
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A molecular electron density theory study of the [3 + 2] cycloaddition reaction of nitrones with strained allenes†
Luis R. Domingo *a,
Mar Ríos-Gutiérreza and
Patricia Pérezb
*a,
Mar Ríos-Gutiérreza and
Patricia Pérezb
aDepartment of Organic Chemistry, University of Valencia, Dr Moliner 50, E-46100 Burjassot, Valencia, Spain. E-mail: domingo@utopia.uv.es; Web: http://www.luisrdomingo.com
bUniversidad Andres Bello, Facultad de Ciencias Exactas, Departamento de Ciencias Químicas, Millennium Nucleus Chemical Processes and Catalysis (CPC), Av. República 498, 8370146, Santiago, Chile
First published on 17th May 2017
Abstract
The [3 + 2] cycloaddition (32CA) reaction of C-phenyl-N-tert-butylnitrone with 1,2-cyclohexadiene (CHDE), a strained allene, has been studied within Molecular Electron Density Theory (MEDT) at the DFT B3LYP/6-311G(d,p) computational level. This non-polar 32CA reaction, which takes place through a non-concerted two-stage one-step mechanism, proceeds with a moderate Gibbs free activation energy of 22.7 kcal mol−1, and presents low stereo- and regioselectivities. The reaction begins by the creation of a pseudoradical center at the central carbon of the strained allene with a relatively low energy cost, which immediately promotes the formation the first C–C single bond. This scenario is completely different from that of the 32CA reaction involving the simplest allene. The strain present in CHDE changes its reactivity to that characteristic of radical species. Consequently, not distortion as previously proposed, but the radical reactivity type of the strained allene is responsible for the feasibility of this 32CA reaction.
1. Introduction
The use of strained species such as benzyne 5 and cyclic alkynes such as cyclopentyne 8 in organic synthesis permits their participation in organic reactions in which linear alkynes do not react (see Scheme 1). Thus, while the non-polar ene reaction between 2-methylpropene 1 and ethylene 2 presents a very high activation energy, 34.7 kcal mol−1,1 the non-polar ene reaction between 2-methylbut-2-en 4 and benzyne 5 has an unappreciable activation energy, 1.2 kcal mol−1.2 Similarly, the [2 + 2] cycloaddition between enol ether 7 and cyclopentyne 8 also presents a low activation energy, 6.2 kcal mol−1.3 | ||
| Scheme 1 Non-polar reactions of ethylene and strained species. Activation energies, in parentheses, are given in kcal mol−1. | ||
One appealing procedure that provides a straightforward connection between the electron density distribution and the chemical structure is the quantum chemical analysis of the Becke and Edgecombe's Electron Localisation Function (ELF).4 An ELF topological analysis of the electronic structure of benzyne 5 made it possible to explain the high reactivity of this strained aromatic alkyne.2 The ELF of benzyne 5 shows the presence of two monosynaptic basins, V(C1) and V(C2), integrating 0.64e each one (see Fig. 1). This electronic characteristic of benzyne 5 allowed associating its reactivity to that of a high reactive pseudodiradical species.2,5 Note that pseudoradicals are closed-shell species topologically characterised by the presence of at least one V(C) monosynaptic basin integrating less than 1.0e at one carbon atom.6
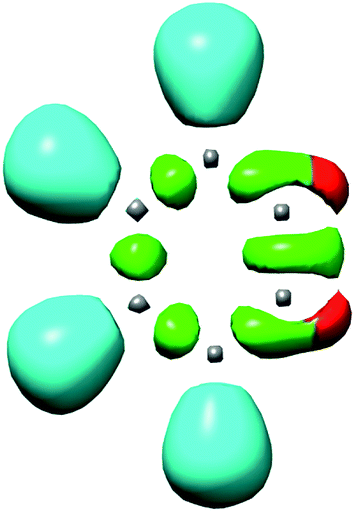 | ||
| Fig. 1 ELF localisation domains of benzyne 5. The two non-bonding V(C1) and V(C2) monosynaptic basins are represented in red. | ||
Unlike arynes and cyclic alkynes, whose structure and reactivity have been widely studied,1–3,7 highly strained allene species have been studied to a much lesser extent. Since 1966, when Wittig reported, for the first time, the existence of 1,2-cyclohexadiene (CHDE) 10,8 the chemistry of this highly strained species has received little attention especially compared to its aryne and alkyne counterparts. Only some theoretical studies devoted to [2 + 2]9–13 and [4 + 2]14,15 cycloadditions of CHDE 10 yielding cycloadducts 12 and 14, respectively, have been reported (see Scheme 2).
 | ||
| Scheme 2 Cycloaddition reactions of CHDE 10. | ||
Very recently, Houk et al. studied experimentally as well as theoretically the [3 + 2] cycloaddition (32CA) reaction of the in situ generated CHDE 10 with nitrone 15 participating as the three-atom-component (TAC), yielding the two stereoisomeric isoxazolidines 16 (Ph/H syn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :anti 9.5:1); regioisomeric isoxazolidines 17 were not observed (see Scheme 3).16
:anti 9.5:1); regioisomeric isoxazolidines 17 were not observed (see Scheme 3).16
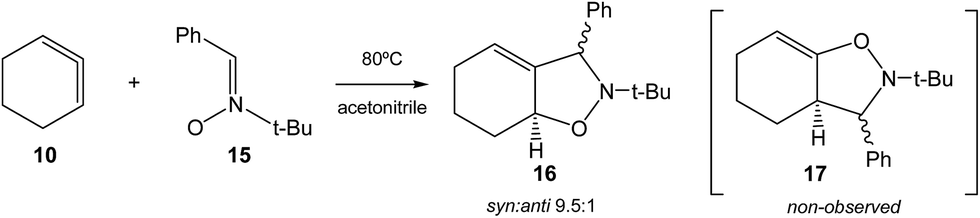 | ||
| Scheme 3 32CA reaction of CHDE 10 with nitrone 15 yielding the stereoisomeric isoxazolidines 16.16 | ||
B3LYP/6-31G(d) calculations for the four competitive channels resulted in a poor stereoselectivity, ΔΔG≠ = 0.1 kcal mol−1, and a poor regioselectivity, ΔΔG≠ = 0.7 kcal mol−1.16 For the stereoisomeric channels giving isoxazolidines 16a, Ph and H syn, and 16b, Ph and H anti, associated with the attack of the C5 carbon of CHDE 10 on the C1 carbon atom of the nitrone (see Scheme 4 for atom numbering), two reaction mechanisms were established: (i) a “concerted” mechanism yielding syn isoxazolidine 16a; and (ii) a stepwise mechanism yielding an open-shell diradical intermediate IN. From this open-shell intermediate, two competitive reaction channels associated to the ring closures yielding isoxazolidines 16a and 16b were characterised (see Scheme 4). Houk proposed that the “concerted” mechanism giving cis isoxazolidine 16a via TSc was easily characterised, while the “concerted” exo transition state structure (TS) could not be located, suggesting that the “concerted” exo reaction was higher in energy. Furthermore, he suggested that the low ∼15 kcal mol−1 barrier for these reactions compared with the >30 kcal mol−1 for the 32CA reaction with the simplest allene 18 can be attributed to the predistortion of CHDE 10 into geometries similar to those of the TSs for cycloadditions.16
 | ||
| Scheme 4 Houk's “concerted” and stepwise mechanisms associated to the formation of the stereosiomeric isoxazolidines 16a and 16b.16 B3LYP/6-31G(d) relative Gibbs free energies, in parentheses, are given in kcal mol−1. | ||
To explain the reactivity of a series of TACs towards ethylene 2 in 32CA reactions, in 2008, Houk introduced the Distortion/Interaction Energy Model (DIEM) in order to interpret the activation energies.17,18 He concluded that the distortion energy of the reagents towards the TS is the major factor controlling the reactivity differences of TACs. However, the distortion energy, which is computed through the partition of the TS geometry into two separated structures, has no physical meaning, since within Density Functional Theory (DFT)19 the energy of a system is a functional of the electron density and the external potential, i.e. the nuclei positions. Consequently, the energy of the two separated fragments cannot be correlated with the energy of the TS as each of them loses the external potential created by the other fragment.20
The theoretical study of strained compounds is of special interest for organic chemists as it allows establishing a relationship between structure and reactivity. As aforementioned, Houk proposed that the predistortion of CHDE 10 into geometries similar to those of the TSs for cycloadditions could be responsible for the low activation energy of these reactions.16 However, similarly to benzyne 5,2 the strain present at the sp hybridised C5 carbon of CHDE 10 could provide some pseudoradical character to the C5 carbon, enabling CHDE 10 to experience a different reactivity pattern to that of linear allenes, and consequently, the corresponding reaction paths will be non-comparable.
These different interpretations about the role of the strain in the reactivity of strained species prompted us to revisit the 32CA reaction of the strained CHDE 10 with nitrone 15 within the recently proposed Molecular Electron Density Theory (MEDT)21 (see Scheme 3). The main purpose of the present theoretical study is to understand how the strain modifies the reactivity of CHDE 10 with respect to the non-strained linear allene 18; this MEDT study provides a different explanation to the one previously reported.16 Thus, in order to establish the special reactivity of the strained allene CHDE 10, an MEDT study of the 32CA reaction of nitrone 15 with the simplest allene 18 is also performed for comparative analysis. This MEDT study is given in ESI.†
2. Computational methods
DFT calculations were performed using the B3LYP functional22,23 together with the 6-311G(d,p) basis set.24 Optimisations were carried out using the Berny analytical gradient optimisation method.25,26 The stationary points were characterised by frequency computations in order to verify that TSs have one and only one imaginary frequency. The IRC paths27 were traced in order to check the energy profiles connecting each TS to the two associated minima of the proposed mechanism using the second order González–Schlegel integration method.28,29 Solvent effects of acetonitrile were taken into account by full optimisation of the gas phase structures using the polarisable continuum model (PCM) developed by Tomasi's group30,31 in the framework of the self-consistent reaction field (SCRF).32–34 Enthalpies, entropies and Gibbs free energies in acetonitrile were calculated with standard statistical thermodynamics at 80 °C and 1 atm from the optimised structures in acetonitrile.24 A comparative analysis of the thermodynamic data, obtained by using the MPWB1K,35 ωB97XD36 and M06-2X37 functionals, indicates that the B3LYP and MPWB1K ones are the more adequate to study this non-polar 32CA reaction (see the comparative analysis in ESI†);38 therefore, the B3LYP functional was selected in the present MEDT study to be consistent with previously reported calculations.16 The global electron density transfer39 (GEDT) is computed by the sum of the natural atomic charges (q), obtained by a natural population analysis (NPA),40,41 of the atoms belonging to each framework (f) at the TSs; GEDT = Σqf. The sign indicates the direction of the electron density flux in such a manner that positive values mean a flux from the considered framework to the other one. All computations were carried out with the Gaussian 09 suite of programs.42ELF studies were performed with the TopMod43 program using the corresponding gas phase B3LYP/6-311G(d,p) monodeterminantal wavefunctions. For the BET study, the corresponding gas phase reaction channel was followed by performing the topological analysis of the ELF for 862 nuclear configurations along the IRC path. ELF calculations were computed over a grid spacing of 0.1 a.u. for each structure and ELF basin isosurfaces were obtained for an ELF value of 0.75.
Conceptual DFT44,45 (CDFT) provides different indices to rationalise and understand chemical structure and reactivity. The global electrophilicity index,46 ω, is given by the following expression, ω = (μ2/2η), in terms of the electronic chemical potential, μ, and the chemical hardness, η. Both quantities may be approached in terms of the one-electron energies of the frontier molecular orbitals HOMO and LUMO, εH and εL, as μ ≈ (εH + εL)/2 and η ≈ (εL − εH), respectively.47,48 The global nucleophilicity index,49,50 N, based on the HOMO energies obtained within the Kohn–Sham scheme,51 is defined as N = EHOMO(Nu) − EHOMO(TCE), where tetracyanoethylene (TCE) is the reference.
3. Results and discussion
The present MEDT study has been divided into four parts: (i) in the first one, an analysis of the CDFT reactivity indices at the ground state (GS) of the reagents involved in the 32CA reactions of nitrone 15 with allenes 10 and 18 is performed; (ii) then, the reaction paths associated the 32CA reaction of nitrone 15 with CHDE 10 are explored and characterised; (iii) in the third part, a topological analysis of the ELF of the simplest allene 18 and strained CHDE 10 is performed in order to characterise their electronic structures; and (iv) finally, a BET study of the 32CA reaction of nitrone 15 with CHDE 10 is carried out in order to establish the origin of the high reactivity of the strained allene CHDE 10.3.1. Analysis of the CDFT reactivity indices of nitrone 15 and allenes 10 and 18
Numerous studies devoted to Diels–Alder and 32CA reactions have shown that the analysis of the reactivity indices defined within the CDFT44,45 is a powerful tool to understand the reactivity in cycloaddition reactions. Recent MEDT studies have shown that the feasibility of the 32CA reactions involving nitrones depends on the nucleophilic character of these TACs and the electrophilic character of the ethylene derivative.52,53 Consequently, an analysis of the CDFT reactivity indices computed in gas phase at the GS of nitrone 15 and allenes 10 and 18 was performed to predict their reactivity in 32CA reactions. The global indices, namely, the electronic chemical potential, μ, chemical hardness, η, electrophilicity, ω, and nucleophilicity, N, at the GS of the reagents involved in these 32CA reactions are given in Table 1.| μ | η | ω | N | |
|---|---|---|---|---|
| Nitrone 15 | −3.29 | 4.27 | 1.26 | 3.70 |
| CHDE 10 | −3.44 | 4.77 | 1.24 | 3.29 |
| Ethylene 2 | −3.37 | 7.77 | 0.73 | 1.87 |
| Allene 18 | −3.30 | 7.72 | 0.70 | 1.97 |
The electronic chemical potential of nitrone 15, μ = −3.29 eV, is close to that of the simplest allene 18, μ = −3.30 eV, and only slightly higher than that of CHDE 10, μ = −3.44 eV. Thus, none of the reagents will have a tendency to exchange electron density with the other along these 32CA reactions, suggesting non-polar reactions.
Nitrone 15 presents an electrophilicity ω index of 1.26 eV and a nucleophilicity N index of 3.70 eV, being classified as a moderate electrophile and as a strong nucleophile according to the electrophilicity54 and nucleophilicity55 scales. Note that nitrones usually behave as nucleophilic zwitterionic TACs participating in polar 32CA reactions towards electrophilic ethylene derivatives.52,53
Polar cycloaddition reactions require the participation of good electrophiles and good nucleophiles. Ethylene 2 is one of the poorest electrophilic, ω = 0.73 eV, and nucleophilic, N = 1.87 eV, species involved in cycloaddition reactions, being classified as a marginal electrophile and a marginal nucleophile. CDFT reactivity indices of the simplest linear allene 18 are very similar to those of ethylene 2 (see Table 1), thus being also classified as a marginal electrophile and on the borderline between marginal and moderate nucleophiles. Otherwise, the angular strain of the allene framework in CHDE 10 causes an increase of both the electrophilicity ω and nucleophilicity N indices to 1.24 eV and 3.29 eV, respectively, being classified as a moderate electrophile and a strong nucleophile. However, in spite of this electrophilic activation with respect to the simplest linear allene 18, this is not sufficient to favour the GEDT,39 in clear agreement with the analysis of the electronic chemical potentials of the reagents. Consequently, it is expected that the corresponding non-polar 32CA reactions will present high activation barriers (see the 32CA reaction between nitrone 15 and the simplest allene 18 in ESI†).
3.2. Study of the reaction paths associated with the 32CA reaction of nitrone 15 with CHDE 10
Due to the non-symmetry of the two reagents, the 32CA reaction of nitrone 15 with CHDE 10 can take place along four isomeric channels: one pair of stereoisomeric channels and one pair of regioisomeric ones. The regioisomeric channels are related to the initial formation of the C1–C5 single bond, channel r1, or to the initial formation of the O3–C5 single bond, channel r2, while the endo and exo stereoisomeric channels are related to the relative position of the allenic H4 hydrogen with respect to the nitrone N2 nitrogen, in such a manner that along the endo channel this hydrogen atom is far away. This 32CA reaction presents a one-step mechanism; only one TS, TS1n, TS1x, TS2n and TS2x, and the corresponding isoxazolidine, 16a, 16b, 17a and 17b, were located and characterised along each reactive channel (see Scheme 5). Relative Gibbs free energies in acetonitrile of the stationary points involved in the 32CA reaction of nitrone 15 with CHDE 10 are given in Scheme 5, while total thermodynamic data are gathered in Table S5 in ESI.† The Gibbs free energy profile is represented in Fig. 2.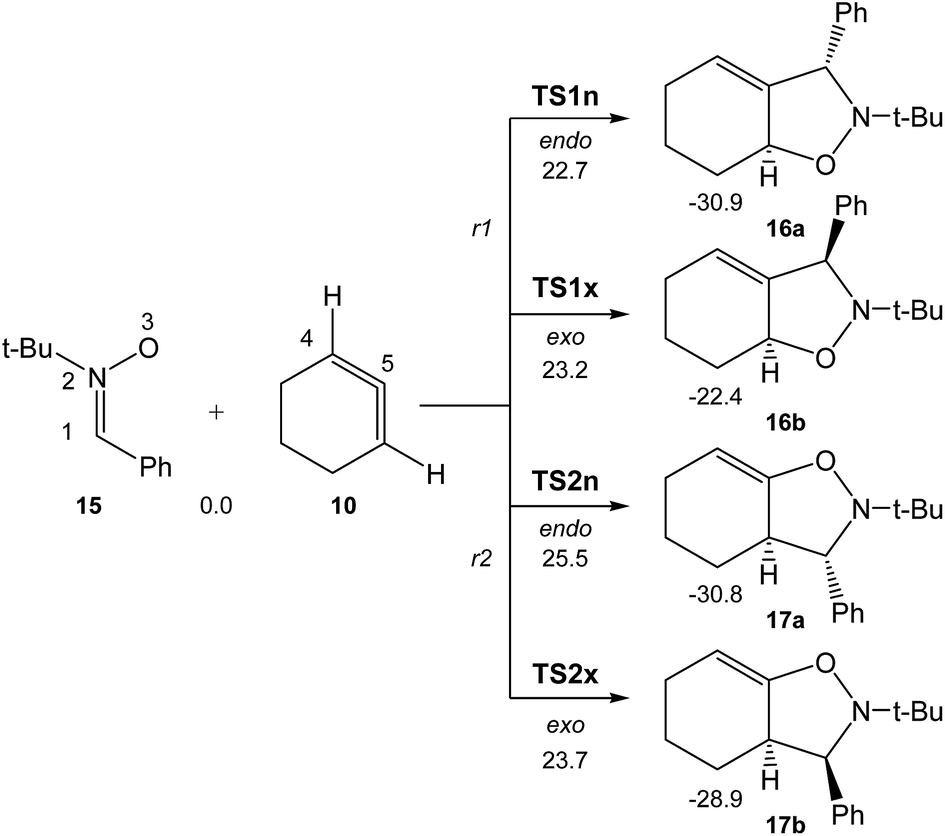 | ||
| Scheme 5 The four competitive reactive channels associated with the 32CA reaction of nitrone 15 with CHDE 10. B3LYP/6-311G(d,p) relative Gibbs free energies, in acetonitrile, are given in kcal mol−1. | ||
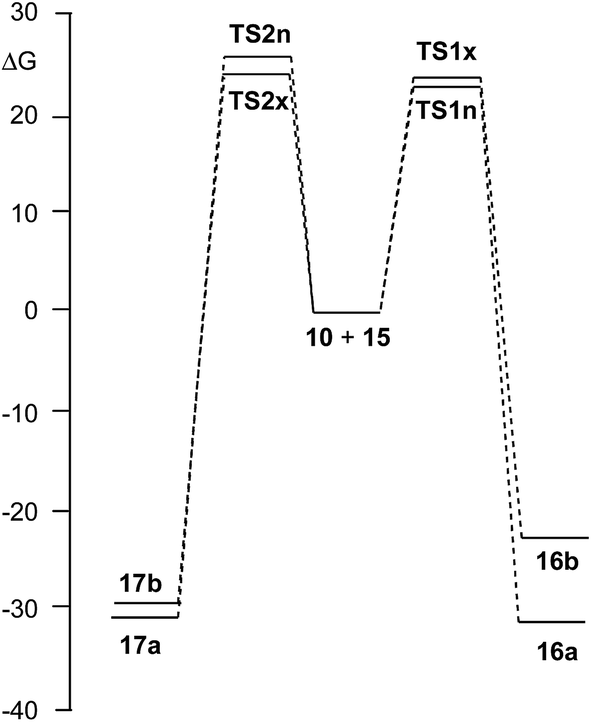 | ||
| Fig. 2 B3LYP/6-311G(d,p) Gibbs free energy profile, in kcal mol−1, of the 32CA reaction of nitrone 15 with CHDE 10. | ||
The activation Gibbs free energies associated with the four competitive channels are 22.7 (TS1n), 23.2 (TS1x), 25.5 (TS2n), and 23.7 (TS2x) kcal mol−1, the reaction being strongly exergonic, between 22–31 kcal mol−1. Some appealing conclusions can be drawn from these relative energies: (i) the activation Gibbs free energy associated with the 32CA reaction of nitrone 15 with CHDE 10 via TS1n is 19.9 kcal mol−1 lower in energy than that associated with the 32CA reaction of nitrone 15 with the simplest allene 18 (see ESI†); (ii) this 32CA reaction presents a low endo selectivity as TS1n is 0.5 kcal mol−1 lower in energy than TS1x; (iii) this 32CA reaction presents a low regioselectivity as TS2x is only 1.0 kcal mol−1 higher in energy than TS1n; (iv) the 32CA reaction of nitrone 15 with CHDE 10 presents an opposed regioselectivity to that found in the reaction with the simplest allene 18 (see ESI†). The most favourable reactive channel corresponds to the initial C1–C5 bond formation; (v) this 32CA reaction is strongly exergonic by 30.9 kcal mol−1 (16a). Note that the most favourable reactive channel associated with the 32CA reaction involving the simplest allene 18 is exergonic by only 4.3 kcal mol−1 (see ESI†). Consequently, the strain present in CHDE 10 does not only affect the kinetics, but also the thermodynamics; and finally, (vii) the Gibbs free energy profile corresponding to the exo/r1 reactive channel in acetonitrile is quite different to that reported by Houk et al. in gas phase (see the stepwise mechanism via an open-shell intermediate given in Scheme 4 as proposed by Houk,16 and the one-step mechanism shown in Fig. 2). It is interesting to note that the gas phase B3LYP/6-311G(d,p) IRC from exo TS1x to exo isoxazolidine 16b discontinues at a species that is not a stationary point; a downhill calculation from this species gives the final exo isoxazolidine 16b in a straightforward manner (see Fig. S10 in ESI†).
The optimised geometries of the TSs in acetonitrile involved in the 32CA reaction of nitrone 15 with CHDE 10, including the distances between the four interacting atoms, are given in Fig. 3. Some appealing conclusions can be drawn from these geometrical parameters: (i) the distances between the interacting atoms at the four TSs indicate that they correspond to highly asynchronous C–C and C–O single bond formation processes; (ii) at the two pairs of stereoisomeric TSs associated with the most favourable regioisomeric r1 channel, the more unfavourable exo TS1x is slightly more advanced and more asynchronous than endo TS1n; (iii) at the four TSs, the formation of the single bond involving the C5 atom of CHDE 10 is more advanced than that involving the C4 atom; (iv) CHDE 10 approaches nitrone 15 perpendicularly, while in the 32CA reaction between nitrone 15 and the simplest allene 18, the two frameworks approach each other in a parallel manner (see Fig. S1 in ESI†). These different approach modes emphasise different reactivities; (v) both geometries and relative energies of the four TSs indicate that the strain present in CHDE 10 notably modifies the reactivity of this strained cyclic allene when it is compared to that of the simplest allene 18. Note that the two regioisomeric TSs associated with the 32CA reaction of nitrone 15 with the simplest allene 18 are poorly asynchronous (see ESI†).
 | ||
| Fig. 3 B3LYP/6-311G(d,p) geometries in acetonitrile of the TSs involved in the 32CA reaction of nitrone 15 with CHDE 10. Distances are given in angstroms, Å. | ||
In acetonitrile, endo TS1n and exo TS1x have a great similitude; they present a similar only imaginary frequency, −297.6918 cm−1 (TS1n) and −311.8100 cm−1 (TS1x), with a similar transition vector, 0.86023 B(C1–C5) and 0.25045 B(C4–O3) (TS1n) and 0.80798 B(C1–C5) and 0.31925 B(C4–O3) (TS1x). The vibration modes of these stereoisomeric TSs are completely different to those of the TSs associated with the 32CA reaction of nitrone 15 with the linear allene 18, indicating again different reactivities; TS1n and TS1x are associated to a two-center interaction in which only the C5 carbon of CHDE 10 and the C1 carbon of nitrone 15 participate. In addition, these stereoisomeric channels, which are associated to a non-concerted two-stage one-step mechanism,56 present analogous IRCs (see Fig. 4). In this mechanism, the formation of the second O3–C4 single bond begins once the first C1–C5 single bond has been completely formed in a single elementary step (see later).
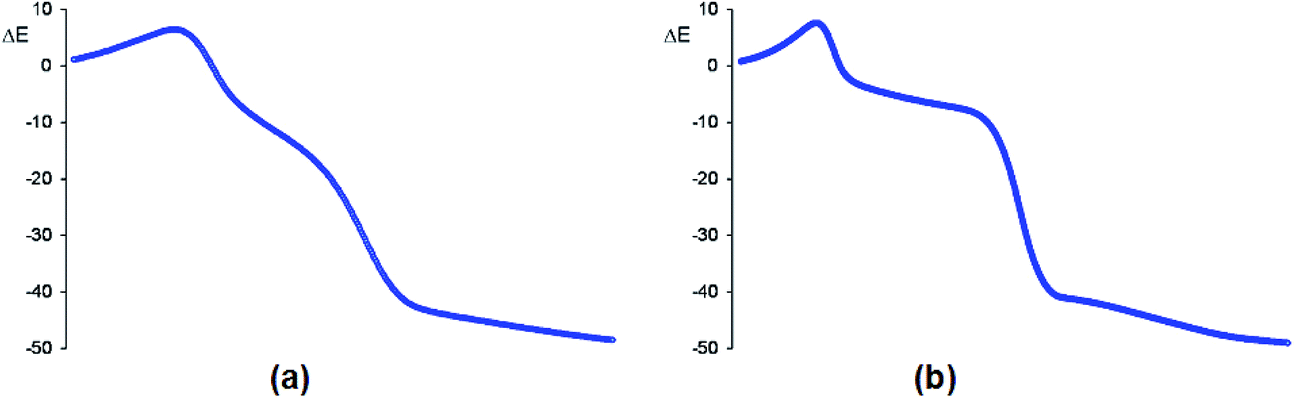 | ||
| Fig. 4 Relative energy (ΔE, in kcal mol−1) variations along the IRC (amu1/2 bohr) associated with the endo/r1 (a) and exo/r1 (b) reactive channels of the 32CA reaction between nitrone 15 and strained allene CHDE 10 in acetonitrile. Relative energies are given with respect to the separated reagents. | ||
In order to evaluate the polar or non-polar electronic nature of these TSs, the GEDT was analysed.39 The GEDT at the four TSs, which fluxes from the nitrone to the CHDE frameworks, is 0.02e at TS1n, 0.02e at TS1x, 0.02e TS2n and 0.09e at TS2x. These negligible values indicate that this 32CA reaction has a non-polar character.
3.3. Topological analysis of the ELF of linear allene 18 and strained CHDE 10
As commented in the introduction, the high reactivity of benzyne 5 was attributed to its pseudodiradical character, which was topologically characterised by the presence of two ELF V(C) monosynaptic basins (see Fig. 1).2 Consequently, a topological analysis of the ELF of linear allene 18 and strained CHDE 10 was performed in order to characterise their electronic structure. ELF attractors, together with the C4–C5 valence basin populations, ELF localisation domains and the proposed ELF-based Lewis structures are shown in Fig. 5. | ||
| Fig. 5 ELF attractors, together with the C4–C5 valence basin populations, in average number of electrons (e), ELF localisation domains and the proposed Lewis structures for allenes 10 and 18. | ||
ELF topological analysis of both allenes 10 and 18 shows the presence of two V(C4,C5) and V′(C4,C5) disynaptic basins within the C4–C5 bonding region, integrating a total population of 3.68e at both allenes, which indicates that the C4–C5 bonding region possesses a strong double bond character (see Fig. 5). However, the topology of the ELF V(Cx,Cy) disynaptic basins associated with the C4–C5 and C5–C6 bonding regions shows a notable difference at the two structures; while these disynaptic basins are clearly differentiated in the linear allene 18 as they are positioned in two perpendicular molecular planes, they are linked in a singular and twisted manner at strained CHDE 10 due to the slight torsion of the C1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2C3 framework (see ELF localisation domains in Fig. 5). This particular feature found in the strained CHDE 10 suggests the participation of the two C–C double bonds of the strained allene in the special reactivity of this strained species.
C2C3 framework (see ELF localisation domains in Fig. 5). This particular feature found in the strained CHDE 10 suggests the participation of the two C–C double bonds of the strained allene in the special reactivity of this strained species.
Consequently, although the strained CHDE 10 does not present a pseudoradical structure such as benzyne 5 (see Fig. 1), the ELF topological analysis of the C4–C5–C6 bonding region suggests that this species will have a different chemical behaviour to that of linear allene 18.
3.4. BET study of the 32CA reaction between nitrone 15 and strained allene CHDE 10 yielding endo isoxazolidine 16a
When trying to achieve a better understanding of bonding changes in organic reactions, the so-called BET57 has proven to be a very useful methodological tool. This quantum-chemical methodology makes it possible to understand the bonding changes along a reaction path and, thus, to establish the nature of the electronic rearrangement associated with a given molecular mechanism.58–60Recently, a BET study of the bonding changes along the 32CA reactions of C-phenyl-N-methyl nitrone 21 with electron-deficient acrolein 22 was carried out in order to understand the molecular mechanism of these 32CA processes.61 Herein, in order to understand the different reactivity of linear or cyclic strained allenes towards nitrones, a BET study of the molecular mechanism of the most favourable endo/r1 reaction channel associated with the 32CA reaction between nitrone 15 and strained allene CHDE 10 is performed; the complete BET study is discussed in ESI.† The characterisation of the molecular mechanism of the 32CA reaction of nitrone 15 with the simplest linear allene 18 is also carried out with the aim of rationalising the origin of the differences between the reactivity of both allenes (see ESI†). The attractor positions of the ELF basins for the points involved in the bond formation processes are shown in Fig. 6.
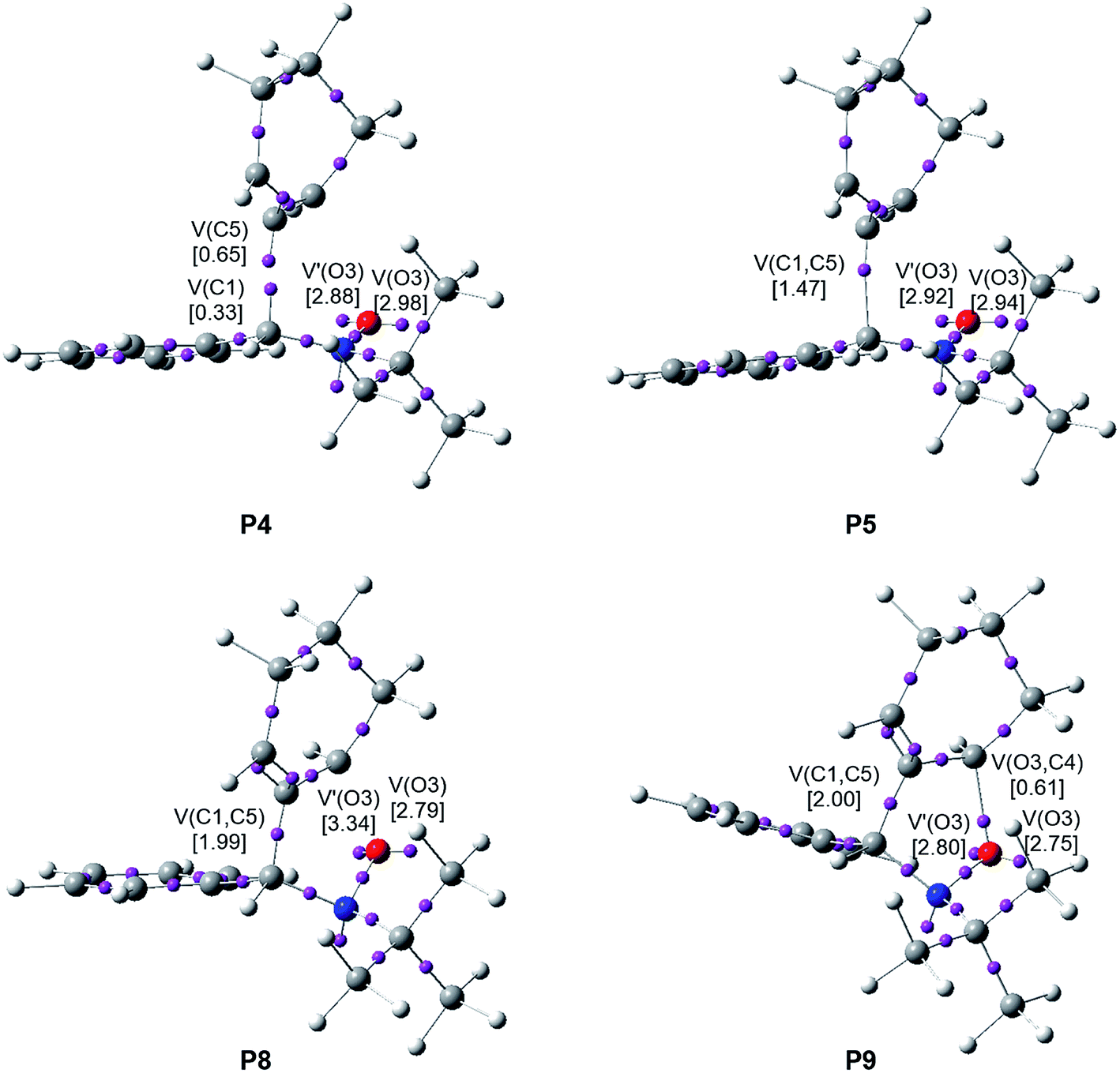 | ||
| Fig. 6 ELF attractor positions for the points of the IRC involved in the formation of the C1–C5 and O3–C4 single bonds along the most favourable endo/r1 reactive channel associated with the 32CA reaction between nitrone 15 and strained allene CHDE 10. The electron populations, in e, are given in brackets. | ||
Some appealing conclusions can be drawn from this BET study: (i) the IRC of the endo/r1 reactive channel is divided in ten differentiated phases related to the disappearance or creation of valence basins, emphasising the non-concertedness of the reaction; (ii) the reaction begins with the depopulation of the allenic C4–C5–C6 bonding region of the strained CHDE 10 in order to permit the creation of a C5 pseudoradical center. Due to the strain present in CHDE 10, this electronic change demands a moderate energy cost of 8.3 kcal mol−1 (see Table S3 in ESI†), which is 15.1 kcal mol−1 lower than that demanded for the creation of the first pseudoradical center at the nitrone 15 in the reaction involving the simplest allene 18 (see Table S2 in ESI†); (iii) thus, the moderate activation energy found in this non-polar 32CA reaction, 8.5 kcal mol−1, can be mainly associated to the creation of a pseudoradical center at the C5 carbon of strained allene CHDE 10; (iv) once the C5 pseudoradical center is formed in the allenic framework, the subsequent rupture of the C1–N2 double bond of the nitrone fragment and creation of the C1 pseudoradical center has an unappreciable energy cost, 0.1 kcal mol−1. Consequently, once the C5 pseudoradical center is created, it induces the easy rupture of the C1–N2 double bond (see Table S3 in ESI†). This reactivity behaviour is characteristic of radical species. (v) Formation of the first C1–C5 single bond begins at a C1–C5 distance of 1.94 Å through a C-to-C coupling of two C1 and C5 pseudoradical centers (see P4 and P5 in Fig. 6).39 Interestingly, the C5 pseudoradical center participates with a high electron density, 0.89e, in the formation of the new C1–C5 single bond; (vi) however, formation of the second O3–C4 single bond begins at an O3–C4 distance of 1.75 Å through the donation of the electron density of one of the two O3 oxygen lone pairs of the nitrone framework to the C2 carbon atom of the allene fragment, similar to previous O–C bond formations,61 (see P8 and P9 in Fig. 6); and (vii) formation of the second O3–C4 single bond takes place at the end of the reaction path once the first C1–C5 single bond has already reached 99% of its the population present at endo isoxazolidine 16a. This fact allows characterising the molecular mechanism of this 32CA reaction as a non-concerted two-stage one-step mechanism56 associated to the attack of the pseudoradical allenic C5 carbon of CHDE 10 on the C1 carbon of the nitrone which, after the complete C1–C5 single bond formation, experiences a rapid ring-closure process.
A comparative analysis of the BET studies of the non-polar 32CA reactions between nitrone 15 and allenes 10 and 18 makes it possible to explain the high reactivity of strained cyclic allene CHDE 10. The bonding changes along the non-polar 32CA reaction between nitrone 15 and the simplest allene 18 are very similar to those along the less favourable ortho/endo regioisomeric channel associated with the low-polar 32CA reaction of nitrone 21 with electron-deficient acrolein 22, which begins with the rupture of the nitrone C1–N2 double bond.61 The lower activation energy associated with the reaction involving acrolein 22 can be related to the slight GEDT, which favours the rupture of the double bonds in the nitrone and ethylene frameworks involved in this 32CA reaction.61 However, a completely different scenario is found along the non-polar 32CA reaction between nitrone 15 and strained allene CHDE 10.
At the beginning of the reaction, a pseudoradical center is created at the central C5 carbon of the strained allene CHDE 10 with an energy cost of 8.3 kcal mol−1. Interestingly, once this C5 pseudoradical center reaches a population of 0.89e, the subsequent bonding changes take place very easily (see the shortness of phases II–IV in the reaction involving CHDE 10 in Fig. S5† in contrast to their extension in the reaction involving the linear allene 18 in Fig. S2†). This behaviour indicates that, at the beginning of the reaction, CHDE 10 becomes a pseudoradical species,5 which explains the high reactivity of strained allenes in cycloaddition reactions.9–15
These findings clearly reveal that the bonding changes demanded to reach the corresponding TSs are completely different and, consequently, they are neither electronically nor geometrically comparable.
4. Conclusions
The 32CA reaction of nitrone 15 with the strained allene CHDE 10 giving isoxazolidines 16 and 17 has been studied within MEDT at the DFT B3LYP/6-311G(d,p) computational level in order to explain the higher reactivity of CHDE 10 with respect to linear allenes.This 32CA reaction can take place along two pairs of regio- and stereoisomeric reaction channels. In acetonitrile, this reaction presents a low activation Gibbs free energy, 22.7 kcal mol−1, as well as low endo stereo- and regioselectivities. The non-polar character of the reaction measured by the GEDT at the TSs agrees with the analysis of the CDFT reactivity indices carried out at the GS of the reagents.
Analysis of the geometries of the endo and exo stereoisomeric TSs associated with the more favourable r1 regioisomeric channels indicates that they are related to highly asynchronous C–C and C–O single bond formation processes. Analysis of the IRCs associated to these TSs in acetonitrile clearly permits to establish that this 32CA reaction takes place through a non-concerted two-stage one-step mechanism initialised by the attack of the central C5 carbon of CHDE 10 on the C1 carbon of nitrone 15.
BET analysis of the most favourable endo/r1 reactive channel allows establishing the molecular mechanism of the 32CA reaction of nitrone 15 with strained allene CHDE 10. At the beginning of the reaction, a pseudoradical center with a population of 0.89e is created at the central C5 carbon of the strained allene framework with a moderate energy cost of 8.3 kcal mol−1, which induces the formation of the C1 pseudoradical center at the nitrone moiety, demanded for the formation of the first C1–C5 single bond through a two-center interaction. Formation of the second O3–C4 single bond takes place at the end of the reaction path when the first C1–C5 single bond is almost completely formed. This behaviour supports the mechanism of this 32CA reaction as a non-concerted two-stage one-step one.
This mechanism is completely different to that associated with the 32CA reaction involving the simplest linear allene 18, which begins with the rupture of the C1–N2 double bond of the nitrone framework in order to create the C1 pseudoradical center required for the formation of the first C1–C5 single bond. This bonding change demands an energy cost of 23.4 kcal mol−1, justifying the high activation energy associated to this non-polar process, 26.2 kcal mol−1.
Consequently, when comparing the mechanisms of the 32CA reactions of nitrone 15 with allenes 10 and 18 characterised from an MEDT perspective, we can conclude that the geometrical predistortion of strained allene CHDE 10 is not responsible for the high reactivity of this species as previously proposed,16 but a change of reactivity of the strained CHDE 10, which behaves as a radical species rather than an ethylene derivative, as occurs in the case of linear allene 18. It is worth mentioning that while the pseudoradical character of a species can be correlated with a molecular strain, the energy associated with the geometrical distortion of a relaxed species cannot give any information about its electronic structure.
Acknowledgements
This work has been supported by the Ministry of Economy and Competitiveness (MINECO) of the Spanish Government, project CTQ2016-78669-P, Fondecyt (Chile) grants 1140341, Millennium Nucleus Chemical Processes and Catalysis (CPC) project No. 120082, and the Universidad Andres Bello (UNAB) by grant DI-793-15/R. Prof L. R. D. thanks FONDECYT for continuous support through Cooperación Internacional. Finally, M. R.-G. also thanks MINECO for a pre-doctoral contract co-financed by the European Social Fund (BES-2014-068258).References
- L. R. Domingo, M. J. Aurell and P. Pérez, Org. Biomol. Chem., 2014, 12, 7581–7590 CAS.
- P. Pérez and L. R. Domingo, Eur. J. Org. Chem., 2015, 2826–2834 CrossRef.
- L. R. Domingo, P. Pérez and R. Contreras, Eur. J. Org. Chem., 2006, 498–506 CrossRef CAS.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- L. A. Errede, J. M. Hoyt and R. S. Gregorian, J. Am. Chem. Soc., 1960, 53, 5224–5227 CrossRef.
- L. R. Domingo, E. Chamorro and P. Pérez, Lett. Org. Chem., 2010, 7, 432–439 CrossRef CAS.
- I. Fernandez and F. P. Cossio, J. Comput. Chem., 2016, 37, 1265–1273 CrossRef CAS PubMed.
- G. Wittig and P. Fritze, Angew. Chem., Int. Ed. Engl., 1966, 5, 846 CrossRef.
- A. T. Bottini, L. L. Hilton and J. Plott, Tetrahedron, 1975, 31, 1997–2001 CrossRef CAS.
- M. Christl, H. Fischer, M. Arnone and B. Engels, Chem.–Eur. J., 2009, 15, 11266–11272 CrossRef CAS PubMed.
- M. Christl and M. Schreck, Angew. Chem., Int. Ed. Engl., 1987, 26, 449–451 CrossRef.
- W. R. Moore and W. R. Moser, J. Org. Chem., 1970, 35, 908–912 CrossRef CAS.
- I. Quintana, D. Pena, D. Perez and E. Guitian, Eur. J. Org. Chem., 2009, 5519–5524 CrossRef CAS.
- M. Balci and W. M. Jones, J. Am. Chem. Soc., 1980, 102, 7607–7608 CrossRef CAS.
- L. M. Tolbert, M. N. Islam, R. P. Johnson, P. M. Loiselle and W. C. Shakespeare, J. Am. Chem. Soc., 1980, 112, 6416–6417 CrossRef.
- J. S. Barber, E. D. Styduhar, H. V. Pham, T. C. McMahon, K. N. Houk and N. K. Garg, J. Am. Chem. Soc., 2016, 138, 2512–2515 CrossRef CAS PubMed.
- D. H. Ess and K. N. Houk, J. Am. Chem. Soc., 2007, 129, 10646–10647 CrossRef CAS PubMed.
- D. H. Ess and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 10187–10198 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- L. R. Domingo, M. Ríos-Gutiérrez, M. Duque-Noreña, E. Chamorro and P. Pérez, Theor. Chem. Acc., 2016, 135, 160 CrossRef.
- L. R. Domingo, Molecules, 2016, 21, 1319 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- H. B. Schlegel, J. Comput. Chem., 1982, 3, 214–218 CrossRef CAS.
- H. B. Schlegel, in Modern Electronic Structure Theory, ed. D. R. Yarkony, World Scientific Publishing, Singapore, 1994 Search PubMed.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef CAS.
- C. González and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef.
- C. González and H. B. Schlegel, J. Chem. Phys., 1991, 95, 5853–5860 CrossRef.
- B. Y. Simkin and I. Sheikhet, Quantum Chemical and Statistical Theory of Solutions – A Computational Approach, Ellis Horwood, London, 1995 Search PubMed.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef CAS.
- V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404–417 CrossRef CAS.
- Y. Zhao and G. D. Truhlar, J. Phys. Chem. A, 2004, 108, 6908–6918 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- L. R. Domingo, M. Ríos-Gutiérrez and P. Pérez, Tetrahedron, 2017, 73, 1718–1724 CrossRef CAS.
- L. R. Domingo, RSC Adv., 2014, 4, 32415–32428 RSC.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. A., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian 09 D.01, Gaussian, Inc, Wallingford CT, 2013 Search PubMed.
- S. Noury, X. Krokidis, F. Fuster and B. Silvi, Comput. Chem., 1999, 23, 597–604 CrossRef CAS.
- P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793–1873 CrossRef CAS PubMed.
- L. R. Domingo, M. Ríos-Gutiérrez and P. Pérez, Molecules, 2016, 21, 748 CrossRef PubMed.
- R. G. Parr, L. von Szentpaly and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
- L. R. Domingo, E. Chamorro and P. Pérez, J. Org. Chem., 2008, 73, 4615–4624 CrossRef CAS PubMed.
- L. R. Domingo and P. Pérez, Org. Biomol. Chem., 2011, 9, 7168–7175 CAS.
- W. Kohn and L. J. Sham, Phys. Rev. [Sect.] B, 1965, 140, 1133–1138 CrossRef.
- L. R. Domingo and S. R. Emamian, Tetrahedron, 2014, 70, 1267–1273 CrossRef CAS.
- L. R. Domingo, M. J. Aurell and P. Pérez, Tetrahedron, 2014, 70, 4519–4525 CrossRef CAS.
- L. R. Domingo, M. J. Aurell, P. Pérez and R. Contreras, Tetrahedron, 2002, 58, 4417–4423 CrossRef CAS.
- P. Jaramillo, L. R. Domingo, E. Chamorro and P. Pérez, J. Mol. Struct.: THEOCHEM, 2008, 865, 68–72 CrossRef CAS.
- L. R. Domingo, J. A. Saéz, R. J. Zaragozá and M. Arnó, J. Org. Chem., 2008, 73, 8791–8799 CrossRef CAS PubMed.
- X. Krokidis, S. Noury and B. Silvi, J. Phys. Chem. A, 1997, 101, 7277–7282 CrossRef CAS.
- J. Andrés, P. González-Navarrete and V. Safont, Int. J. Quantum Chem., 2014, 114, 1239–1252 CrossRef.
- S. Berski, J. Andrés, B. Silvi and L. R. Domingo, J. Phys. Chem. A, 2003, 107, 6014–6024 CrossRef CAS.
- V. Polo, J. Andrés, S. Berski, L. R. Domingo and B. Silvi, J. Phys. Chem. A, 2008, 112, 7128–7136 CrossRef CAS PubMed.
- M. Ríos-Gutiérrez, P. Pérez and L. R. Domingo, RSC Adv., 2015, 58464–58477 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Study of the reaction paths associated with the 32CA reaction between nitrone 15 and the simplest allene 18. BET study of the regiosiomeric r1 reactive channel associated with the 32CA reaction between nitrone 15 and the simplest allene 18 and the endo/r1 reactive channel associated with the 32CA reaction between nitrone 15 and strained allene CHDE 10. ELF topological analysis along the exo/r1 reactive channel associated with the 32CA reaction between nitrone 15 and strained allene CHDE 10. Theoretical background of the ELF and BET. B3LYP/6-311G(d,p) thermodynamic data. B3LYP/6-311G(d,p) thermodynamic data of the stationary points involved in the 32CA reaction of nitrone 15 with CHDE 10. Analysis of the dependence of the thermodynamic data of the stationary points involved in the 32CA reaction between nitrone 15 and CHDE 10 with the DFT functional. B3LYP/6-311G(d,p) total and relative energies, in gas phase and in acetonitrile, of the stationary points involved in the 32CA reaction of nitrone 15 with the simplest allene 18 and CHDE 10 and finally, the computed total energies, the only imaginary frequency and Cartesian coordinates, in acetonitrile, of the structures involved in the 32CA reaction between nitrone 15 and strained allene CHDE 10. See DOI: 10.1039/c7ra01916e |
| This journal is © The Royal Society of Chemistry 2017 |