
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Poly(vinyl alcohol)–cationic cellulose copolymer encapsulated SiO2 stationary phase for hydrophilic interaction liquid chromatography†
Yahui Peng,
Feifang Zhang,
Xiao Pan,
Yanjie Hou and
Bingcheng Yang *
*
School of Pharmacy, East-China University of Science and Technology, Shanghai 200237, China. E-mail: bcyang@ecust.edu.cn; Tel: +86-21-64250627
First published on 19th April 2017
Abstract
A poly(vinyl alcohol)–cationic cellulose (PVA–CC) copolymer encapsulated SiO2 stationary phase (PVA–CC–Sil) for hydrophilic interaction liquid chromatography (HILIC) is described. It is prepared simply by combining SiO2 with a preformed PVA–CC solution, then filtering and curing thermally, finally generating a thin PVA–CC copolymer coating onto SiO2. The preparation is easy to implement and no organic solvent is involved in. The phase shows obvious positive electrostatic character and typical HILIC character, exhibiting different selectivity relative to twelve commercial HILIC phases. Good running stability and a wide pH tolerance range (at least 10) are observed for PVA–CC–Sil. Excellent separation efficiency is obtained for model analytes (e.g. plate counts are ∼114![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 plates per m and ∼150000 plates per m for ribose and 4-hydroxybenzoic acid, respectively). Good separation performance for saccharides of low or high degrees of polymerization by PVA–CC–Sil is also observed.
000 plates per m and ∼150000 plates per m for ribose and 4-hydroxybenzoic acid, respectively). Good separation performance for saccharides of low or high degrees of polymerization by PVA–CC–Sil is also observed.
1. Introduction
Since being coined by Alpert in 1990,1 hydrophilic interaction liquid chromatography (HILIC) has been an effective technique for the separation of polar analytes by using a polar stationary phase and an organic-rich aqueous mobile phase. To meet various separation needs, many types of novel polar stationary phases have been developed with versatile functional groups such as amide,2 amino,3 zwitterionic groups,4 ionic liquids5 and macromolecular carbohydrates/saccharides.6–9 This is different from common reversed-phase liquid chromatography (RPLC), in which just the C18 phase is most commonly used and is a standard choice for a large number of developed methods.Saccharides have attracted some attention due to their important biological functions.10 However, due to their high polarity, much similar chemical compositions and lack of chromophores for optical detection, their separation or detection remains challenging. Common RPLC is not a good option for the separation of saccharides owing to their much poor retention onto C18. Besides ion chromatography equipped with pulsed amperometric detector,11,12 HILIC has also received much interest for the analysis of saccharides in recent years.8,9,13 Some silica-based stationary phases with saccharide functional groups have been accordingly developed. Via a click chemistry approach, a number of saccharide-functionalized HILIC phases have been reported,6,7,9 such as β-cyclodextrin (β-CD),6 maltose.7 In addition, Sheng et al. described a cationic cellulose agglomerated sulfonated silica phase prepared by a three-step synthetic strategy, including the initial modification of silica with γ-mercaptopropyltriethoxysilane, subsequent sulfonation via oxidation and final agglomeration between cationic cellulose and negatively sulfonated silica (termed as CC-300).13 Their subsequent work reported a dextran-bonded stationary phase for saccharide separation by immobilizing polysaccharide onto the silica gel.14 To prepare chemical bonded stationary phases, multi-stage reactions and the use of organic solvents are always required, which are both tedious and environmentally-unfriendly. Thus a facile and green synthetic approach would be very desirable for developing saccharide-modified HILIC phases while avoiding these issues.
As a highly hydrophilic polymer, polyvinyl alcohol (PVA) has found many applications in many fields.15–17 Recently, we described a PVA encapsulated silica gel stationary phase for HILIC by generating one (or more) thin layer(s) of coating onto silica particles to form a water-like hydrogel.18,19 The PVA coating displayed strong affinity to the silica gel and the obtained phase exhibited good HILIC character and a wider pH tolerance range in comparison to bare silica gel.18 The ability of PVA to entrap various gels or be doped with a desired ingredient can be extended to prepare a wide variety of stationary phases with different supports or exchangeable functionalities. This is accessible even to those without the skills of a synthetic chemist. More recently, the former has been proved by preparing a PVA encapsulated porous graphic carbon phase for HILIC, which showed good hydrophilic character and excellent chromatographic efficiency.20 The latter will be explored in the present report to prepare a saccharide-functionalized stationary phase. Cationic cellulose (CC) is a nontoxic macromolecule rich with repeated sugar units and an ammonium group, thus bearing superior hydrophilicity, solubility and is compatible with anionic, cationic, nonionic and zwitterionic surfactants. By generating a copolymer of PVA and cationic cellulose (CC) onto silica, it is supposed to yield a positively charged hydrophilic phase with the potential for separating polar analytes including acids and saccharides. Owing to the protection of the generated copolymer coating, pH tolerance range of the obtained phase should also be extended relative to bare silica gel. More importantly, such method avoids the drawback of lengthy preparation procedures of chemical bonding method.
2. Experimental
2.1 Chemicals and materials
Spherical silica was obtained from Fuji Silysia Chemical Ltd. (Kasugai, Japan; 5 μm dia.; 100 Å pore size; 300 m2 g−1 surface area). PVA (DP 1750 ± 50) was from Ling Feng Chemical Reagent Corp. (Shanghai, China). Cationic cellulose JR-400 (Polyquaternium-10, MW 1.5 × 106 to 2.0 × 106) was purchased from Weixi Chem. Co. Ltd. (Guangzhou, China). Ammonium formate (NH4FA, 99%) and formic acid (FA, 99%) were purchased from Acros (Fair Lawn, USA). Ammonium acetate (AcONH4, 98%) was from J&K Scientific Ltd. (Beijing, China). HPLC grade acetonitrile (ACN, 99.9%) and methanol (MeOH, 99.8%) were available from Tedia (Ohio, USA). The used model analytes were from Sigma and their chemical structures were shown in ESI-Scheme 1.† Ultrapure water was provided by a Milli-R04 purification system from Millipore Corp. (Billerica, MA, USA).2.2 Apparatus
Chromatographic experiments were conducted on a Waters Alliance HPLC system (Milford, MA, USA) that equipped with a 2695 separation module, an autosampler, a column oven, a 2489 UV-VIS detector and a 2424 evaporative light-scattering detector (ELSD). Chromatograms were recorded on a computer with Empower 3 workstation software. Unless otherwise stated, the separation was conducted at the flow rate of 1 mL min−1 and the column temperature of 30 °C.2.3 Preparation and column packing
The preparation procedure of PVA–CC–Sil was same to that of previous description with some small modifications.18 Briefly, PVA (3 g) and cationic cellulose (1 g) were mixed in 50 mL water, and then heated in an oil bath at 95 °C with agitation until a homogeneous solution was obtained. Followed by the addition of 4 g silica gel, the solution was treated by sonication for 5 min. The mixture was magnetically stirred at 250 rpm for 30 min and then filtered through a G3 sand core funnel. The obtained residue was finally heated at 140 °C for 1 h to form a partially cross-linked PVA–CC copolymer coating onto silica. The preparation route was schematically illustrated in Fig. 1.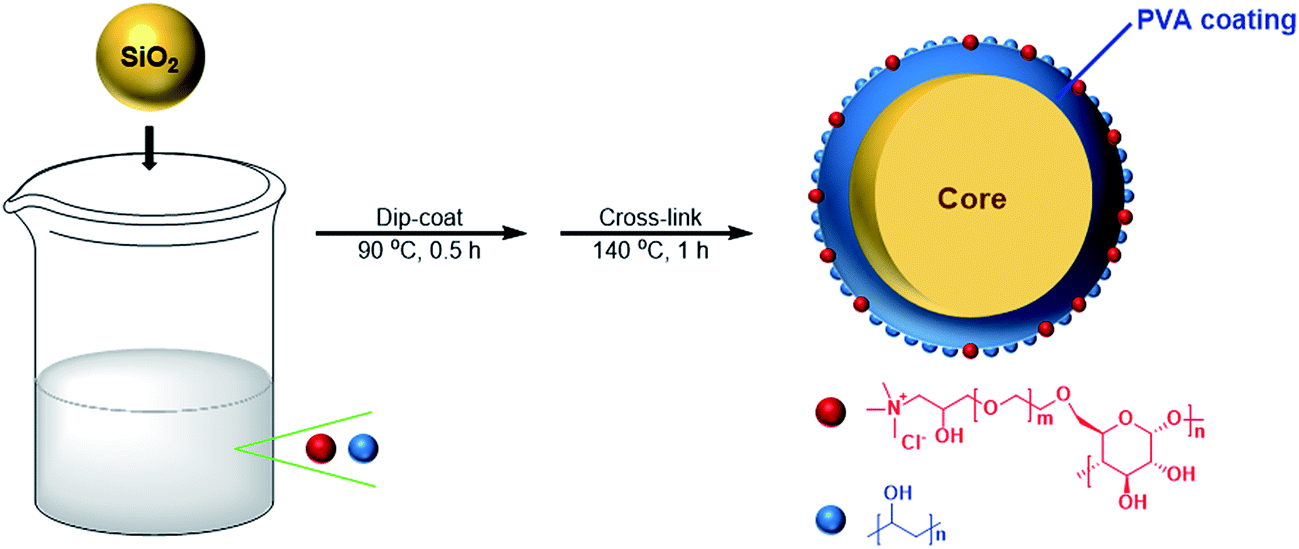 | ||
| Fig. 1 Schematic diagram of the preparation route of PVA–CC–Sil. | ||
PVA–CC–Sil (2.5 g) was slurry-packed into a stainless steel tube (4.6 mm i.d. × 150 mm length) using methanol as slurry and propulsion solvent. For comparison, 15 columns were used, including a homemade packed PVA-coated silica gel one described previously, seven domestic and seven imported ones. Among which, an XAmide, a Unitary Diol, a Unitary NH2, a Click XIon, a Click SAX, an XAqua C18 and an XCharge C18 were available from ACCHROM Corp. (Zhejiang, China). Besides, a Fuji silica gel column, a Waters Spherisorb NH2, a Thermo Scientific Synchronis HILIC, an Accucore HILIC, an Accucore-150-Amide-HILIC, an Acclaim HILIC-10 and an Acclaim Mixed-Mode HILIC-1 were available from popular column manufacturers. All columns tested shared a same size of 4.6 mm i.d. × 150 mm length and a same particle size (5 μm dia.) except for 2.6 μm of Accucore HILIC and Accucore-150-Amide-HILIC, and 3 μm of Click XIon and Acclaim HILIC-10. Further detail information of the domestic columns was given in ESI-Table 1.†
3. Results and discussion
3.1 Characterization of PVA–CC–Sil
The morphology of PVA–CC–Sil was analyzed by scanning electron micrographs (SEM), as can be seen in ESI-Fig. 1.† Results showed the monodispersity and spherical shape of silica particles were well maintained after being encapsulated by PVA/CC copolymers and a slight increase of surface roughness was also observed. Treatments of N2 adsorption–desorption isotherms with the Brunauer–Emmett–Teller (BET) equation and pore size distribution with Barrett–Joyner–Halenda (BJH) equation manifested decreases of surface area, pore volume and pore diameter in PVA–CC–Sil (data shown in ESI-Table 3 and ESI-Fig. 2†), indicating the successful modification of PVA/CC copolymers onto silica particles.The characterization of elemental analysis was listed in ESI-Table 2.† Relative to bare silica, an obvious increase of N% in PVA–CC–Sil was observed. The C/N mass ratio in PVA–CC–Sil was ∼4.1 times higher than that of CC. These clearly indicated the successful incorporation of CC into the formed copolymer. The loading amount of CC for PVA–CC–Sil was calculated to be ∼53.9 mg g−1.
3.2 The surface charge of PVA–CC–Sil
The surface charge of PVA–CC–Sil was characterized via zeta-potential measurement, as shown in Fig. 2. Bare silica was positively charged in the solutions at pH < 3.5. PVA–Sil showed higher positive potentials than bare silica while both presented a similar variation tendency. By contrast, PVA–CC–Sil remained obviously positively charged in the pH until 6.7, indicating its positive surface charge state within this range.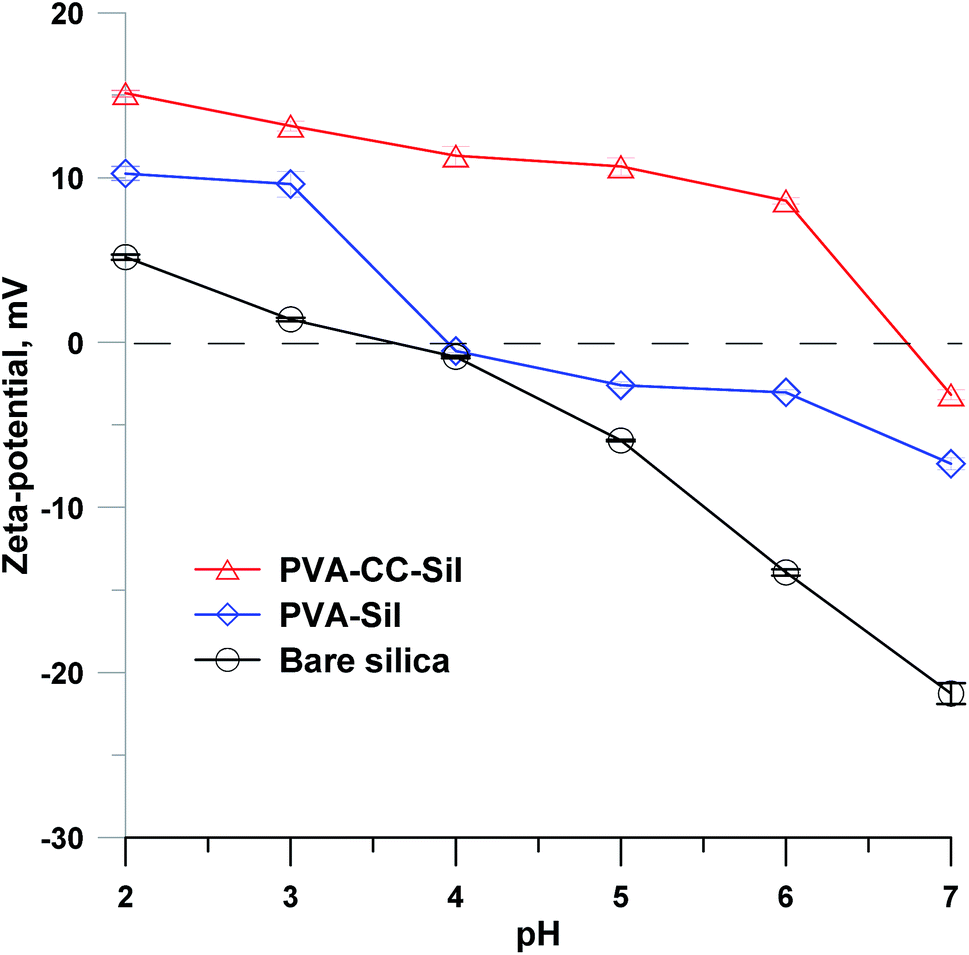 | ||
| Fig. 2 Zeta-potentials of bare silica, PVA–Sil and PVA–CC–Sil in 20 mM NH4FA solutions at different pH values. Measurement temperature, 25 °C. | ||
3.3 Chromatographic evaluation of PVA–CC–Sil under HILIC mode
To evaluate the chromatographic performance of PVA–CC–Sil, three kinds of polar analytes were chosen as models, including nucleosides, nucleotides and aromatic acids (their chemical structures were shown in ESI-Scheme 1†). The chromatogram of seven nucleosides on PVA–CC–Sil is shown in Fig. 3b. Good separation could be achieved and their elution order is consistent with their polarity. Comparative separations on three commercial HILIC phases resulted in insufficient selectivity, as can be seen in ESI-Fig. 3.† Since nucleosides are neutral compounds, the positively charged character of PVA–CC–Sil cannot be effectively reflected. Thus five aromatic acids were also used to evaluate the possible anion exchange and hydrophilic characters of PVA–CC–Sil. Fig. 3a shows the separation of aromatic acids with pKa values ranging from 2.97–4.44. Five aromatic acids were well separated in less than 4 min and high efficiency was achieved, e.g. the plate count was ∼150000 plates per m for 4-hydroxybenzoic acid. Their elution order was basically opposite to their pKa values, in which the acid with lower pKa had a higher dissociation degree at the tested pH (3.48), then leading to stronger interaction due to electrostatic attraction between the negatively charged acid and positively charged PVA–CC–Sil. Comparative chromatograms on commercial columns are given in ESI-Fig. 4,† in which inferior separation selectivity and efficiency were observed.
 | ||
| Fig. 3 Chromatograms of (a) aromatic acids and (b) nucleosides onto PVA–CC–Sil. Conditions: (a) 90% ACN/10% H2O (250 mM HCOONH4, pH 3.48), (b) 90% ACN/10% H2O; analytes, 1, cinnamic acid; 2, benzoic acid; 3, 4-hydroxybenzoic acid; 4, salicylic acid; 5, p-nitrobenzoic acid; 6, uracil; 7, 5-methyl uridine; 8, uridine; 9, adenosine; 10, adenine; 11, cytosine; 12, cytidine; injection volume, 5 μL; flow rate, 1.0 mL min−1; absorbance wavelength, 254 nm. | ||
Compared to nucleosides, nucleotides contain additional phosphate groups (chemical structures shown in ESI-Scheme 1†), which render them highly polar, then resulting in stronger retention on the polar stationary phase. Fig. 4 presents the separation of nucleotides onto PVA–CC–Sil and five nucleotides can be well separated at an elevated pH value (10), which is beyond the typical pH operation range of silica. Comparatively, poor retention was observed on three commercial HILIC columns at pH 8 (see ESI-Fig. 5†). The existence of PVA–CC copolymer offers a wider pH tolerance range for silica gel since it shields silica gel inside from erosion of solution outside, which has been proved in our previous report.18 Such has been further confirmed by the fact that no obvious change was found after ∼6900-fold column volumes of continuously operation using the mobile phase at pH 10 (data not shown).
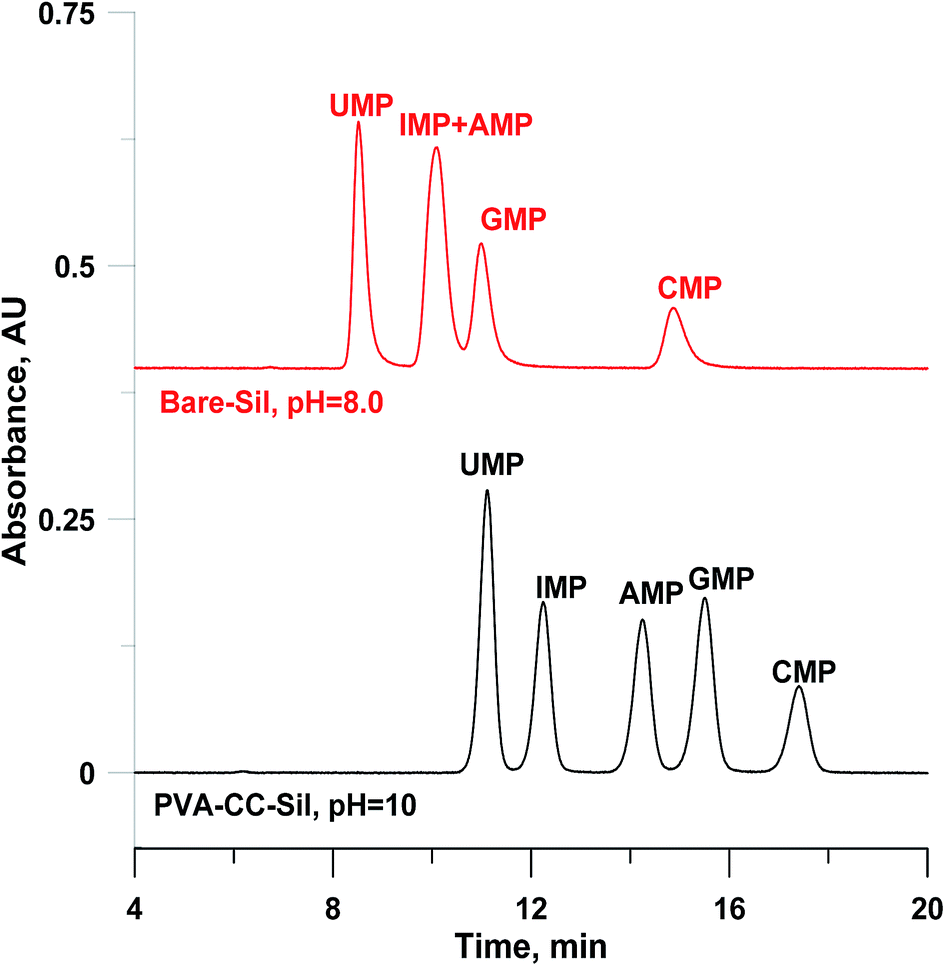 | ||
| Fig. 4 Chromatograms of nucleotides onto silica and PVA–CC–Sil. Conditions: mobile phase, A, H2O; B, ACN; C, 200 mM HCOONH4, pH as indicated, 0–30 min, A/B/C (v/v/v), 20/70/10 → 25/65/10; injection volume, 3 μL; other conditions same as Fig. 3. | ||
The existence of Si–OH may cause unwanted electrostatic interaction when separating alkaline compounds, then leading to peak tailing. The introduced PVA–CC–Sil coating would reduce such interaction and here such potential has been explored by separating four small molecular bases. As shown in ESI-Fig. 6,† peak tailing on PVA–CC–Sil was much improved compared with bare silica gel, demonstrating the shielding effect of the copolymer coating on silica particles.
Good running stability of PVA encapsulated SiO2 or porous graphic carbon has been well proved previously.18,20 Here the running stability of PVA–CC–Sil was also investigated. PVA–CC–Sil exhibited excellent inter-day and intra-day repeatability, indicated by the fact that inter-day and intra-day relative standard deviations (RSDs) of retention time (tR) achieved for all five nucleotides (eluent pH 10) were in the range of 0.21–0.38% (n = 9) and 2.39–2.74% (n = 3), respectively. Moreover, the running stability of PVA–CC–Sil was also studied by the retention change of three model analytes (cytidine, dirophyline, and 4-aminobenzoic acid, representing neutral, positively, and negatively charged, respectively) over 40 h (∼1850-fold column volume) of continuous operation using the eluent of 75% ACN/25% 20 mM NH4FA (pH 6.8). Three consecutive injections were performed every 5 h. As shown in ESI-Fig. 7,† the tR and plate count (N) for all three model analytes were almost constant throughout the test (0.27–1.39% RSDs), demonstrating its good stability. Good batch-to-batch repeatability was also observed, as indicated by the RSDs of tR for the five above-mentioned nucleotides (n = 3) were in the range of 1.04–5.03%.
3.4 Selectivity of PVA–CC–Sil
Based on ref. 20–22, three pairs of analytes were chosen to probe the primal interactions involved in PVA–CC–Sil phase, in which kcytosine/kuracil, ksalicylic acid/kcytosine and kadenosine acid/kadenine were used to measure the hydrophilicity, anion exchange and H-bonding characters, respectively (here k standing for the retention factor). The selectivity of PVA–CC–Sil was depicted in a 3D plot along with those of fifteen phases including thirteen HILIC and two RPLC ones (Fig. 5). As can be seen, PVA–CC–Sil occupies a unique position in having strong HILIC character with a similar index (∼2.42) to those of Accucore-150-Amide-HILIC (∼2.32), Spherisorb NH2 (∼2.43), Click XIon (∼2.54) and PVA–Sil (∼2.55), while Accucore HILIC (∼2.98) and Accucore-150-Amide-HILIC (∼3.18) exhibit the highest hydrophilicity. Strong anion exchange character is also displayed by PVA–CC–Sil (index ∼0.56), which is only lower than two pure strong anion exchangers Click-SAX and Spherisorb NH2 (indexes ∼0.821 and ∼1.02, respectively). The H-bonding index of PVA–CC–Sil is mediate among the phases studied. The results indicate that PVA–CC–Sil has above average HILIC, anion exchange and H-bonding mixed characters.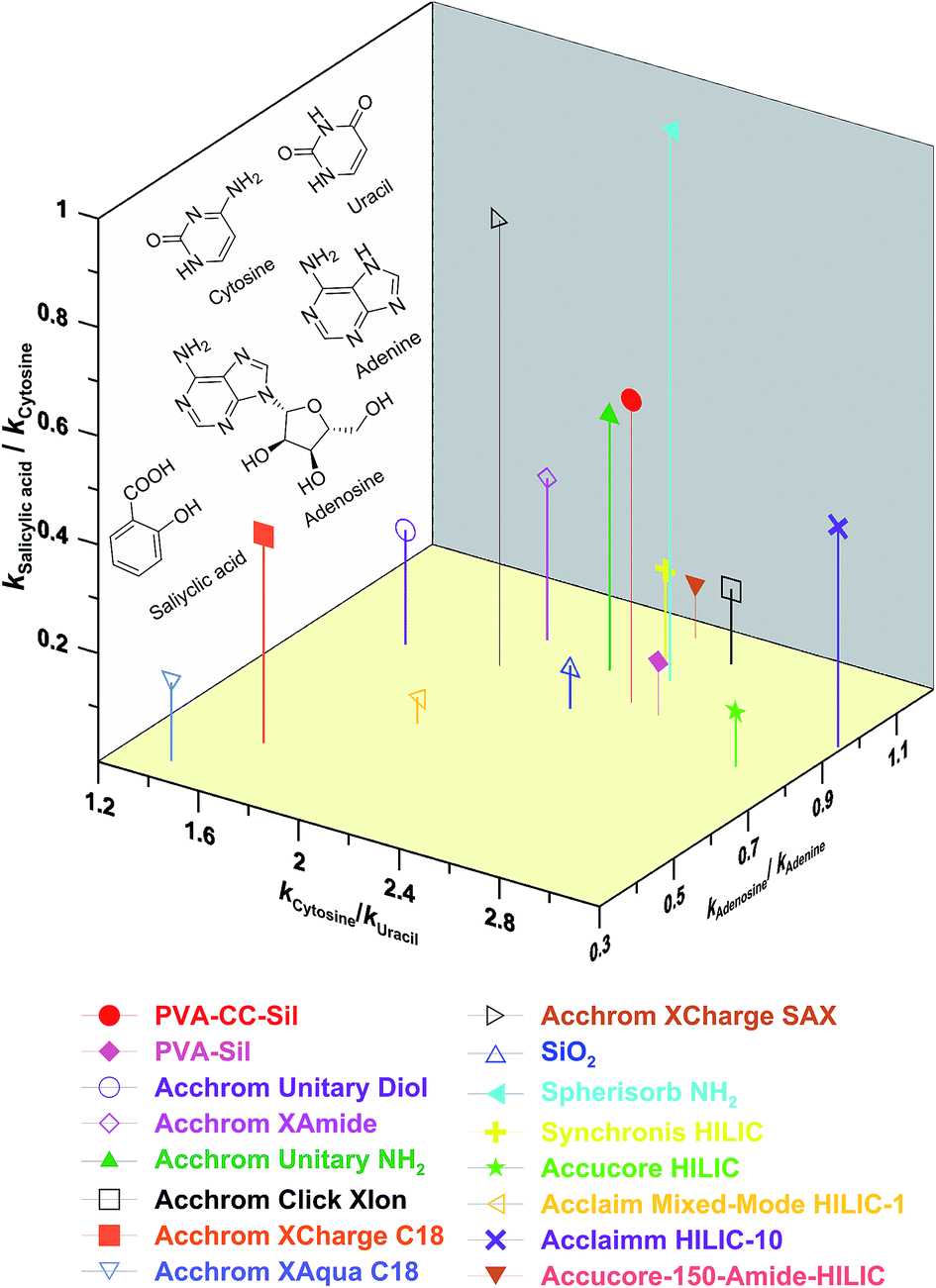 | ||
| Fig. 5 Selectivity plot of PVA–CC–Sil and other columns. Conditions, mobile phase, 80% ACN/20% H2O (25 mM NH4OAc, pH 6.81); flow rate, 0.5 mL min−1; other conditions same as Fig. 3. | ||
3.5 Retention mechanism investigation
The possible retention mechanism of PVA–CC–Sil was investigated as follows.Firstly, the effect of water content in the mobile phase on the retention of five nucleosides was studied by varying the volume fraction of water from 5–25% (see ESI-Fig. 8†). The natural logarithm of retention factor (lnk) was plotted against the volume fraction of water (φ). The plots were highly fitted (r2 0.9998–0.9999) with the quantitative HILIC retention model proposed previously23 (lnk = a + blnφ + cφ where a, b, and c are constants).
Secondly, since the addition of electrolyte in the mobile phase used in HILIC is found to be helpful to increased retention or improved peak shape,24 here such effect was explored by choosing aromatic acids as models (see ESI-Fig. 9†), aiming to investigate the possible anion exchange and HILIC mechanism. Plots of k vs. 1/M+ (1/M+ standing for the reciprocal of electrolyte concentration) show the retention decreases as M+ increases, suggesting a greater contribution from weakened electrostatic interaction than the increase in polarity of the water-rich layer. But non-linear plots indicate other mechanisms also influences the retention of these acids to a certain extent besides electrostatic interaction.25
Thirdly, the thermodynamics of PVA–CC–Sil was also explored by choosing nucleosides as models (ESI-Fig. 10†). Clearly, their retention decreases with the decrease of column temperature and the plots are well fitted with van't Hoff equation 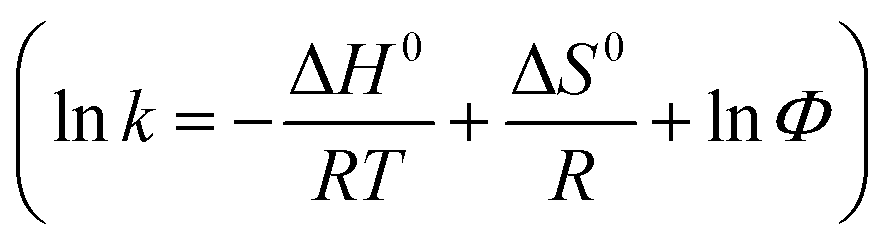 where ΔH0 and ΔS0 are retention enthalpy and entropy, respectively; R is gas constant and Φ represents phase ratio. Good linearity is obtained for all plots (r2 0.990–0.999), indicating a partitioning-dominated retention for nucleosides in HILIC as proposed by Alpert.1 The calculated ΔH0 were in the range of −(4.1–6.4) kJ mol−1, revealing an exothermic process of transferring solutes from the mobile phase to the stationary phase (ESI-Table 4†). Generally, the plate count has less dependence on the temperature although higher plate count is observed at higher column temperature.
where ΔH0 and ΔS0 are retention enthalpy and entropy, respectively; R is gas constant and Φ represents phase ratio. Good linearity is obtained for all plots (r2 0.990–0.999), indicating a partitioning-dominated retention for nucleosides in HILIC as proposed by Alpert.1 The calculated ΔH0 were in the range of −(4.1–6.4) kJ mol−1, revealing an exothermic process of transferring solutes from the mobile phase to the stationary phase (ESI-Table 4†). Generally, the plate count has less dependence on the temperature although higher plate count is observed at higher column temperature.
3.6 Illustrative separation of saccharides
The structure of CC contains repeated sugar units, thus leading to a potential of PVA–CC–Sil for separation of saccharide. Such potential has been demonstrated by a CC agglomerated sulfonated SiO2 HILIC phase (CC-300) described previously.13 Here PVA–CC–Sil was explored to separate saccharides (their chemical structures are also shown in ESI-Scheme 1†). Firstly, six sugars and sugar alcohols including mono-, di- and tri-saccharides were chosen as models. The analytes were well separated and eluted in accordance with their polarity order from weak to strong (Fig. 6a). A direct comparison was made with CC-300. Under the same elution condition, PVA–CC–Sil exhibited a slight higher selectivity and much stronger retention for the models. E.g., the retention time of raffinose on PVA–CC–Sil is ∼2-fold that on CC-300.13 Moreover, higher efficiency was also achieved on the present phase with calculated plate count ∼114000 plates per m for ribose (∼70000 plate per m on CC-300).
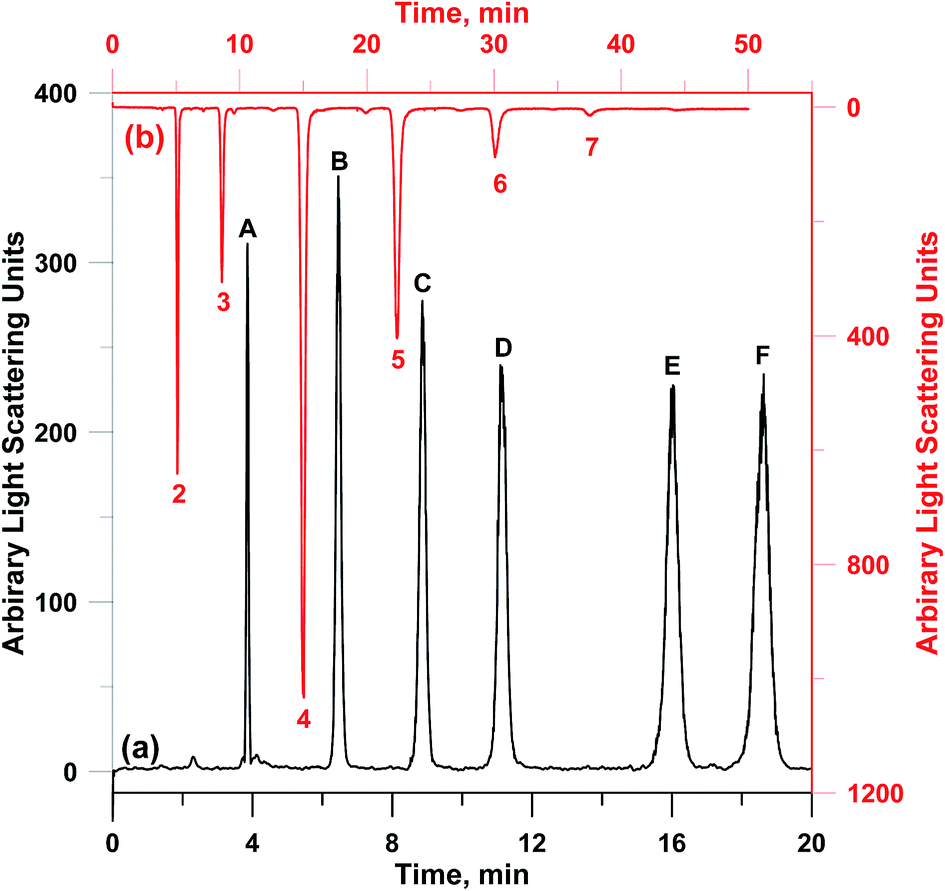 | ||
| Fig. 6 Separation of (a) non-reducing sugars plus sugar alcohols, and (b) galacto-oligosaccharides and onto PVA–CC–Sil. Condition: mobile phase, (a) 20% H2O/80% ACN; (b) A, 100 mM HCOONH4; B, ACN; C, MeOH, 0–60 min, A/B/C (v/v/v), 10/70/20 → 10/30/60. Analytes, (a) A, ribose; B, mannitol; C, maltitol; D, sucrose; E, melezitose; F, raffinose and (b) galacto-oligosaccharides (DP identified from 2 to 7; injection volume, (a) 10 μL; (b) 5 μL; ELSD, nitrogen nebulizer gas 30 psi, tube temperature, 80 °C, gain, 10). | ||
Two medium molecular weight oligosaccharides including neutral galacto-oligosaccharides and acidic sodium alginate could be well separated on PVA–CC–Sil, as shown in Fig. 6b and ESI-Fig. 11,† respectively. As can be seen in ESI-Fig. 6,† separation comparisons of galacto-oligosaccharides were made among PVA–CC–Sil, Acclaim HILIC-10 and Accucore HILIC. Although baseline separation could be achieved with Acclaim HILIC-10 and Accucore HILIC, poor peak shapes were also obtained. In addition, the separation of two oligosaccharides with higher degree of polymerization (DP) (gluco-oligosaccharides and fructo-oligosaccharides) was also performed, as shown in Fig. 7. Clearly, good separation was achieved for gluco-oligosaccharides and fructo-oligosaccharides with DPs ranging from 3 to 15 and from 4 to 20, respectively. These results clearly exhibit the utility of PVA–CC–Sil for efficient separation of saccharides.
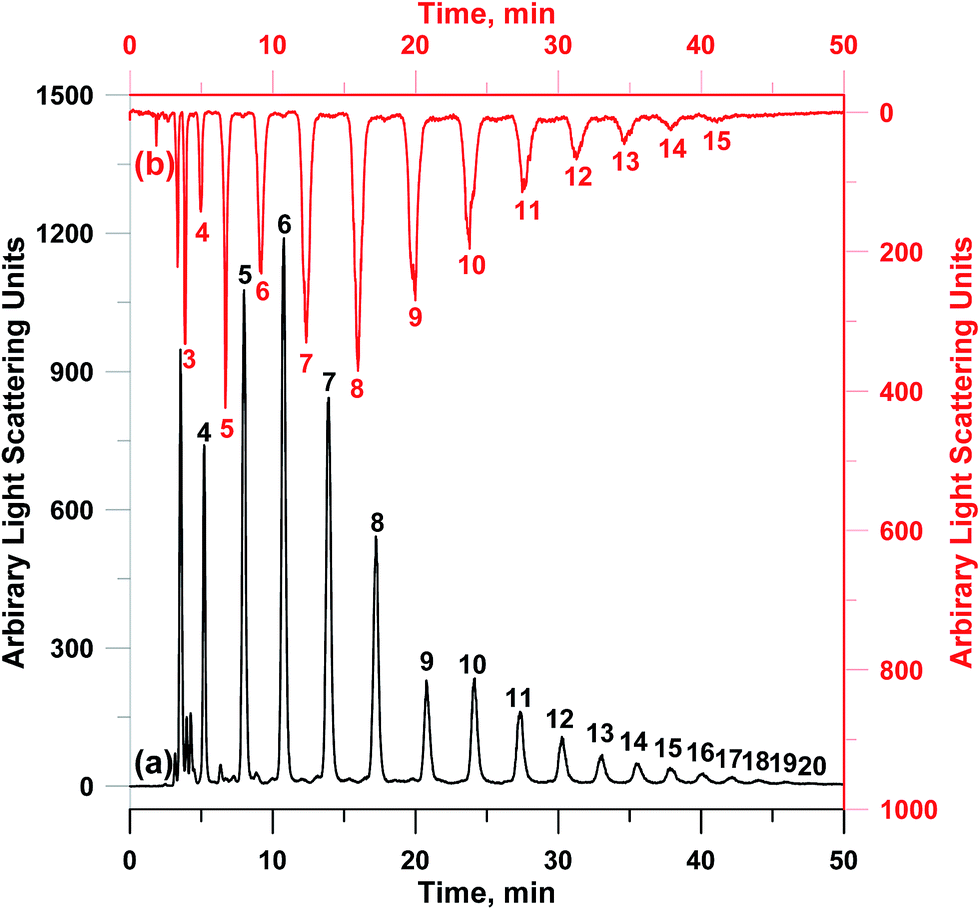 | ||
| Fig. 7 Chromatograms of (a) fructo-oligosaccharides and (b) gluco-oligosaccharides on PVA–CC–Sil. Condition: mobile phase, A, 100 mM HCOONH4; B, ACN; C, MeOH, 0–60 min, A/B/C (v/v/v), 10/70/20 → 10/30/60; injection volume, 5 μL; other conditions same as Fig. 6. | ||
4. Conclusions
A poly(vinyl alcohol)–cationic cellulose polymer encapsulated SiO2 stationary phase for HILIC is described. The preparation way is easy to implement simply by soaking silica particles in aqueous solution of an inexpensive, nontoxic chemical PVA doped with additive cationic cellulose, then filtering and drying/curing thermally. The phase shows obvious positive electrostatic character and typical HILIC character, exhibiting different separation selectivity and good stability. It exhibits good separation towards aromatic acids, nucleosides, nucleotides and saccharides. By replacing SiO2 particles with other hydrolytic-stable gels such as TiO2, ZrO2 or porous graphite carbon, the above way can be used for preparation of more hydrolytic stable phases for HILIC.Acknowledgements
The research was sponsored by National Natural Science Foundation of China (No. 21322502, 21477037). BCY gratefully acknowledges support from the outstanding young talent cultivation fund of ECUST.References
- A. J. Alpert, J. Chromatogr. A, 1990, 499, 177–196 CrossRef CAS PubMed.
- G. Shen, F. Zhang, B. Yang, C. Chu and X. Liang, Talanta, 2013, 115, 129–132 CrossRef CAS PubMed.
- Y. Liu, Q. Du, B. Yang, F. Zhang, C. Chu and X. Liang, Analyst, 2012, 137, 1624–1628 RSC.
- S. Wu, X. Li, F. Zhang, G. Jiang, X. Liang and B. Yang, Analyst, 2015, 140, 3921–3924 RSC.
- L. Qiao, S. Wang, H. Li, Y. Shan, A. Dou, X. Shi and G. Xu, J. Chromatogr. A, 2014, 1360, 240–247 CrossRef CAS PubMed.
- Z. Guo, A. Lei, Y. Zhang, Q. Xu, X. Xue, F. Zhang and X. Liang, Chem. Commun., 2007, 24, 2491–2493 RSC.
- Q. Fu, Z. Guo, T. Liang, X. Zhang, Q. Xu and X. Liang, Anal. Methods, 2010, 2, 217–224 RSC.
- Q. Fu, T. Liang, X. Zhang, Y. Du, Z. Guo and X. Liang, Carbohydr. Res., 2010, 345, 2690–2697 CrossRef CAS PubMed.
- L. Moni, A. Ciogli, I. D'Acquarica, A. Dondoni, F. Gasparrini and A. Marra, Chem.–Eur. J., 2010, 16, 5712–5722 CrossRef CAS PubMed.
- T. Best, E. Kemps and J. Bryan, Nutr. Rev., 2005, 63, 409–418 CrossRef PubMed.
- T. R. Cataldi, G. Margiotta, L. Lasi, B. D. Chio, C. Xiloyannis and S. A. Bufo, Anal. Chem., 2000, 72, 3902–3907 CrossRef CAS PubMed.
- A. Caseiro, I. L. Marr, M. Claeys, A. Kasper-Giebl, H. Puxbaum and C. A. Pio, J. Chromatogr. A, 2007, 1171, 37–45 CrossRef CAS PubMed.
- Q. Sheng, Y. Ke, K. Li, D. Yu and X. Liang, J. Chromatogr. A, 2013, 1291, 56–63 CrossRef CAS PubMed.
- Q. Y. Sheng, X. D. Su, X. L. Li, Y. X. Ke and X. M. Liang, J. Chromatogr. A, 2014, 1345, 57–67 CrossRef CAS PubMed.
- M. Gilges, M. H. Kleemiss and G. Schomburg, Anal. Chem., 1994, 66, 2038–2046 CrossRef CAS.
- B. Bolto, T. Tran, M. Hoang and Z. L. Xie, Prog. Polym. Sci., 2009, 34, 969–981 CrossRef CAS.
- J. M. Yang and H. C. Chiu, J. Membr. Sci., 2012, 419–420, 65–71 CrossRef CAS.
- S. Ji, Y. Zheng, F. Zhang, X. Liang and B. Yang, Analyst, 2015, 140, 6250–6253 RSC.
- S. Ji, F. Zhang, S. Wu, B. Yang and X. Liang, Analyst, 2014, 139, 5594–5599 RSC.
- Y. Hou, F. Zhang, X. Liang, B. Yang, X. Liu and P. K. Dasgupta, Anal. Chem., 2016, 88, 4676–4681 CrossRef CAS PubMed.
- N. P. Dinh, T. Jonsson and K. Irgum, J. Chromatogr. A, 2011, 1218, 5880–5891 CrossRef CAS PubMed.
- M. E. A. Ibrahim, Y. Liu and C. A. Lucy, J. Chromatogr. A, 2012, 1260, 126–131 CrossRef CAS PubMed.
- G. Jin, Z. Guo, F. Zhang, X. Xue, Y. Jin and X. Liang, Talanta, 2008, 76, 522–527 CrossRef CAS PubMed.
- P. Jandera, Anal. Chim. Acta, 2011, 692, 1–25 CrossRef CAS PubMed.
- D. V. McCalley, J. Chromatogr. A, 2010, 1217, 3408–3417 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01958k |
| This journal is © The Royal Society of Chemistry 2017 |