Results
3.1 Co-treatment of MT and sorafenib decreases the cell viability and alters morphology in HCC cell lines
To determine whether MT could potentiate the sorafenib-induced inhibition of hepatoma cell lines survival, we first investigated the effect of MT or sorafenib alone on cell viability in HCC cell lines Bel-7402 and SMMC-7721 using a CCK-8 assay. Treatments with MT alone [200–2000 μM] or sorafenib alone [1–12 μM] for 48 or 72 hours have shown to considerably suppress hepatoma cell viability in a dose and time-dependent manner (Fig. 1A and B). Combined treatment of MT [1.0 mM] and sorafenib [4–8 μM] for 48 hours significantly enhanced the sorafenib-mediated inhibition of cell viability in both Bel-7402 and SMMC-7721 cells as compared with those treated with either MT or sorafenib alone (Fig. 1C). Additionally, we analyzed the changes in cell spreading and morphology of Bel-7402 and SMMC-7721 cells cultured in 6-well plates (Fig. 1D; 8 μM sorafenib and/or 1 mM MT for Bel-7402; 4 μM sorafenib and/or 1 mM MT for SMMC-7721). The combined treatment showed a significant reduction in cell density as well as changes in cell morphology, where cells become smaller and granulated in shape when compared with those treated either with sorafenib or MT alone. These results demonstrated that combined treatment of sorafenib and MT induces changes in cell spreading and morphology of hepatoma cells.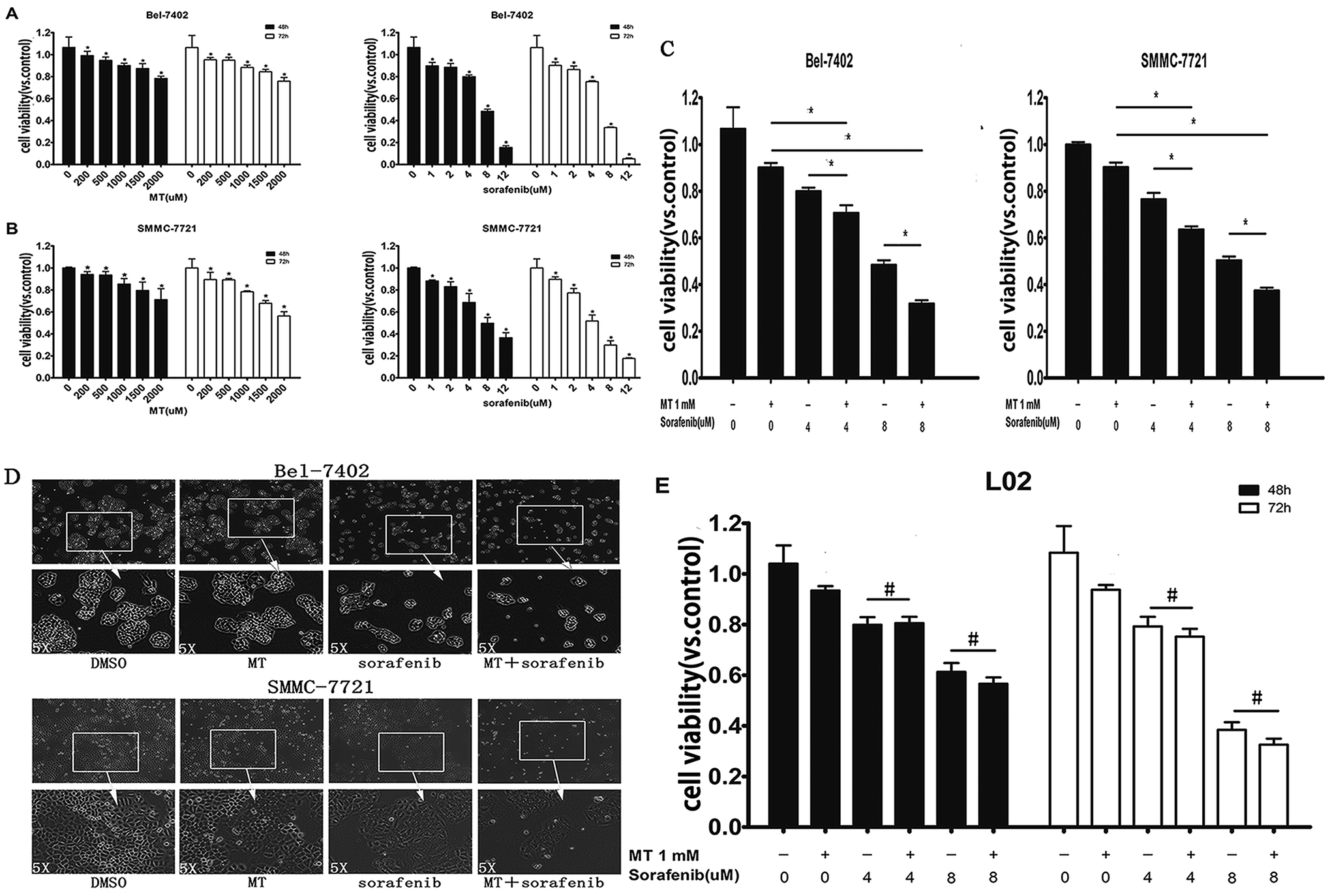 | ||
| Fig. 1 Melatonin (MT) potentiated the inhibitory effect of sorafenib on the viability of hepatocarcinoma cells but does not affect normal liver cells. (A and B) Bel-7402 and SMMC-7721 cells were treated with MT or sorafenib from 0–72 hours at the indicated doses. 48 and 72 hours after treatment, cell viability was determined using a CCK-8 assay. Cells treated with vehicle (DMSO) were set as 100% cell viability. The percentage of cell viability in each treatment group was calculated relative to cells treated with vehicle control. (C) Bel-7402 and SMMC-7721 cells were treated with MT or/and sorafenib during 48 hours at the indicated doses. 48 hours after treatment, cell viability was determined using a CCK-8 assay. (D) Changes in cell morphology and spreading of Bel-7402 and SMMC-7721 cells treated with sorafenib [8 μM] for Bel-7402 and [4 μM] for SMMC-7721 and/or MT [1 mM] for 48 hours in 6-well plates were observed and photographed using an Olympus inverted microscope with digital camera. Each cell line was photographed with a magnification of 4 × 10 fold (the picture above) and 20 × 10 fold (the picture below) (E) L02 cells (a normal hepatocyte cell lines) were treated with MT or/and sorafenib from 0–72 hours at the indicated doses. After 48 and 72 hours after treatment, cell viability was determined using a CCK-8 assay. The data are presented as mean ± SD of three separate experiments. P < 0.05 indicates significant differences between treatment groups and control groups or among treated groups. #P > 0.05 represents no significant differences among groups. | ||
It has been reported that MT exerts a protective effect in normal cells.27,28 Therefore, we investigated whether co-treatment of MT and sorafenib had an impact on normal L02 cells. Treatment with sorafenib [4–8 μM] for 48–72 hours significantly reduced cell viability compared with untreated controls (Fig. 1E). Interestingly, in contrast to hepatocarcinoma cells, MT had no significant effect on cell viability when it is combined to sorafenib compared to single drug treatment.
3.2 MT potentiates the inhibitory effect of sorafenib on the colony formation of hepatocarcinoma cells
To further confirm the effect of MT in enhancing the sorafenib-mediated inhibition of cell growth in HCC cell lines. We employed a colony formation assay where Bel-7402 and SMMC-7721 cells were treated with MT and/or sorafenib at specific concentrations for 2 weeks. Both MT and sorafenib caused an observable decrease in the numbers of colonies in both cell lines (Fig. 2A and B). Dramatically, the combination of MT and sorafenib triggered a more pronounced inhibition of colony formation compared to single treatment.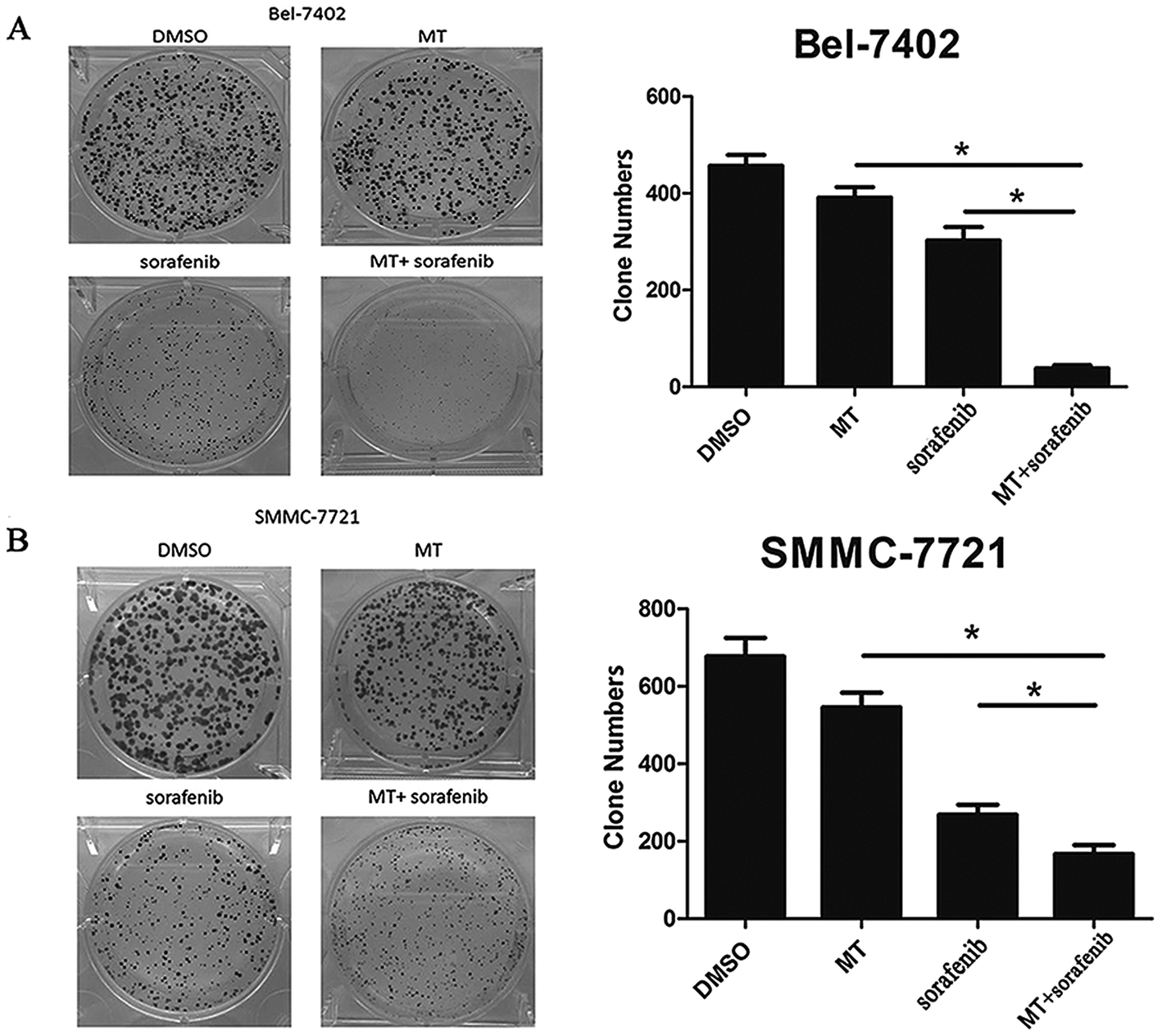 | ||
| Fig. 2 MT potentiated the inhibitory effect of sorafenib on colony formation of hepatocarcinoma cells. (A) Bel-7402 cells were grown in 6-well plates in quadruplicate (800 cells per well). After 48 hours, cultures were replaced with fresh medium containing MT [1 mM] and/or sorafenib [8 μM] for 2 weeks. After 2 weeks, the colonies were stained with 1% crystal violet and were counted. Clone numbers are shown in the bar graph (right panel). (B) SMMC-7721 cells were grown in 6-well plates in quadruplicate (800 cells per well). After 48 hours, cultures were replaced with fresh medium containing MT [1 mM] and/or sorafenib [4 μM] for 2 weeks. After 2 weeks, the colonies were stained with 1% crystal violet and were counted. Clone numbers are also shown in the bar graph (right panel). Bars represent mean ± SD from 3 independent experiments (*P < 0. 05 indicates significant difference among the groups). | ||
3.3 MT enhances the antitumor effect of sorafenib via G0/G1 phase arrest
To explore whether MT could enhance the antitumor effect of sorafenib in Bel-7402 cells through the alteration of the cell cycle, we examined cell cycle progression using FACS analysis. MT or sorafenib arrested the cell cycle at the G0/G1 phase in Bel-7402 cells compared with the control groups. However, combined treatment of MT and sorafenib further enhanced cell cycle arrest at G0/G1 phase compared with single treatment with sorafenib (Fig. 3A and B).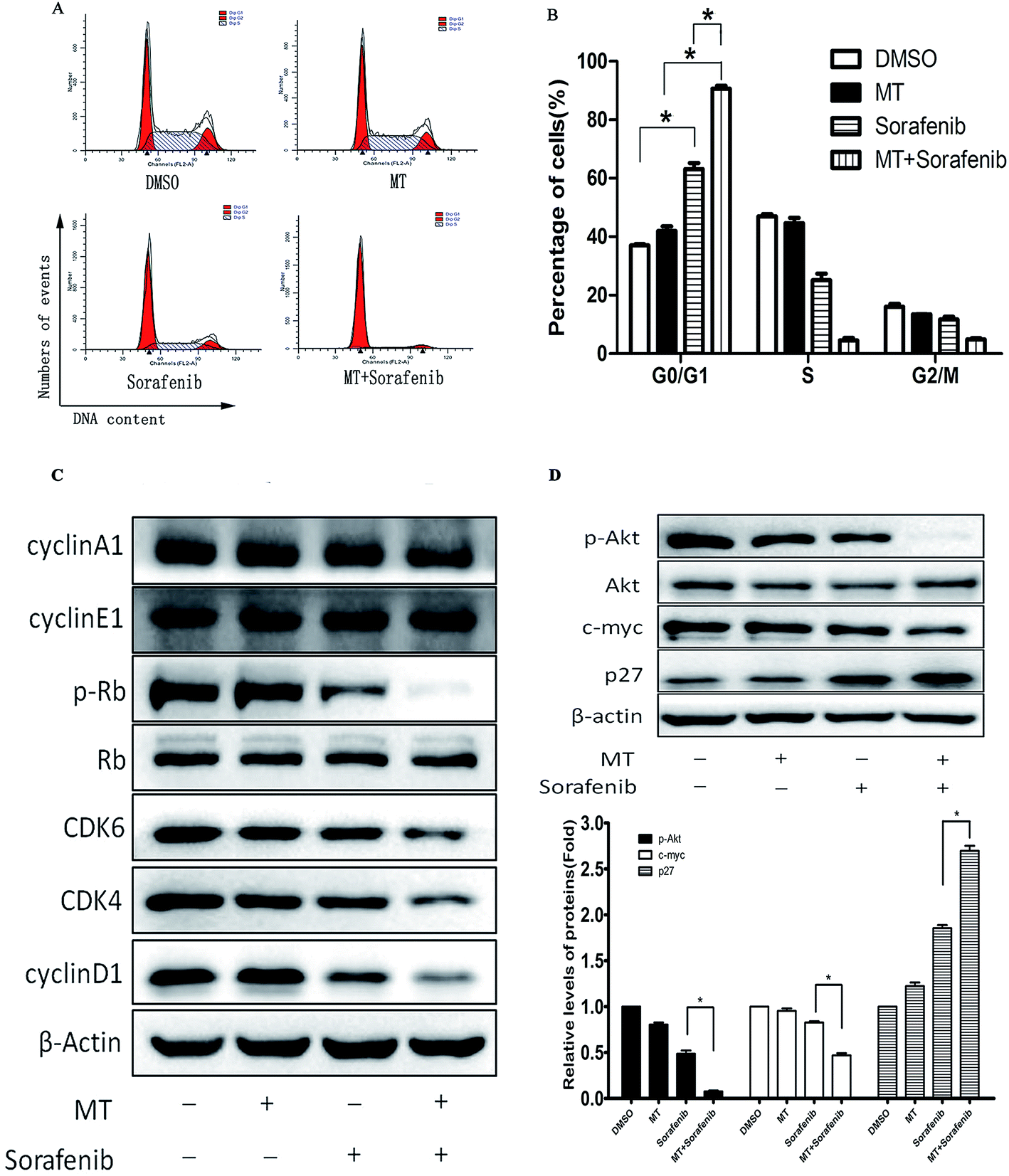 | ||
| Fig. 3 MT enhanced the antitumor effects of sorafenib via G0/G1 phase arrest. Bel-7402 cells were treated with MT [1.0 mM] or/and sorafenib [8.0 μM] for 48 hours. (A and B) Flow cytometry was performed to analyse the progression of the cell cycle in Bel-7402. The percentage of cells (%) is shown in the bar graph. (C and D) Expression of cycle-related proteins and relevant regulatory proteins in cell lysates were detected using western blot. (*P < 0.05 indicates significant difference among the groups). | ||
Alterations in the checkpoint mechanisms of the cell cycle lead to cell proliferation. The Cyclin D1/CDK4/6 complex increases the phosphorylation of Rb (p-Rb) followed by cell entry into the S phase from the G1 phase.29 Since cells were arrested at the G0/G1 phase, the protein expression of Cyclin D1, CDK4, CDK6, cyclin E1 and p-Rb, responsible for the transition from G0/G1 to S phase were analyzed using western blot. As expected, Cyclin D1, CDK4, CDK6 and the downstream p-Rb were down-regulated following combined treatment of MT and sorafenib compared with the other groups, despite no changes were observed in Cyclin E1 and Cyclin A1 expression (Fig. 3C). Thus, these results demonstrated that the combined treatment of MT and sorafenib caused cell cycle arrest at the G0/G1 phase in Bel-7402 cells via downregulation of Cyclin D1, CDK4, CDK6, and p-Rb.
The p27Kip1 gene is located on chromosome 12p13.30 The CDK-inhibitory domain resides in the N-terminal portion of p27Kip1 and is sufficient to arrest cells at G0/G1.31 In early G1, phosphorylation of p27Kip1 at Ser10 by AKT facilitates its binding to chromosomal maintenance 1 (CRM1) and its transport from the nucleus to the cytoplasm.32 Furthermore, in the early G1 phase, p27Kip1 phosphorylation at Thr157 and Thr198 by AKT causes a delay in nuclear import.33 MYC can also suppress p27Kip1 expression through several pathways.32 Thus, we examined the expression of p27, c-myc and AKT. Co-treatment with MT and sorafenib downregulated p-AKT and c-myc and up-regulated p27 protein levels (Fig. 3D).
3.4 Overexpression of p-AKT reverses the effect of MT and sorafenib co-treatment on cell viability in HCC cell lines
To explore the impact of p-AKT on the growth of HCC cells, we assessed the effect of reactivation of p-AKT on MT and sorafenib-induced inhibition of cell viability. Interestingly, up-regulation of p-AKT by SC79 [8.0 μg ml−1] abrogated the G0/G1 phase arrest (Fig. 4A) and the inhibition of cell viability (Fig. 4B) induced by the combination of MT and sorafenib. In addition, the reactivation of p-AKT reversed the combined treatment-induced increase of p27 and decrease of c-myc (Fig. 4C). Together, these results suggested that the inhibition of AKT pathway might be the underlying mechanism by which MT and sorafenib combination induces cell cycle arrest and enhances the cytotoxicity in HCC cells.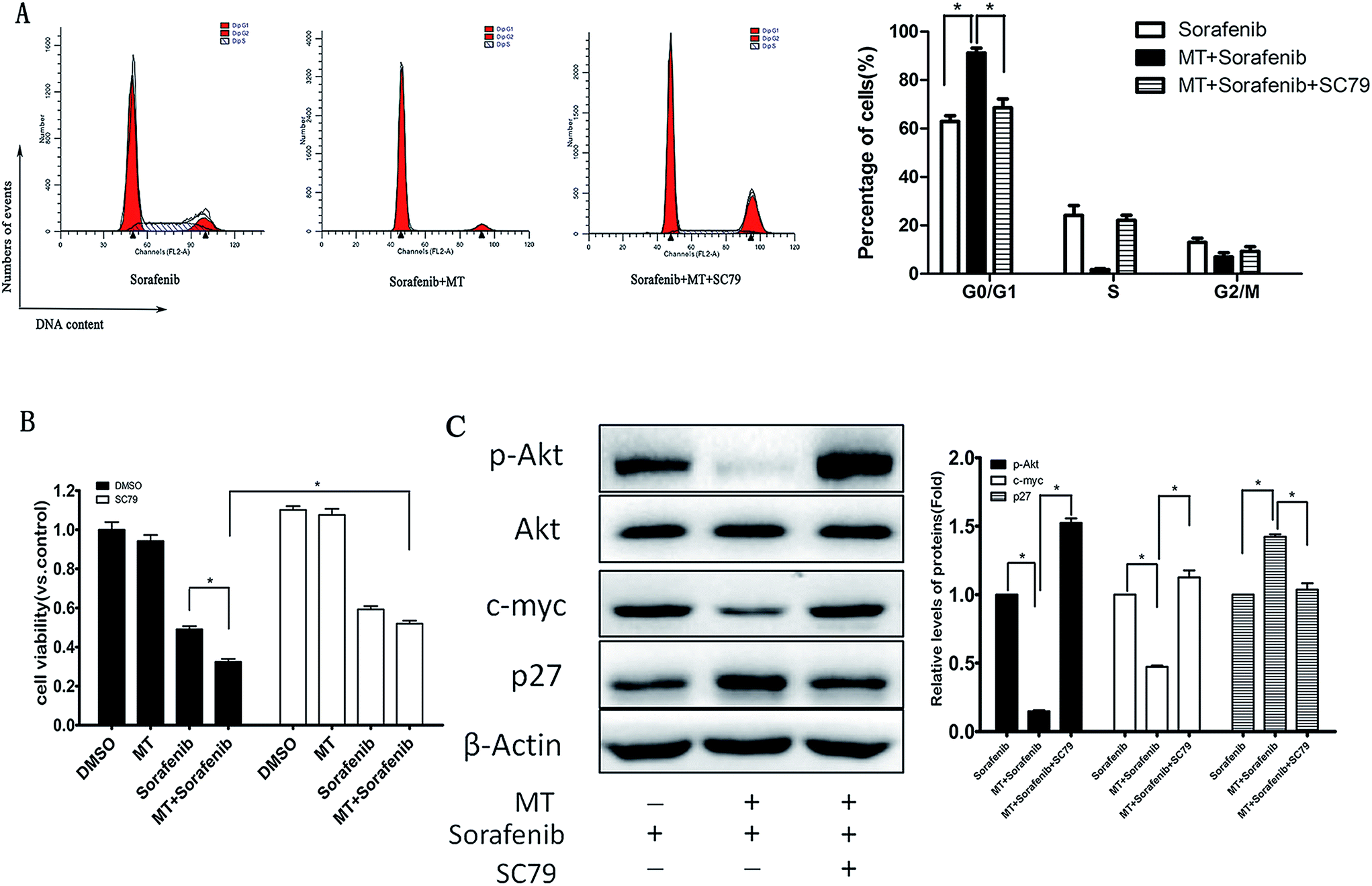 | ||
| Fig. 4 Reactivation of p-AKT abrogated the G0/G1 phase arrest and the inhibitory effect on cell viability as well as the expression of relevant regulatory proteins, induced by the combined treatment of MT and sorafenib in Bel-7402 cells. Cells were treated with MT [1.0 mM] or/and sorafenib [8.0 μM] for 48 hours in the presence or absence of SC79 [8.0 μg ml−1] for 48 hours. (A) Cell cycle distribution in Bel-7402 cells was analysed by flow cytometry with PI staining. The percentage of different cell cycle phases was drawn in a histogram to reflect the alteration of cell cycle phases (right panel). The mean and SD obtained from three independent experiments are plotted. (B) Cell viability was measured using CCK-8 assay after 48 hours. (C) After the corresponding treatment p-AKT, AKT, p27 and c-myc protein levels were examined using western blotting. (*P < 0.05 indicates significant difference among the groups). | ||
3.5 MT enhances the sorafenib-mediated inhibition of liver cancer growth in a xenograft tumor model in vivo
To validate the enhanced sorafenib-mediated inhibition of liver cancer growth exerted by MT, we analyzed the effects of MT and sorafenib treatment in vivo using SMMC-7721 xenograft nude mouse model. Single or combined treatment of MT (25 mg kg−1)34 and sorafenib (10 mg kg−1)35 were administered for 18 days, where both the tumor volume and tumor weight were measured. As shown in Fig. 5A–C, treatment with MT and sorafenib together markedly enhanced the inhibitory effect on tumor growth compared with the sorafenib treatment alone in the xenograft model. Furthermore, no significant weight loss (Fig. 5D) or other signs of acute or delayed toxicity were observed during treatment. These data implied that combined treatment of MT and sorafenib may be a potential therapeutic approach with low toxicity for the treatment of HCC.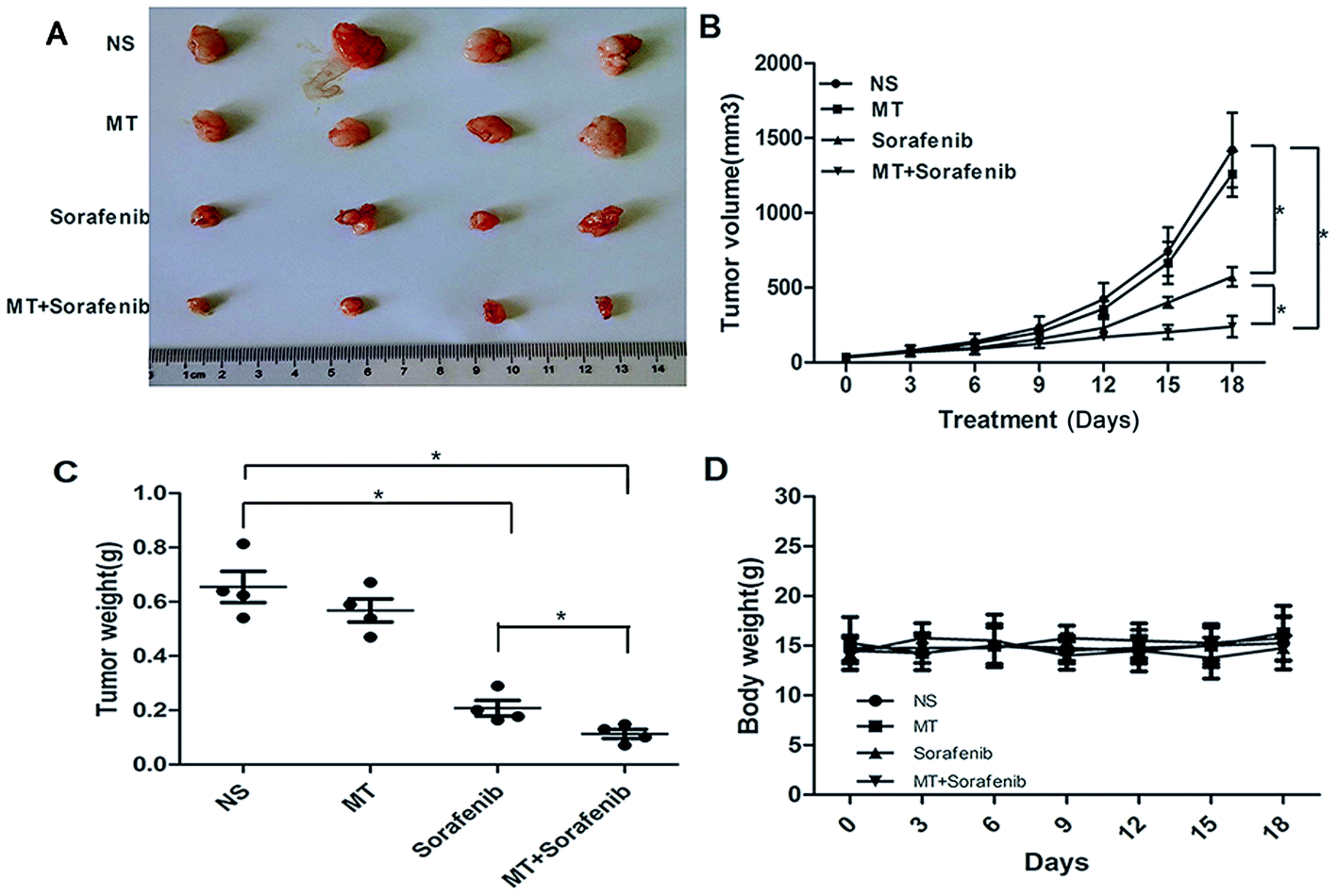 | ||
| Fig. 5 MT enhanced the sorafenib-mediated inhibition of liver cancer growth in a xenograft tumor model in vivo. SMMC-7721 cells were injected subcutaneously into nude mice to evaluate the effects of MT and sorafenib. (A) Tumor pictures. (B) Tumor volumes at different times. (C) Tumor weight. (D) Body weight of mice (*P < 0.05 indicates significant difference among the groups. Four mice were used for each group). | ||
4. Discussion
Hepatocellular carcinoma (HCC) is one of the most common causes of cancer mortality worldwide.36,37 Currently, there are few effective chemotherapy drugs to combat this highly malignant cancer.38 Sorafenib, the first oral multikinase inhibitor approved for the treatment of HCC, was reported to inhibit proliferation of cancer cells and block angiogenesis.39,40 Although sorafenib showed survival benefits in large randomized phase III studies, the response rate is rather low.2,41 In addition, sorafenib has been reported to have several side effects and many patients have to use it at a reduced dose or even stop the treatment.42 In order to increase sorafenib sensitivity and reduce its associated toxicity in advanced HCC, many research groups have explored combination therapies.43–46 MT exhibits an inhibition of cell proliferation, invasiveness and metastatic properties in cancer cell cultures.47,48 Indeed, MT can enhance the activity of many chemotherapeutic agents allowing the use of lower doses of these agents and thus reducing their side effects.49,50In this study, we demonstrated not only the combined anti-tumor effects of MT and sorafenib against HCC in vitro, but also a novel mechanism by which MT sensitized HCC cells to sorafenib. In addition, our study evaluated the anticancer activity of MT-sorafenib combined treatment on tumor growth in xenograft-bearing nude mice. Furthermore, the cytotoxicity of the co-treatment showed no significant difference from that treated with sorafenib, consistent with previous reports.28
There are four phases in the eukaryotic cell cycle. At the G1 phase, cells begin to grow and prepare for protein synthesis, a phase related to DNA replication. Then cells enter the S phase when nuclear DNA is replicated. Cells continue to grow in the G2 phase preparing for mitosis (M). Finally, sister chromatids are separated and distributed to the newly forming daughter cells in the M phase.51 However, the cell cycle of cancer cells is often deregulated as a result of genetic mutations, which lead to uncontrolled cell proliferation.52,53 Therefore, the inhibition of the cell cycle might be a suitable strategy for the treatment of cancer as well as other proliferative diseases.54–56
In the present study, we examined the cell cycle and investigated the inhibition of cell proliferation. Interestingly, we found that treatment of cells with sorafenib alone induced G1/G0 phase arrest in HCC cells, which was further induced by sorafenib and MT combination, consistent with other publications.57 These results suggest that cell arrest at the G1/G0 phase is the biochemical basis for the anticancer effect of the MT-sorafenib combination.
Progression through the eukaryotic cell cycle is governed by a complex regulatory system whose central component is the cyclin-dependent kinase (CDK). CDK activation requires association with a cyclin subunit, as well as phosphorylation of the CDK subunit by the CDK-activating kinase (CAK). The three major classes of cyclins, called G1/S, S, and M cyclins, oscillate in the cell cycle to generate a series of cyclin-CDK complexes that are abruptly switched on at specific cell-cycle transitions.58 The general procedure in mammals is that CDK4 and CDK6 together with cyclin D initiate the phosphorylation of Rb proteins which can release the transcription factor E2F protein, leading to the induction of the transition from G1 to S phase.51 Next, we examined the markers related to cell cycle arrest at G1. Interestingly, we found significant differences between sorafenib treatment alone and combined treatment of sorafenib plus MT regarding the expression of cyclin D1, CDK4, CDK6, and p-Rb. The activities and effects of CDK/cyclin complex at different conditions including stress, DNA damage, and others, are regulated by p27Kip1 which can inhibit CDK4/6/cyclin D, CDK2/cyclin E, CDK2/cyclin A, CDK1/cyclin A, as well as CDK1/cyclin B activity.59,60 It has been reported that the effect of p27Kip1 on the activity of cyclin D-CDK4/6 is determined by the growth state of the cells.61,62 In proliferating cells, p27 can bind to cyclin D-CDK4/6 complex without affecting its activity, whereas p27 can inactivate cyclin D-CDK4/6 complex in quiescent or resting cells.61 p27Kip1 is an inhibitor of a broad spectrum of CDKs, whose transcription and post-translation is regulated by MYC. MYC is an oncogenic transcription factor that can also suppress p27Kip1 expression through several pathways.32 MYC exerts its function at least in part through antagonizing p27 function as CDK inhibitor.63 Previous reports showed that the expression of MYC could be regulated by the AKT pathway.64 Moreover, in the early G1 phase, phosphorylation at Thr157 and Thr198 by AKT caused a delay in nuclear import of p27Kip1,33 which functions in the nucleus. Vanesa Martín and colleagues showed that MT inhibits AKT activity in human glioma cells without modifying MAPKs (ERK, p38, and JNK) activity.50 Bo Zhai et al. showed that inhibition of AKT reversed the acquired resistance to sorafenib.65 Therefore, we next questioned whether p-AKT could regulate p27 via c-myc, and thus affecting cell cycle progression. Consistent with the expression of these proteins, combined treatment of sorafenib and MT plus SC79, a p-AKT activator, restored a lower G0/G1 percentage of cells and cell viability, as well as the expression of c-myc, p27 and cell cycle-related proteins. This indicates that AKT can regulate c-myc, p27 and, subsequently, cell cycle. Thus AKT is a crucial factor in the enhanced antitumor effect of co-treatment of sorafenib and MT compared with sorafenib treatment alone. Also, ERK has been reported that it is related to cell proliferation66 and MT could inhibit the expression of p-ERK in SK-OV-3 cells.28 Moreover, as a CDK inhibitor, p21 can inhibit multiple CDK/cyclin complexes through various ways.67 Accordingly, we examined the expressions of p-ERK and p21. To our disappointment, combination of MT and sorafenib could not change the expressions of the two proteins compared to sorafenib group (ESI Fig. S1†). Therefore, we guess that MT modulation of cell cycle seems to be cell-type and context-dependent, similar to its effects on other signaling pathways. The proposed model elucidating the role of MT in regulating the proliferation of hepatocellular carcinoma (HCC) cells treated with sorafenib is shown in Fig. 6. However, further studies need to be performed in order to investigate this interesting phenotype and pinpoint the precise molecular mechanism.
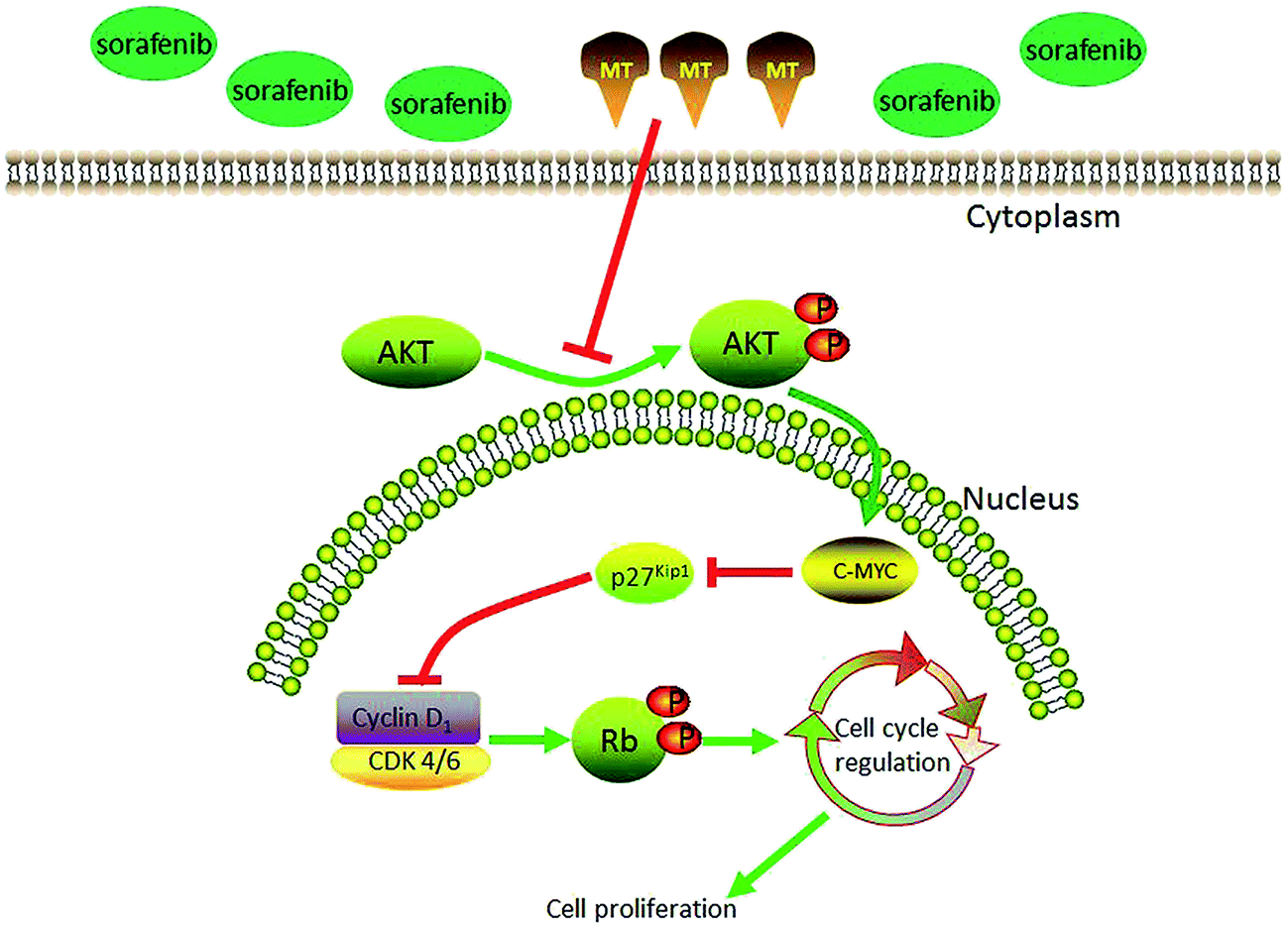 | ||
| Fig. 6 Proposed model elucidating the role of MT in regulating the proliferation of hepatocellular carcinoma (HCC) cells treated with sorafenib. | ||
5. Conclusions
In summary, our results indicate that co-treatment of MT and sorafenib potentiated the antitumor activity and arrested the cell cycle via regulation of p-AKT. Therefore, our findings suggest that MT, which has demonstrated no significant toxicity to normal liver cells treated with sorafenib, can be a useful chemotherapeutic agent for the improvement of the therapeutic effect of sorafenib in liver cancer.Acknowledgements
This work was supported by funds from the National Natural Science Foundation of China (81471755) and the clinical capability construction project for Liaoning provincial hospitals (LNCCC-B03–2014).References
- H. B. El-Serag and K. L. Rudolph, Gastroenterology, 2007, 132, 2557–2576 CrossRef CAS PubMed.
- J. M. Llovet, S. Ricci, V. Mazzaferro, P. Hilgard, E. Gane, J. F. Blanc, A. C. de Oliveira, A. Santoro, J. L. Raoul, A. Forner, M. Schwartz, C. Porta, S. Zeuzem, L. Bolondi, T. F. Greten, P. R. Galle, J. F. Seitz, I. Borbath, D. Haussinger, T. Giannaris, M. Shan, M. Moscovici, D. Voliotis, J. Bruix and S. I. S. Group, N. Engl. J. Med., 2008, 359, 378–390 CrossRef CAS PubMed.
- Y. H. Hwang, J. Y. Choi, S. Kim, E. S. Chung, T. Kim, S. S. Koh, B. Lee, S. H. Bae, J. Kim and Y. M. Park, Hepatol. Res., 2004, 29, 113–121 CrossRef CAS PubMed.
- M. A. Avila, C. Berasain, B. Sangro and J. Prieto, Oncogene, 2006, 25, 3866–3884 CrossRef CAS PubMed.
- M. A. Worns and P. R. Galle, Nat. Rev. Gastroenterol. Hepatol., 2014, 11, 447–452 CrossRef PubMed.
- C. Berasain, Gut, 2013, 62, 1674–1675 CrossRef CAS PubMed.
- C. Zavaglia, A. Airoldi, A. Mancuso, M. Vangeli, R. Viganò, G. Cordone, M. Gentiluomo and L. S. Belli, Eur. J. Gastroenterol. Hepatol., 2013, 25, 180–186 CrossRef CAS PubMed.
- R. J. Reiter, Endocr. Rev., 1991, 12, 151–180 CrossRef CAS PubMed.
- J. Vanecek, Physiol. Rev., 1998, 78, 687–721 CAS.
- D. X. Tan, L. C. Manchester, L. Fuentes-Broto, S. D. Paredes and R. J. Reiter, Obes. Rev., 2011, 12, 167–188 CrossRef CAS PubMed.
- A. Carrillo-Vico, J. M. Guerrero, P. J. Lardone and R. J. Reiter, Endocrine, 2005, 27, 189–200 CrossRef CAS PubMed.
- E. Mills, P. Wu, D. Seely and G. Guyatt, J. Pineal Res., 2005, 39, 360–366 CrossRef CAS PubMed.
- B. Jung-Hynes, R. J. Reiter and N. Ahmad, J. Pineal Res., 2010, 48, 9–19 CrossRef CAS PubMed.
- S. Y. Shiu, B. Pang, C. W. Tam and K. M. Yao, J. Pineal Res., 2010, 49, 301–311 CrossRef CAS PubMed.
- P. T. Ram, J. Dai, L. Yuan, C. Dong, T. L. Kiefer, L. Lai and S. M. Hill, Cancer Lett., 2002, 179, 141–150 CrossRef CAS PubMed.
- L. Mao, Q. Cheng, B. Guardiola-Lemaitre, C. Schuster-Klein, C. Dong, L. Lai and S. M. Hill, J. Pineal Res., 2010, 49, 210–221 CrossRef CAS PubMed.
- O. Trubiani, R. Recchioni, F. Moroni, J. Pizzicannella, S. Caputi and R. Di Primio, J. Pineal Res., 2005, 39, 425–431 CrossRef CAS PubMed.
- S. Rubio, F. Estevez, J. Cabrera, R. J. Reiter, J. Loro and J. Quintana, J. Pineal Res., 2007, 42, 131–138 CrossRef CAS PubMed.
- G. Garcia-Santos, I. Antolin, F. Herrera, V. Martin, J. Rodriguez-Blanco, M. del Pilar Carrera and C. Rodriguez, J. Pineal Res., 2006, 41, 130–135 CrossRef CAS PubMed.
- G. Z. Omurtag, A. Tozan, A. O. Sehirli and G. Sener, J. Pineal Res., 2008, 44, 432–438 CrossRef CAS PubMed.
- M. Valko, H. Morris and M. T. Cronin, Curr. Med. Chem., 2005, 12, 1161–1208 CrossRef CAS PubMed.
- R. J. Reiter, D.-X. Tan, R. M. Sainz, J. C. Mayo and S. Lopez-Burillo, J. Pharm. Pharmacol., 2002, 54, 1299–1321 CrossRef CAS PubMed.
- E. Esposito, A. Iacono, C. Muia, C. Crisafulli, G. Mattace Raso, P. Bramanti, R. Meli and S. Cuzzocrea, J. Pineal Res., 2008, 44, 78–87 CAS.
- F. Herrera, V. Martin, G. Garcia-Santos, J. Rodriguez-Blanco, I. Antolin and C. Rodriguez, J. Neurochem., 2007, 100, 736–746 CrossRef CAS PubMed.
- E. Peschke, J. Pineal Res., 2008, 44, 26–40 CAS.
- P. Lissoni, S. Barni, M. Mandala, A. Ardizzoia, F. Paolorossi, M. Vaghi, R. Longarini, F. Malugani and G. Tancini, Eur. J. Cancer, 1999, 35, 1688–1692 CrossRef CAS PubMed.
- S. Klongpanichapak, P. Phansuwan-Pujito, M. Ebadi and P. Govitrapong, J. Pineal Res., 2007, 43, 65–73 CrossRef CAS PubMed.
- J. H. Kim, S. J. Jeong, B. Kim, S. M. Yun, D. Y. Choi and S. H. Kim, J. Pineal Res., 2012, 52, 244–252 CrossRef CAS PubMed.
- Z. A. Stewart, M. D. Westfall and J. A. Pietenpol, Trends Pharmacol. Sci., 2003, 24, 139–145 CrossRef CAS PubMed.
- J. A. Pietenpol, S. K. Bohlander, Y. Sato, N. Papadopoulos, B. Liu, C. Friedman, B. J. Trask, J. M. Roberts, K. W. Kinzler and J. D. Rowley, et al., Cancer Res., 1995, 55, 1206–1210 CAS.
- K. Polyak, J. Y. Kato, M. J. Solomon, C. J. Sherr, J. Massague, J. M. Roberts and A. Koff, Genes Dev., 1994, 8, 9–22 CrossRef CAS PubMed.
- S. S. Hnit, C. Xie, M. Yao, J. Holst, A. Bensoussan, P. De Souza, Z. Li and Q. Dong, Int. J. Biochem. Cell Biol., 2015, 68, 9–14 CrossRef CAS PubMed.
- D. Morishita, R. Katayama, K. Sekimizu, T. Tsuruo and N. Fujita, Cancer Res., 2008, 68, 5076–5085 CrossRef CAS PubMed.
- J. J. Lu, L. Fu, Z. Tang, C. Zhang, L. Qin, J. Wang, Z. Yu, D. Shi, X. Xiao, F. Xie, W. Huang and W. Deng, Oncotarget, 2016, 7, 2985–3001 Search PubMed.
- Y. Liang, T. Zheng, R. Song, J. Wang, D. Yin, L. Wang, H. Liu, L. Tian, X. Fang, X. Meng, H. Jiang, J. Liu and L. Liu, Hepatology, 2013, 57, 1847–1857 CrossRef CAS PubMed.
- S. Waly Raphael, Z. Yangde and C. Yuxiang, ISRN Oncol., 2012, 2012, 421673 Search PubMed.
- A. Forner, J. M. Llovet and J. Bruix, Lancet, 2012, 379, 1245–1255 CrossRef.
- R. Cabrera and D. R. Nelson, Aliment. Pharmacol. Ther., 2010, 31, 461–476 CrossRef CAS PubMed.
- A. Galmiche, B. Chauffert and J. C. Barbare, Cancer Lett., 2014, 346, 159–162 CrossRef CAS PubMed.
- V. Di Marco, F. De Vita, J. Koskinas, D. Semela, P. Toniutto and C. Verslype, Ann. Oncol., 2013, 24(2), 30–37 Search PubMed.
- A. L. Cheng, Y. K. Kang, Z. Chen, C. J. Tsao, S. Qin, J. S. Kim, R. Luo, J. Feng, S. Ye, T. S. Yang, J. Xu, Y. Sun, H. Liang, J. Liu, J. Wang, W. Y. Tak, H. Pan, K. Burock, J. Zou, D. Voliotis and Z. Guan, Lancet Oncol., 2009, 10, 25–34 CrossRef CAS PubMed.
- J. T. Hartmann, M. Haap, H. G. Kopp and H. P. Lipp, Curr. Drug Metab., 2009, 10, 470–481 CrossRef CAS PubMed.
- B. Zhai and X. Y. Sun, World J. Hepatol., 2013, 5, 345–352 CrossRef PubMed.
- C. H. Hsu, Y. C. Shen, Z. Z. Lin, P. J. Chen, Y. Y. Shao, Y. H. Ding, C. Hsu and A. L. Cheng, J. Hepatol., 2010, 53, 126–131 CrossRef CAS PubMed.
- A. Y. Chang and M. Wang, Anti-Cancer Drugs, 2013, 24, 251–259 CrossRef CAS PubMed.
- C. Hsu, Y. C. Shen and A. L. Cheng, Expert Rev. Clin. Pharmacol., 2009, 2, 129–136 CrossRef CAS PubMed.
- S. Cos, R. Fernandez, A. Guezmes and E. J. Sanchez-Barcelo, Cancer Res., 1998, 58, 4383–4390 CAS.
- M. M. Marelli, P. Limonta, R. Maggi, M. Motta and R. M. Moretti, Prostate, 2000, 45, 238–244 CrossRef CAS PubMed.
- S. Casado-Zapico, J. Rodriguez-Blanco, G. Garcia-Santos, V. Martin, A. M. Sanchez-Sanchez, I. Antolin and C. Rodriguez, J. Pineal Res., 2010, 48, 72–80 CrossRef CAS PubMed.
- V. Martín, G. García-Santos, J. Rodriguez-Blanco, S. Casado-Zapico, A. Sanchez-Sanchez, I. Antolín, M. Medina and C. Rodriguez, Cancer Lett., 2010, 287, 216–223 CrossRef PubMed.
- H. Harashima, N. Dissmeyer and A. Schnittger, Trends Cell Biol., 2013, 23, 345–356 CrossRef CAS PubMed.
- G. I. Evan and K. H. Vousden, Nature, 2001, 411, 342–348 CrossRef CAS PubMed.
- L. H. Hartwell and M. B. Kastan, Science, 1994, 266, 1821–1828 CAS.
- K. W. Kohn, J. Jackman and P. M. O'Connor, J. Cell. Biochem., 1994, 54, 440–452 CrossRef CAS PubMed.
- B. T. Hill and R. Baserga, Cancer Treat. Rev., 1975, 2, 159–175 CrossRef CAS PubMed.
- W. J. Chen and J. K. Lin, J. Biol. Chem., 2004, 279, 13496–13505 CrossRef CAS PubMed.
- Q. Cao, Y. Jiang, J. Shi, X. Liu, J. Chen, T. Niu and X. Li, Clin. Exp. Pharmacol. Physiol., 2015, 42, 502–509 CrossRef CAS PubMed.
- D. O. Morgan, Cell, 2008, 135, 764 CrossRef CAS PubMed.
- H. Toyoshima and T. Hunter, Cell, 1994, 78, 67–74 CrossRef CAS PubMed.
- O. Aprelikova, Y. Xiong and E. T. Liu, J. Biol. Chem., 1995, 270, 18195–18197 CrossRef CAS PubMed.
- M. K. James, A. Ray, D. Leznova and S. W. Blain, Mol. Cell. Biol., 2008, 28, 498–510 CrossRef CAS PubMed.
- A. Ray, M. K. James, S. Larochelle, R. P. Fisher and S. W. Blain, Mol. Cell. Biol., 2009, 29, 986–999 CrossRef CAS PubMed.
- G. Bretones, M. D. Delgado and J. Leon, Biochim. Biophys. Acta, 2015, 1849, 506–516 CrossRef CAS PubMed.
- M. A. Dominguez-Caceres, J. M. Garcia-Martinez, A. Calcabrini, L. Gonzalez, P. G. Porque, J. Leon and J. Martin-Perez, Oncogene, 2004, 23, 7378–7390 CrossRef CAS PubMed.
- B. Zhai, F. Hu, X. Jiang, J. Xu, D. Zhao, B. Liu, S. Pan, X. Dong, G. Tan, Z. Wei, H. Qiao, H. Jiang and X. Sun, Mol. Cancer Ther., 2014, 13, 1589–1598 CrossRef CAS PubMed.
- M. Cully and J. Downward, Cell, 2008, 133, 1292 CrossRef CAS PubMed.
- Y. Wang, J. C. Fisher, R. Mathew, L. Ou, S. Otieno, J. Sublet, L. Xiao, J. Chen, M. F. Roussel and R. W. Kriwacki, Nat. Chem. Biol., 2011, 7, 214–221 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02113e |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |