
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in photoinduced glycosylation: oligosaccharides, glycoconjugates and their synthetic applications
Rekha Sangwan
ab and
Pintu Kumar Mandal
 *ab
*ab
aMedicinal and Process Chemistry Division, CSIR-Central Drug Research Institute, BS-10/1, Sector 10, Jankipuram extension, Sitapur Road, P.O. Box 173, Lucknow, 226 031, India. E-mail: pintuchem06@gmail.com; Fax: +91 522 2623405; Tel: +91-522-2772450 extn 4657
bAcademy of Scientific and Innovative Research, New Delhi – 11000, India
First published on 17th May 2017
Abstract
Carbohydrates have been demonstrated to perform crucial tasks in biological processes. However, the advancement in carbohydrate research is relatively slow due to the problems associated with the complexity of carbohydrate structures and the lack of general synthetic methods. Considering that the unique process of photoinduced glycosylation is rapidly emerging as a promising tool in carbohydrate chemistry, this academic review inspects the recent evolution in the chemistry of carbohydrates, including mostly synthetic, and to a lesser extent mechanistic aspects, by examining the strategies that apply photoinduced glycosylation in the synthesis of oligosaccharides and glycoconjugates. We have chosen to present several representative examples that illustrate the diverse and advance uses of photoinduced glycosylation in carbohydrate chemistry for the synthesis of oligosaccharides, thiosugars, glycoconjugates and glycoproteins. As simple techniques for obtaining carbohydrate targets, these methods are mild and effective for the construction of glycosidic bonds via photoinduced promoted glycosylation, which is environmentally friendly. We mainly highlight the symbiotic cooperation of photoinduced glycosylation via electron transfer, hydrogen atom transfer and energy transfer with or without photocatalyst for diverse arrays in the field of total synthesis of carbohydrate based vaccines, thiyl radical mediated clustering, chemical biology and material chemistry.
 Rekha Sangwan | Rekha Sangwan graduated from the University of Pt. B.D. Sharma, Lord Shiva College in 2012 with a B. Pharmacy Degree in Pharmaceutical Science and post-graduate M.S (Pharm.) in Medicinal Chemistry from NIPER, Raebareli in 2014. She is currently undertaking her PhD at the CSIR-Central Drug Research Institute (CDRI), Lucknow, India under the supervision of Dr Pintu Kumar Mandal. Her research focuses on the synthesis of biological relevant oligosaccharides and glycoconjugates. |
 Pintu Kumar Mandal | Pintu Kumar Mandal obtained B. Sc. and M.Sc. Degrees in Chemistry from Vidyasagar University. He completed his Ph.D. in 2009 at the Jadavpur University, India under the supervision of Prof. Anup Kumar Misra, working in the field of synthetic carbohydrate chemistry. He was selected as a Newton International Fellow (Royal Society of Chemistry, 2009–2011) for Postdoctoral studies at the University of Leeds, UK with Prof. W. Bruce Turnbull. After completion, he moved to continue his postdoctoral studies at Georgia State University (Georgia, USA; 2011–2013) with Prof. Peng G. Wang. He started his independent career at the CSIR-Central Drug Research Institute (CDRI), Lucknow (India) as a Sr. Scientist in 2013 and Assistant Professor at the Academy of Scientific and Innovative Research (AcSIR), New Delhi. His research interests include the synthesis of biologically relevant oligosaccharides, glycoconjugates, new methods for glycosidic bond formation, and the supramolecular chemistry of carbohydrates. |
1. Introduction
Oligosaccharides and glycoconjugates are essential biological compounds which have various versatile functions, such as immune responses,1 structural modification of proteins, lipids, and secondary metabolites, molecular recognition elements in cellular adhesion, cell–cell recognition, and cellular transport, and along with glycolipids help to form mammalian cell surfaces.2 They also play a critical role in vaccine therapeutics,3 which stimulates investigations to characterize the role of glycoconjugates in biological processes. Recently, the interest in the synthesis of these compounds has been increased with the main focus on new approaches for the glycosidic bond.4 Glycosylations connect the anomeric carbon of a glycosyl donor with one of the hydroxyl groups of a glycosyl acceptor, which is a crucial process in the chemical synthesis of oligosaccharides.5 From a synthetic standpoint, the efficiency of the glycosylation reaction is generally evaluated based on high chemical yield, regioselectivity, and α/β-stereoselectivity. A number of methods have already been developed to achieve the synthesis of complex oligosaccharides and glycoconjugates. Classical methods involving the formation of glycosidic bonds generally demand time consuming protective chemistry and the activation of a glycosyl donor. Furthermore, some of these activating agents are air and moisture sensitive and must be used under strict anhydrous and low temperature conditions. Much of the focus of glycosylation reactions has shifted toward improving their efficiency by maximizing yields, regioselectivity and stereoselectivity. The ongoing progress in the synthesis of complex oligosaccharides and glycoconjugates is closely related to the development of new glycosylation methods. Chemical glycosylation refers to the cleavage of the glycosidic bond of a donor using chemical or physical means to generate a glycosyl oxocarbenium species, which undergoes the subsequent coupling reaction with an acceptor. Despite the numerous elegant strategies and methods developed for the efficient formation of glycosidic linkages, stereoselective construction of α- and β-glycosides remains challenging. The formation of each glycosidic linkage results in the creation of a new stereogenic center, in contrast to the situation for amide and phosphate diester linkages in peptides and oligonucleotides. Stereochemical control of glycosidic linkage formation is a key challenge. However, little attention has been paid to the ecological efficiency of glycosylation. From an ecological point of view as well as that of energy considerations, reusable photocatalysts are greatly advantageous. Numerous recent studies have resulted in the development of an impressively diverse range of photocatalytic transformations,6 the applications of which range from natural product7 synthesis to late-stage pharmaceutical functionalization8 and polymer synthesis.9The recent development of photoinduced carbohydrate synthesis under mild, room-temperature, and less anhydrous reaction conditions, in conjunction with substoichiometric amounts of activating agents, could further advance the field of carbohydrate chemistry.10 The use of photocatalysts is conducive to achieving “greener” chemistry, where air and moisture tolerance, performance at room temperature, and enhanced synthetic efficiency through the reduction of unnecessary waste are attained.11 The interaction between an electronically excited photocatalyst and an organic molecule can result in the generation of a diverse array of reactive intermediates that can be manipulated in a variety of ways to result in synthetically useful bond constructions.12 Recently, photocatalysis has attracted great attention due to its virtue of eco-friendliness and versatility in organic synthesis, but has barely been addressed in carbohydrate chemistry. Catalysis plays a central role in the development of all major areas of contemporary synthetic chemistry. Catalysts are capable of providing a high level of control over the stereo and regioselectivity of complexity-building reactions. Photocatalysts are powerfully enabling in synthetic applications because they absorb light with greater efficiency and at longer wavelengths than simple organic small molecules. These species operate by converting the energy of an absorbed photon into chemical potential which can be used to transform organic substrates in a number of ways. Thus, many of the most practical synthetic strategies involving radical ions, diradicals, and electronically excited organic compounds rely upon the use of photocatalysts. The use of light as clean energy for promoting the glycosylation reaction would also be beneficial.13 The activation of glycosyl donors largely depends on the redox potential of a photosensitizer or photocatalyst. The existing methods require a careful adjustment of anomeric leaving groups to ensure successful light-induced activation and exhibit an extremely limited substrate scope. There are three main requirements for an efficient glycosylation method:14 (1) small amounts of reagents must be used; that is, the glycosyl donor must be generated in a simple process and the donor activated by a catalytic amount of reagent; (2) the glycosylation step must be stereoselective and high-yielding; and (3) the method must be applied on a large scale. The methods presented in this review largely satisfy these requirements. We have organized this review around three common photocatalytic activation steps and mechanistically distinct modes of photocatalysis are discussed, including photoinduced electron transfer, hydrogen atom transfer and energy transfer. The purpose of this review is to provide an overview of the ways in which photoinduced strategies have been applied to synthesis of carbohydrates and the contemporary developments in carbohydrate chemistry. These methods are mild and effective for constructing glycosidic bonds with reduced levels of waste through the utilization of substoichiometric amounts of photocatalyst to promote glycosylation as well as from the viewpoint of not only a useful and selective protocol but also green chemistry. The diverse applications of efficient photocatalysts in carbohydrate chemistry are summarized in this review, giving special attention to the most recent and outstanding contributions in this area.
2. Mechanisms involved in photocatalysis
Photoenergy may result in unique reactivity, where the generation of a highly active catalytic species on demand by the light irradiation of an inactive precatalyst presents several key benefits, such as: (1) handling metal species that are stable under ambient conditions; (2) switching on catalysis at a given time and place; and (3) regulation of the generated active catalyst by controlling the photoactivation process. It has been identified that irradiation with light is an intriguing approach for the activation of thio, chalcogenoglycosides and selenoglycosides under mild, user friendly reaction conditions. Photocatalysts and photosensitizers act as receivers to collect light and transfer it to the substrate via sensitization, which can occur via energy or electron transfer. The structures15,16 (Fig. 1) of this family of catalysts are dominated by highly conjugated systems, as expected for a class of molecules designed to interact with light, which include simple aromatic chromophores, both neutral and charged, functionalized organic dyes, inorganic clusters, and transition metal complexes whose properties can be easily tuned via ligand modification.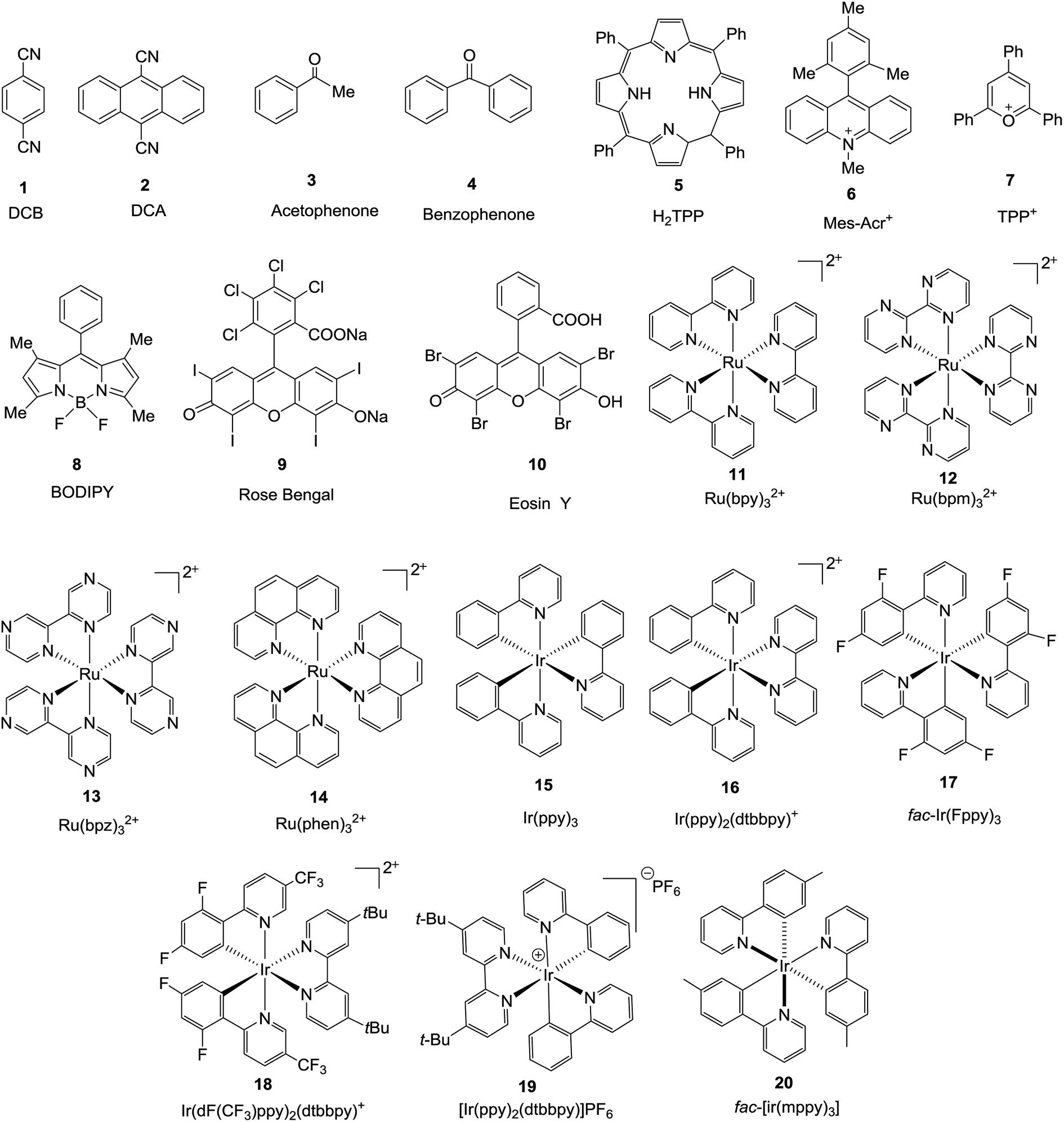 | ||
| Fig. 1 Structures of different photocatalysts. | ||
The potential role of photocatalysis in carbohydrate chemistry has been validated due to its significant applications such as the synthesis of heteroglycoclusters, vaccines and oligosaccharides. This section is divided into three sections on the basis of the common photocatalytic activation steps. The various photoactivation processes often lead to similar reactive intermediates and due to their short lifetimes, it is often difficult to unambiguously determine which mechanism of photoactivation is operative in a given transformation.
The first approach is the activation of glycosyl donors by single electron transfer using electrochemical and photochemical methods. Photoinduced electron transfer is an essential step in the conversion of solar energy to chemical energy. Photochemical synthesis is based on the photoexcitation of molecules to participate in electron transfer (or “photoredox”) processes.17 During this process, the absorption of light generates molecules in excited electronic states which are susceptible to accepting or donating electrons. Thus, reactions involve activation via either a one-electron reduction or one-electron oxidation of an organic substrate by the photocatalyst (Fig. 2), and the resulting organic radical ion species can directly react in a number of different bond-forming reactions.
 | ||
| Fig. 2 Mechanism of electron transfer via a photocatalyst. | ||
The second approach is photoinduced hydrogen atom transfer, which includes radical intermediates. This results from the reaction of excited-state photocatalysts via direct hydrogen atom abstraction, rather than by stepwise electron transfer (Fig. 3).18 It is based on the principle called persistent radical effect (PRE), which is the highly selective cross-coupling between a persistent and a transient radical when both species are formed at equal rates.19 The PRE is a general principle that explains the highly specific formation of the cross-coupling product between two radicals when one species is persistent (long lived) and the other transient and the two radicals are formed at equal rates.
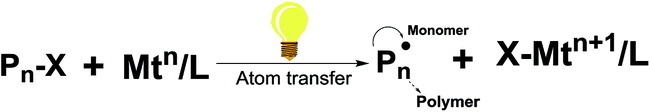 | ||
| Fig. 3 Mechanism of atom transfer via a photocatalyst. | ||
Third, organic substrates can also be activated via energy transfer (Fig. 4).20 The transfer of excited-state energy from a photocatalyst to an organic substrate commonly occurs through Dexter energy transfer. Transfer of excited-state energy from the photocatalyst to the substrate must be thermodynamically feasible. Photosensitization of reactions via this mode of activation is quite common in synthesis. The ability to absorb and convert the energy of a photon into useful chemical potential does not necessarily require strong bonding interactions with the substrate. Thus, the presence of potentially reactive binding site of photocatalysts is not required.
 | ||
| Fig. 4 Mechanism of energy transfer via a photocatalyst. | ||
3. Photoinduced electron transfer
Extensive research on the photophysics of complex formation and electron-transfer processes in the excited state has stimulated many investigations on the potential role of such intermediates in photochemical reactions.11,21,22 Photoinduction involves the promotion of an electron from the HOMO to the LUMO of a molecule. If the lifetime of this excited state is sufficiently long to engage in subsequent intermolecular reactions, it can either donate its high-energy electron to an appropriate acceptor (A) or fill its partially occupied orbital from a suitable electron donor (D). Thus, due to the formation of electron–hole pairs, an excited photocatalyst can be involved in both reductive and oxidative chemistry and the electron–hole pairs act as both a stronger reductant and a stronger oxidant than their ground-state.Fig. 5 outlines the generalized photoredox processes of photocatalysts. Metal complexes are generally referred to as visible light photocatalysts or photoredox catalysts. The absorption of light results in the generation of an electronically excited state. The redox properties of the excited state can be exploited upon fast electron transfer to an electron-deficient acceptor species (A) or from an electron-rich donor species (D). These species are referred to as oxidative and reductive quenchers, respectively, because they result in the formation of a different oxidation state of a photocatalyst in its electronic ground-state configuration. Regeneration of the photochemically active state requires a second electron-transfer process from a complementary donor or acceptor species. Thus, photoredox catalysis typically results in the formal transfer of an electron from one reagent to another, which produces a pair of reactive radical ions made up of the oxidized donor (D˙+) and reduced acceptor (A˙−).
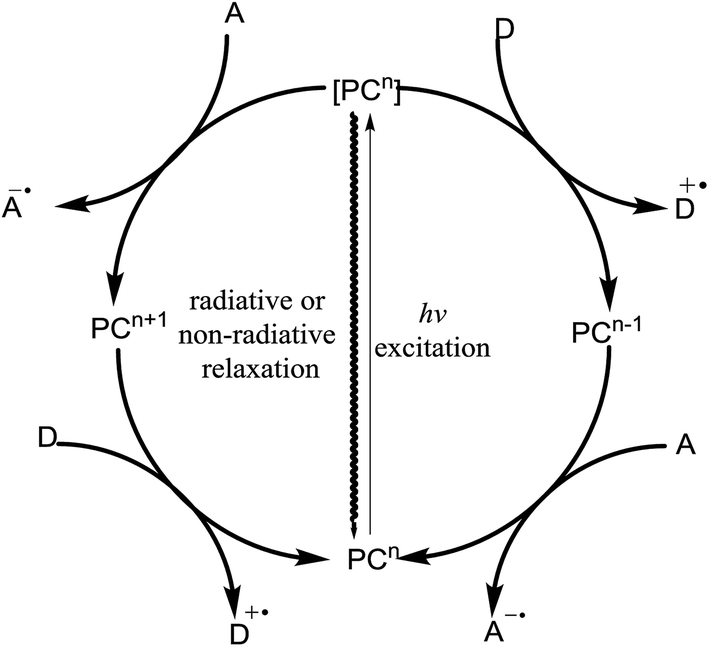 | ||
| Fig. 5 General cycle for photoinduced electron transfer (PET). | ||
These ions play an important role as the central intermediates and products in the dissociation reactions of many ionized organic molecules, the best-known example of which is the McLafferty rearrangement of the molecular ions of carbonyl compounds. As a result, the most important feature of a photocatalytic species is its ability to convert the energy of an incident photon into synthetically useful electrochemical potential.
Radical ion chemistry began in the context of a broader interest in the use of visible light in synthetic transformations.23 Photoinduced electron transfer reactions that occur between neutral electron donor molecules and neutral electron acceptor molecules have the following characteristics: (1) a radical cation and a radical anion are produced in a pair, (2) radical ion species are produced under neutral conditions and (3) the polarity inversion (umpolung) of the original electron donor and electron acceptor molecules commences through their conversion into radical ion species.24
There are two possible pathways by which this can occur. The first is the substrate may directly quench the excited state of the photocatalyst, in which a balanced redox equivalent is required to regenerate the photocatalyst. This pathway is workable when electron transfer between the substrate and the excited photocatalyst is thermodynamically feasible (Fig. 6). If it is not, then, the excited state can also be engaged by a secondary quencher. This produces an oxidized or reduced ground state catalyst that is much more redox active than the excited state (Fig. 6).25
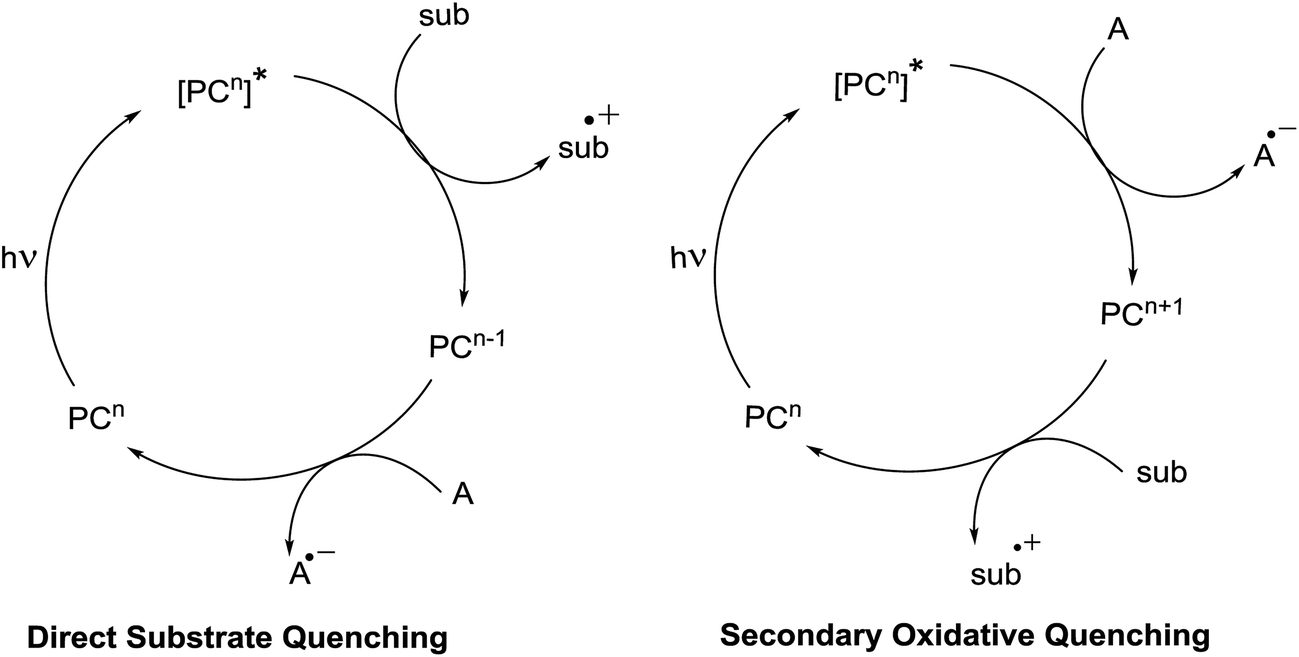 | ||
| Fig. 6 General schematic of photoredox cycles. | ||
3.1. Photocatalytic O-glycosylation with various thioglycoside donors
Significant advancement has been made to the elaboration of PET from 1990 by Griffin et al.26 They proposed that the thioglycoside path could be oppressed using the lability of photosensitive aryl thioglycoside donors in conjunction with their alkyl analogs as acceptors (Fig. 7). Thus, larger alkyl thioglycoside oligosaccharides were generated, activated and transformed into glycosyl donors.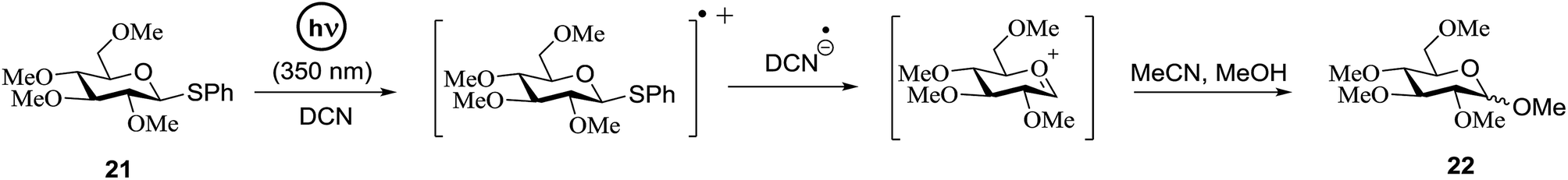 | ||
| Fig. 7 Example of the photoinduced formation of a glycosyl cation from a thioglycoside. | ||
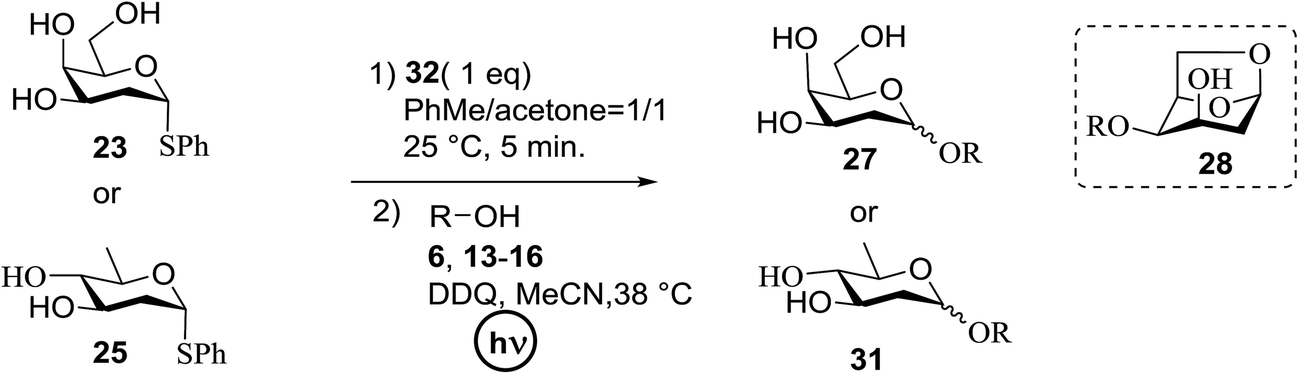
A novel photo-induced O-glycosylation reaction using an unprotected deoxythioglycosyl donor was developed by Nakanishi and co-workers.27 The O-glycosylation was executed using a single electron transfer (SET) mechanism with a variety of unprotected deoxythioglycosides and alcohols, and the use of 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) under long-wavelength UV irradiation. They proved that self-coupling or the formation of 1,6-anhydroglycoside can be efficiently prevented by the use of boronic acid as a temporary protecting group for the glycosyl donors. Diverse unprotected 2-deoxythioglycosides (Table 1) as glycosyl donors, including 2-deoxythiogalactoside (23),28 2-deoxythioglucoside (24),29 2,6-dideoxythioglucoside, thioolivoside,30 4-methoxyphenylboronic acid (32) as a temporary cyclic protecting group and DDQ as a single electron transfer, were chosen (Scheme 1) due to the reactivity of 2-deoxyglycosyl donors. These donors are easily activated compared to fully hydroxylated glycosyl donors due to the absence of an electron-withdrawing group at the C2 position31 which is also beneficial for regioselective glycosylation using an unprotected glycosyl donor.
| Entry | Donor | DDQ (equiv.) | Boronic acid | hν | % yield (α/β) |
|---|---|---|---|---|---|
| 1 | 23 | 1.5 | — | − | NR |
| 2 | 23 | — | — | + | NR |
| 3 | 23 | — | 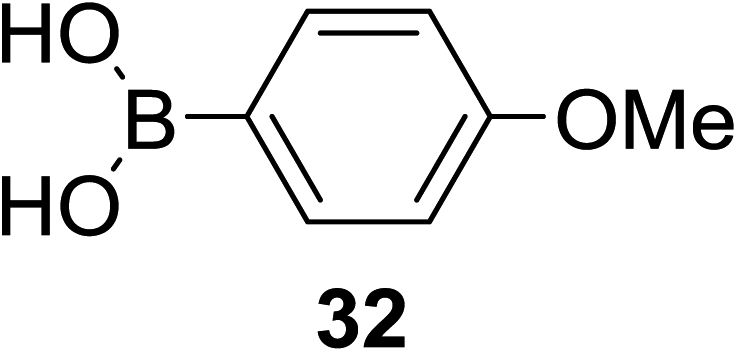 |
− | NR |
| 4 | 23 | 1.5 | — | + | 27 38 (81![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19) :19) |
| 5 | 23 | 1.5 | 32 | + | 27 74 (76:24) |
| 6 | 23 | 1.5 |  |
+ | 27 57 (73:27) |
| 7 | 24 | 1.5 | — | + | 30 57 (78:22) |
| 8 | 24 | 1.5 | 32 | + | 30 72 (74:26) |
| 9 | 25 | 1.5 | — | + | 31 82 (73:27) |
| 10 | 25 | 1.5 | 32 | + | 31 32 (80:20) |
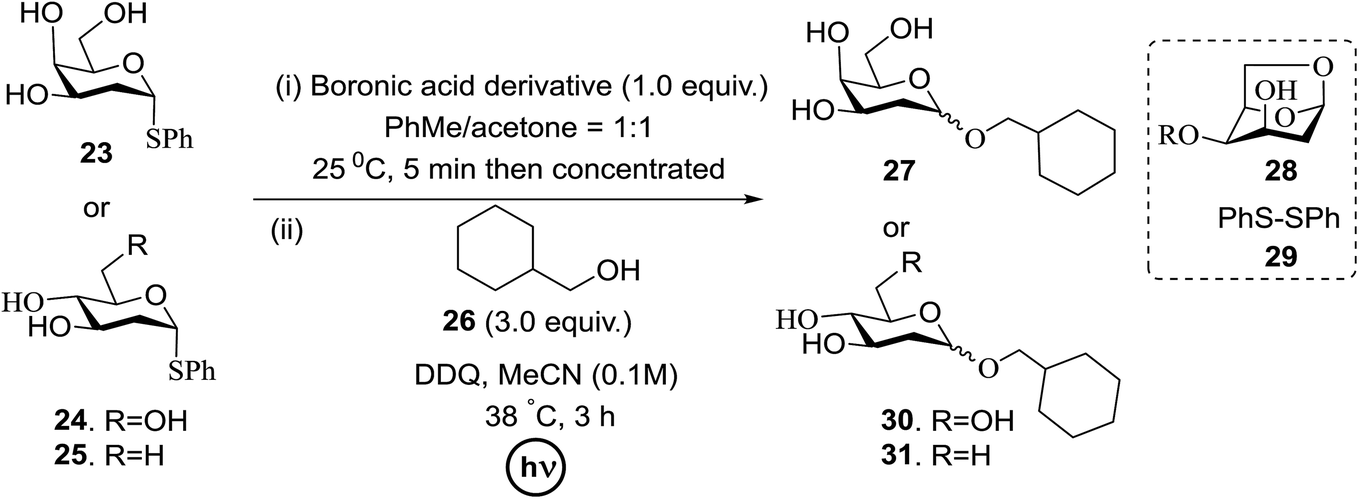 | ||
| Scheme 1 Glycosylation of 23–25 with 26 using DDQ under photo-irradiation conditions. | ||
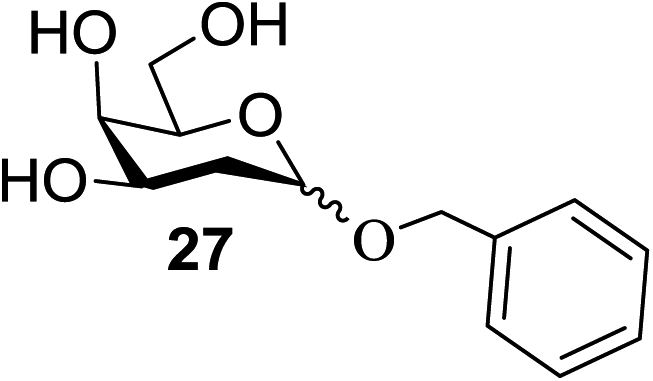
With the use of DDQ with photoirradiation via an SET mechanism, the transformation of the dithioacetal functionality into the respective ketone functionality in the presence of water has been recorded.32 On the basis of this a thioglycosyl donor and alcohol were used in the place of dithioacetal and water, respectively, and the O-glycoside was efficiently formed in a similar way. The glycosylation of 23 and cyclohexylmethanol (26) using DDQ under photo-irradiation conditions (365 nm, 100 W) were examined. The glycosylation of 23 and 26 using DDQ under photoirradiation for 3 h provided the glycoside 27 in 38% yield (entry 4, Table 1) and in contrast, in the presence of 32 under photoirradiation for 3 h the reaction smoothly proceeded to give 27 in 74% yield (entry 4, Table 1). This result clearly indicates the utility of temporary protection of the 4,6-diol of 23 by 32. On the contrary, the glycosylation of 23 and 26 using DDQ in the presence of 32 under photoirradiation for 3 h smoothly proceeded to give the glycoside 27 in 74% yield, (entry 5, Table 1). This result definitely confirms the application of temporary protection of the 4,6-diol of 23 by 32. On the other hand, the yield of 27 decreased when phenylboronic acid (33)33 was used instead of 32. The reason for this is due to the low activation of 23 resulting from the stronger electron-withdrawing nature of 33 compared to 32 (entry 6, Table 1). Under these reaction conditions no epimerization of 27 appeared which indicates that glycosylation proceeded under kinetic control and high α-stereoselectivity was provided due to the kinetic anomeric effect. These results are compiled in Table 1.
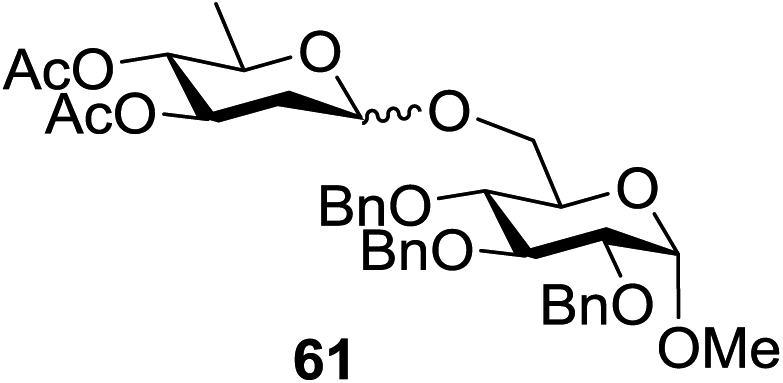
Similar type of behaviour was observed with 24, where the glycosylation of 24 and 26 using DDQ in the absence of 32 under photoirradiation conditions afforded 30 in moderate (57%) yield (entry 7 in Table 1) but in the presence of 32 under photo-irradiation conditions afforded 30 in good (72%) yield (entry 8 in Table 1). On the other hand, when 25 was used as a glycosyl donor, the opposite pattern was detected; the glycosylation of 25 and 26 using DDQ in the absence of 32 under photoirradiation conditions gave 31 in high yield (82%, entry 9, Table 1), whereas the presence of 32 gave 31 in low yield (32%, entry 10, Table 1). This may be due to the fact that 25 selectively reacts with 26 even without 32 due to the lack of a highly reactive primary hydroxyl group at the C6 position. Furthermore, the results suggest that in the presence of 32, the free 32, which cannot bind to 26, trapped 26 to give the corresponding boronic ester.
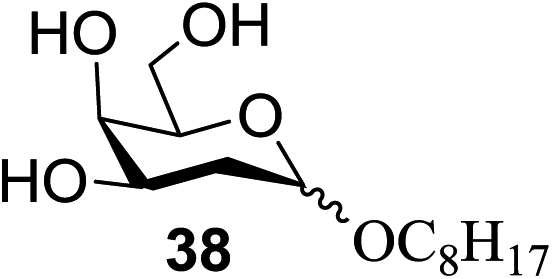
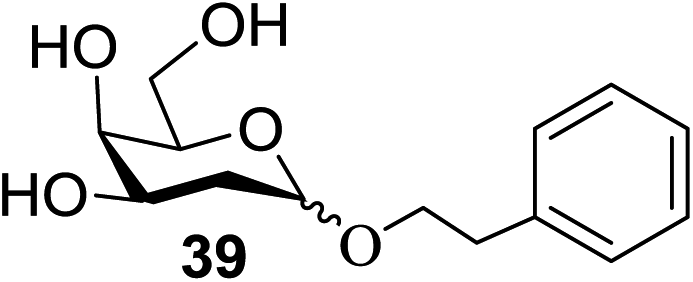
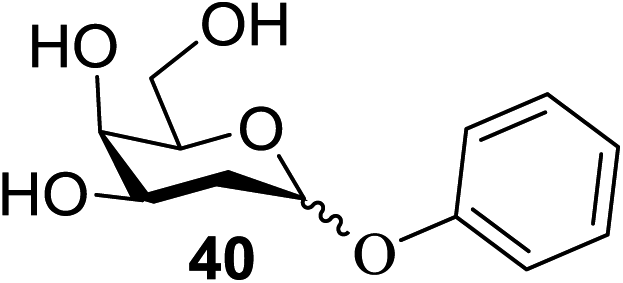
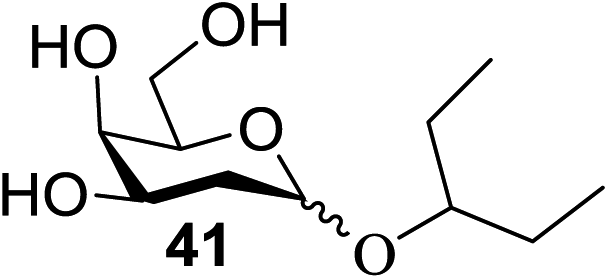
The universality of the present glycosylation method using several primary and secondary alcohols, 34–37, was explored (Scheme 2). These results are summarized in Table 2. It was found that good to high yields of the unprotected glycosides 38–41 were obtained from the glycosylation of 23 and 34–37, as well as 26, using DDQ in the presence of 32 for 3 h under photoirradiation (entries 4, 6, 8 and 10, Table 2). It was also proven that these results are completely opposite to that achieved in the absence of 4 (entries 3, 5, 7 and 9, Table 14). On the other hand, the glycosylation of 25 and 34–37, as well as 26, using DDQ in the absence of 32 for 3 h under photoirradiation resulted in the unprotected glycosides 42–45, respectively, in good to high yield (entries 12–15, Table 2). The generality, appropriateness and applicability of the present glycosylation method are represented by these results.
 | ||
| Scheme 2 O-Glycosylation under photoirradiation condition using different alcohols. | ||
| Entry | Donor | Alcohol | Boronic acid (32) | Product | % yield (α:β) |
% yield 28 |
|---|---|---|---|---|---|---|
| 1 | 23 |  |
— |  |
38 (81:19) |
61 |
| 2 | 23 | 26 | 32 | 27 | 74 (76:24) |
— |
| 3 | 23 |  |
— |  |
39 (75:25) |
47 |
| 4 | 23 | 34 | 32 | 38 | 80 (75:25) |
— |
| 5 | 23 |  |
— |  |
36 (74:26) |
43 |
| 6 | 23 | 35 | 32 | 39 | 84 (81:19) |
— |
| 7 | 23 |  |
— |  |
15 (82:18) |
61 |
| 8 | 23 | 36 | 32 | 40 | 71 (86:14) |
— |
| 9 | 23 |  |
— |  |
9 (85:15) |
53 |
| 10 | 23 | 37 | 32 | 41 | 70 (85:15) |
— |
| 11 | 25 | 26 | — |  |
82 (81:19) |
— |
| 12 | 25 | 34 | — | 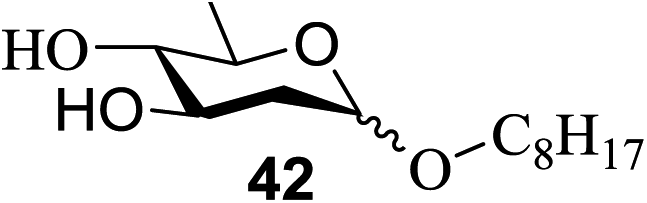 |
79 (76:24) |
— |
| 13 | 25 | 35 | — | 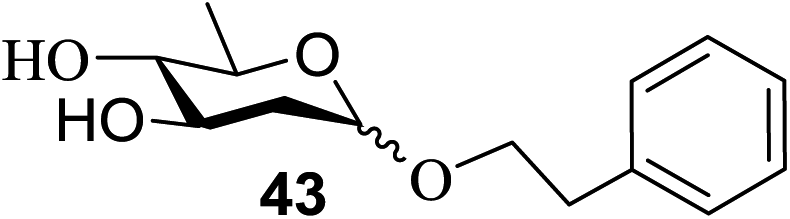 |
72 (70:30) |
— |
| 14 | 25 | 36 | — | 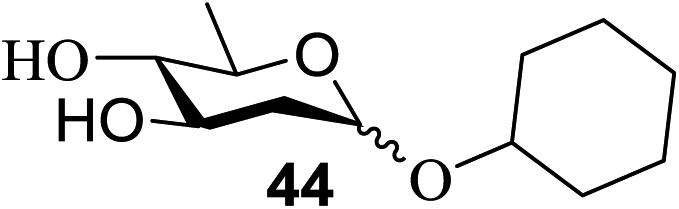 |
70 (71:29) |
— |
| 15 | 25 | 37 | — | 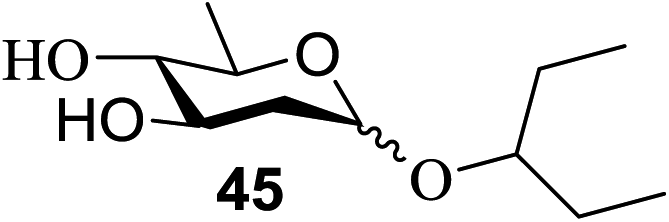 |
70 (70:30) |
— |
As further proof, Bowers and co-workers34 developed the O-glycosylation of thioglycosides activated via visible light. Visible light catalysis permits the efficient formation of single electron transfer (SET) redox cycles. Mechanistic studies indicate that the full reaction is light sensitive and it occurs through a mechanism involving the decomposition of an oxidatively formed sulfur radical cation and propagation via the reduction of the thiol as a side product. They determined that the metal–ligand charge complex is generated by oxidative quenching of an excited state visible light catalyst, such as Ru(bpy)3Cl2 or Ir[dF(CF3)ppy]2(dtbbpy)PF6. These catalysts in the presence of visible light are sufficiently energetic to oxidize an electron-rich aryl thioglycoside, which generates a stabilized oxocarbenium ion intermediate via fragmentation of the radical cation.35 Subsequent condensation with an alcohol acceptor results in an O-glycosidic linkage with the formation of an acid and the symmetric disulfide as a side product. This is another example of a visible light mediated glycosylation from thioglycosides, although photoinitiated glycosylations via UV irradiation of selenoglycosides were known. The priority of a protic solvent to favour solvation and the separation of the charge transfer complex have been well documented for visible light catalysts such as Ru(bpy)3Cl2 and Ir[dF(CF3)ppy]2(dtbbpy)PF6.
All primary, secondary, and tertiary alcohols (Table 3) were acceptable nucleophiles and yielded a mixture of O-glycosides anomers. The majority of work was carried out with the tetramethyl donor 48 and tetrabenzyl donor 46 due to their good yields, whereas tetra-acetyl thioglycoside (47) did not activate under the present conditions, presumably due to its higher oxidation potential. A non-nucleophilic, protic solvent effects glycosylation with ≤2 equivalent of acceptor alcohol. Hexafluoroisopropanol (HFIP) proved an effective alternate and 51% yield of methyl glycoside was obtained. However, a more significant increase in yield was obtained when the co-oxidant was charged in stoichiometric quantities. 0.25 equivalent of either CBr4 or BrCCl3 contributed to good yields of methyl glycosides and decreased side products. The reaction is completely dependent on light and ceases in its absence.
|
||||
|---|---|---|---|---|
| Entry | Donor | R1OH (2 equiv.) | Product | Yield (%) (α:β) |
| 1 | 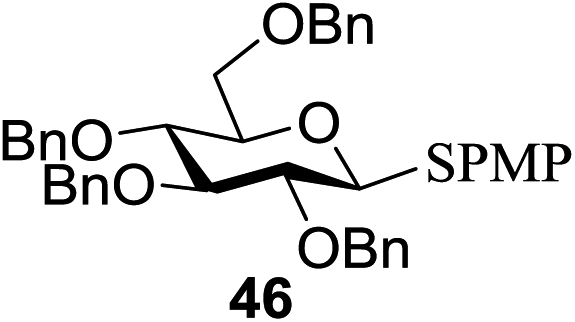 |
 |
 |
65 (0.9:1) |
| 2 | 46 |  |
 |
66 (1:1.5) |
| 3 | 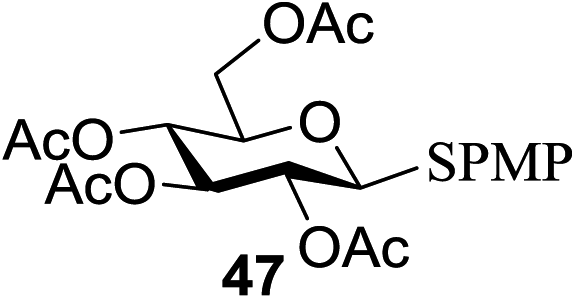 |
50a | N/A | NR |
| 4 | 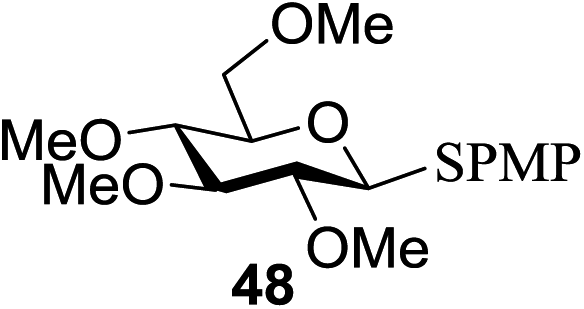 |
50a | 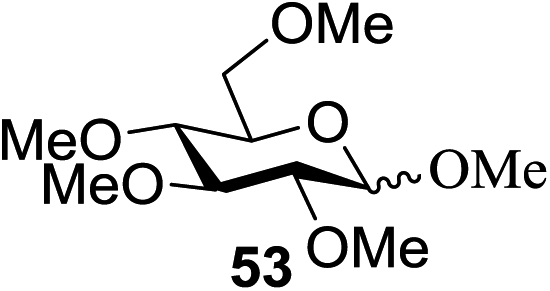 |
71 (1.6:1) |
| 5 | 48 |  |
 |
77 (0.75:1) |
| 6 | 48 | 50b | 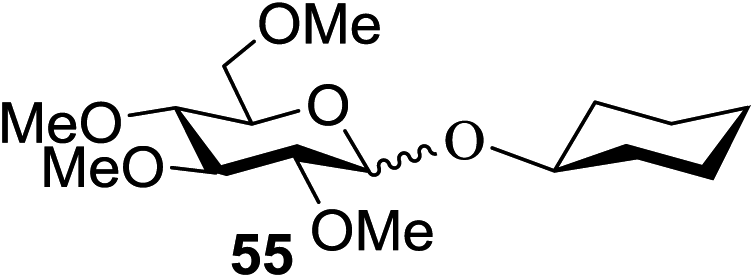 |
87 (1.6:1) |
| 7 | 48 |  |
 |
71 (1.1:1) |
| 8 | 48 |  |
 |
71 (1.6:1) |
| 9 | 48 |  |
 |
23 (1.2:1) |
| 10 | 48 |  |
 |
61 (1.6:1) |
| 11 |  |
50a |  |
82 (1.4:1) |
| 12 | 49 |  |
 |
42 (1.2:9) |
The mechanism involves a visible light activated single-electron oxidation on sulfur, which potentiates formation of an oxocarbenium ion. The catalytic cycle is initiated by the cooxidant and propagated by the reduction of a thiol radical generated from the collapse of the thioglycoside radical cation. The latter thiol radical may be present as a naked radical or thiol radical cation formed by combination with in situ generated acid (Fig. 8). Separation of the disulfide could be achieved by the oxidation of 2 equiv. of thiol, possibly with the concomitant formation of molecular hydrogen.
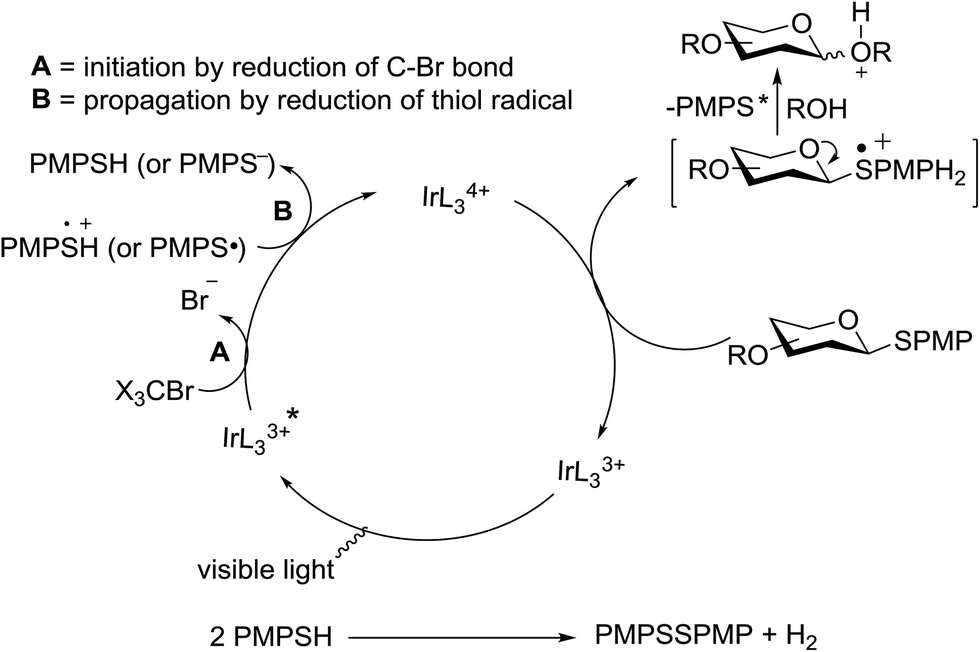 | ||
| Fig. 8 Proposed mechanism of O-glycosylation of thioglycosides under visible light. | ||
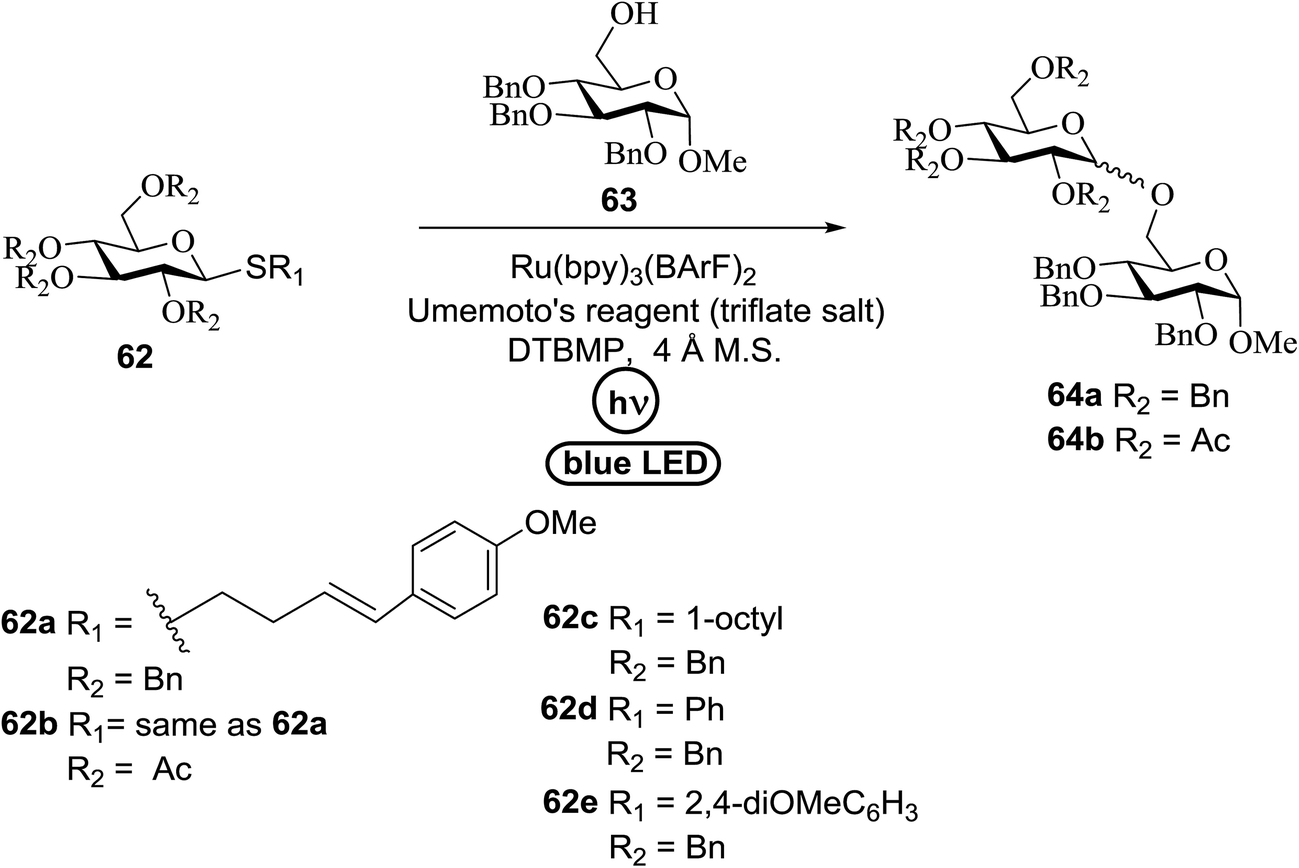
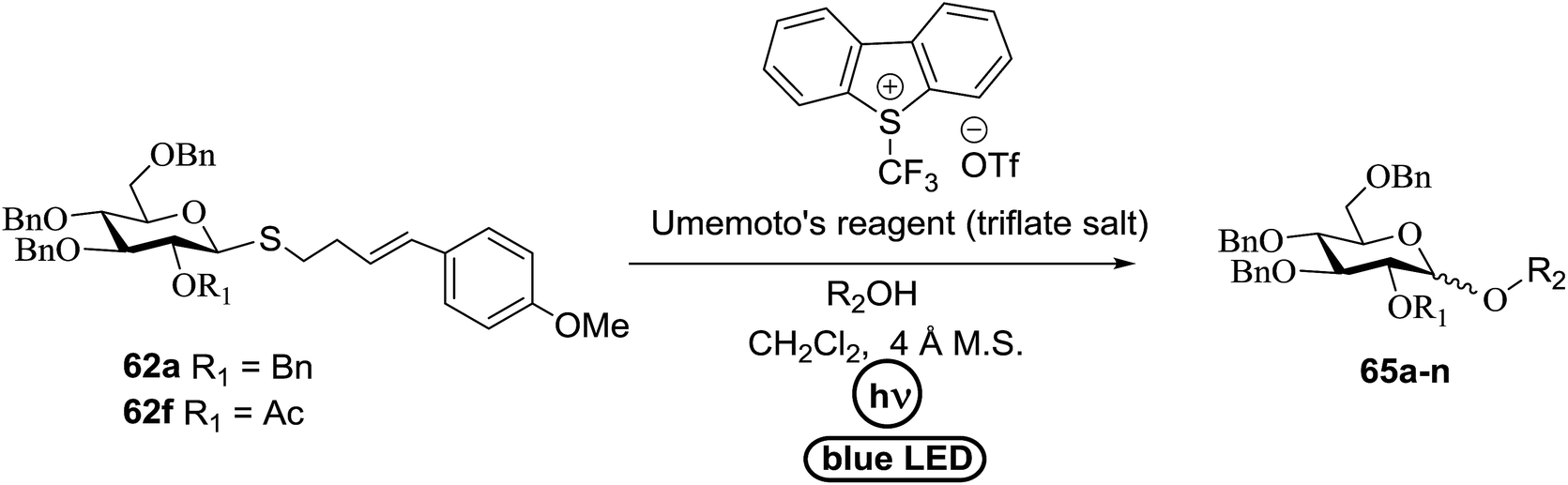
An attempt to enhance the utility of visible-light irradiation of 4-p-methoxyphenyl-3-butenylthioglucoside donors in the presence of Umemoto's reagent and alcohol acceptors serves as a soft approach to O-glycosylation. Ragains J. R. et al.36 utilized irradiation with visible light as an absorbing method for the activation of thio- and selenoglycosides under mild, user-friendly reaction conditions (Scheme 3). This reaction represents the first example of a visible-light promoted glycosylation not requiring a photocatalyst/photosensitizer.37 The reaction conditions involved irradiation with blue LEDs (λmax = 455 nm) in the presence of Umemoto's reagent, 2,6-di-tert-butyl-4-methylpyridine (DTBMP), and the acceptor 63 with 4 Å molecular sieves in CH2Cl2. O-Glycosylations progress from moderate to high (44–93%) yields and complete β-selectivity can be achieved. They synthesized a series of thioglycoside (Table 4), including the benzyl protected 62a, acetyl protected 62b and the aryl- and alkyl thioglycosides 62c–e.38 Irradiation with the donor 62a resulted in 55% yield of the product 64a as a mixture of anomers.
 | ||
| Scheme 3 [Ru(bpy)3] catalyzed glycosylation with Umemoto's reagent under visible light. | ||
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Donor | Yield | α/β | Entry | Donor | Yield | α/β |
| 1 | 62a | 55 | 1.4:1 |
6 | 62a | 75 | 1.6:1 |
| 2 | 62a | 0 | — | 7 | 62b | 0 | — |
| 3 | 62a | 0 | — | 8 | 62c | 0 | — |
| 4 | 62a | 50 | 1.3:1 |
9 | 62d | 0 | — |
| 5 | 62a | 76 | 1.1:1 |
10 | 62e | 0 | — |
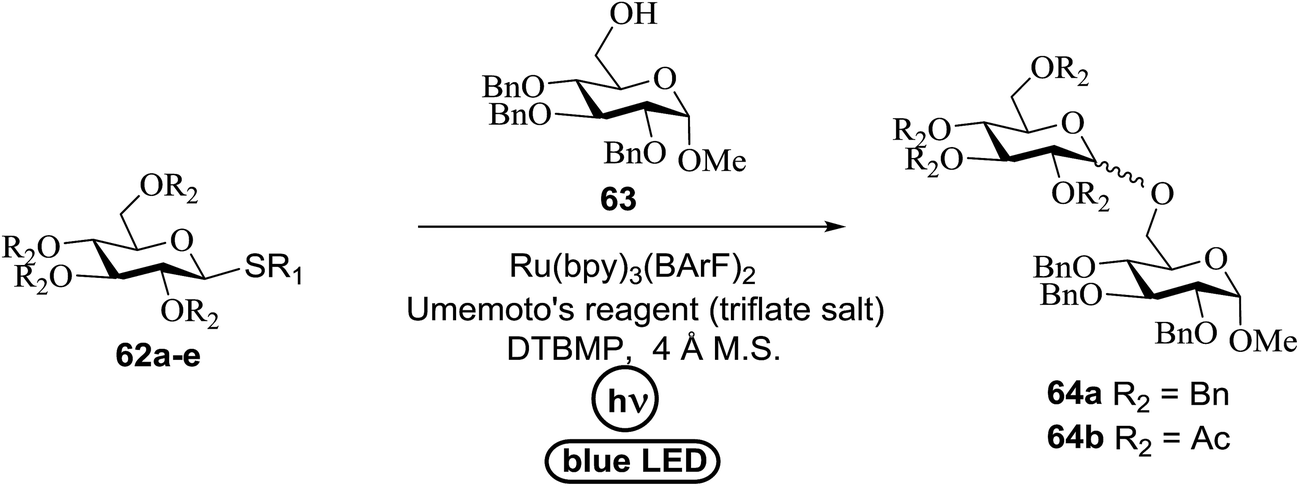
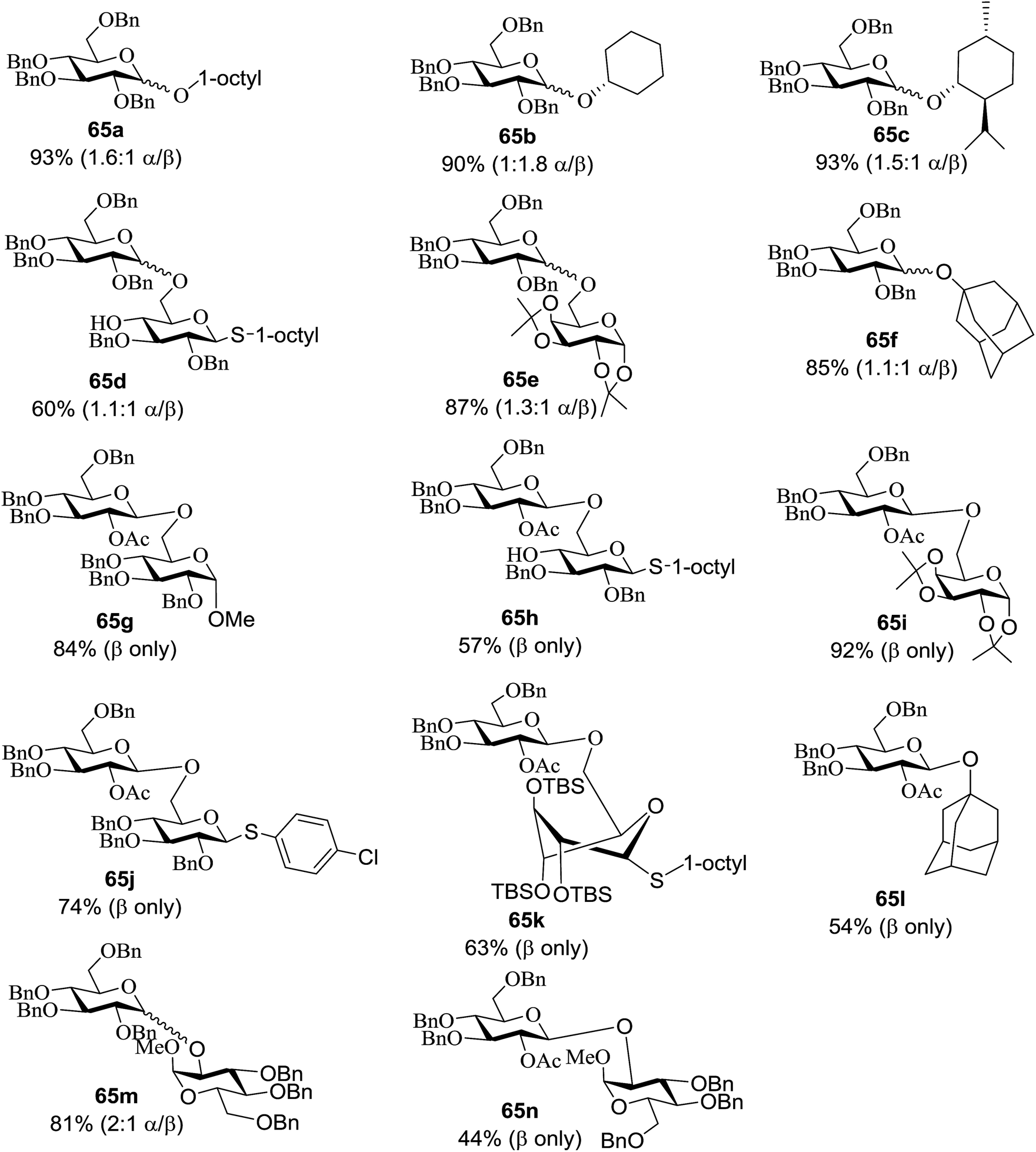
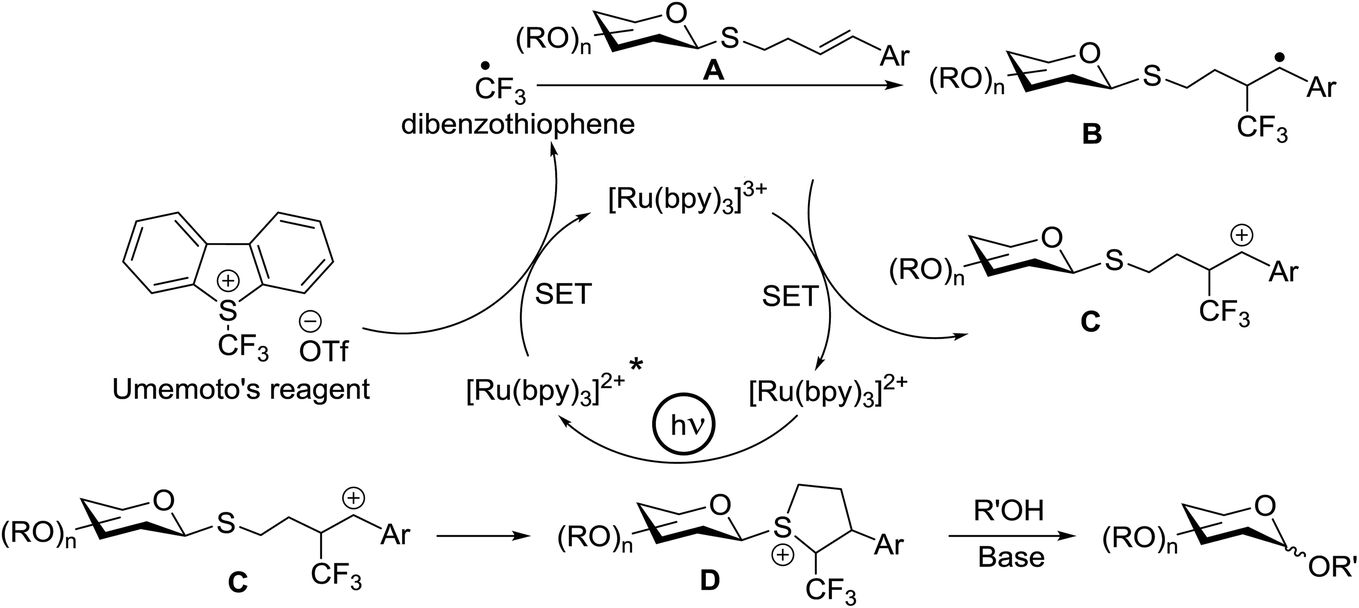
Optimization of the reaction conditions showed that using 62a in excess and increasing the concentration results in an increase in yield (entry 5, Table 4). The yields were not improved by increasing the concentration, but using the acceptor 63 in excess, as in entries 1–4 in Table 4. The sulfur atom in 62b is much more resistant to oxidation, but species such as 62c are highly reactive toward activation by thiophilic electrophiles; whereas the electron-rich nature of the aryl thio moiety of 62e is particularly responsible for oxidation by SET. Reaction of the sulfur atom with an activated intermediate was inhibited with 62b. The substrate scope of the reaction is shown in Table 5. Glycosylation of 1-octanol, cyclohexanol, and (−)-menthol with 62a proved to be high-yielding. Stereoselectivity was low with the benzyl group present at the 2-position of 62a, but the donor 62f, which bears acetate at the 2-position identical to donor 62f, provided complete β-selectivity (Table 5, entries 7–12) due to neighbouring group participation. This reaction condition contributed to extraordinary yields of the glycosidic product and no detection of oligomerization in the silylated acceptor, which verified the novelty of this method. The visible-light promoted excitation of [Ru(bpy)3]2+ precedes single-electron transfer to Umemoto's reagent to generate a trifluoromethyl radical, dibenzothiophene, and [Ru(bpy)3] (Fig. 9). Further attack of the trifluoromethyl radical on the styrene portion of glycosyl donors 62a results in the benzylic radical B, which could be oxidized by [Ru(bpy)3]3+ to generate the carbocation C and regenerate [Ru(bpy)3]2+.39 Cyclization of sulfur onto the cation of C would result in the intermediate D, which is expected to be competent as an activated donor for O-glycosylation. Experimental and computational evidence showed the intervention of an electron donor–acceptor (EDA) complex.40
|
|---|
 |
 | ||
| Fig. 9 Proposed catalytic cycle of [Ru(bpy)3] catalyzed O-glycosylation. | ||
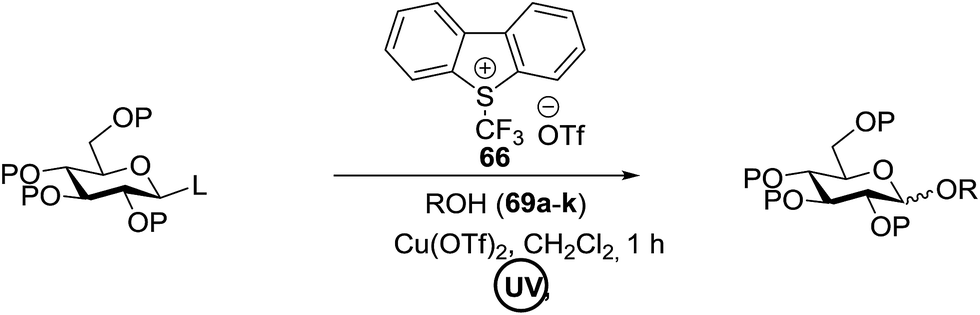
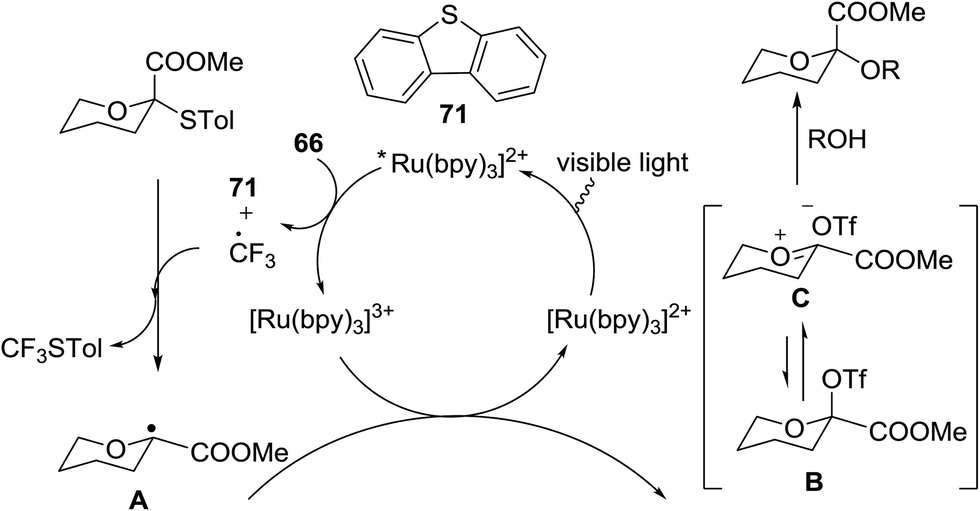
The activation of thioglycosides using a light driven strategy via a consolidated CF3 radical process and subsequent glycosylation with glycosyl acceptors was explored by Mao and co-workers.41 This protocol effectively activates thioglycosides, which increases the substrate scope of reaction. Glycosylation reactions that were completely non-reactive under the existing photo-induced glycosylation conditions occurred efficiently in high yields. Many common protective groups were applicable under the coupling conditions. Both UV and visible light and even sunlight were used as the source of light for the reactions.
This glycosylation was based on the assumption that the merger of an additional radical pathway with light-mediated glycosylation would lead to a potentially interdependent pathway, which activates a glycosyl donor regardless of its redox potential (Fig. 10, path II). This scheme requires the radical precursor 66 to immediately bear an electrophilic radical R under illumination. For this objective, two main requirements must be fulfilled for 66. (1) It must be inert under normal conditions, whereas reactive under light; and (2) the radical must be generated in a controllable manner. It has been found that light can be employed to form the highly electrophilic trifluoromethyl radical by the use of commercially available CF3 sources under mild conditions.42,43 The trifluoromethyl radical reacted with thiols to form trifluoromethyl sulfides via a radical mechanism.44,45 Thus, trifluoromethylating reagents46 such as Umemoto's reagent47 and Togni's reagent48 (Fig. 10) might be ideal radical precursors for light-induced glycosylation.
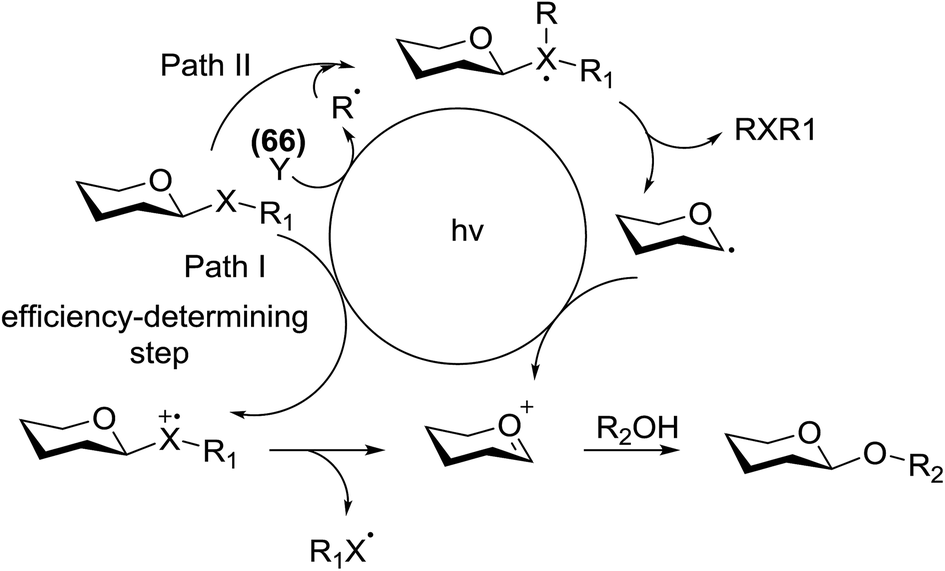 | ||
| Fig. 10 Mechanistic path for light induced glycosylation. | ||
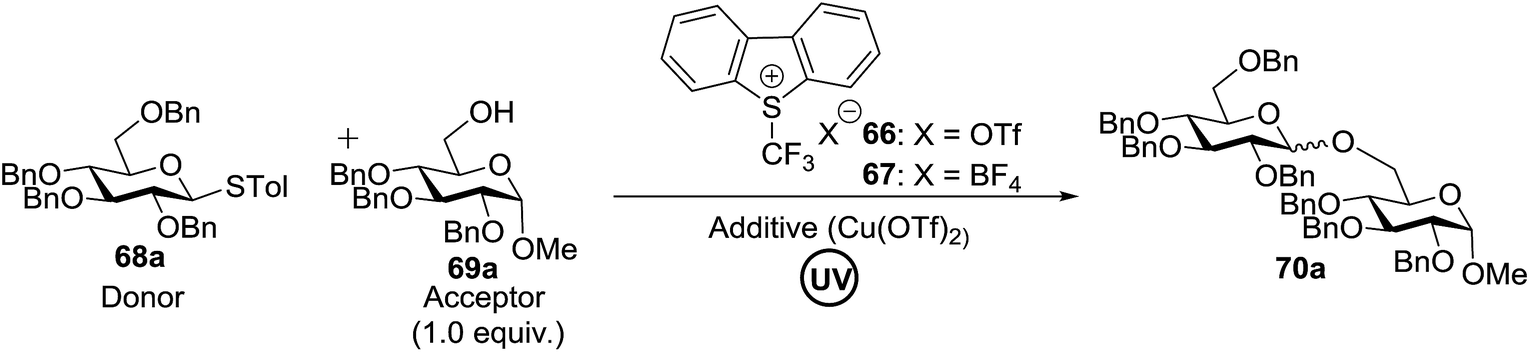
The reaction of donor 68a with acceptor 69a under UV light irradiation was investigated under different reaction conditions. In the absence of photoirradiation, no desired product was detected with the use of either Umemoto's reagent or Togni's reagent (Table 6, entry 1). In the presence of both photoirradiation and trifluoromethylating reagents, only a small amount (not more than 9% yield) of the required product 70a was formed in various solvents (Table 6, entries 2–5). Additives including bases, photosensitizers, and metal salts improved the coupling yield in a trace amount. The additive Cu(OTf)2 afforded a good yield (Table 6, entry 6, 39%) which was promoted to 72% by decreasing the reaction temperature (Table 6, entries 7 and 8). Further standardization of the equivalent of Cu(OTf)2 (Table 6, entries 8–10) and Umemoto's reagent 66 (Table 6, entries 12–15) as well as the ratio of 68/69a (Table 6, entries 10–12) determined the optimized conditions of donor 68a (1.3 equiv.), acceptor 69a (1.0 equiv.), 166 (1.5 equiv.), 4 Å MS and CH2Cl2 in a quartz flask under UV light irradiation for one hour at −72 °C (Table 6, entry 16, 85%). Control experiments showed that though Cu(OTf)2 seemed to be not essential for the yield of 70a, it led to a higher yield and better reproducibility (Table 6, entries 16 and 17).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | 68a (equiv.) | Activator (equiv.) | Additive (equiv.) | Yield (%) | Entry | 68a (equiv.) | Activator (equiv.) | Additive (equiv.) | Yield (%) |
| 1 | 1.2 | 66 (1.8) | — | 0 | 11 | 1.3 | 66 (2.0) | 2.0 | 79 |
| 2 | 1.2 | 66 (1.8) | — | 5 | 12 | 1.4 | 66 (2.0) | 1.5 | 78 |
| 3 | 1.2 | 67 (1.8) | — | 5 | 13 | 1.3 | 66 (1.0) | 1.5 | 69 |
| 4 | 1.2 | 66 (1.8) | — | 9 | 14 | 1.3 | 66 (1.3) | 1.5 | 77 |
| 5 | 1.2 | 67 (1.8) | — | 6 | 15 | 1.3 | 66 (1.4) | 1.5 | 83 |
| 6 | 1.2 | 66 (1.8) | — | 39 | 16 | 1.3 | 66 (1.5) | 1.5 | 85 |
| 7 | 1.2 | 67 (1.8) | 1.0 | 43 | 17 | 1.3 | — | — | 80 |
| 8 | 1.2 | 66 (1.8) | 1.0 | 72 | 18 | 1.3 | 66 (1.5) | 1.5 | 93 |
| 9 | 1.2 | 66 (1.8) | 1.0 | 75 | 19 | 1.3 | 66 (1.5) | 1.5 | 81 |
| 10 | 1.2 | 66 (1.8) | 1.5 | 67 | |||||
Under the optimized high-yielding reaction conditions, a further investigation was conducted using the disarmed donor 68b and various acceptors (Fig. 11). The glycosyl donor 68b was completely non-reactive under the conventional photo-induced glycosylation conditions. The reaction of 68b with the glycosyl acceptors 69a–e having a free hydroxyl group at the C-6, C-4, C-2, and C-3 positions, respectively, continued in good to excellent yields under the standard conditions. However, the coupling yields were 6–17% higher with the use of the disarmed donor. This might be due to the formation of less reaction by-products. The tough coupling reaction of the disarmed 68b with the hindered 69c was successful and isopropylidene, benzoyl and benzyl protective groups were well accepted under these conditions.
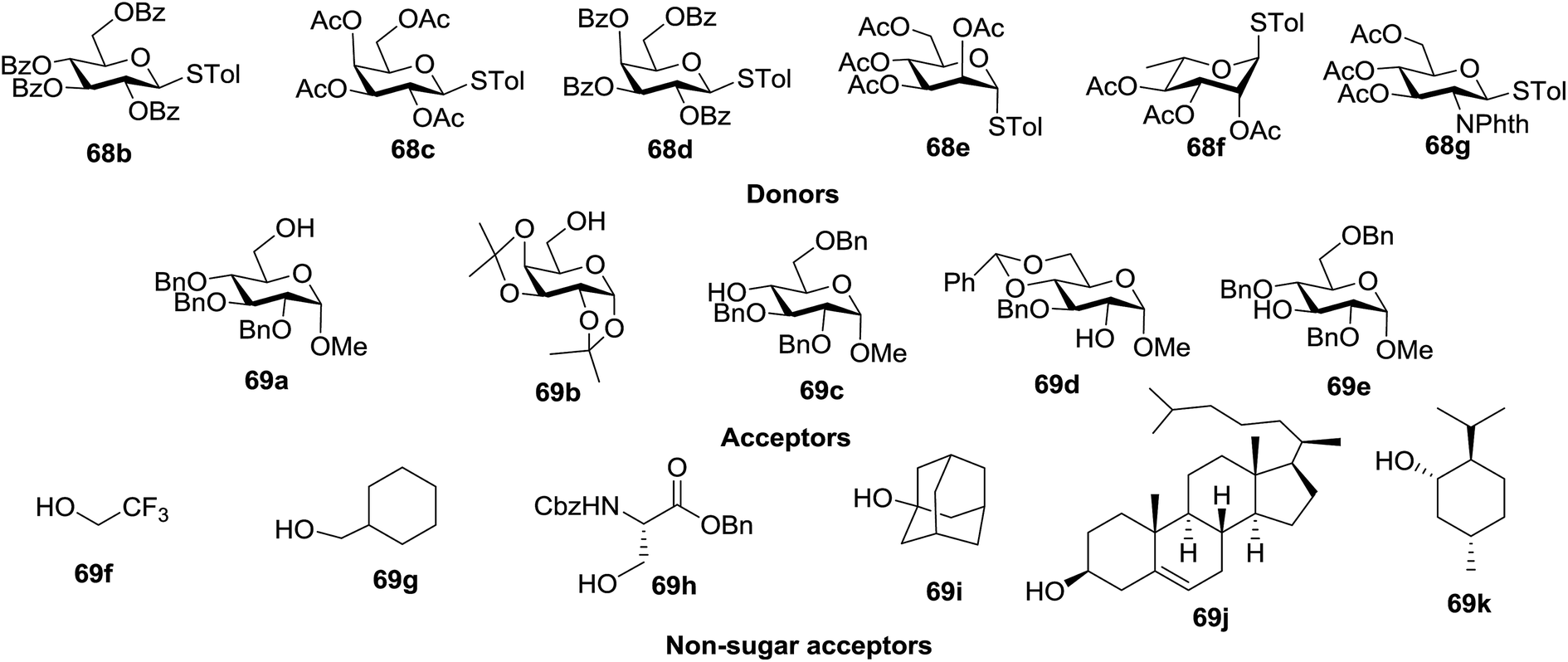 | ||
| Fig. 11 Various glycosyl donors, acceptors and non-sugar acceptors. | ||
On the basis of the optimized results a broad range of glycosyl donors (68a–g) and acceptors (69a–k) was further evaluated for glycosylation using the pre-activation protocol under UV light. Glycosyl acceptors with a free hydroxyl group at different positions of the glucose moiety were properly glycosylated with disarmed galactosyl, mannosyl, rhamnosyl or glucosaminyl donors in good to excellent yields (Fig. 11).
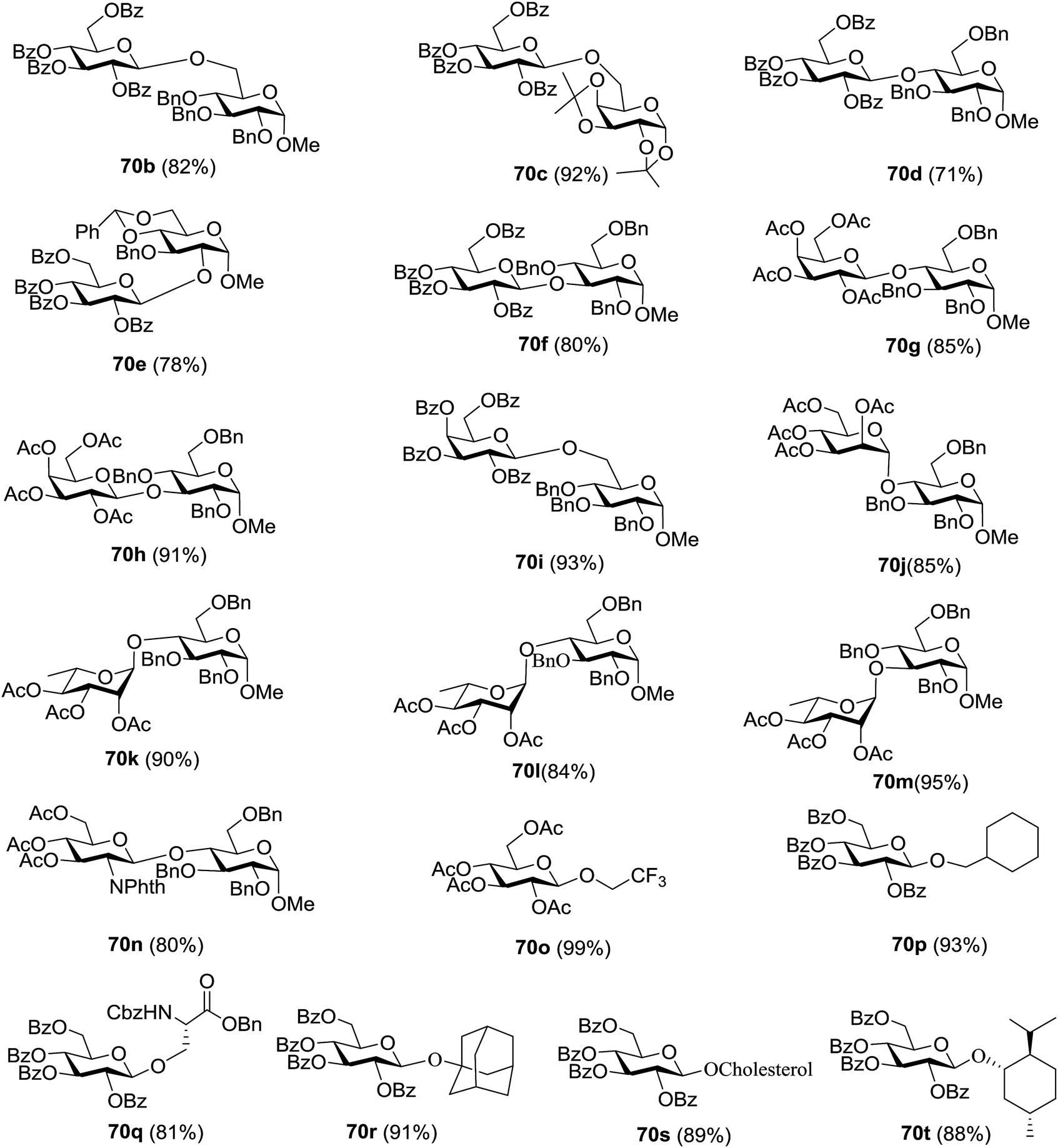
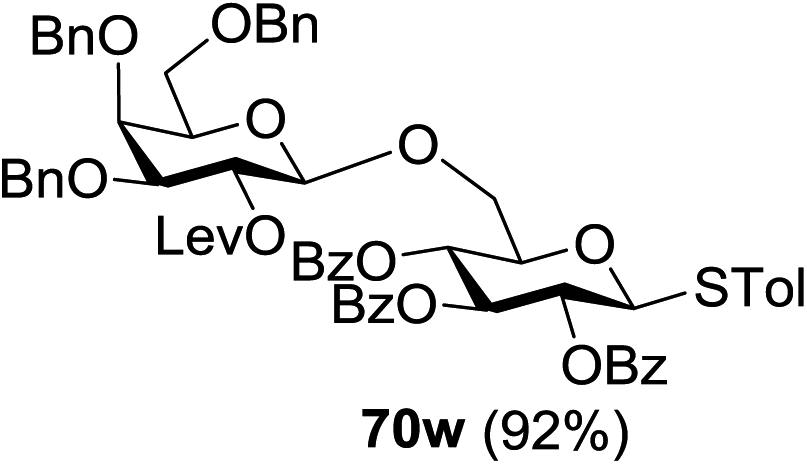
The coupling reactions of the disarmed donors with non-sugar alcohols, such as trifluoroethanol, cyclohexyl methanol, protected serine, adamantanol, cholesterol, and menthol, also proceeded smoothly in high yields (Table 7). Except for the SBox group, thioglycosides with an anomeric leaving group of 2-methylphenylthio, 2,6-dimethylphenylthio, ethylthio (SEt), or 2-pyridylthio (SPy) were also investigated as glycosyl donors (Table 8). Acceptor 69l with a p-tolylthio leaving group was also glycosylated to generate disaccharide 70w in 92% isolated yield, which explored the potential application of this protocol in the preactivation-based one-pot synthesis of oligosaccharides (Table 8, entry 6). The high efficiency of the light-promoted glycosylations was further exemplified by the one-pot assembly of trisaccharide 70x and tetrasaccharide 70y. As described in Fig. 12, the synthesis of trisaccharide 70x under visible-light irradiation was examined via the reactivity-based one-pot strategy which provide trisaccharide 70x in 52% isolated yield. Under UV light irradiation, the preactivation-based one-pot synthesis of oligosaccharides was also studied and 61% isolated yield of tetrasaccharide 70y and tetrasaccharide 70y was finally obtained without any intermediate isolation.
|
|---|
 |
| Entry | Donor | Acceptor | Disaccharide and yield |
|---|---|---|---|
| 1 |  |
69a | 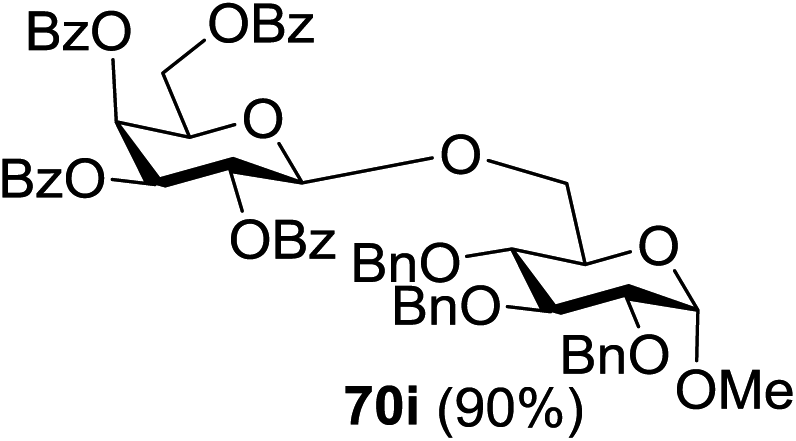 |
| 2 |  |
69a |  |
| 3 | 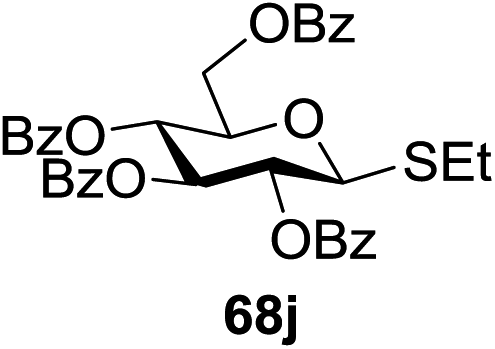 |
69a | 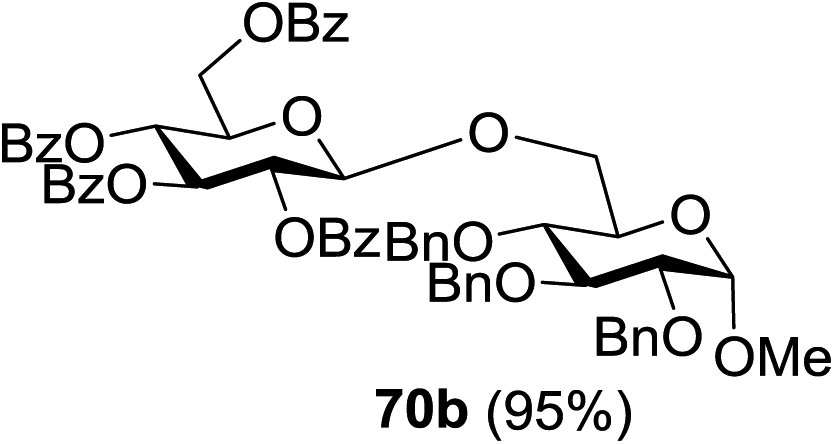 |
| 4 | 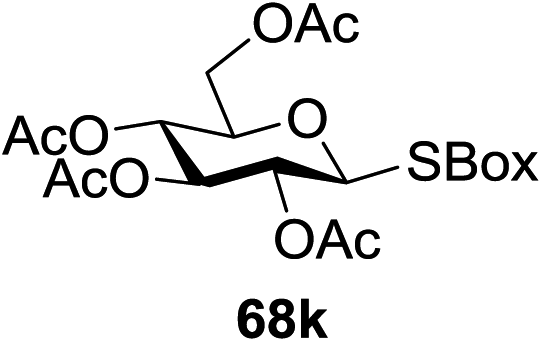 |
69a | 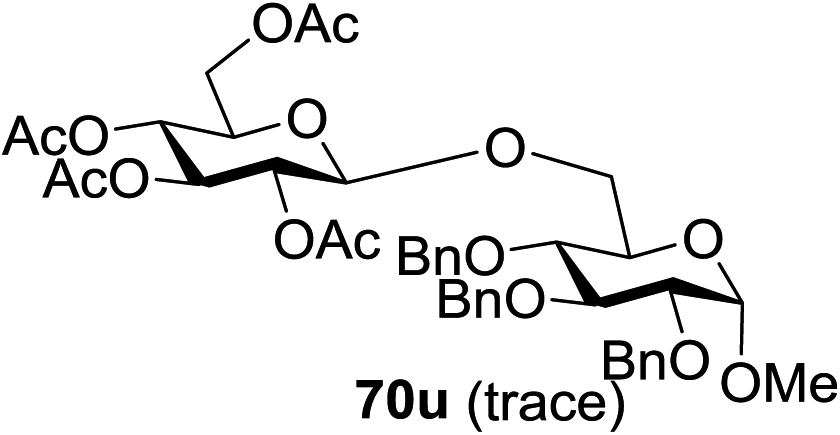 |
| 5 | 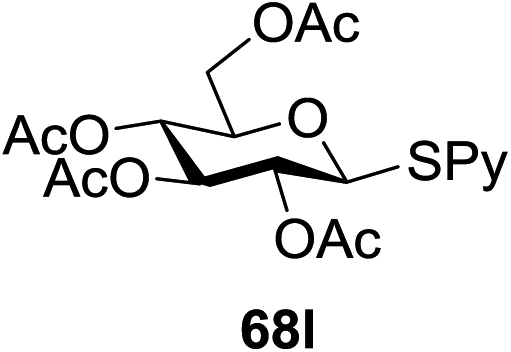 |
69c | 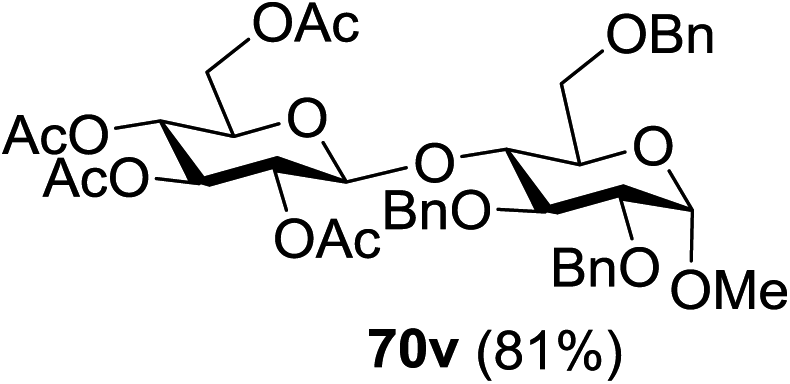 |
| 6 | 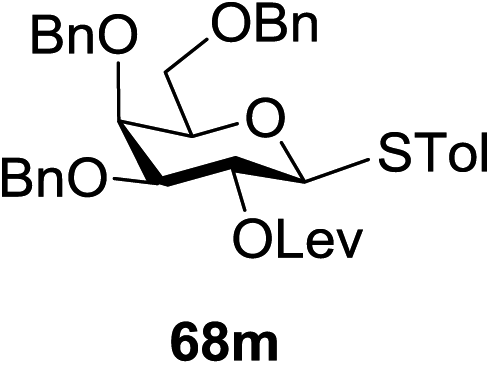 |
 |
 |
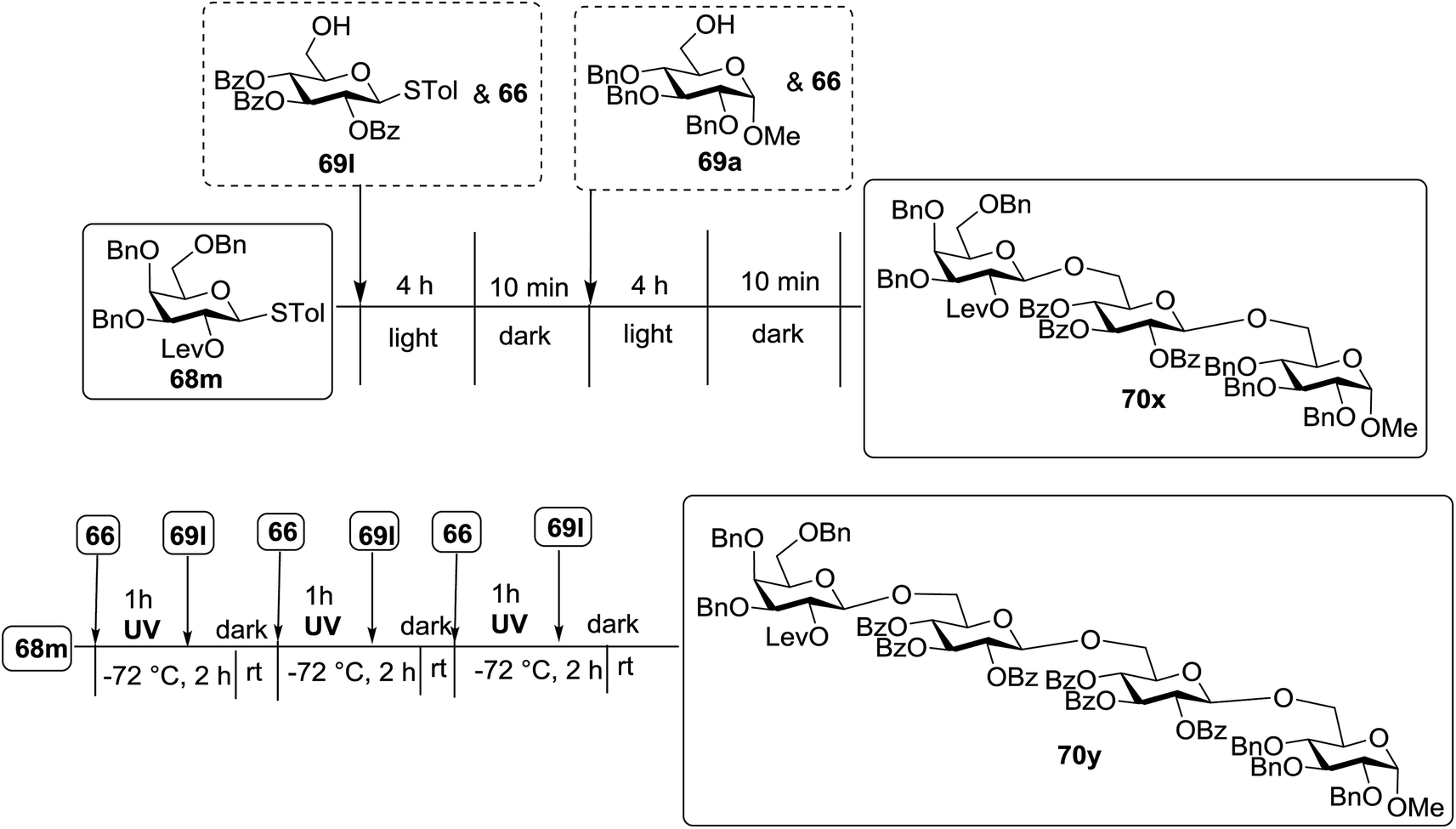 | ||
| Fig. 12 One-pot synthesis of trisaccharide 70x and tetrasaccharide 70y. | ||
On the basis of the reaction observations and literature the proposed possible mechanism is explained (Fig. 13).49–51 First, in the presence of visible light (path II), Umemoto's reagent 66 is reduced by the excited state *[Ru(bpy)3]2+ to give a CF3 radical, which further reacts with the sulfur atom in the thioglycoside donor to generate the radical intermediate D. The formation of p-tolyl(trifluoromethyl)sulfide from D produces the glycosyl radical intermediate E, which is then oxidized by [Ru(bpy)3]3+ to afford the glycosyl cation C. Under UV light irradiation, the hemolytic generation of the CF3 radical and dibenzothiophene radical cation (A) is followed by a conventional glycosylation process (path I) and/or a CF3 radical-participated process (path II). The recombination of the CF3 radical and TolS radical or dimerization of the TolS radical results in p-tolyl(trifluoromethyl)sulfide or TolSSTol.
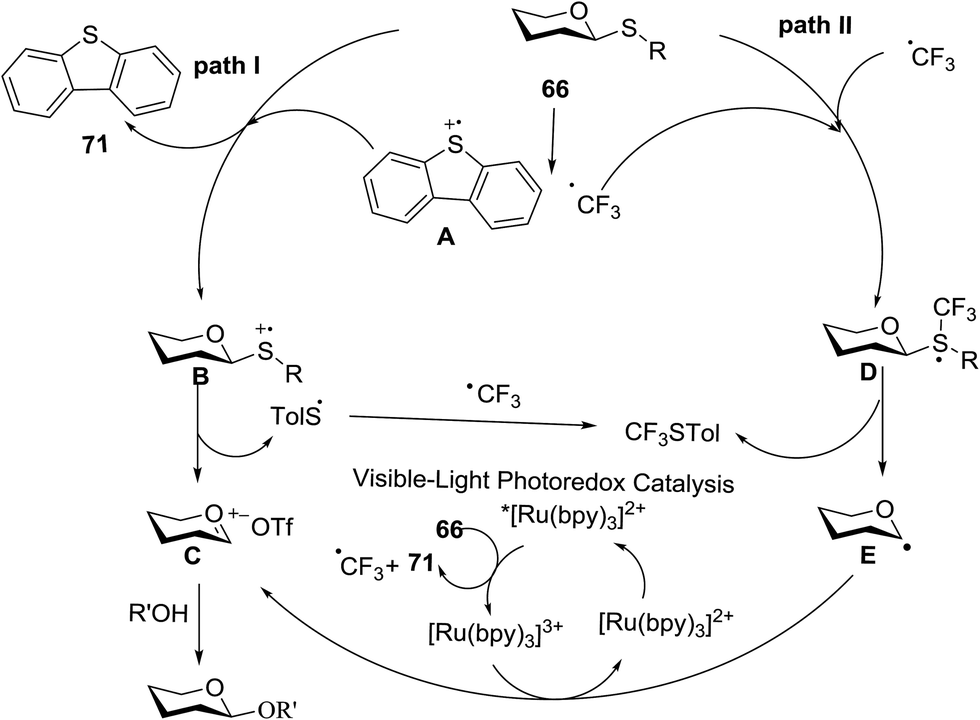 | ||
| Fig. 13 Proposed mechanism for the light-driven glycosylation. | ||
In continuation, Ye and co-workers developed an efficient use of Umemoto's reagent for O-sialylation using thiosialoside donors under visible light using a photocatalyst.52 Thiosialosides were energized under irradiation with blue light in the presence of Umemoto's reagent as the CF3 radical source, Ru(bpy)3(PF6)2 as the photocatalyst, and Cu(OTf)2 as the additive source in acetonitrile/dichloromethane at −30 °C (Table 9).
|
|||||
|---|---|---|---|---|---|
| Entry | 73 ROH (equiv.) | Activator (equiv.) | Solvent | Tempr (°C) | Yield (%) (α:β) |
| a No Cu(OTf)2, no light.b No light. | |||||
| 1 | 0.8 | 66 (1.5) | DCM | 25 | 0 (—) |
| 2 | 0.8 | 66 (1.5) | CH3CN | 25 | 23 (1.3:1) |
| 3 | 0.8 | 66 (1.5) | CH3CN/DCM (2/1) | 25 | 42 (1:1.4) |
| 4 | 0.8 | 66 (1.5) | CH3CN/DCM (1/2) | 25 | 33 (1:1.8) |
| 5 | 0.8 | 66 (1.5) | THF | 25 | 0 (—) |
| 6 | 0.8 | 66 (1.5) | Et2O | 25 | 0 (—) |
| 7 | 0.8 | 66 (1.5) | CH3CN/DCM (2/1) | 0 | 50 (1:1.2) |
| 8 | 0.8 | 66 (1.5) | CH3CN/DCM (2/1) | −20 | 65 (1:1.5) |
| 9 | 0.8 | 66 (1.5) | CH3CN/DCM (2/1) | −30 | 74 (1:1.3) |
| 10 | 0.8 | 66 (1.5) | CH3CN/DCM (2/1) | −40 | 0 (—) |
| 11 | 1.3 | 66 (1.5) | CH3CN/DCM (2/1) | −30 | 82 (1:1.4) |
| 12 | 1.3 | 66 (1.5) | CH3CN/DCM (2/1) | −30 | 83 (1:1.4) |
| 13 | 1.3 | 66 (1.5) | CH3CN/DCM (2/1) | −30 | 0 (—) |
| 14 | 1.3 | — | CH3CN/DCM (2/1) | −30 | 0 (—) |
| 15a | 1.3 | 66 (1.5) | CH3CN/DCM (2/1) | −30 | 0 (—) |
| 16b | 1.3 | 66 (1.5) | CH3CN/DCM (2/1) | −30 | 0 (—) |
| 17 | 1.3 | 66 (1.5) | CH3CN/DCM (2/1) | −30 | 56 (1:1.5) |
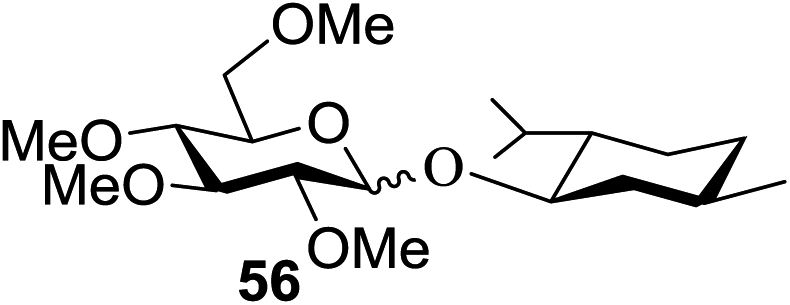
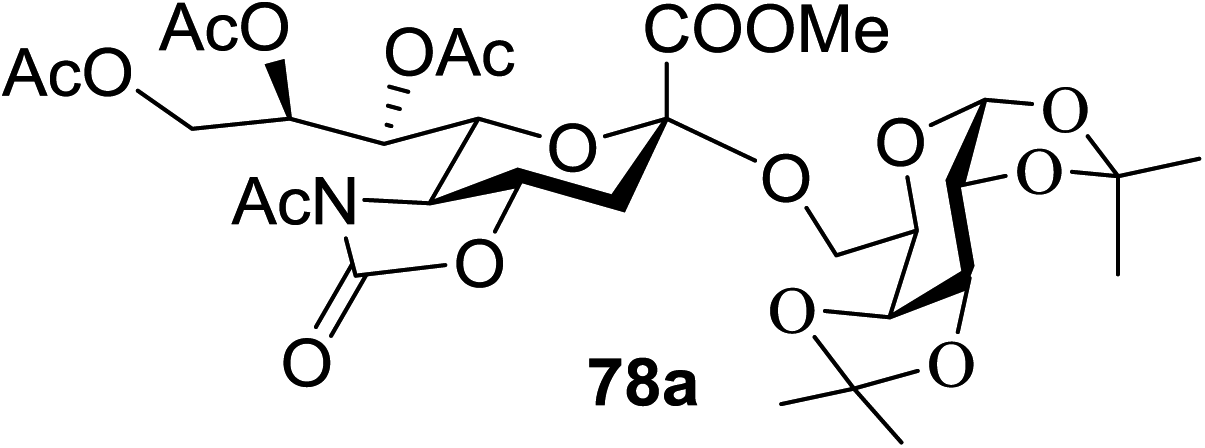
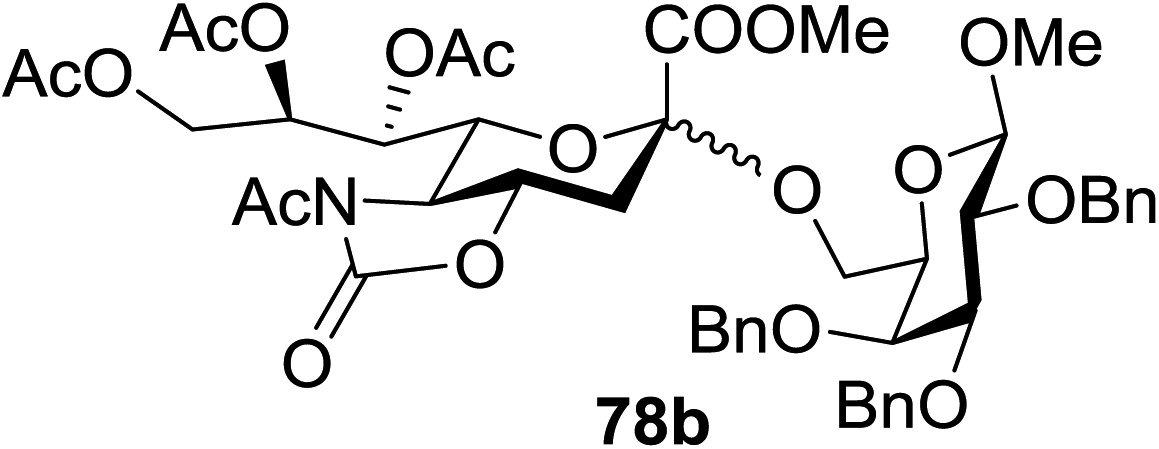
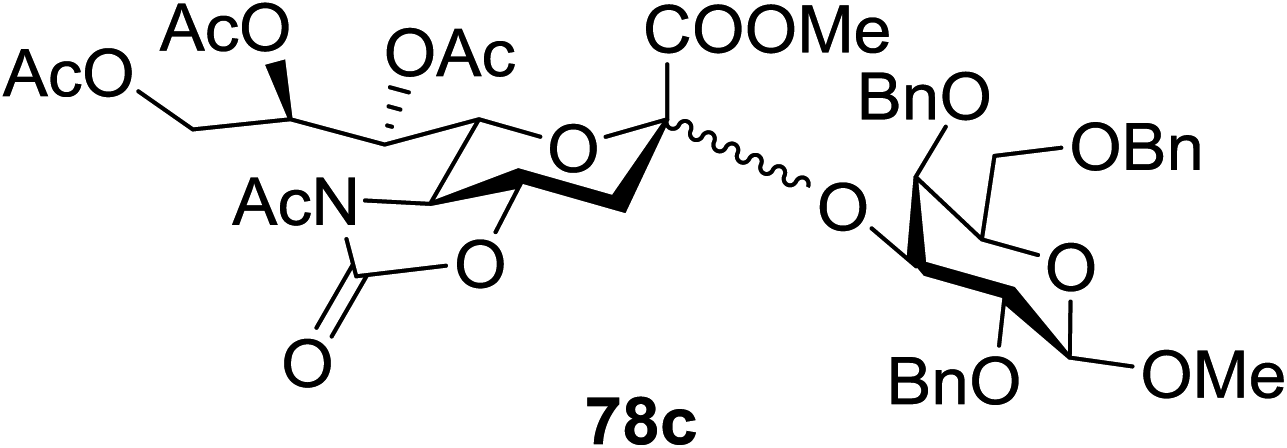
When the reaction was implemented at room temperature in CH2Cl2 in the presence of Umemoto's reagent 66, only the glycal side-product was obtained. Increasing the amount of Umemoto's reagent 66 did not increase the yield. When the reaction was performed in CH3CN, the coupled product 74 was isolated in only 23% yield (entry 2), probably due to the effect of acetonitrile participation to maintain the oxocarbenium ion.53,54 The reaction of the thiosialoside donor 72 with acceptor 73 was analysed under UV light irradiation, but no enhancement was observed. On the other hand, the target product was not produced in the absence of Umemoto's reagent 66, Cu(OTf)2, or irradiation from blue LEDs (entries 14–16). The optimized conditions (Table 9) included Umemoto's reagent 66, Ru(bpy)3(PF6)2, Cu(OTf)2 in CH3CN/CH2Cl2 (2:1) and blue LED irradiation. The scope of this sialylation reaction was explored by altering both the donors and acceptors (Table 10). The coupling reactions of donor 72 with 1,2:3,4-di-O-isopropylidene-α-D-galactopyranoside (77a)55 or methyl 2,3,4-tri-O-benzyl-β-D-galactopyranoside (77b)56 gave the corresponding disaccharides 78a and 78b in good yields with excellent α-stereoselectivity (α only, entry 1). The coupling reaction of donor 72 with the secondary alcohol methyl 2,4,6-tri-O-benzyl-β-D-galactopyranoside (77c) furnished the disaccharide 78c in 72% isolated yield with the β-anomer as the major product.
|
||||
|---|---|---|---|---|
| Entry | Donor | Acceptor | Product | Yield (%) (α/β) |
| 1 | 72 | 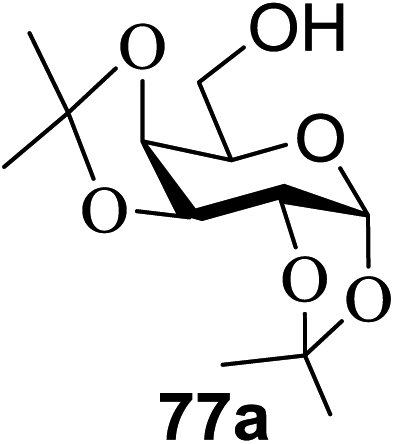 |
 |
92 (α only) |
| 2 | 72 | 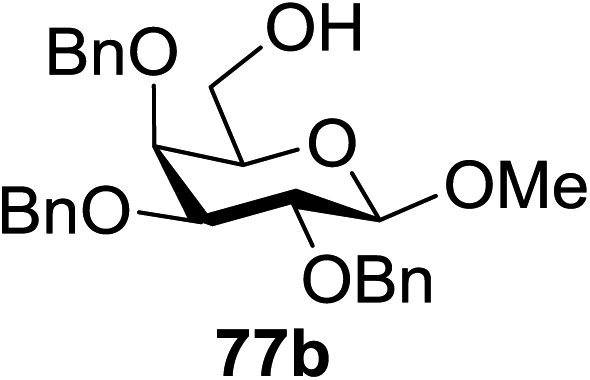 |
 |
85 (>10:1) |
| 3 | 72 | 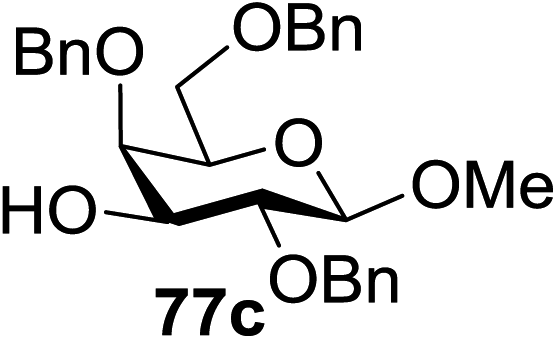 |
 |
72 (1:6) |
| 4 | 72 |  |
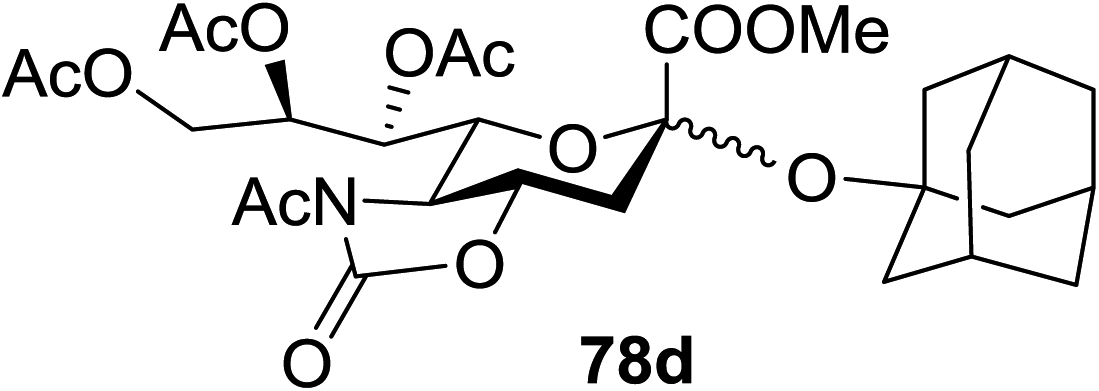 |
85 (2.6:1) |
| 5 | 75 | 77a |  |
90 (4.2:1) |
| 6 | 72 |  |
 |
75 (α only) |
| 7 | 76 |  |
78a | 0 (—) |
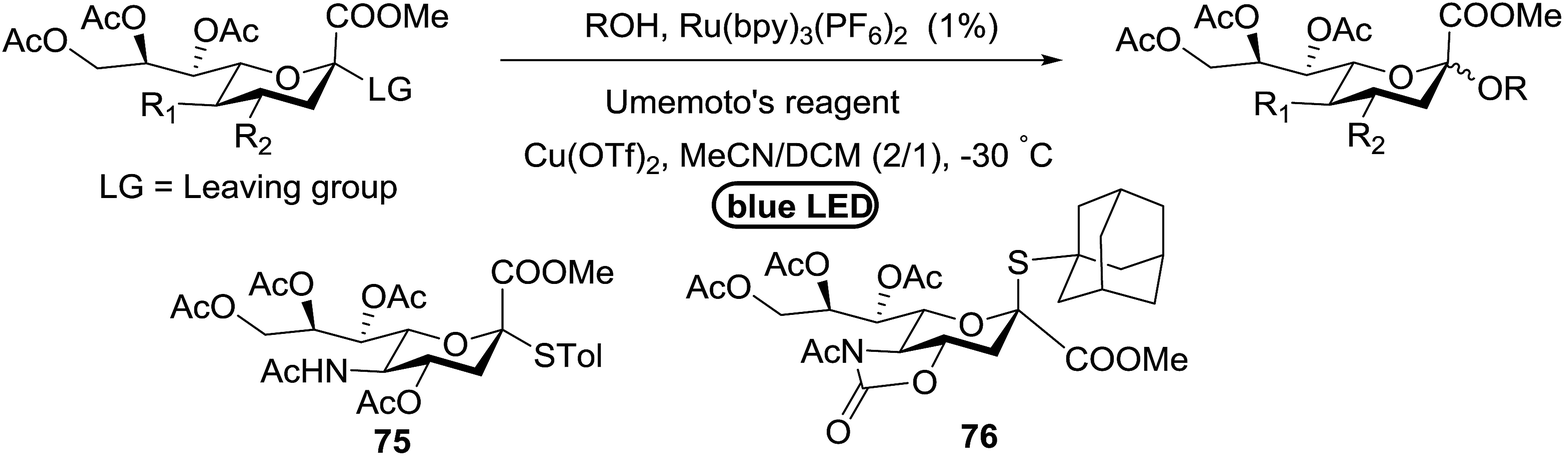
The mechanism of this reaction is presented in Fig. 14. In the presence of visible light, Umemoto's reagent 66 is reduced by the excited state *[Ru(bpy)3]2+ to afford a CF3 radical, which reacts with the sulfur atom in the thiosialoside donor to generate the radical intermediate A and p-tolyl-(trifluoromethyl)sulfide. [Ru(bpy)3]3+ oxidises the glycosyl radical intermediate A to the glycosyl cation C. Finally, the glycosylation reaction of the glycosyl cation C with the acceptor occurs. It is suggested that Cu(OTf)2 not only plays a beneficial role in increasing the amount of CF3 radical in the thiosialoside activation, but also acts as an extra triflate source to stabilize the oxocarbenium ion intermediate. In the absence of Cu(OTf)2, only the glycal product was detected (Table 9, entry 15).
 | ||
| Fig. 14 Proposed mechanism for visible light photoredox-catalyzed O-sialylation. | ||
Direct activation of thioglycosides via photoinduced glycosyl coupling and subsequent O-glycosylation in the absence of photosensitizer was developed for the first time by Ye and co-workers.57 The activation of thioglycosides by light usually requires a photosensitizer or photocatalyst. Armed (p-methoxy)phenyl thioglycosides could undergo a visible light promoted photoredox O-glycosylation reaction, whereas disarmed (p-methoxy)phenyl thioglycosides were unreactive. It was noticed that UV light had the capability to break the C–S bond. The activation of thioglycosides and glycosylation required that UV light directly break the C–S bond in thioglycosides without the use of any photosensitizer. This was confirmed by irradiating β-thiogalactopyranoside 79 for 22 h, where the C–S bond was broken which produced β-thiogalactopyranoside 79 (48% yield), α-thiogalactopyranoside 80 (26% yield), 1,5-anhydro-galactitol 81 (25% yield) and TolSSTol (12% yield) (Scheme 4).
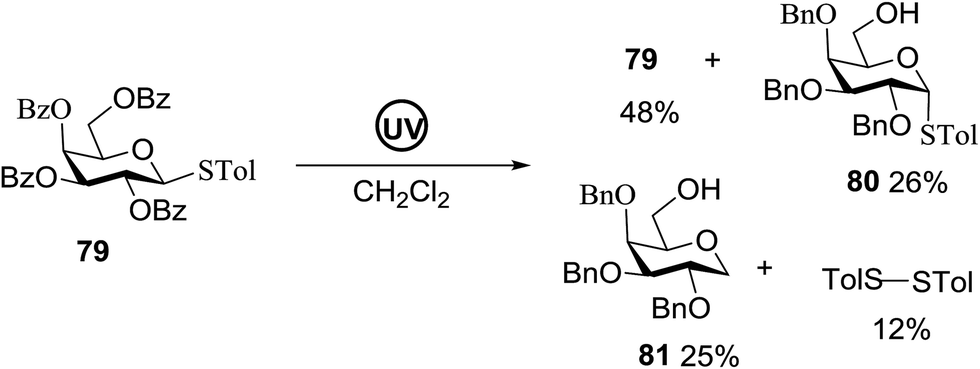 | ||
| Scheme 4 Irradiation of thioglycoside 79 with UV light. | ||
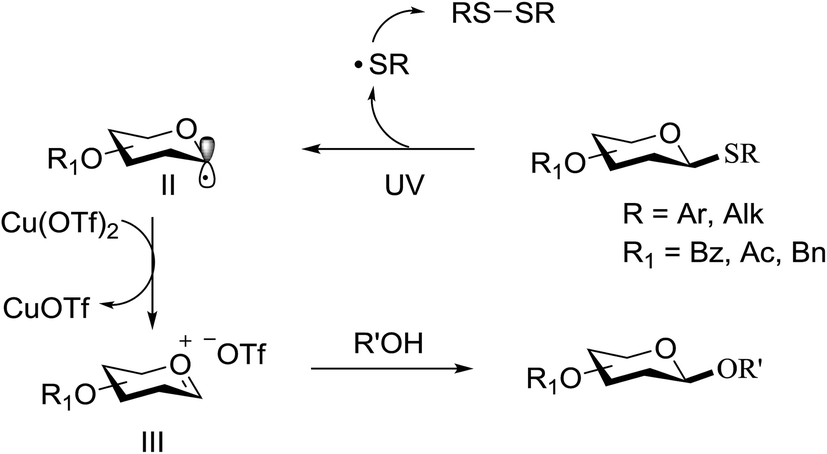
This suggests the feasible hemolytic cleavage of 79 to the form the galactosyl radical and p-tolylthiyl radical, which underwent recombination, dimerization, or hydrogen atom abstraction to form the above-mentioned products. From this it can be concluded that the glycosylation of a thioglycoside with an acceptor would proceed upon ultraviolet illumination in the presence of a proper oxidant, which oxidizes the glycosyl radical (II) to a glycosyl oxocarbenium ion (III), as shown in Scheme 5. This glycosylation protocol is different from the conventional light-mediated glycosylation involving an S-radical cation (I). It is a new O-glycosylation protocol via the photoinduced direct C–S bond cleavage of thioglycosides and subsequent oxidation by Cu(OTf)2. The reaction was almost inhibited by the addition of 2,2,6,6-tetramethylpiperidinooxy (TEMPO, 3.0 equiv.), which is a radical trapper. These observations indicated that the presence of a glycosyl radical and p-tolylthiyl radical resulted from the direct homolytic cleavage of the C–S bond upon UV irradiation. In the reaction process, the impressive decrease in the amount of Cu(OTf)2 during reaction confirmed that Cu(OTf)2 acts as the oxidant.
 | ||
| Scheme 5 Strategy for the light-induced activation of thioglycosides and glycosylation. | ||
The optimized conditions for the reaction were analyzed using the exemplary reaction between perbenzoylated thioglucopyranoside 82 and the glucosyl acceptor 83 with the C-6 hydroxyl. The feasibility of the reaction was revealed by varying different oxidants and conditions (Table 11). In the absence of oxidants or UV, no glycosylated product was observed (entries 1, 13 and 14). Disaccharide 84 was accessed by the addition of oxidants such as metal salts (entries 2–7, 0–28% yields). Among the metal salts, Cu(OTf)2 resulted in the highest yield (entry 8, 75% yield). The yield of 84 was improved gradually as the equivalent of Cu(OTf)2 was increased (entries 8–10, 75–90%). The glycosylation yields reduced when the amount of glycosyl acceptor 83 (entries 10–12) was increased and dichloromethane was the best solvent (entries 15–16). Thus, the optimized conditions constituted a donor (1.0 equiv.), an acceptor (0.5 equiv.) and Cu(OTf)2 (1.7 equiv.) in CH2Cl2 (0.01 M) under the UV light irradiation at room temperature.
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | 82/83 (equiv.) | Oxidant (equiv.) | Solvent | Yield (%) | Entry | 82/83 (equiv.) | Oxidant (equiv.) | Solvent | Yield (%) |
| 1 | 1/0.5 | — | CH2Cl2 | 0 | 9 | 1/0.5 | Cu(OTf)2 (1.5) | CH2Cl2 | 79 |
| 2 | 1/0.5 | InBr3 (1.0) | CH2Cl2 | 13 | 10 | 1/0.5 | Cu(OTf)2 (1.7) | CH2Cl2 | 90 |
| 3 | 1/0.5 | CuCl2 (1.0) | CH2Cl2 | Trace | 11 | 1/0.75 | Cu(OTf)2 (1.7) | CH2Cl2 | 80 |
| 4 | 1/0.5 | CuSO4 (1.0) | CH2Cl2 | 28 | 12 | 1/1 | Cu(OTf)2 (1.7) | CH2Cl2 | 67 |
| 5 | 1/0.5 | Cu(OAc)2 (1.0) | CH2Cl2 | Trace | 13 | 1/0.5 | Cu(OTf)2 (1.0) | CH2Cl2 | 0 |
| 6 | 1/0.5 | RuCl3 (1.0) | CH2Cl2 | Trace | 14 | 1/0.5 | Cu(OTf)2 (1.0) | CH2Cl2 | 0 |
| 7 | 1/0.5 | InCl3 (1.0) | CH2Cl2 | 20 | 15 | 1/0.5 | Cu(OTf)2 (1.7) | CH3CN | 86 |
| 8 | 1/0.5 | Cu(OTf)2 (1.0) | CH2Cl2 | 75 | 16 | 1/0.5 | Cu(OTf)2 (1.7) | Et2O | 62 |
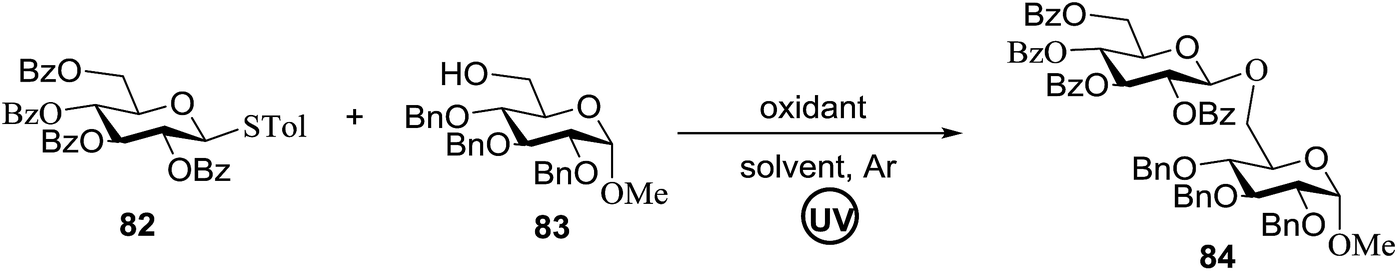
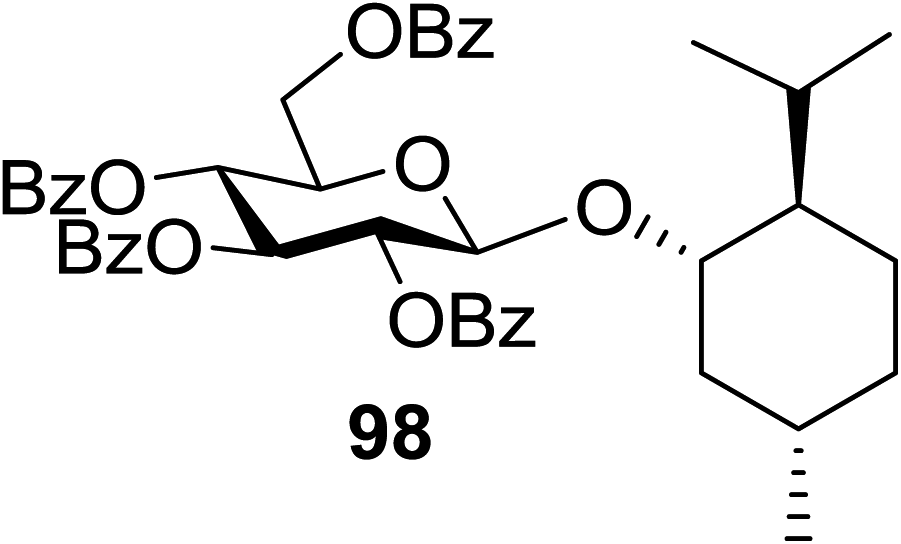
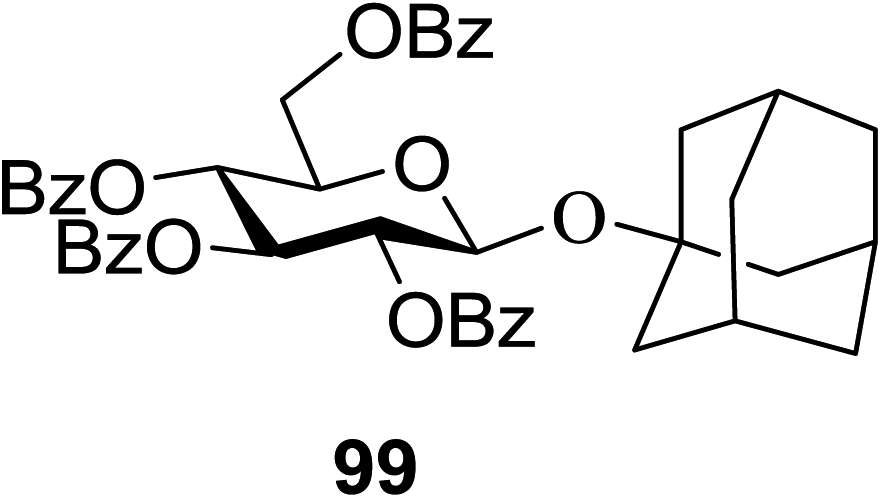
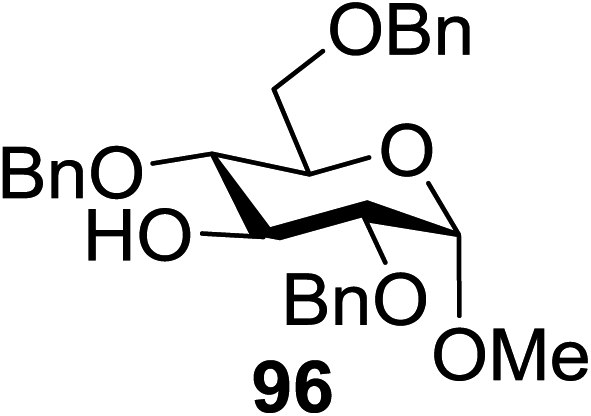
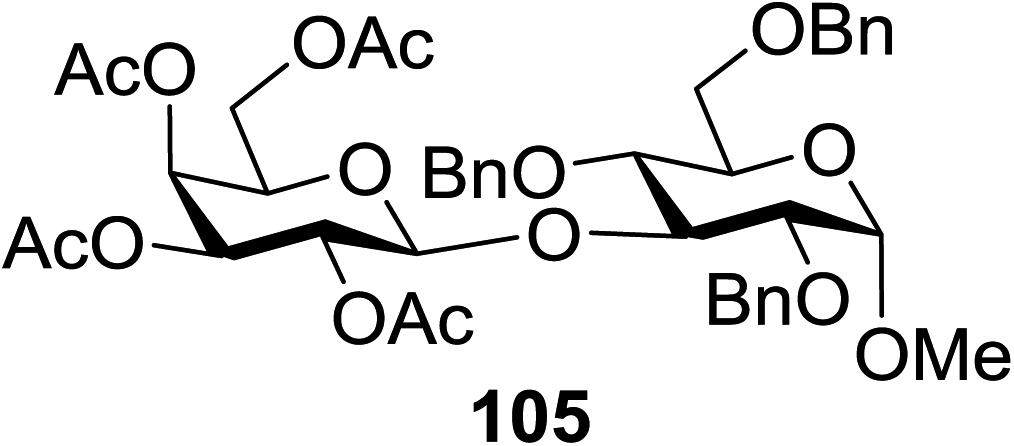
The scope of glycosylation of the donor with various acceptors is outlined under the optimal conditions in Table 12. The reactions of 82 with primary alcohol 89 (entry 1, 70%), secondary alcohols 90 and 92 (entry 2, 88% and entry 4, 52%, respectively) or tertiary alcohol 91 (entry 3, 81%) resulted in the respective glycosides. Cholesterol 93 (entry 5, 75%) or protected amino acid 94 (entry 6, 86%) could be utilized as glycosyl acceptors. On the basis of these results further glycosylations of acylated galactosyls (84 and 85, entries 8–9, 76% and 69%, respectively) or rhamnosyl donor (86, entry 10, 80%) with the glucosyl acceptors 83 or 96, having a free hydroxyl group at the C-6 or C-3 position, respectively, was performed which generated the product in good yield. Further exploration of the protocol involved the reactions of the benzylated glucosyl donor 87 with acceptors 83 or 95 which produced the desired disaccharides in moderate (entry 11, 65%, α/β = 1.2/1) to excellent (entry 12, 95%, α/β = 1.7/1) yields, but with a lack of stereoselectivity.
|
||||
|---|---|---|---|---|
| Entry | Donor | Acceptor | Product | Yield (%) |
| 1 | 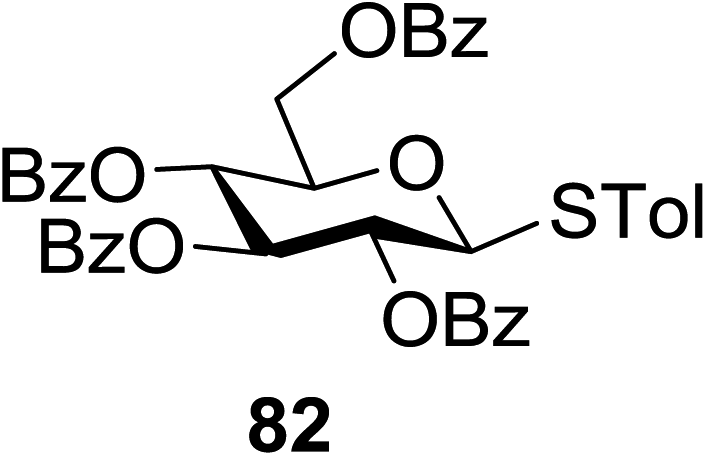 |
 |
 |
70 |
| 2 | 82 |  |
 |
88 |
| 3 | 82 |  |
 |
81 |
| 4 | 82 |  |
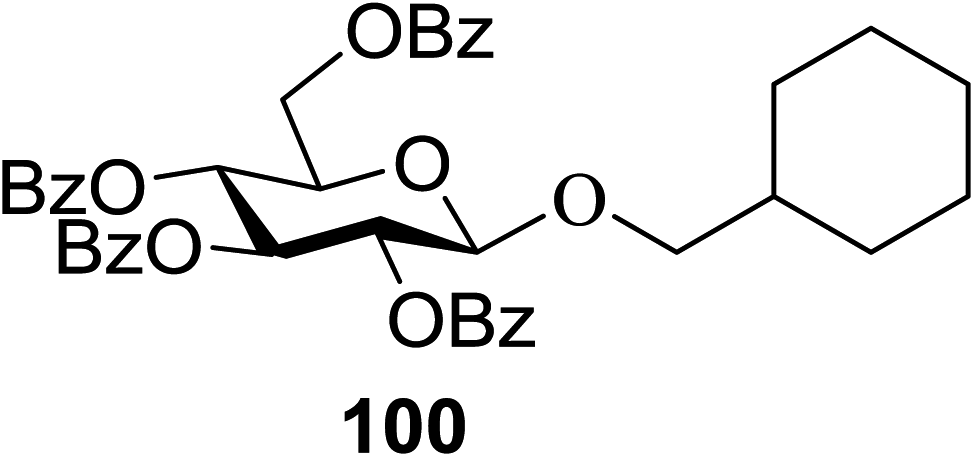 |
52 |
| 5 | 82 |  |
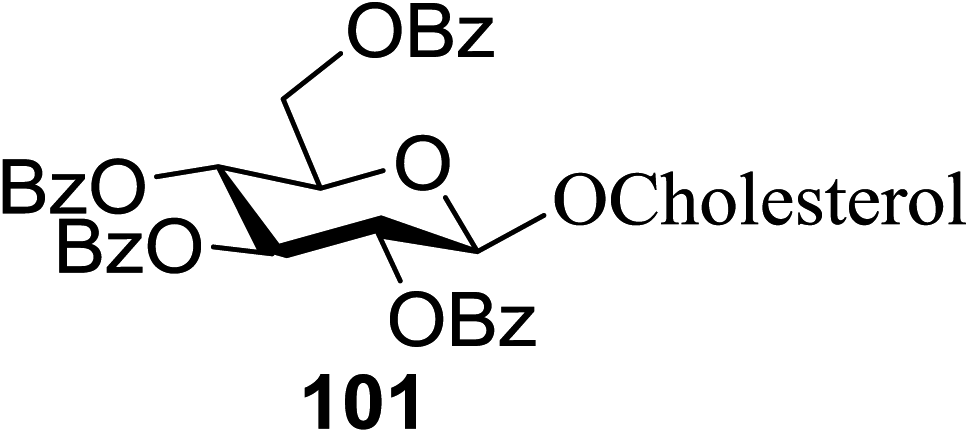 |
75 |
| 6 | 82 | 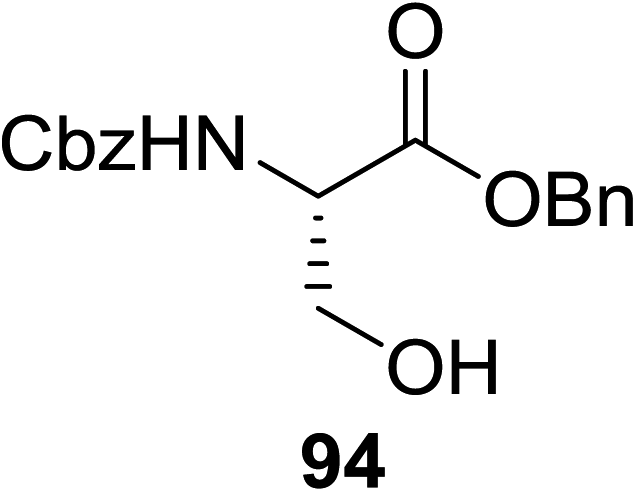 |
 |
86 |
| 7 | 82 |  |
 |
55 |
| 8 | 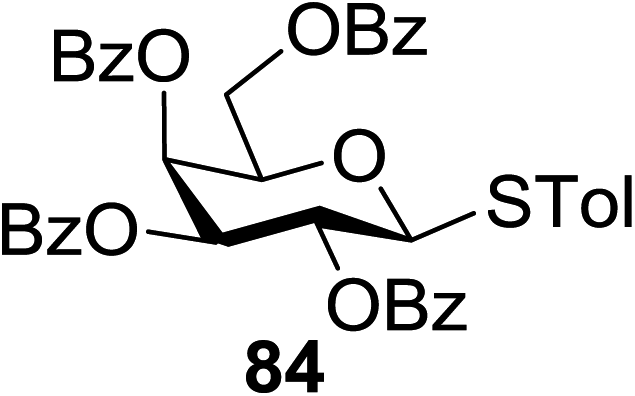 |
83 | 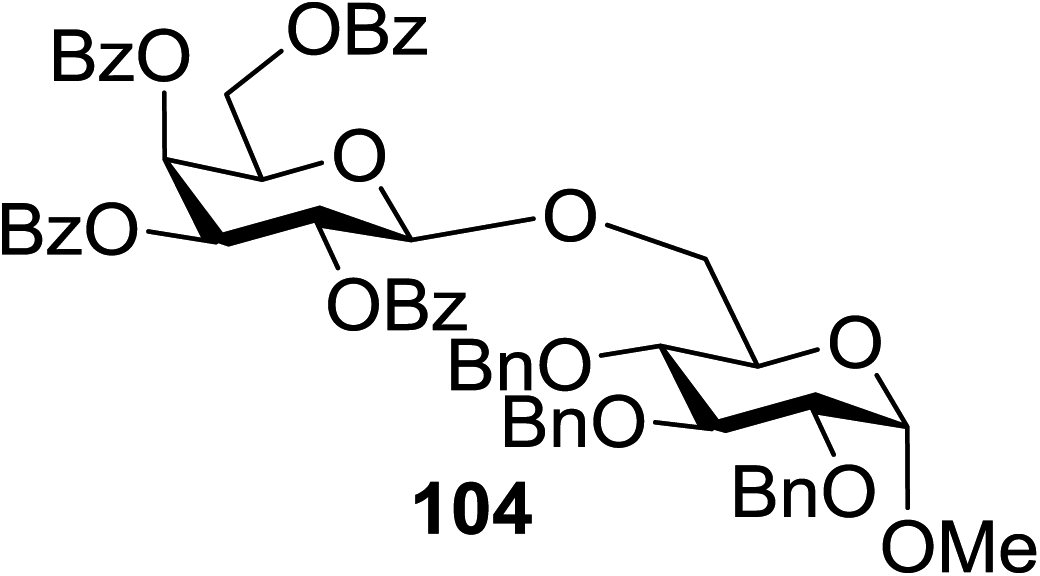 |
76 |
| 9 | 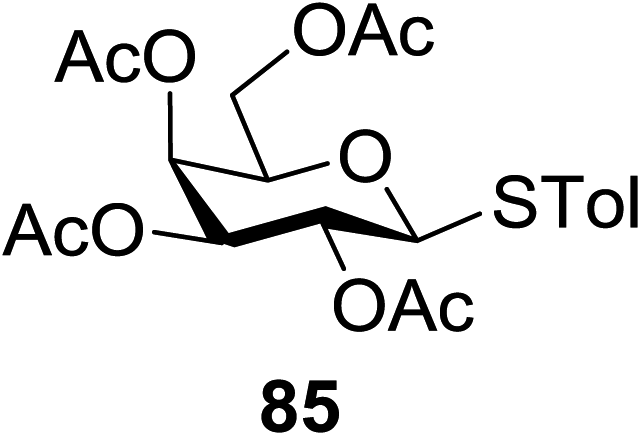 |
 |
 |
69 |
| 10 | 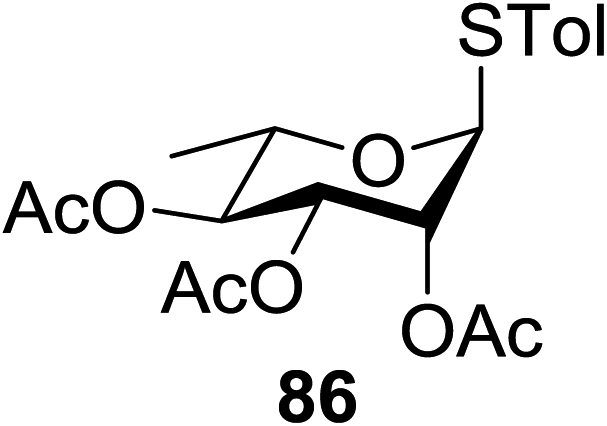 |
96 | 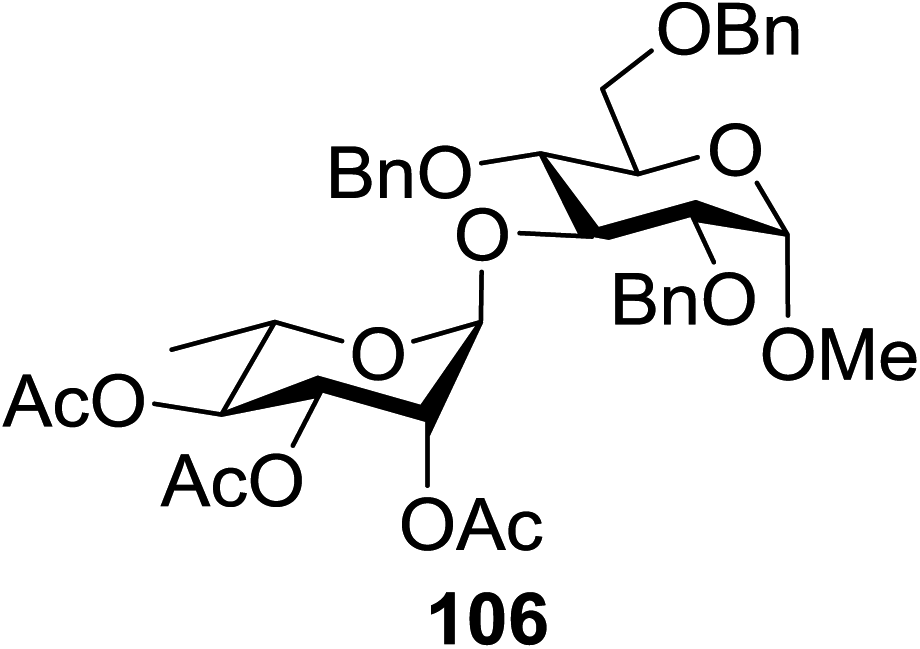 |
80 |
| 11 | 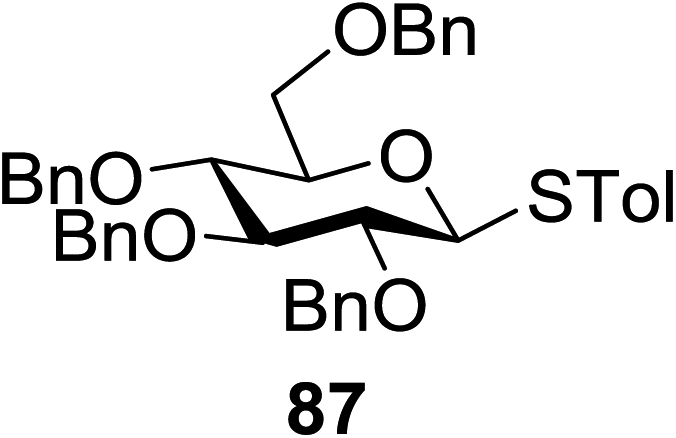 |
95 | 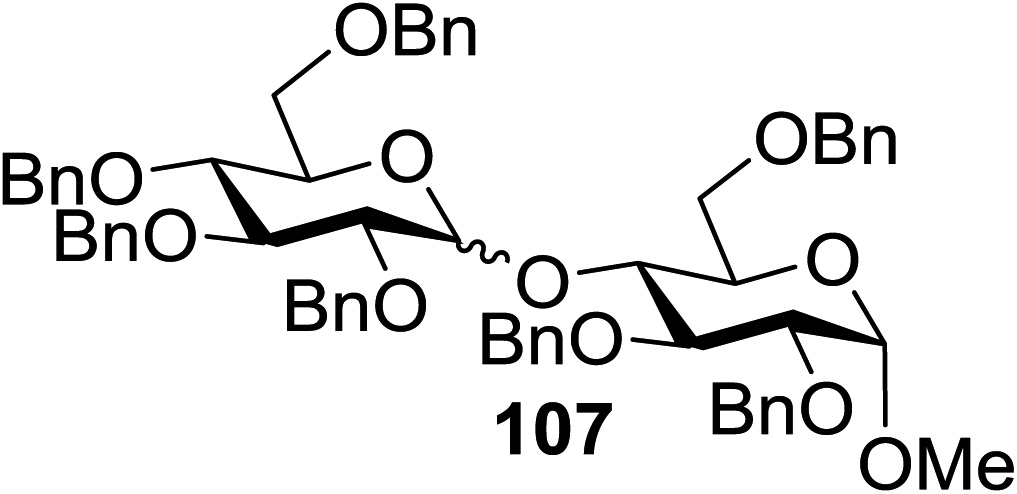 |
65, (α/β = 1.2/1) |
| 12 |  |
83 | 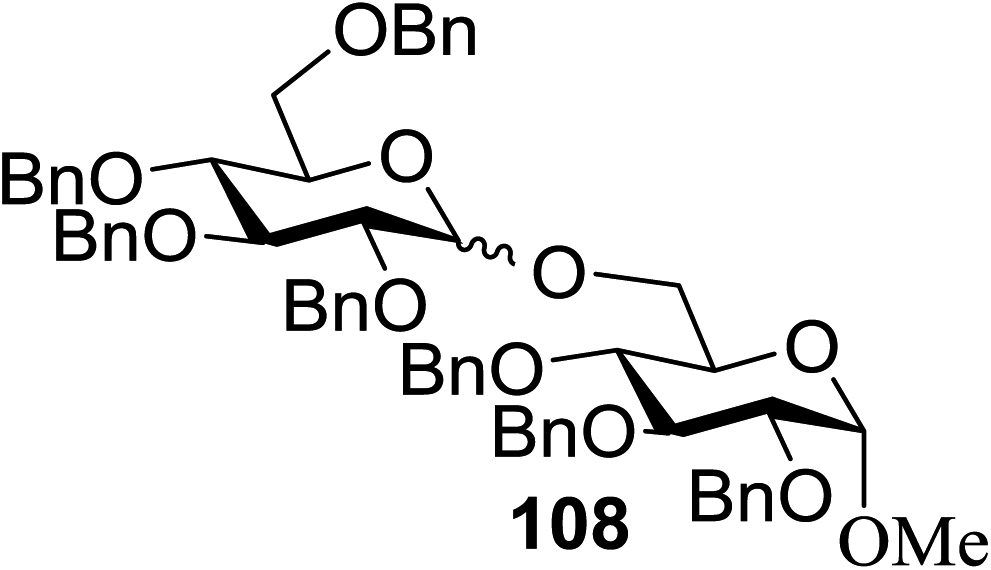 |
95, (α/β = 1.7/1) |
| 13 | 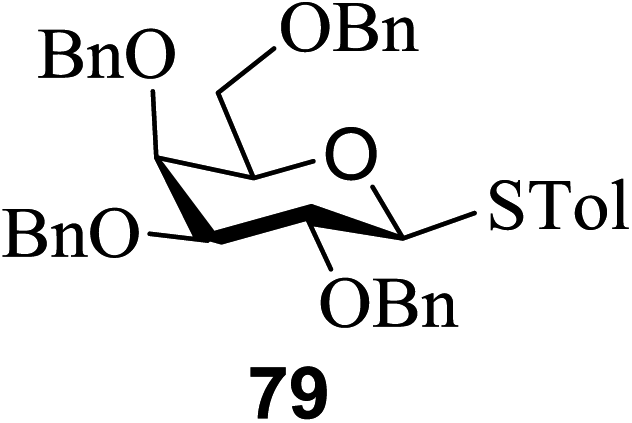 |
83 | 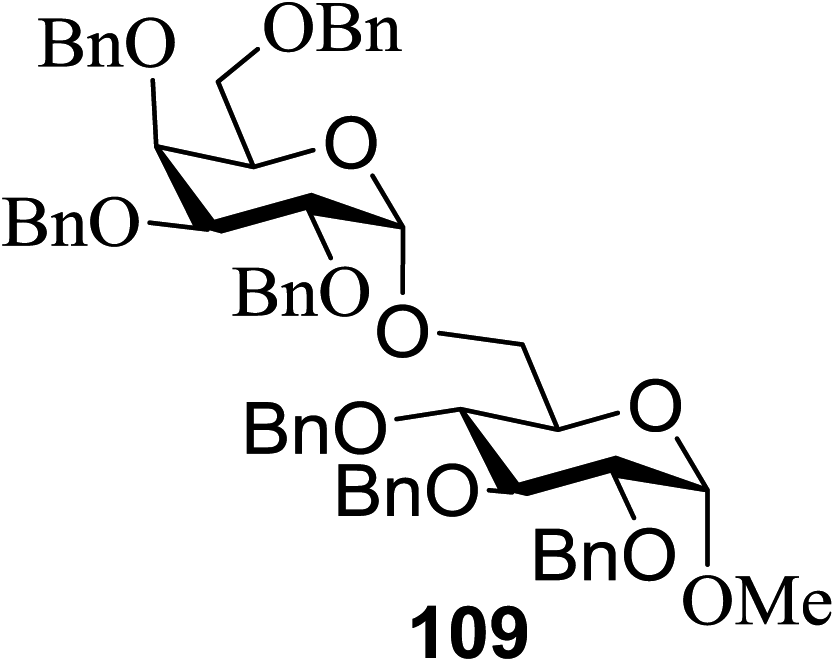 |
87 |
| 14 | 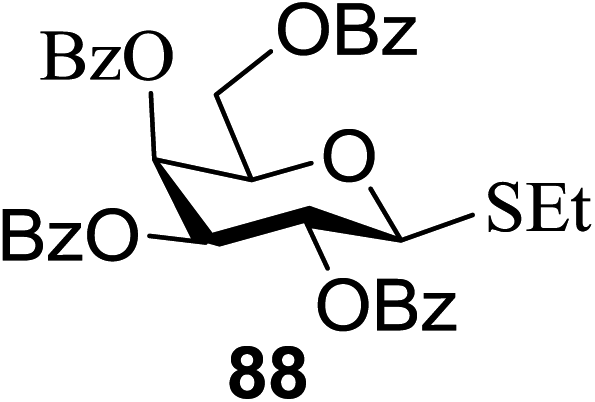 |
83 | 104 | 85 |
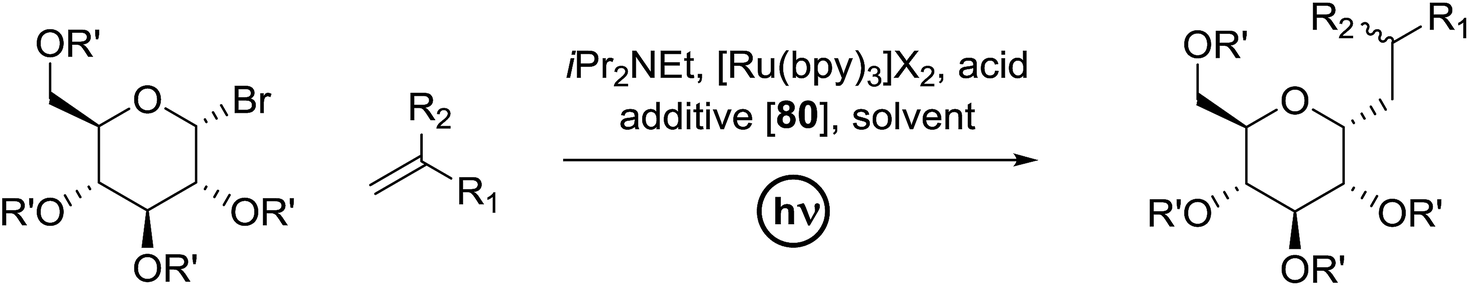
3.2. Photocatalytic C-glycoside formation via glycosyl halide donors
C-Glycosides are an important class of bioactive compounds; therefore, improved methods for their synthesis are in demand. Transition metal photocatalysts have generated significant interest as powerful single electron transfer (SET) reagents that operate under mild conditions. Using this approach, it was found that C-glycosides with outstanding C1 diastereoselectivities could be obtained in yields approaching or exceeding that previously obtained, and it provided promising yields of α-C-glycosides. This is because α-glucosyl bromides generate the C1 α-radical which can be reduced or added to the alkene. Termination of the electron-deficient α radical leads to fully saturated C-glycosides with exclusive α selectivity. Ru(bpy)32+ was developed as a catalyst for the capture and conversion of solar energy. In 2010 Gagne and co-workers58 expressed that the combination of visible light and [Ru(bpy)3]2+ yields nucleophilic C1 sugar radicals59 that react intermolecularly with electron-deficient alkenes to afford C-glycosides in yields exceeding previous bests and with dominating C1 diastereoselectivities. In these reactions the initiating event is the reduction of the photogenerated MLCT state, *[RuIII(bpy)2(bpy˙−)]2+, by amine to produce a potent ligand centered reducing equivalent (Table 13).
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Acid | Additive | Solvent | X | Yield | Entry | Acid | Additive | Solvent | X | Yield |
| 1 | 112-HBr | — | DMA | Cl | 0 | 5 | 112-HBF4 | — | MeCN | BF4 | 61 |
| 2 | 112-HBr | — | MeCN | Cl | 26 | 6 | 112-HBF4 | 111 | MeCN | BF4 | 72 |
| 3 | 112-HBF4 | — | MeCN | Cl | 44 | 7 | 112-HBF4 | 111 | CH2Cl2 | BF4 | 80 |
| 4 | 112-HBF4 | — | MeCN | Cl | 50 | 8 | — | 111 | CH2Cl2 | BF4 | 92 |
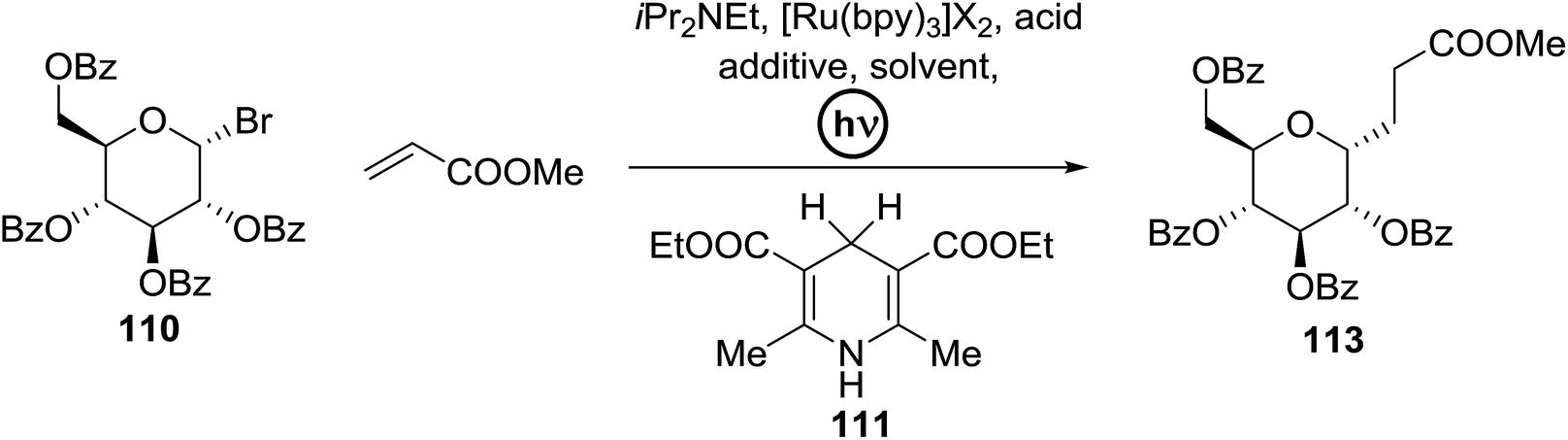
The reaction of α-glucosyl bromide 110 and methyl acrylate with [Ru(bpy)3]X2 (5 mol%) and an acid source to protonate enolates was explored. N,N-Diisopropylethylamine (112) was verified as the perfect reductant, since it afforded encouraging yields of the α-C-glycoside (113) and did not require highly polar solvents (Table 13, entries 2 and 7). In this preliminary α-radical reduction, subsequent conjugate addition to an additional acrylate was competitive, which reveals that the mass balance was dominated by conjugate addition.
The optimisation conditions resulted in the Hantzsch ester (111) successfully terminating the oligomerization and enhanced yields. The scope of the coupling is presented in Table 14. No products were formed in the absence of [Ru(bpy)3]2+ or visible light, and when 112 was absent, 111 rapidly changed into the pyridine. In the presence of an electrophilic hydrogen atom source (tBuSH), C1 reductive debromination was the major product accompanied by small amounts of the C-glycoside.
|
|||||
|---|---|---|---|---|---|
| Entry | Product | Yield | Entry | Product | Yield |
| 1 |  |
114a R = COOMe: 94% | 4 | 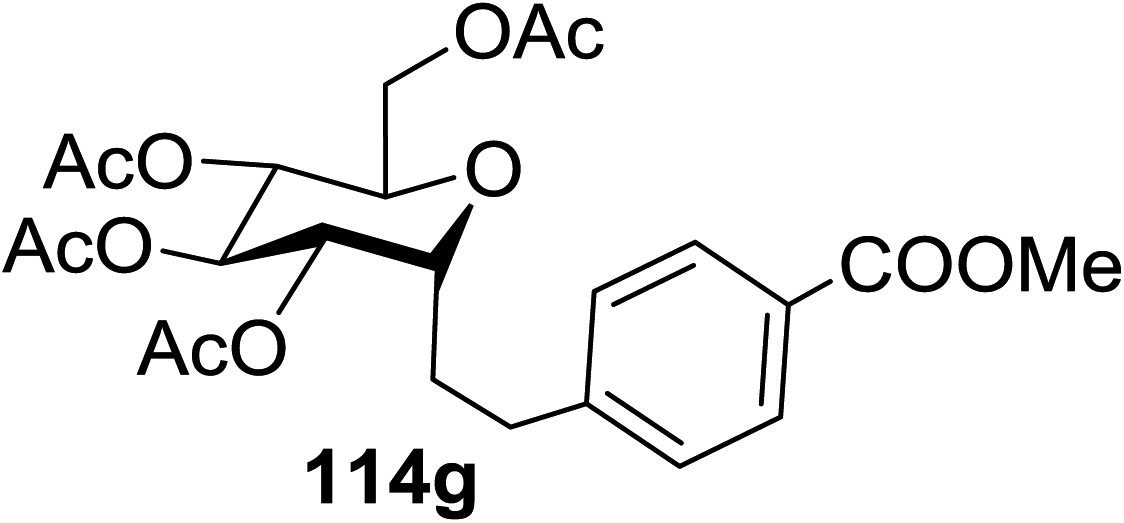 |
51% |
| 114b R = COMe: 86% | |||||
| 114c R = CHO: 85% | |||||
| 114d R = CN: 85% | |||||
| 5 | 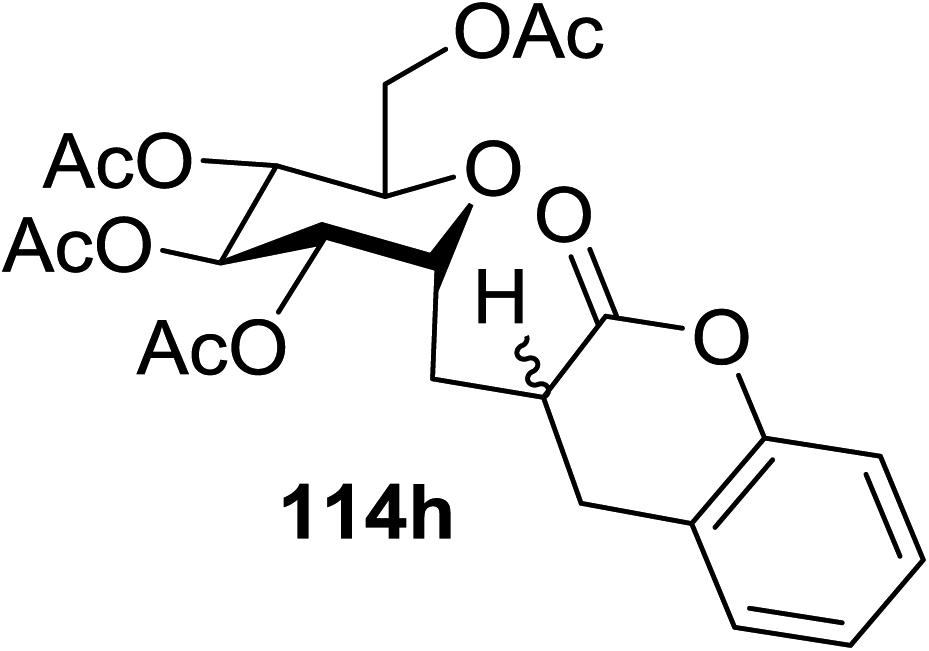 |
98% | |||
| 2 | 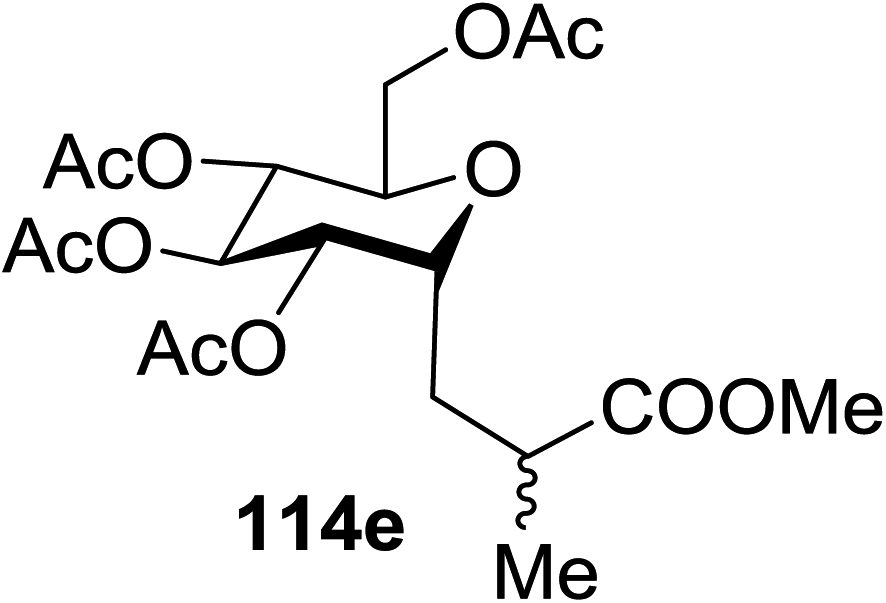 |
98% d.r. = 1.5:1 |
6 | 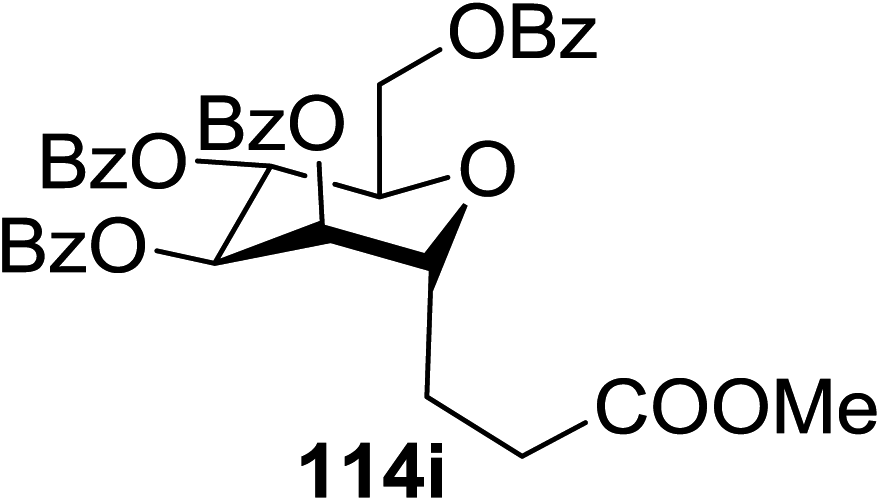 |
81% |
| 3 | 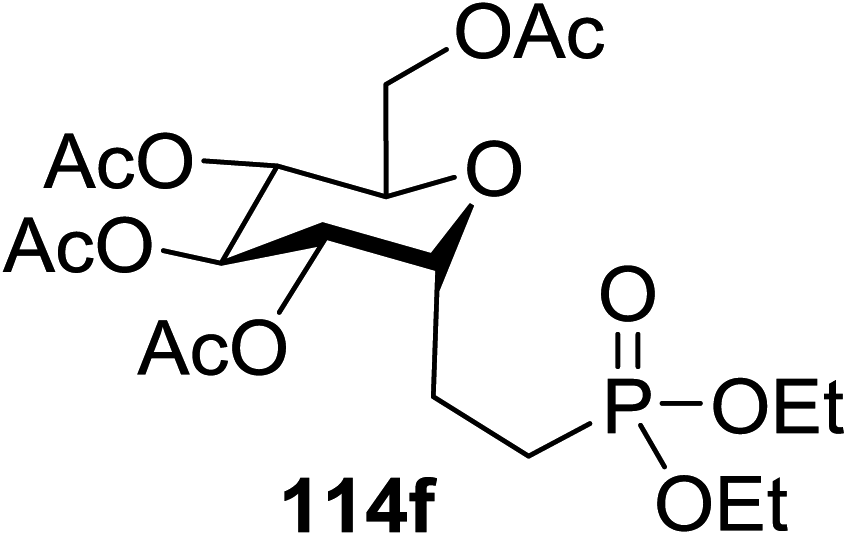 |
42% | 7 | 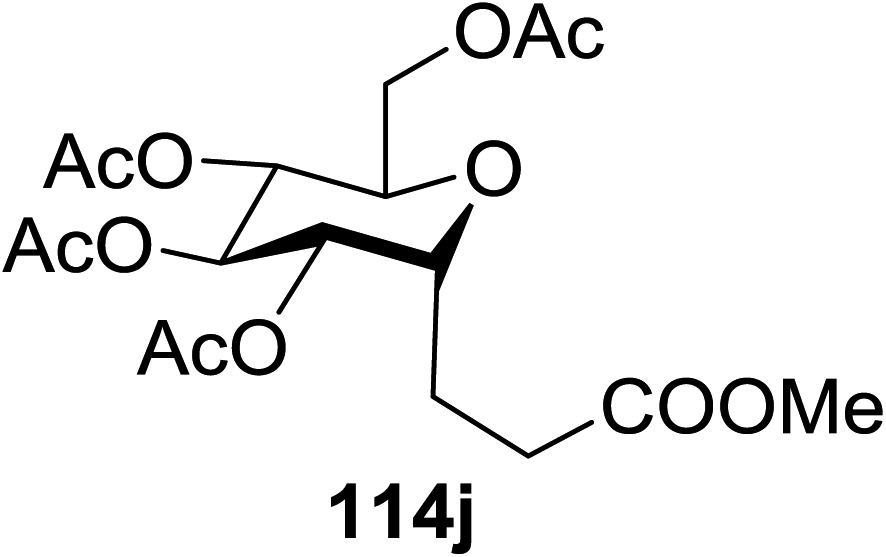 |
80% |
The proposed mechanism shows that ET from the photogenerated [RuII(bpy)3]+ to the glycosyl bromide generates the C1 radical, which can be reduced (k1) or added to the alkene (k2) (Fig. 15). For electron deficient alkenes k2 > k1. When k1 = k2, reductive debromination is competitive, although it can be somewhat satisfied with an increase in alkene concentration. Termination of the electron-deficient α radical is subject to the relative rates of over-conjugate addition (k3) and reduction (k4), the latter of which is accelerated by Hantzsch ester.60
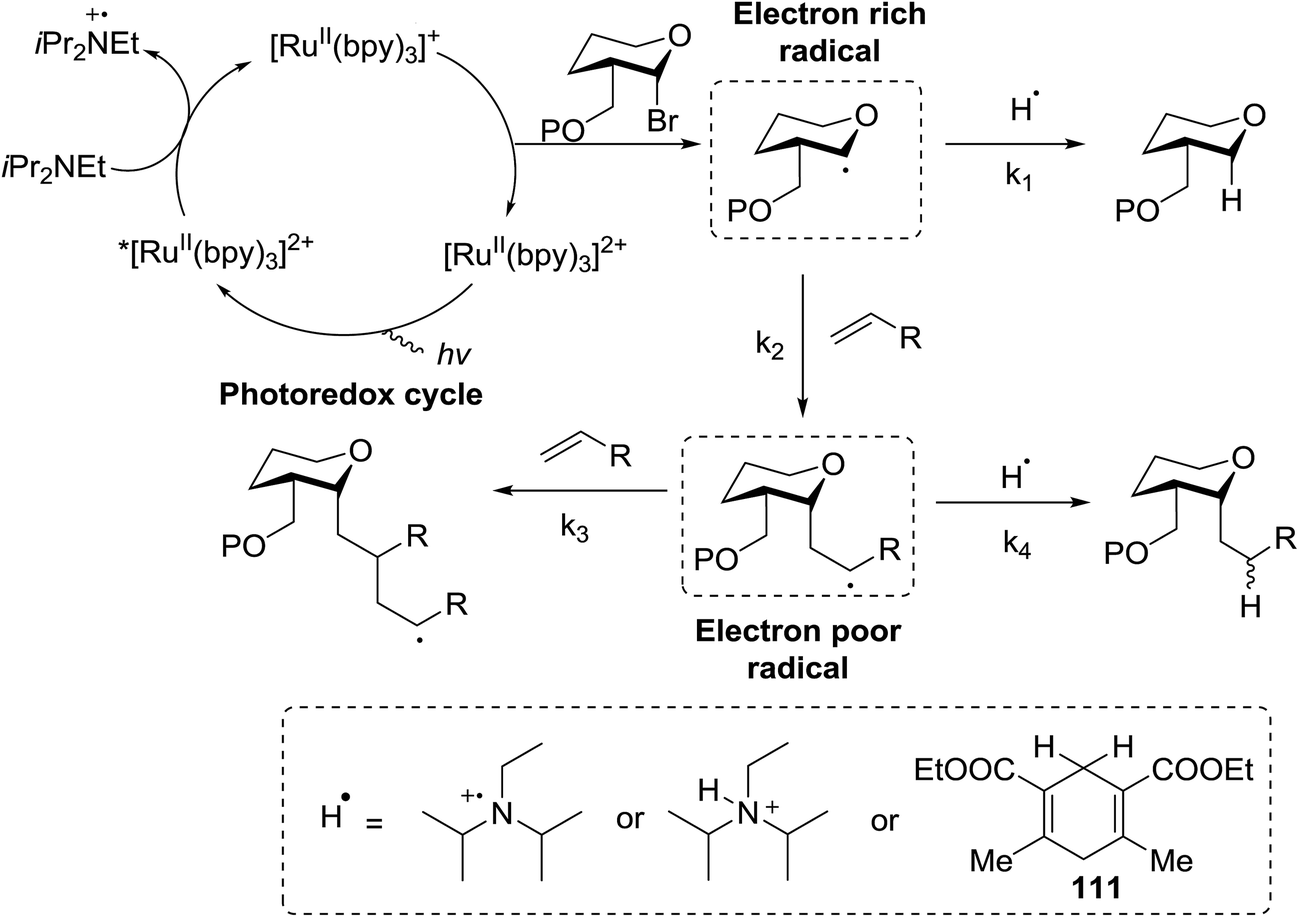 | ||
| Fig. 15 Plausible mechanism for the reactions described herein. | ||
Using a similar principal, Andrews R. S. et al. investigated the photoreduction of glucosyl halide to generate glucosyl radicals in 2011 (Scheme 6).61 A reductive trap was set for the radical (i.e., tBuSH) in place of the alkene. Conditions similar to the previously described reactions (glucosyl bromide (115), [Ru(bpy)3]2+, and N,N-diisopropylethylamine as the stoichiometric reductant) were found to readily debrominate glucosyl bromide to 116 (Scheme 6). Varying the catalyst concentration under dilute conditions (<4 mM) resulted in the rate of the reaction being proportional to [Ru(bpy)32+].
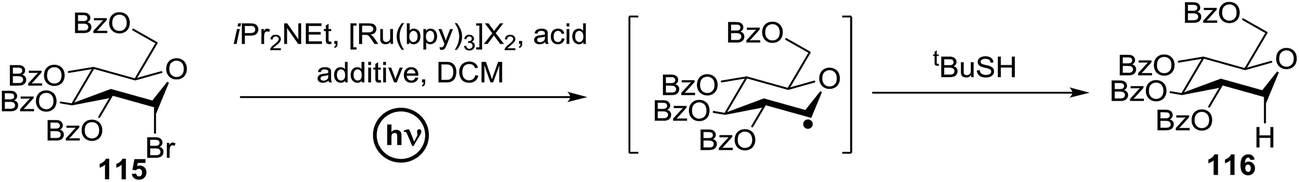 | ||
| Scheme 6 Photoreduction of glucosyl radical. | ||
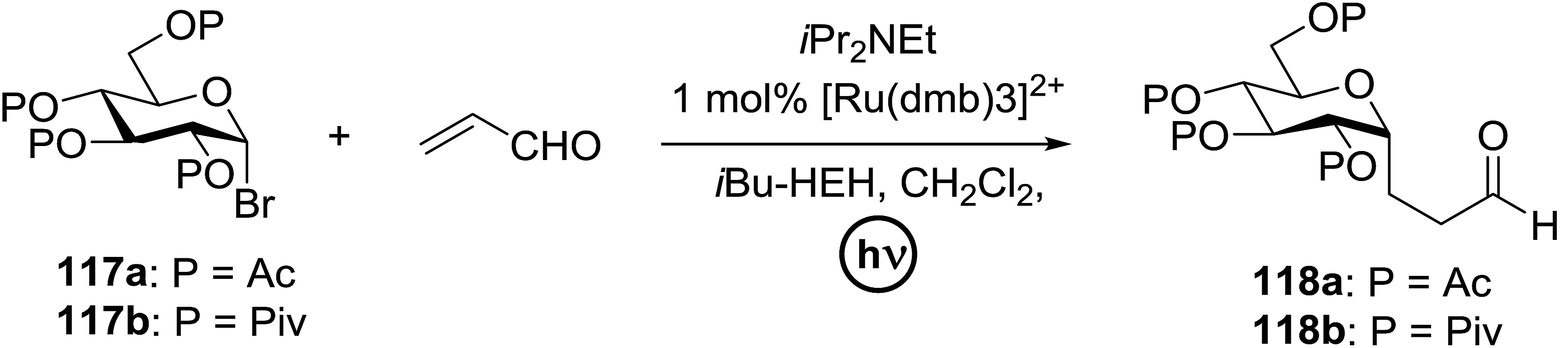
In 2012 Gagne and co-workers focused on a more efficient alternative, the light-mediated conjugate addition of glycosyl radicals into acrolein using [Ru(dmb)3]2+ (Table 15).62 Previously reported syntheses required multiple synthetic steps and used costly or toxic reagents, harsh reaction conditions (strong acid or base) and chiral starting materials (chiral auxiliaries or amino-acid-derived). The yield of 118a increased to 85% by increasing the concentration of acrolein. The pivaloate-protected substrate was slower to react; only 75% conversion took place using two modules (Table 15). However, full conversion was obtained by attaching a third module. Highly absorbing [RuL3]2+ catalysts create strong concentration gradients that localize photoactivated catalysts near the surface of the vessel, thus requiring thinner reaction vessels to maximize efficiencies.
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Acrolein (equiv.) | No. of modules | Product | Yield (%) |
| 1 | 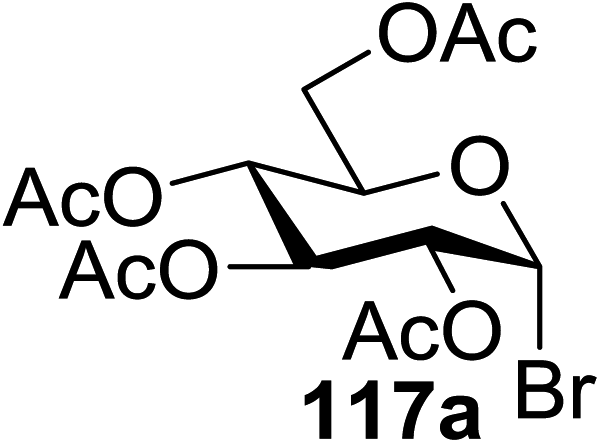 |
2 | 2 | 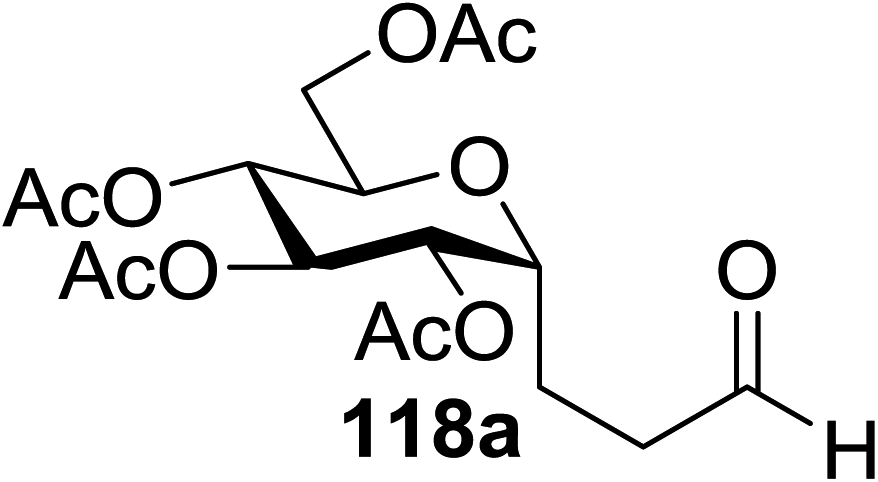 |
70 |
| 2 | 117a | 4 | 2 | 118a | 85 |
| 3 | 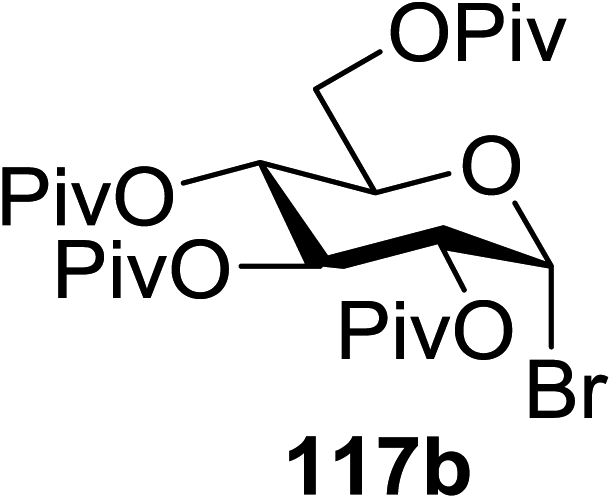 |
4 | 2 | 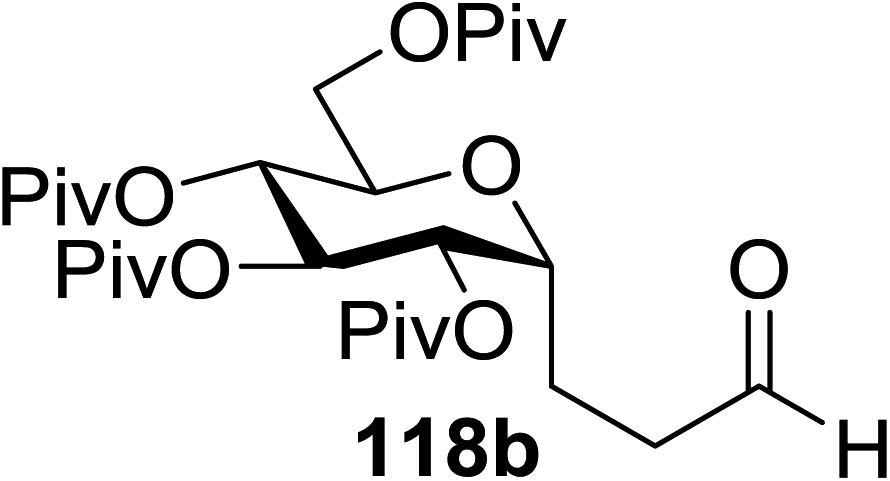 |
— |
| 4 | 117b | 4 | 3 | 118b | 85 |
This protocol was further used for the development of an efficient route for the synthesis of 118a, in which the synthesis of a C-serine glycoamino acid (Scheme 7) was taken under consideration. One-pot asymmetric Strecker cyanation produced the aminonitrile in high yields and diastereoselectivity for both the acetate (119a) and pivaloate protected (119b) glycosides. However, conventional reaction conditions for the hydrolysis or alcoholysis of the nitrile failed to produce the required product, and hydration with Parkin's catalyst (120) formed the amide intermediate with no observable epimerization.63 Alcoholysis of the primary amide afforded the protected C-serine amino ester 122 in excellent yields, although these reaction conditions were questionable with acetate-protected sugars.
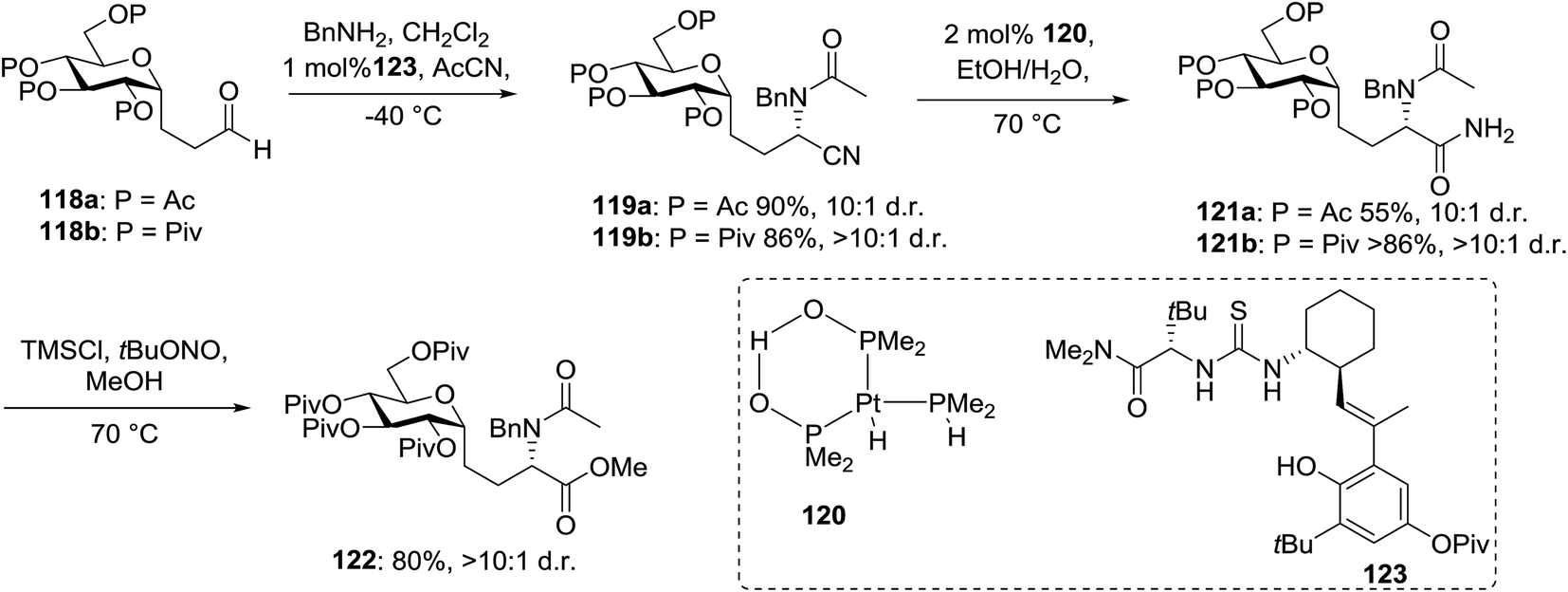 | ||
| Scheme 7 Synthesis of C-linked isostere of glucosyl-serine. | ||
The versatility of 118a was further demonstrated by glycoconjugate synthesis (Scheme 8). A series of derivatizations of the aldehyde was performed. Selective α-chlorination with L-prolinamide and NCS and subsequent oxidation gave the chloroester as a single diastereomer. Azide substitution led to the protected C-glucosylalanine derivative as the azidoester 124 in a short period with an 82% overall yield from 118a. Proline-catalyzed addition of 118a to the iminoglyoxalate and subsequent protection provided 125 as a single diastereomer.64 Reduction of 118a and acylation with dodecanoyl chloride afforded the model C-glycolipid 126.
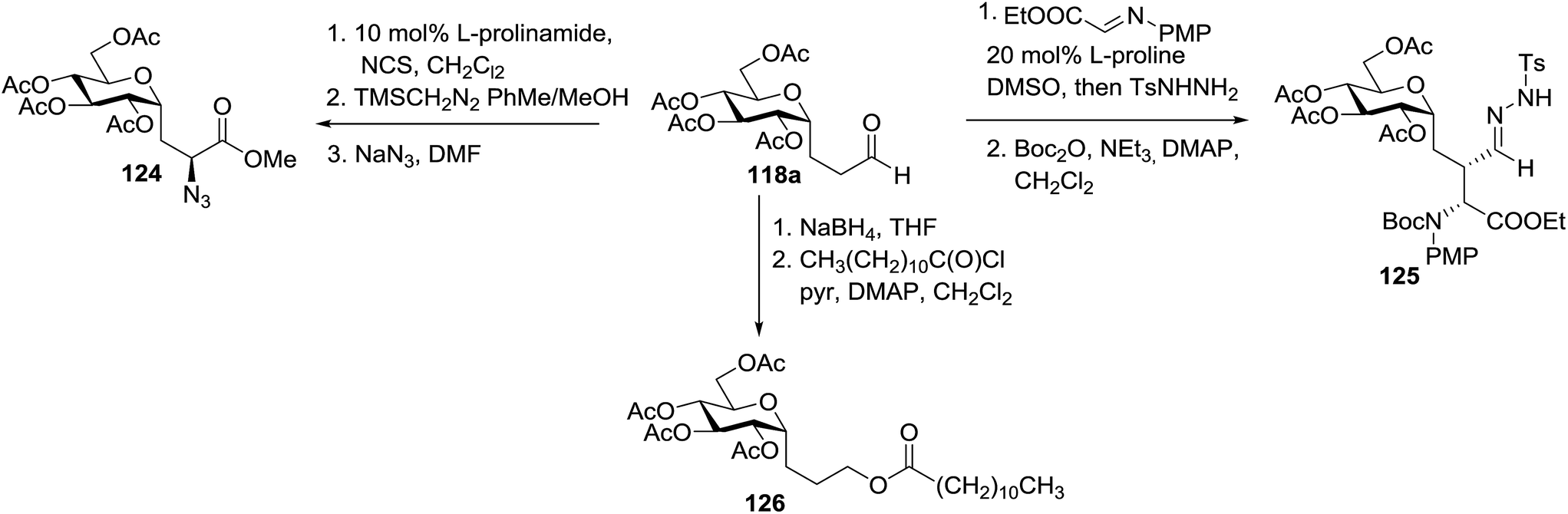 | ||
| Scheme 8 Synthesis of several glycoconjugates. | ||
3.3. Photocatalytic glycosylation using selenosugars as donors
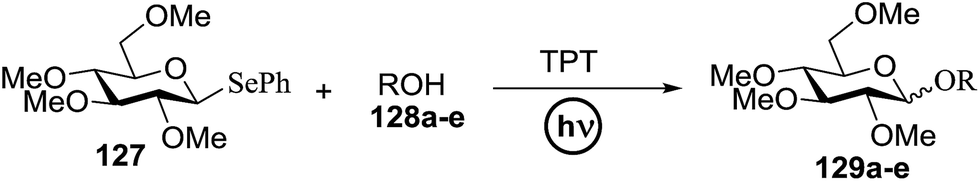
Chalcogenoglycosides, such as thioglycosides and selenoglycosides, have been used widely in the synthesis of oligosaccharides due to their stability during the activation of other glycosyl donors. In 1996 Furuta and co workers achieved the activation of selenoglycosides by photoinduced electron transfer between the phenylselenyl group and aromatic compounds.65 They reported a permethylated glucose derivative as the glycosyl donor and simple alcohols as the glycosyl acceptors in the presence of aromatics, which act as good electron acceptors in their excited states.The photoinduced electron transfer between selenoglycoside and the excited 2,4,6-triphenylpyrylium tetrafluoroborate (TPT) was expected because the fluorescence quenching of TPT. The possible mechanism of the glycosylation reaction is shown in Scheme 9. After the excitation of TPT [or other aromatics (A)], the electron transfer generated the radical cation of 96. Heterolytic breakage of the C–Se bond achieved a glycosyl cation which reacts with a variety of glycosyl acceptors. This indicates that photosensitized oxidation of the selenoglycoside occurs in order to generate the glycosyl cation and produce O-glycoside. This was the first example of a photochemical disaccharide synthesis.
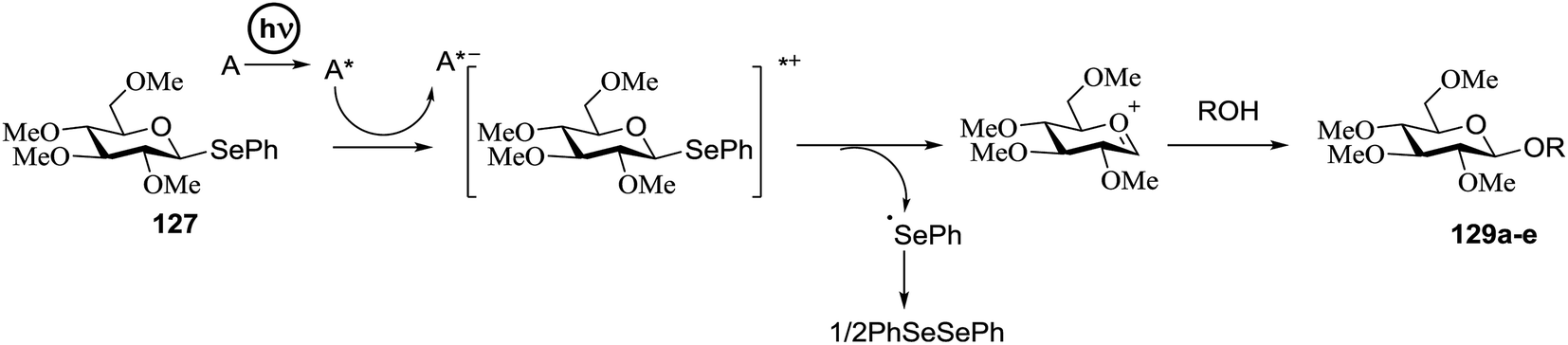 | ||
| Scheme 9 Activation of selenoglycosides by photoinduced electron transfer. | ||
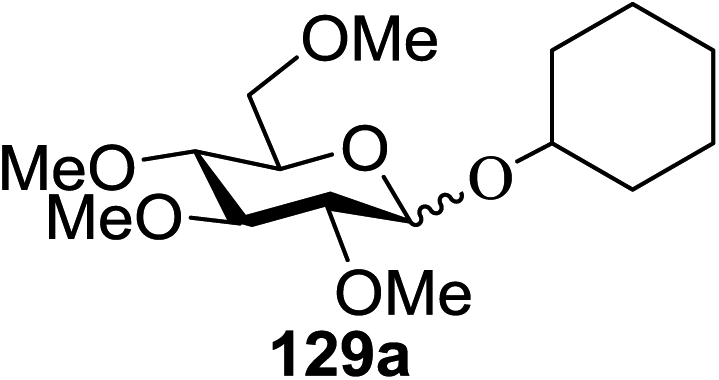
Irradiation of the acetonitrile solution (0.1 mol dm−3) of 2,3,4,6-tetra-O-methyl-1-phenylseleno-β-D-glucopyranoside 127, cyclohexanol 128a (20 equiv.) and TPT (1 equiv.) produced cyclohexylglucoside 129a and 1-hydroxy-2,3,4,6-tetra-O-methyl-D-glucopyranose. The product yields of the irradiation reactions are summarized in Table 16. The use of 2,4,6-triphenylpyrylium tetrafluoroborate (TPT) as a sensitizer gave a 75% isolated yield of the desired glucoside 129a as a stereoisomeric mixture (α/β = 25/75) and hydrolysed product (25%, α/β = 75/25) (entry 1).
|
|||
|---|---|---|---|
| Entry | Acceptor | Product | % yield (α/β) |
| 1 |  |
 |
75 (25:75) |
| 2 |  |
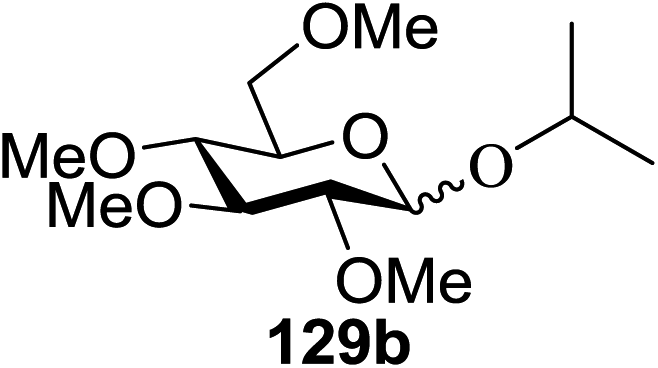 |
71 (30:70) |
| 3 | 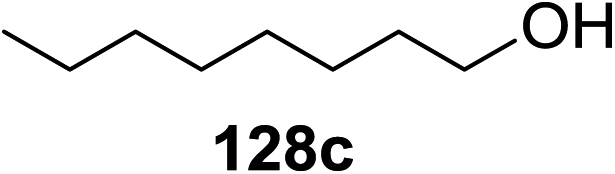 |
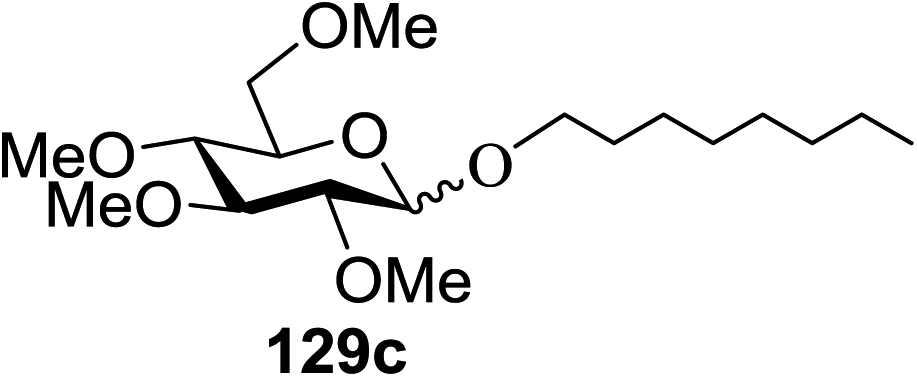 |
68 (30:70) |
| 4 |  |
 |
59 (14:86) |
| 5 |  |
 |
8 (0:100) |
The reactions of 127 and other alcohols (128a–e) in the presence of 2,4,6-triphenylpyrylium tetrafluoroborate (TPT) are presented in Table 16. Simple alcohols (isopropanol and octanol) as well as sugar derivatives were used as glycosyl acceptors. The observed β selectivity of the products was similar to that of the Lewis acid promoted reactions of various glycosyl donors in acetonitrile.
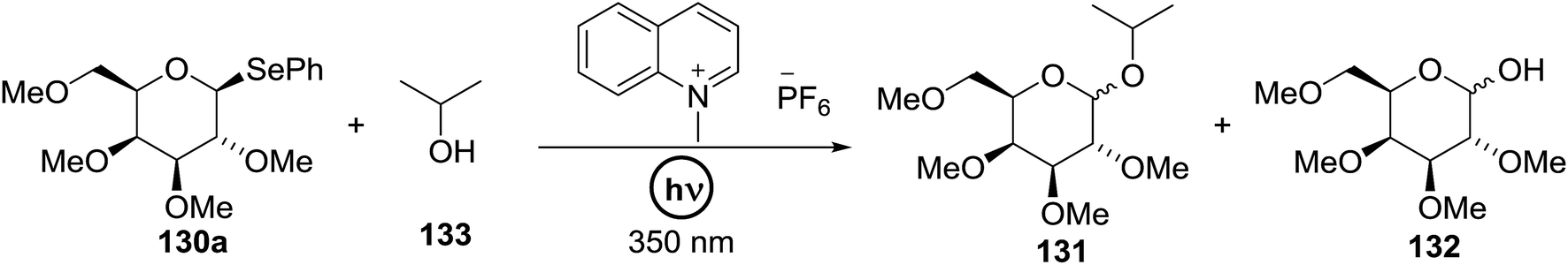
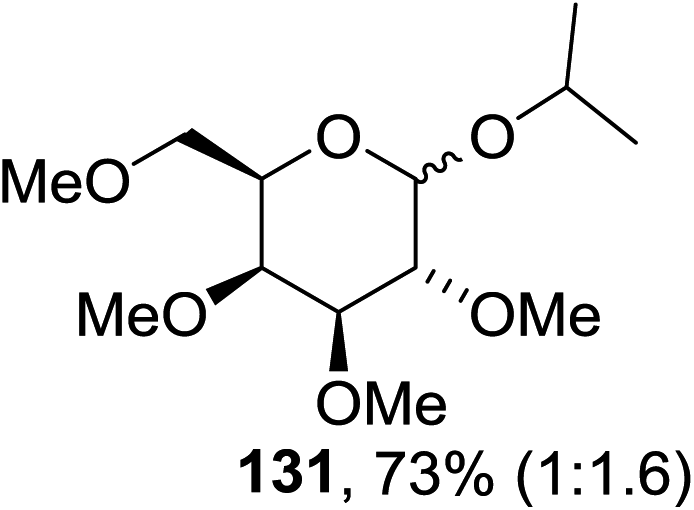
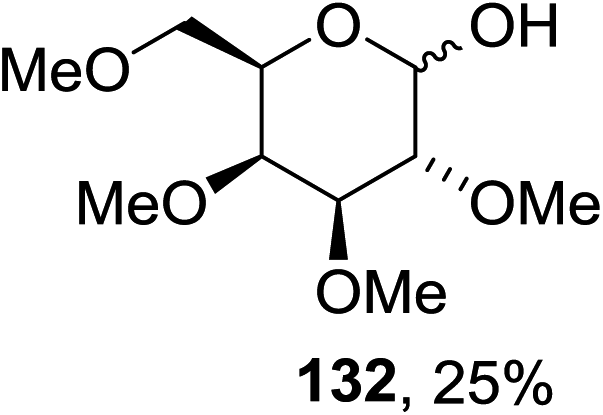
Cumpstey I. and Crich D developed a method for the photochemical activation of selenoglycosides, in which selenoglycosides were activated by single-electron transfer using a photooxidant, N-methylquinolinium hexafluorophosphate as a photosensitizer, and toluene cosolvent as the cosensitizer under 350 nm irradiation.66 N-Methyl quinolinium hexafluorophosphate (NMQ-PF6) is used for breaking the C–Si bond or for acetal formation by oxidative C–C bond cleavage.67,68 Using this photosensitizer, activation of selenoglycosides as glycosyl donors was possible, even under mild activation conditions.69 For optimization of the glycosylation conditions, a methyl ether-protected phenyl selenogalactoside 130a as the glycosyl donor and isopropanol 133 as the acceptor were tested first. Using a mixture of acetonitrile, dichloromethane and toluene as the solvent, 1.5 equiv. of the sensitizer NMQ-PF6, and 2 equiv. of the acceptor alcohol, in air, under light irradiation (350 nm) after 2 h 20 min the glycosides 131 (54%) and hemiacetal 132 (40%) were isolated (Table 17, entry 1). No result was obtained for the same reaction in the dark after 48 h (Table 17, entry 2) and performing the reaction without the NMQ-PF6 sensitizer afforded mostly (>90%) starting material (Table 17, entry 3). This is consistent with the predicted mechanism for the glycosylation reaction involving photochemical activation of the sensitizer salt and consecutive activation of the selenoglycoside donor by single-electron transfer. Diphenyl diselenide, which is the yellow by-product from the glycosylation reaction, was produced. Entry 4 shows the requirement of oxygen for this reaction, since very little reaction was observed when it was performed under Ar (Table 17). This is due to the fact that the catalytic NMQ-PF6 in combination with oxygen can be used as a photooxidant, where oxygen reoxidizes the reduced NMQ radical. Further prolonging the reaction under these conditions (0.3 equiv. NMQ-PF6) resulted in the complete conversion of the selenoglycoside 130a into a mixture of glycosides 131 and hemiacetal 132 (Table 17, entry 5). Performing the reaction in dichloromethane and acetonitrile without toluene gave a somewhat slower reaction, which is consistent with the expected role of toluene as a cosensitizer. Acetonitrile was needed as a cosolvent because the sensitizer NMQ-PF6 is insoluble in toluene.
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | NMQ-PF6 (equiv.) | Atmosphere | Time | Outcome |
| 1 | MeCN/CH2/PhMe, 2:4:1 |
1.5 | Air | 2 h 20 min | 131 (α/β = 1:1.6) 54%, 132 = 40% |
| 2 | MeCN/CH2/PhMe, 2:4:1 |
1.5 | Air | >48 h | No reaction |
| 3 | MeCN/CH2/PhMe, 2:4:1 |
0 | Air | 2 h 20 min | Very little reaction |
| 4 | MeCN/CH2/PhMe, 2:4:1 |
1.5 | Argon | 1 h 30 min | Very little reaction |
| 5 | MeCN/PhMe, 5:2 |
0.3 | Air | 2 h | 131 (α/β = 1:1.8), 132 (α/β = 2:1) |
| 6 | MeCN/PhMe, 5:2 |
0.3 | Air | 3 h | 131 (α/β = 1:1.9), 132 (α/β = 2.7:1.6) |
| 7 | MeCN/PhMe, 5:2 |
0.3 | Air | 3 h | 131 (α/β = 1:1.8), 132 (α/β = 2.3:1) |
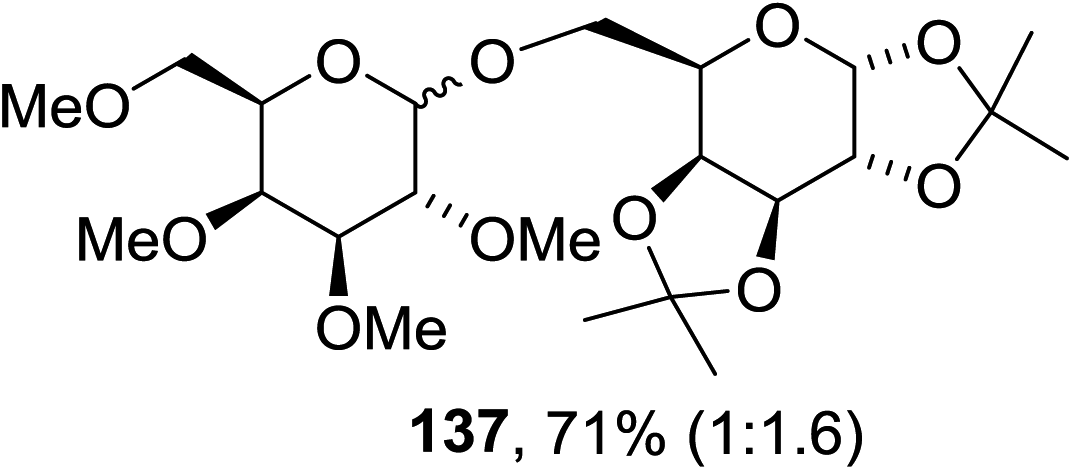
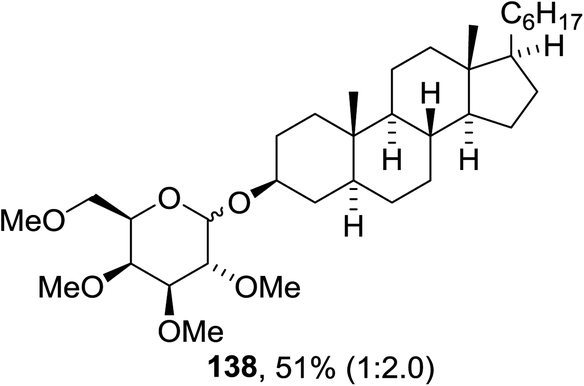
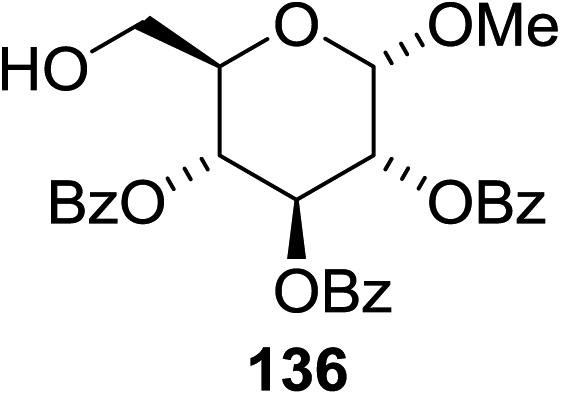
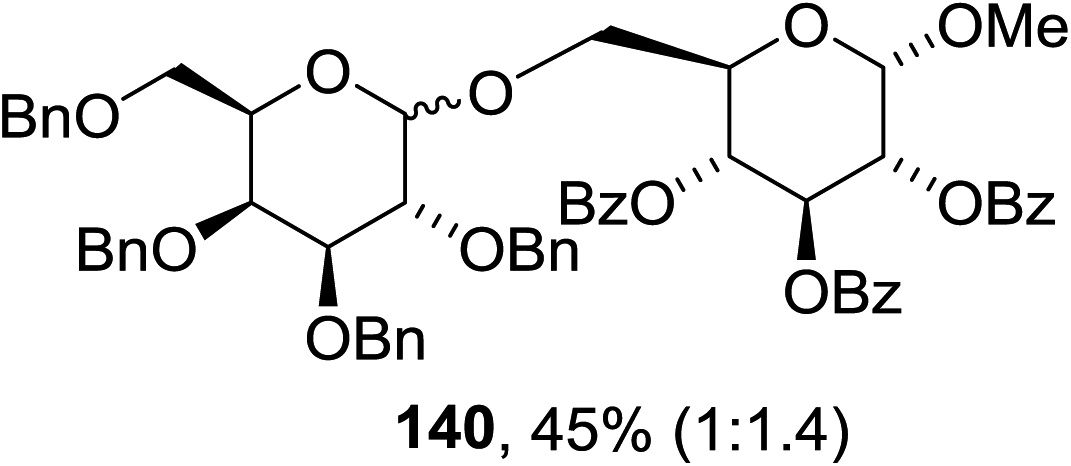
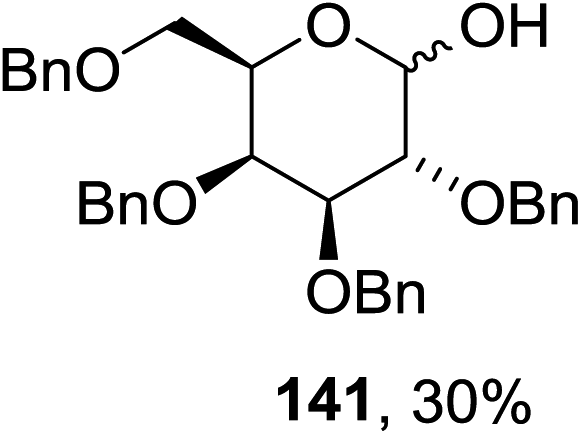
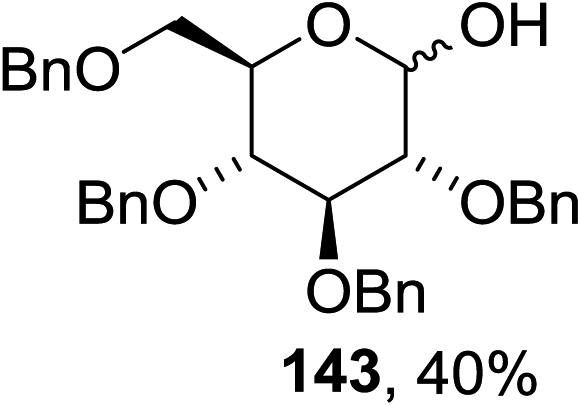
The scope of the reaction with other glycosyl donors and acceptors are given in Table 18. Benzylether-protected galacto 2- and gluco 4-configured selenoglycosides were used as donors without any breaking of the benzyl ethers. This serves as a step forward in photochemical glycosylation since previously only methyl ethers were used, which are more difficult to deprotect. The benzoate esters on the acceptor were also unaffected. The synthesis of the (1→6)-linked disaccharides 137, 139, 140, 142, and 144 and carbohydrate–steroid conjugate 138 was also possible. The hemiacetals 132, 141 and 143 derived from the glycosyl donors were generated as by-products. The glycosidic α/β ratios favored the β-configured products, which is due to the presence of acetonitrile as a cosolvent. The reactions required a catalytic amount of photosensitizer and oxygen or air.
| Entry | Donor | Acceptor | Glycosides yield (α/β) | Hemiacetal yield |
|---|---|---|---|---|
| 1 | 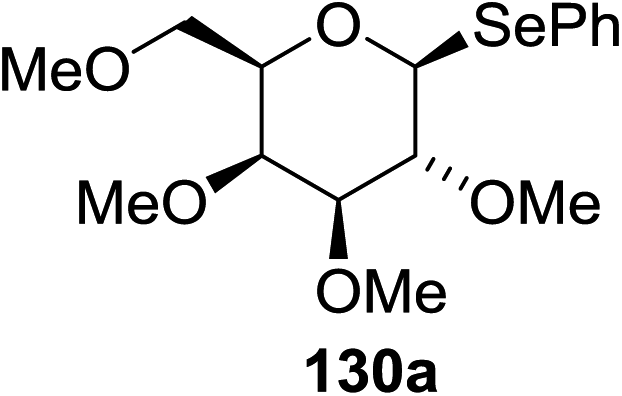 |
 |
 |
 |
| 2 | 130a | 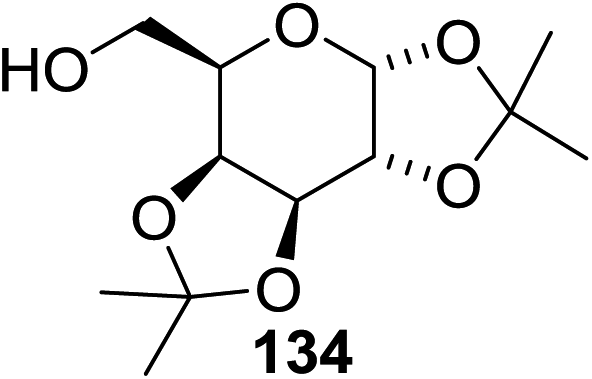 |
 |
Not isolated |
| 3 | 130a | 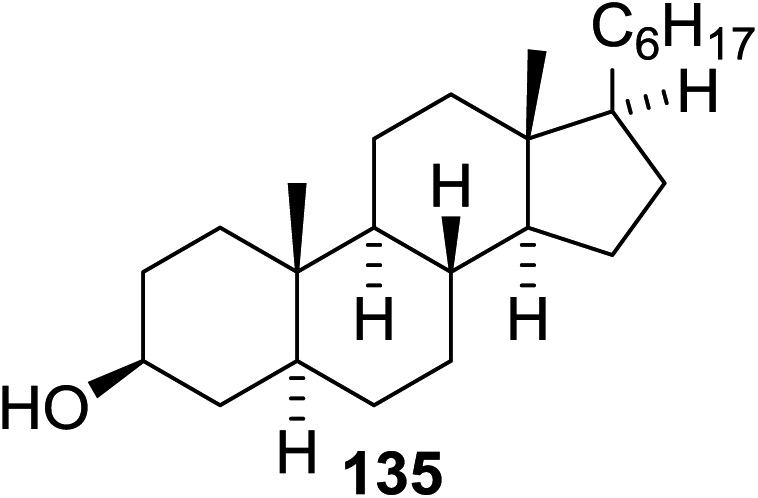 |
 |
Not isolated |
| 4 |  |
134 | 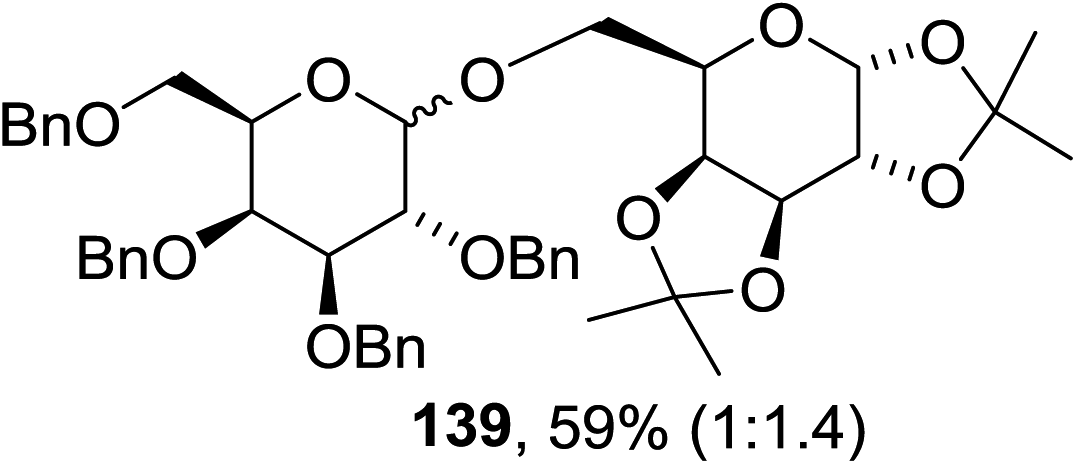 |
Not isolated |
| 5 |  |
 |
 |
 |
| 6 | 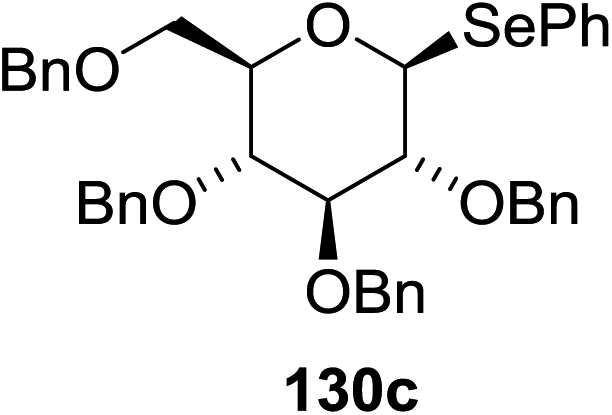 |
134 | 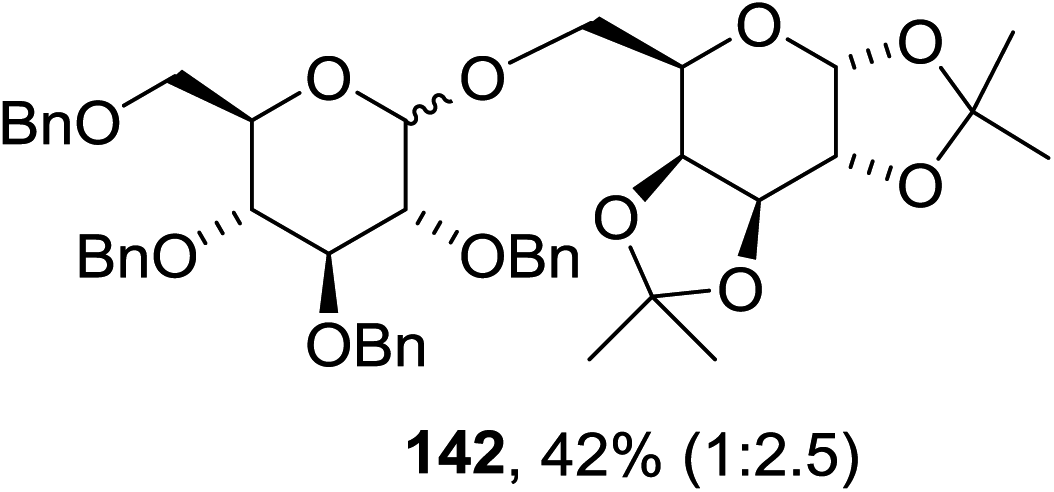 |
 |
| 7 | 130c | 136 | 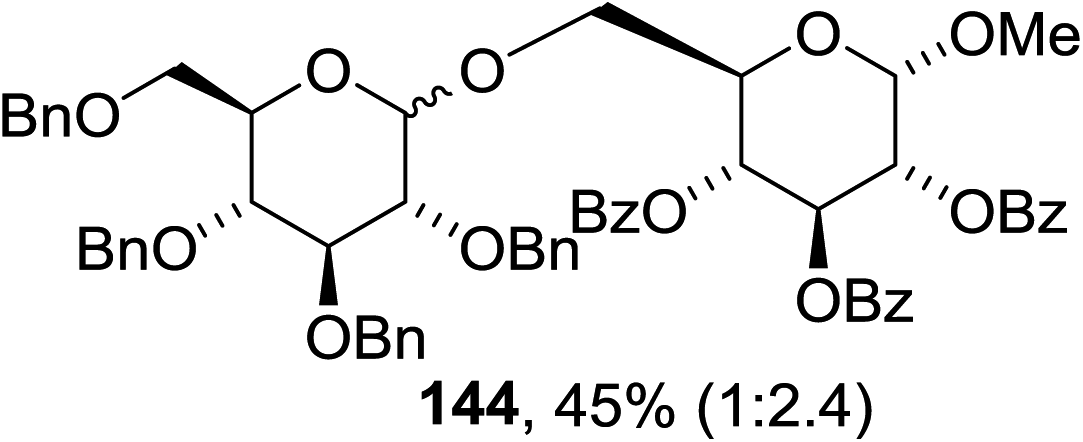 |
143, 47% |
The visible light photocatalytic activation of selenoglycoside donors in the presence of alcohol acceptors was developed by Ragains and co workers.70 This process was demonstrated with both 1-phenylselenyl-2,3,4,6-tetra-O-benzyl glucoside (114a) and 1-phenylselenyl-2,3,4,6-tetra-O-benzyl galactoside (114b), which represents the first example of a visible light-promoted O-glycosylation using a selenoglycoside. To display this concept, structures of selenoglycoside donors, alcohol acceptors, and glycosidic products are shown in Table 19.
|
|||||
|---|---|---|---|---|---|
| Entry | Donor | Alcohol | Product | Yield in presence of | |
| Ru(bpy)3(PF6)2 | PhSeSePh | ||||
| 1 | 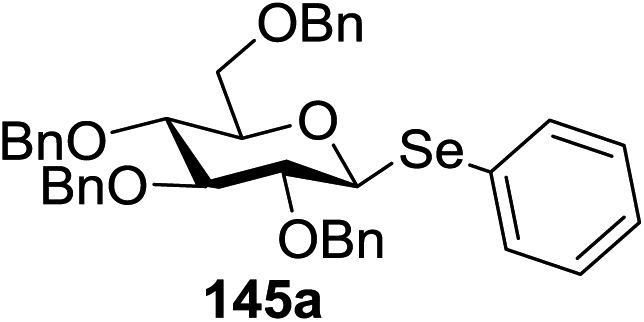 |
MeOH | 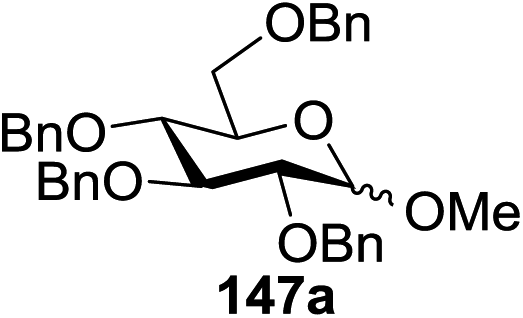 |
75% | — |
| 146a | |||||
| 2 | 145a |  |
 |
81% | 0 |
| 3 | 145a |  |
 |
53% | 65% |
| 4 | 145a | 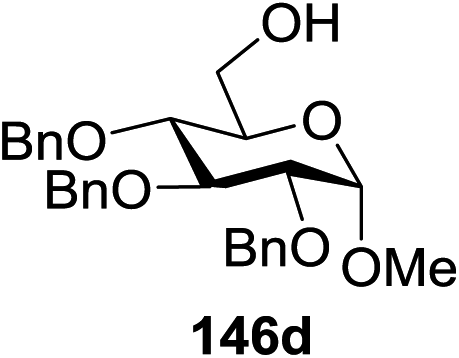 |
 |
43% | 33% |
| 5 | 145a | None | 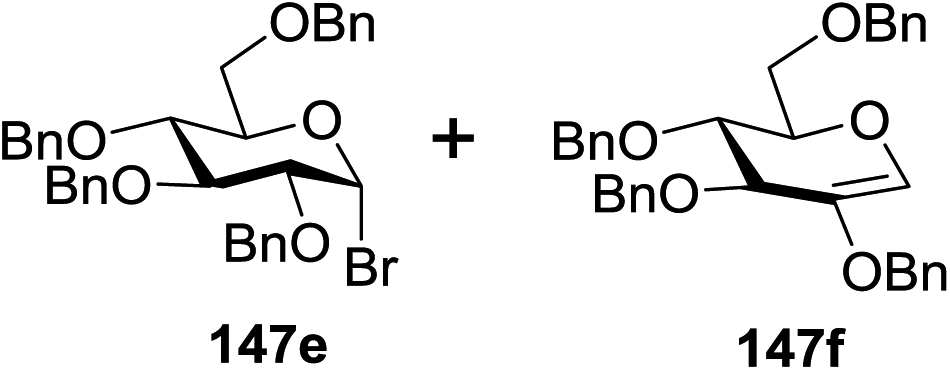 |
147f (1.4:1) not isolated |
|
| 6 | 145a |  |
 |
57% | |
| 7 | 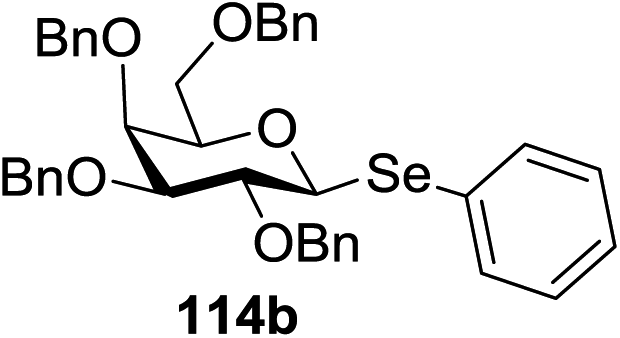 |
115b |  |
65% | |
| 8 | 114b | 115d |  |
50% | |
| 9 | 114b | 115e |  |
55% | |
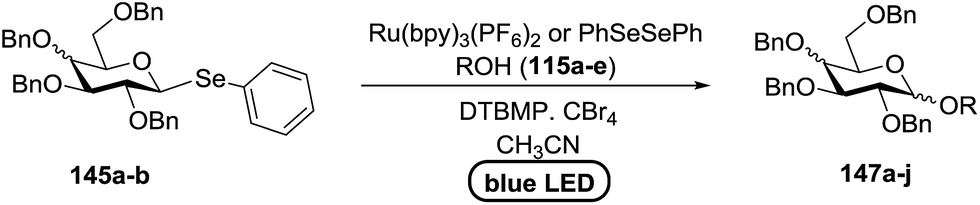
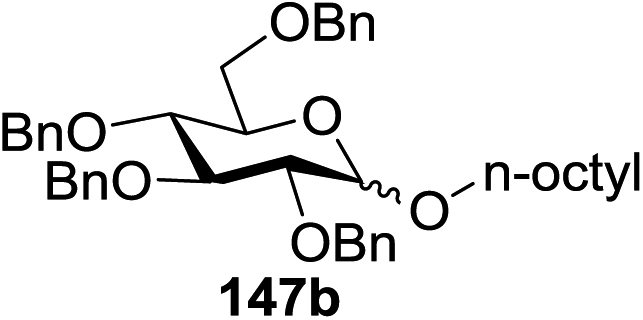
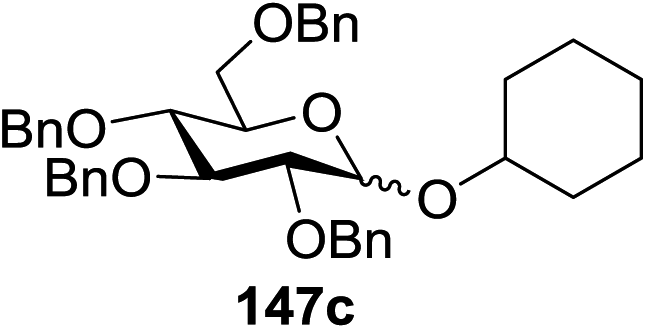
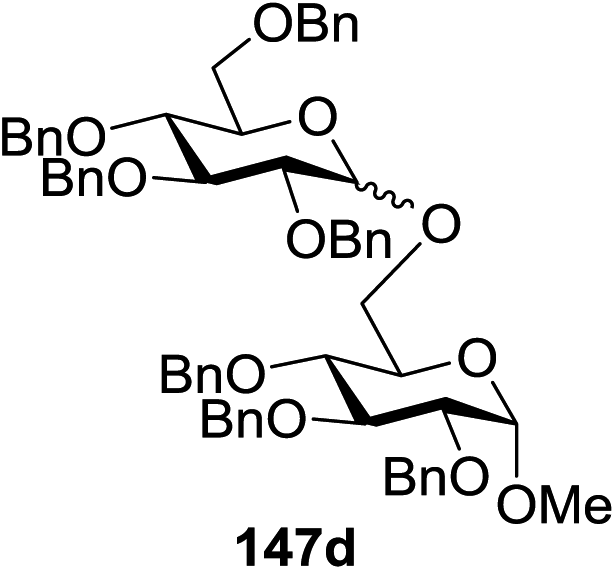
Blue LED irradiation of 145a in the presence of 5 mol% Ru(bpy)3(PF6)2, 1.1 equiv. of the electron acceptor tetrabromomethane and 2,6-di-tert-butyl-4-methylpyridine (DTBMP, 1.2 equiv.) as the base and MeOH (3 equiv.) in CH3CN resulted in the complete utilization of 145a and 75% yield of the glycosylation product 147a (2.5:1 α/β) after 5 h irradiation. Different experiments determined that this method was sufficiently effective for 1-octanol, cyclohexanol and the glucosyl acceptor 146d (Table 19, entries 2–4). The formation of the α-anomer was favored in presence of a methanol acceptor. In the absence of an alcohol acceptor (Table 19, entry 5) the formation of α-glucosyl bromide 147e and glycal 147f (1.4:1, respectively) was observed. The optimal conditions indicate that this process is not possible in the absence of either light or CBr4. In that case no conversion of 145a was observed.
The mechanism describes a process in which oxidative quenching of a photoexcited Ru(II) species takes place, which was derived by the visible light irradiation of tris(2,20-bipyridyl)ruthenium(II) (Ru(bpy)3, Fig. 16), for the 1-electron oxidation of selenoglycosides. The formed selenoglycoside radical cations were unstable and were converted into glycosyl cations which reacted with the glycosyl acceptors for the formation of the glycosylated products.71 It has been noted that similar processes to this have been explained electrochemically (Ru(III) is replaced by an anode), photochemically (with various UV-absorbing photosensitizers)72 and chemically.
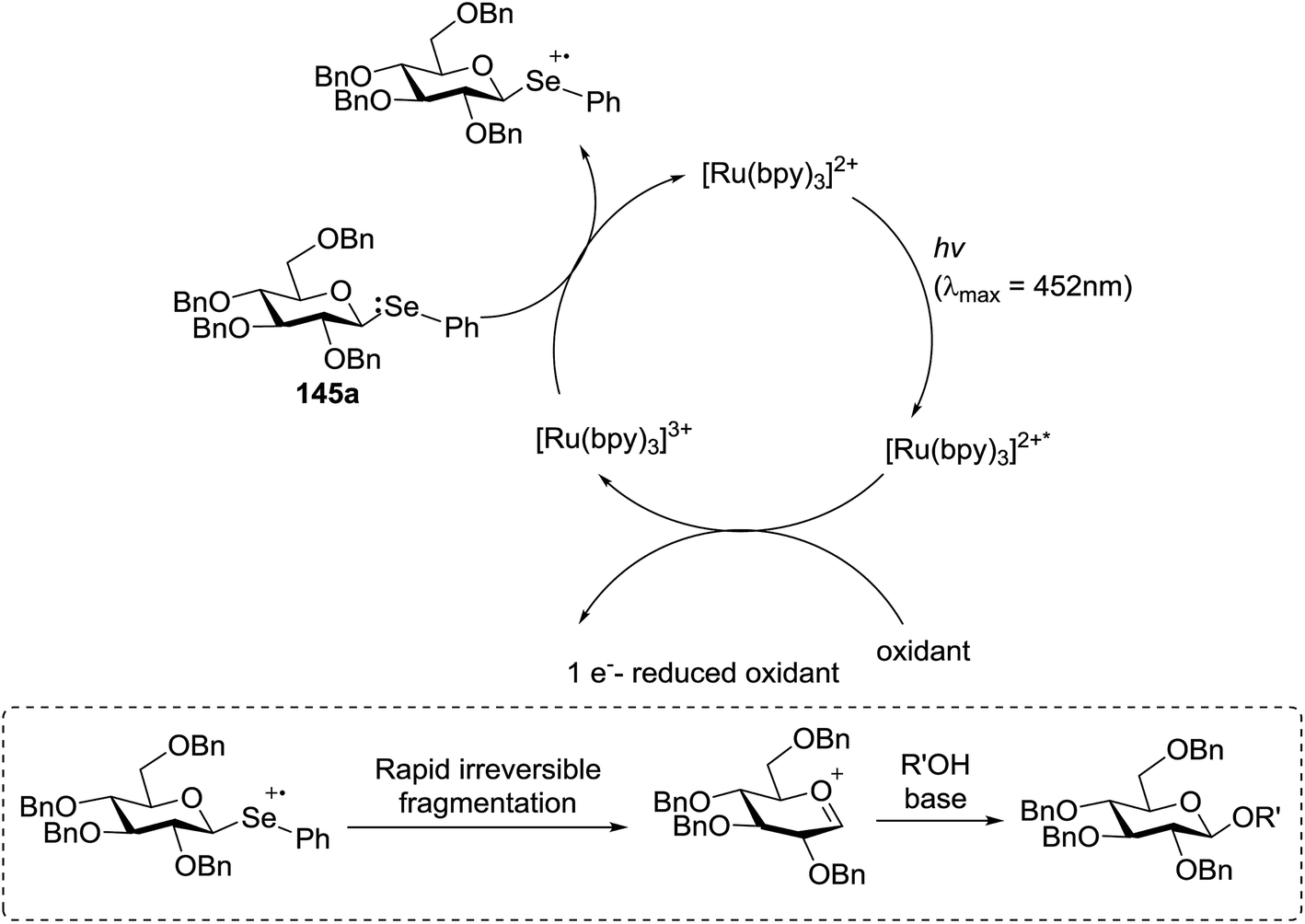 | ||
| Fig. 16 Mechanism for photocatalytic glycosylation by visible light-promoted Ru(bpy)3 catalysis. | ||
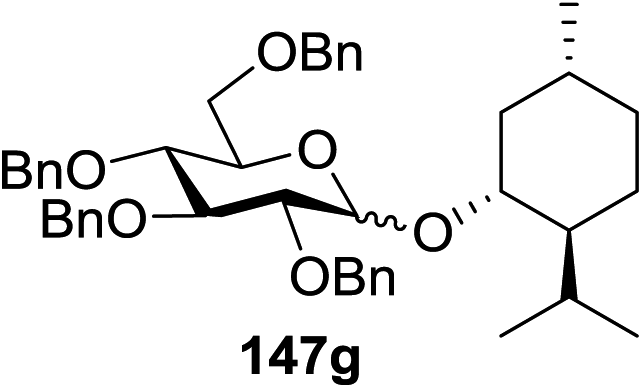
Due to the low cost of diphenyldiselenide compared to Ru(bpy)3(PF6)2, its wide solubility profile in different type of solvents and remarkable reactivity as a visible light photocatalyst, the diphenyldiselenide-catalyzed reaction was improved as a low cost and more adaptable alternative to the Ru(bpy)3-catalyzed reaction. The scope of this organocatalytic glycosylation was further examined via many different experiments under different conditions (Table 19) using 10 mol% diphenyldiselenide, 1.1 equiv. of CBr4, and 1.2 equiv. of DTBMP. Low to moderate yields of the glycosidic products were obtained using 3 equiv. of cyclohexanol, (−)-menthol and glucosyl acceptor 146d with donor 145b (Table 19, entries 7–9). All of these reactions showed higher selectivity for the α-anomer.
The mechanism indicates that the homolysis of the Se–Se bond73 of diphenyldiselenide is promoted with visible light which results in the formation of phenylselenyl radicals (Fig. 17). These radicals react with CBr4 to generate PhSeBr. The reaction of PhSeBr with the Se atom in donors 145a and 145b results in the formation of onium species,74 which directly participate in the glycosylation.
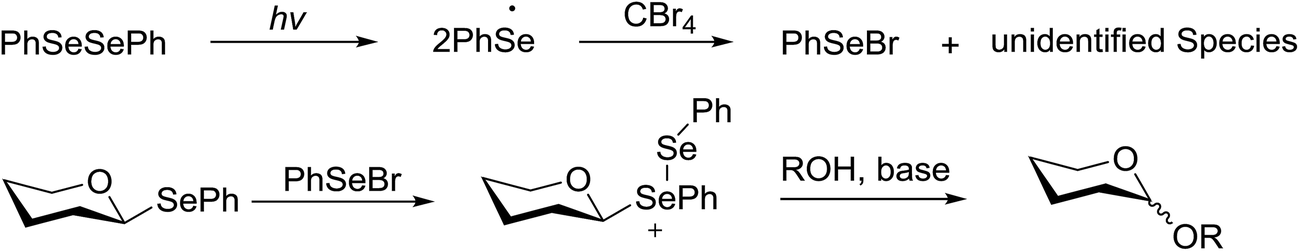 | ||
| Fig. 17 Proposed mechanism for photocatalytic glycosylation by diphenyldiselenide catalysis. | ||
Visible light promotion with Ru(bpy)3 and diphenyldiselenide catalysis contributes an effective approach for O-glycosylation with phenylselenoglycosides and different alcohol acceptors. This approach employs inexpensive reagents, mild conditions and non-hazardous light sources. Glycosylation of both complex primary and secondary alcohol acceptors with glucosyl and galactosyl donors 145a and 145b has been established. This process was selective for the α-anomer with benzyl-protected glucosyl and galactosyl donors.
3.4. Preparation of 2-deoxy-α-glycosides via photoinduced reductive deiodination
Many 2-deoxy sugar linkages show poor tolerance during multistep syntheses. As a result, the usage of direct glycosylation in the preparation of complex 2-deoxy glycosides or natural products are rarely reported in the literature.75 Wang and co-workers76 described an efficient way to prepare 2-deoxy-α-glycosides by the glycosylation of 2-iodo-glycosyl acetate and subsequent visible-light mediated tin-free reductive deiodination (Scheme 10).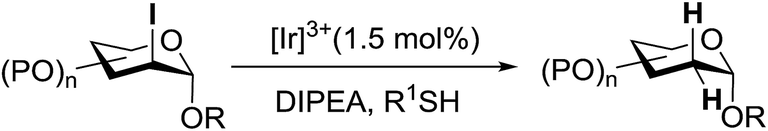 | ||
| Scheme 10 Reductive deiodination. | ||
2-Deoxy sugars and their derivatives widely appear in many pharmaceutically important natural products.77 The development of specific 2-deoxyglycosidic bonds remains a challenge. An efficient way to prepare 2-deoxy-α-glycoside is by the glycosylation of 2-iodo-glycosyl acetate and subsequent visible-light mediated tin-free reductive deiodination. An inspiring result was obtained upon subjecting 148 to irradiation under a 1 W blue LED with fac-[Ir(mppy)3] (fac-[Ir(mppy)3] = fac-tris[2-(p-tolyl)pyridinato-C2,N′]iridium(III)) (1.5 mol%) as a photosensitizer (Table 20).78 Early studies revealed that the combination of nBu3N (2 equiv.) and Hantzsch ester (2 equiv.) in the presence of fac-[Ir(mppy)3] (1.5 mol%) in CH3CN might decrease secondary iodide efficiently within a couple of hours (Table 20, entry 6). Successful optimization showed that the replacement of the expensive Hantzsch ester with the inexpensive p-toluene thiol resulted in an excellent yield (Table 20, entries 7 and 8). In the absence of the reducing reagent (amine), the reduction became less efficient and the 2-deoxyl glycoside 149 was isolated in 39% yield (Table 20, entry 10).
|
||
|---|---|---|
| Entry | Conditions | Yield [%] |
| 1 | nBu3N (10 equiv.), 1.5 h | 84 |
| 2 | nBu3N (2.0 equiv.), 3 h | 69 |
| 3 | DIPEA (10 equiv.), 1.5 h | 80 |
| 4 | DIPEA (10 equiv.), 1.5 h | 60 |
| 5 | nBu3N (10 equiv.), HOOH (10 equiv.), 1.5 h | 86 |
| 6 | nBu3N (2 equiv.), Hantzsch ester (2 equiv.), 1.5 h | 91 |
| 7 | nBu3N (2 equiv.), p-toluenethiol (2 equiv.), 1.5 h | 93 |
| 8 | DIPEA (2 equiv.), p-toluenethiol (2 equiv.), 1.5 h | 95 |
| 9 | DIPEA (2 equiv.), p-toluenethiol (2 equiv.), 1.5 h | 42 |
| 10 | p-Toluenethiol (2 equiv.), 4 h | 39 |
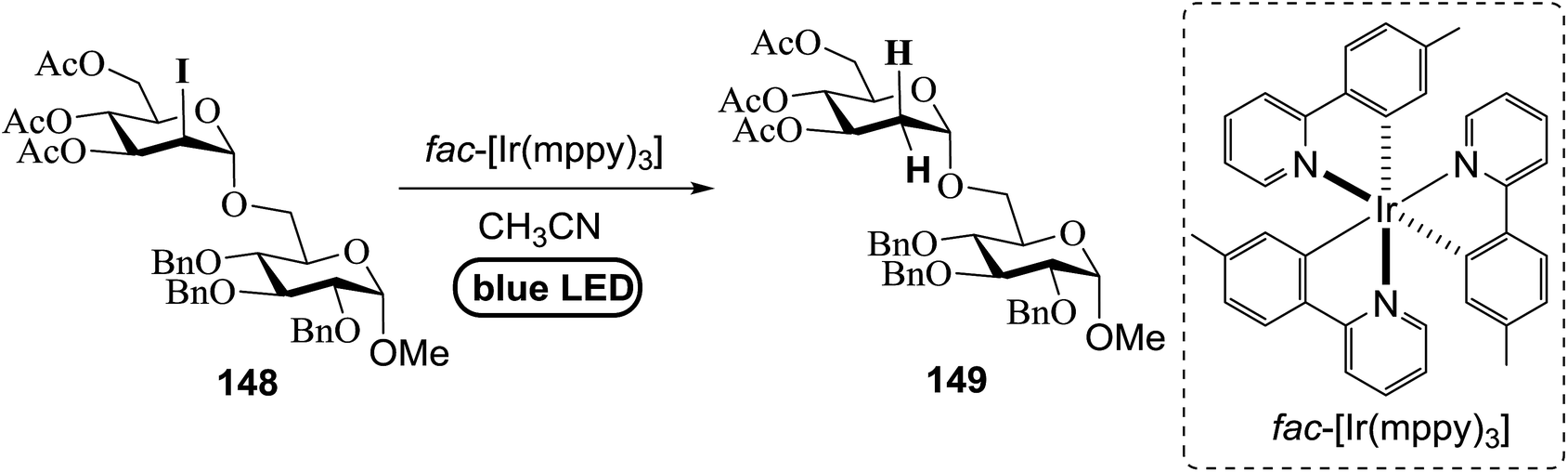
Fig. 18 summarizes the scope of this newly uncovered protocol in 2-deoxy-α-glycoside synthesis. From the known corresponding glycosyl acetates 150a–d,79 α-linked 2-iodo-2-deoxy glycosides were achieved under the altered Roush conditions in good to excellent yield with unique stereochemistry. The optimal deiodination conditions from Fig. 18 (entry 8) were used to examine the scope of functionalization of the C-2 position of newly designed glycosides (Fig. 18).
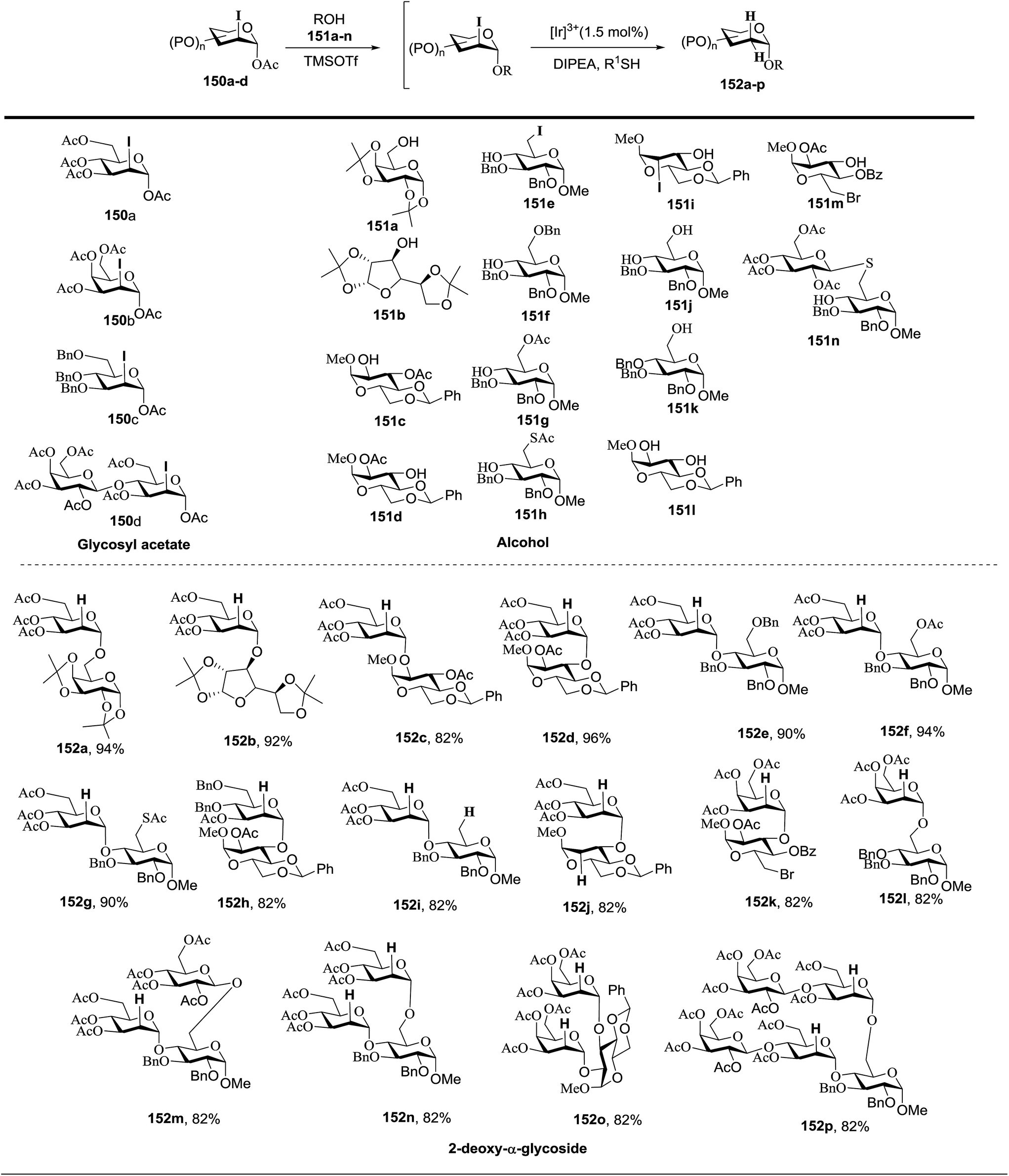 | ||
| Fig. 18 Product scope of α-linked-2-deoxy-glycosides. | ||
3.5. Synthesis of 2-deoxy and 2,3-unsaturated glycosides via photocatalysis
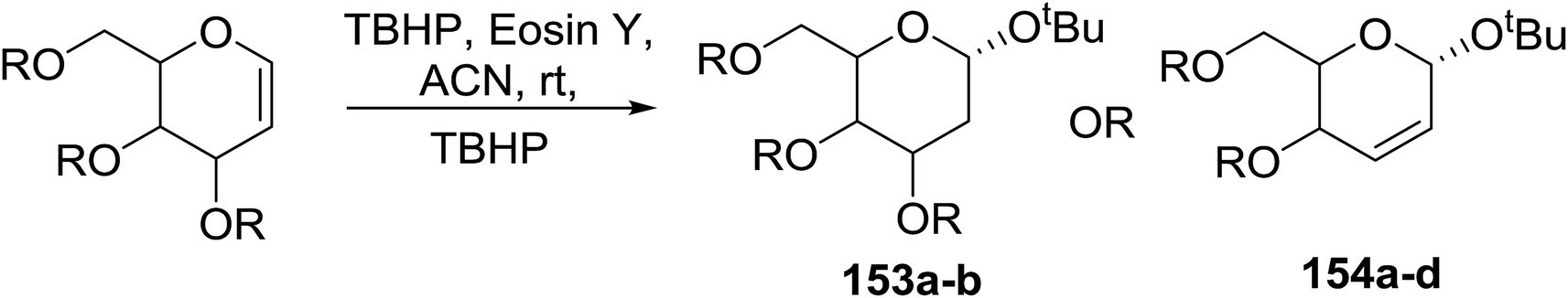
Synthesis of 2-iodo, 2-deoxy and 2,3-unsaturated glycosides were reported by Mukherjee and co-workers.80 Reaction of peroxides with glycals, when carried out in the presence of NIS generates 2-deoxy-2-iodo-α-glycosides in good yields with predominant trans-diaxial selectivity at room temperature. Under photolytic conditions in the presence of a catalytic amount of eosin Y,81 the reaction leads to 2-deoxyglycosides or 2,3-unsaturated-α-glycosides (Scheme 11).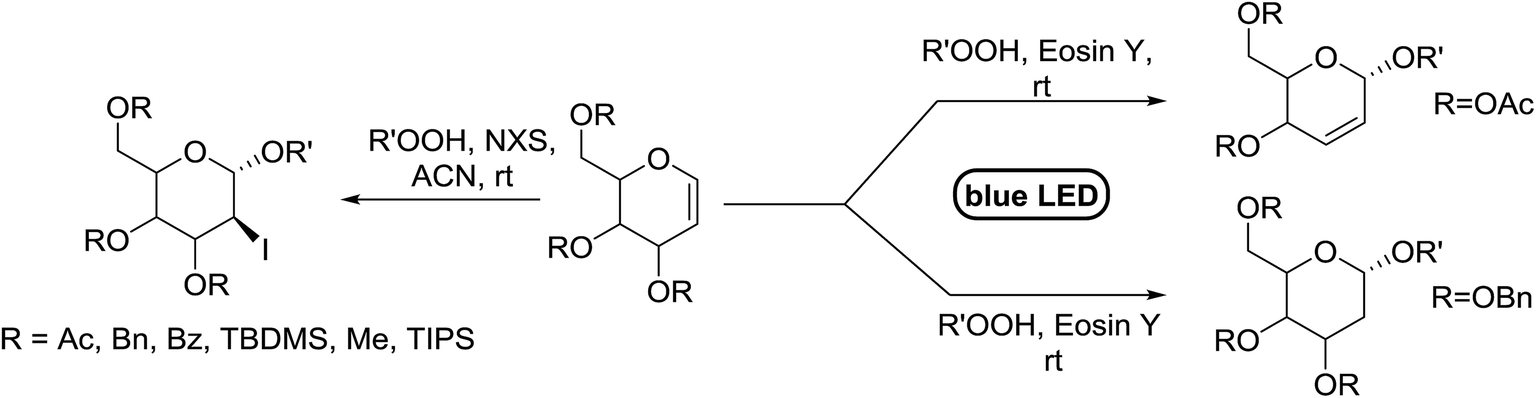 | ||
| Scheme 11 Synthesis of 2-iodo, 2-deoxy and 2,3-unsaturated glycosides. | ||
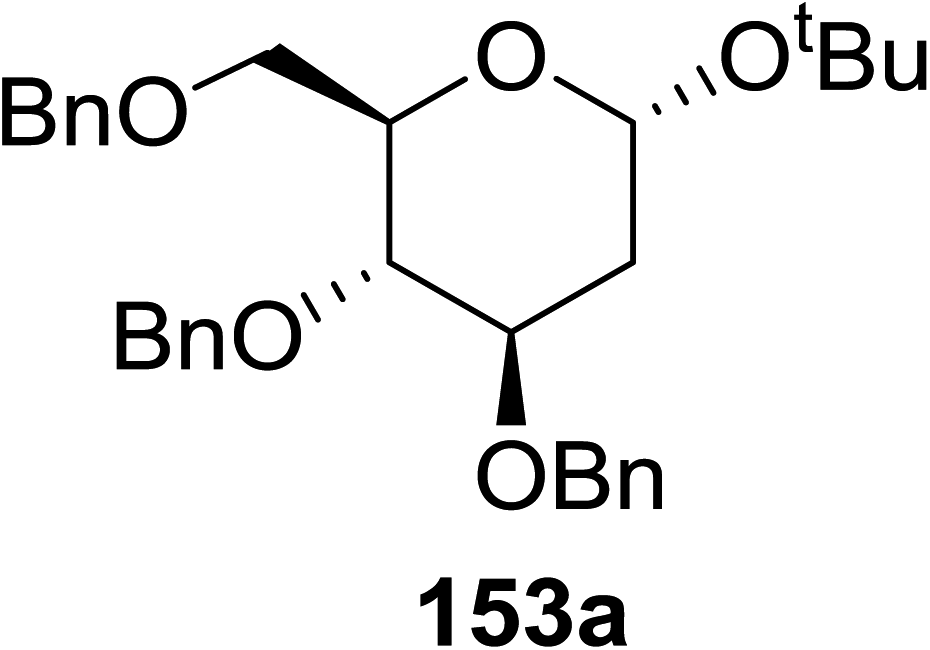
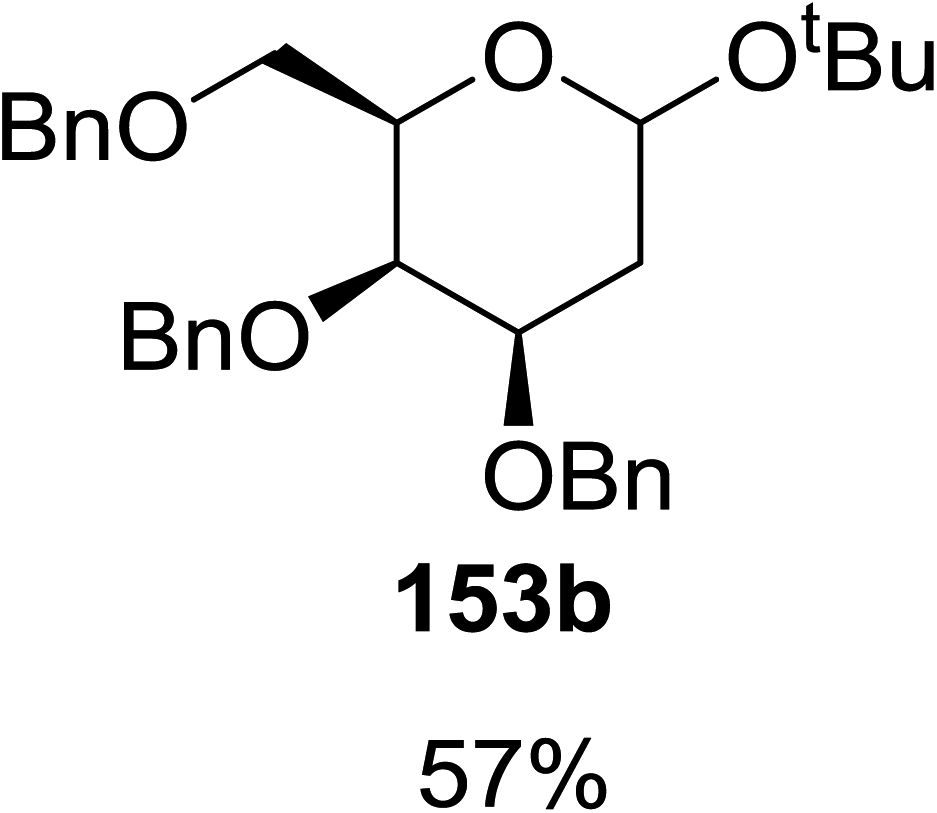
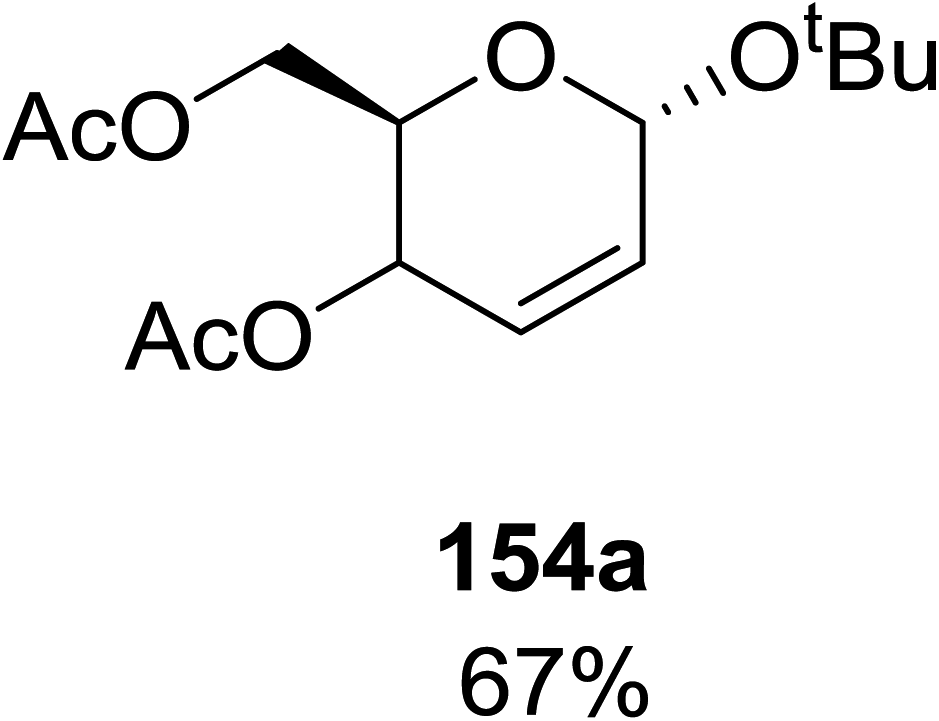
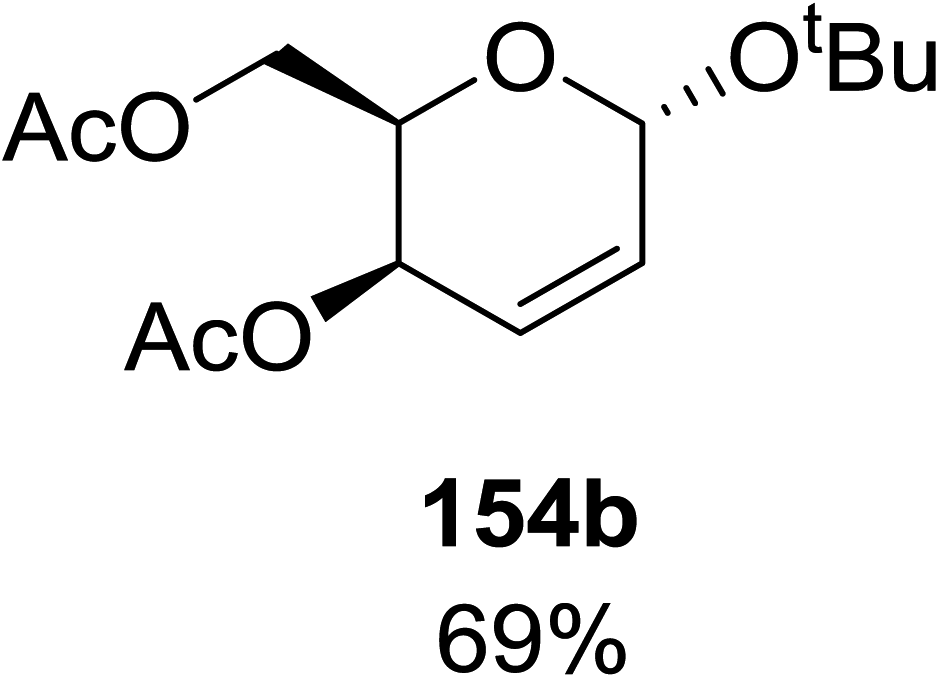
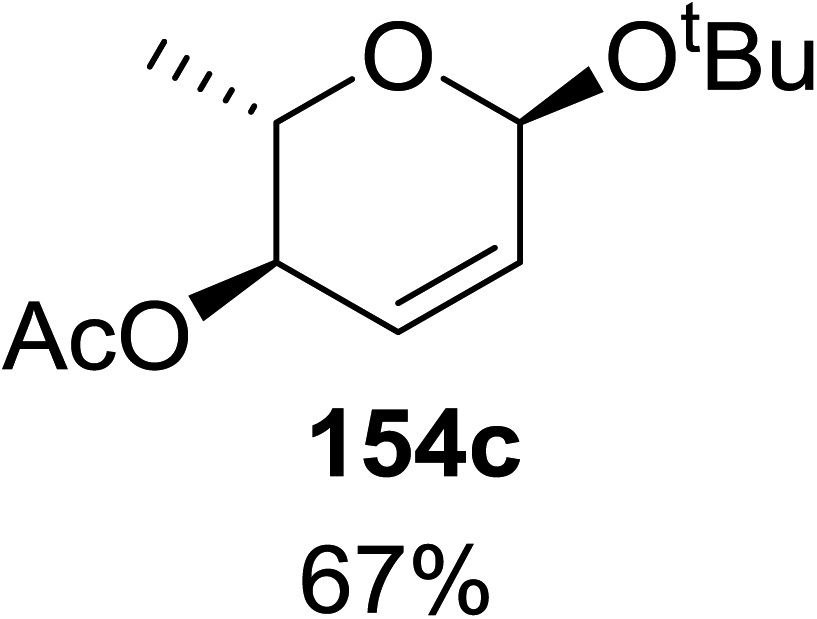
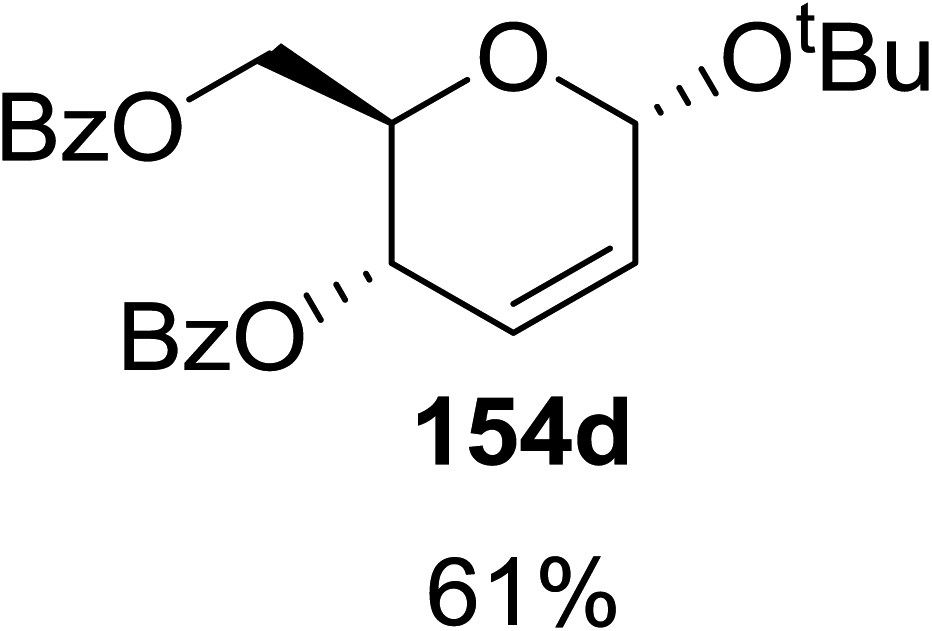
When tri-O-benzyl-D-glucal was reacted with TBHP under photolytic conditions using the eco-friendly, organic dye eosin Y as a photocatalyst (1 mol%) and CFL as a light source (Table 21), the generation of 2-deoxy α-glycoside was observed as a mixture of α and β anomers, but in low yield. In order to further increase the yield of the product, the reaction was performed under various conditions to optimize the parameters (highlighted, Table 21) for the formation of the product in good yield (59%) and with high stereoselectivity giving predominantly the α-glycoside with little β-anomer (9:1). Further screening with different light sources was done which revealed that the use of blue LED improved the yield. Increasing the catalyst loading to 10 mol% did not have a significant effect on the yield of the product and acetonitrile was demonstrated as the solvent of choice. In the absence of light or eosin Y, no reaction took place. The same type of product was noticed from tri-O-benzyl-D-galactal (Table 21, 153b).
|
||||||
|---|---|---|---|---|---|---|
| Product | Light source | Eosin Y (mol%) | TBHP (equiv.) | Solvent | Yield (%) | |
 |
CFL | 1 | 2 | CH3CN | 20 | |
| Blue LED | 1 | 2 | CH3CN | 51 | ||
| Green LED | 1 | 2 | CH3CN | 45 | ||
| Blue LED | 1 | 3 | CH3CN | 59 | ||
| Blue LED | 10 | 3 | CH3CN | 55 | ||
| Blue LED | 1 | 3 | DMSO | No reaction | ||
| Blue LED | 1 | 3 | DMF | No reaction | ||
| Blue LED | 0 | 3 | CH3CN | No reaction | ||
| No light | 1 | 3 | CH3CN | No reaction | ||
| Scope of product | ||||||
 |
 |
 |
||||
 |
 |
|||||
On the basis of a literature study a possible interpretation for the reaction indicates the formation of two different products under photolytic conditions (Fig. 19). The homolytic cleavage of TBHP by photoexcited eosin Y* (Fig. 19) through energy transfer results in the formation of tert-butoxyradical (t-BuO˙) and hydroxyl radical (OH˙). The tert-butoxy radical (t-BuO˙) formed, then attacks the anomeric peroxides under environmentally benign photolytic conditions.
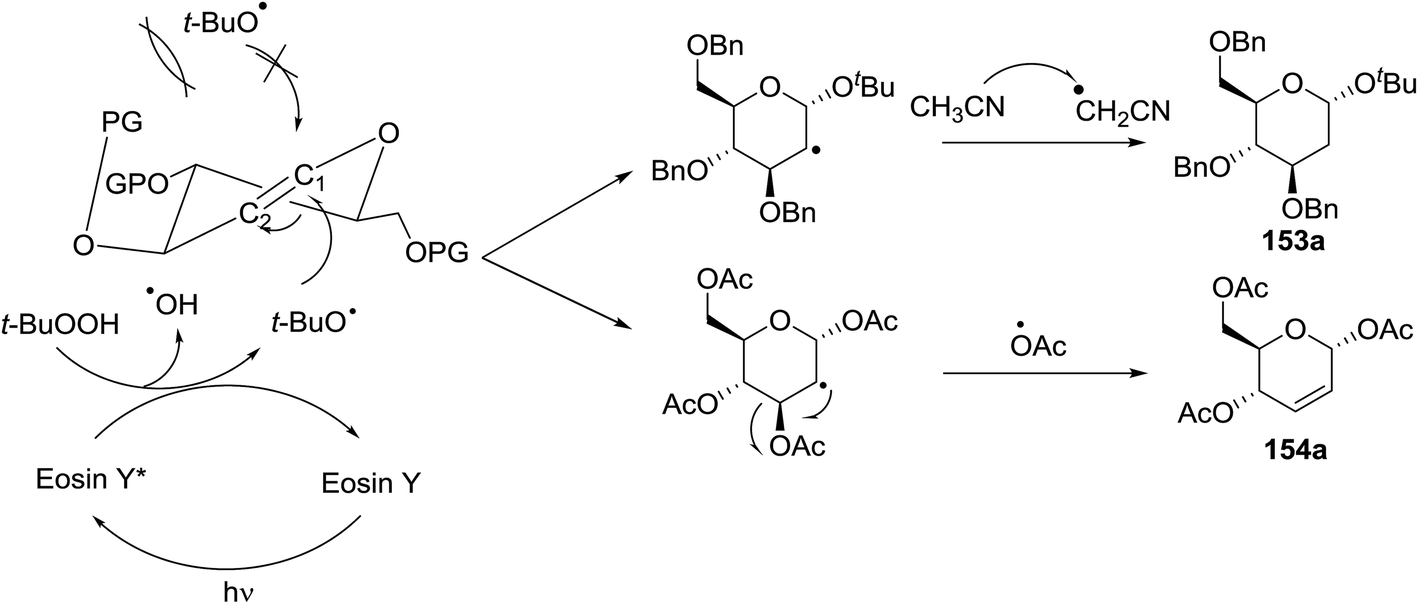 | ||
| Fig. 19 Proposed mechanism for the formation of 2-deoxy and 2,3-unsaturated glycosides. | ||
4. Photoinduced hydrogen atom transfer
The photocatalytic activation mechanism involves the production of radical intermediates via the transfer of a hydrogen atom from an organic substrate directly to a photoexcited chromophore. The feasibility of concerted hydrogen atom transfer is generally determined by the bond strength of reaction centres, such as C–H, O–H and N–H bond strengths, whereas redox potential is the applicable thermodynamic parameter in photoinduced electron-transfer processes. The conversions discussed in this section involve direct cleavage of the O–H and N–H bonds by the photocatalyst and do not require a secondary catalyst to generate the key radical intermediate. Some molecules show different properties in their excited states with respect to acidity. Although certain phenols and naphthols have been known as organophotoacids, their use in organic synthesis has been reported to a lesser extent due to the very short lifetimes of their excited states. In this study, increased acidity in the excited state induced by photoirradiation was sufficient for the deprotection of several acid-sensitive protecting groups widely used in organic synthesis.82 From the viewpoint of this review, the use of organophotoacids would create a good environmentally benign chemical glycosylation reaction.On the other hand, the most widely investigated thiol–ene/yne ligation reactions in synthetic carbohydrate chemistry are the applications of hydrogen abstraction which display an anomeric thiol in addition reactions to terminal olefins and alkynes. Thiol–ene/yne reactions often take place in a highly regioselective manner, which allows the selective introduction of a sulfur atom onto the sugar backbone.
4.1. Photoinduced O-glycosylation using glycosyl trichloroacetimidate donors
Toshima and co-workers reported glycosylation reactions of glycosyl trichloroacetimidates with a variety of alcohols using an organophotoacid. They selected glucosyl trichloroacetimidate 155 and phenol and naphthol derivatives 159–162 as a glycosyl donor and organophotoacids, respectively, as given in Table 22.83 The glycosylation reaction of 155 with alcohol 156 using organophotoacid 159 with and without photoirradiation was studied using a Blackray 100 W lamp irradiating at 365 nm. The glycosylation reactions for optimization were carried out under different conditions, as outlined in Table 22. It was found that the glycosylation reaction of 155 and alcohol 156 using organophotoacid 159 in Et2O under photoirradiation continued to produce glycoside 157 in good yield (entry 2, Table 22). In the reaction without photoirradiation, glycosylation was not successful. These results clearly showed the utility of organophotoacid 159 together with photoirradiation for the glycosylation. After choosing the proper organophotoacids, the solvent effect on the glycosylation reaction was examined. The glycosylation reaction of 155 and 156 using 159 in MeCN and i-Pr2O was found to be stable under the photoirradiation conditions and moderate yields of 157 were obtained along with a considerable amount of hydrolyzed product 158 (entries 3 and 4, Table 22). Based on these results, the standardized solvent conditions were examined with other organophotoacids 160–162 in the glycosylation reaction of 155 and 156 in Et2O. It was found that when 162 was used, the result was similar to that obtained using 159 (entry 8, Table 22), whereas 160 and 161 were found to be less effective in the photoinduced glycosylation reaction (entries 6 and 7, Table 22).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Organophotoacid | Light strength (mW cm−2) | Solvent | Temp. (°C) | Time (h) | Yield | |
| 157 (α:β) |
158 | ||||||
| 1 | 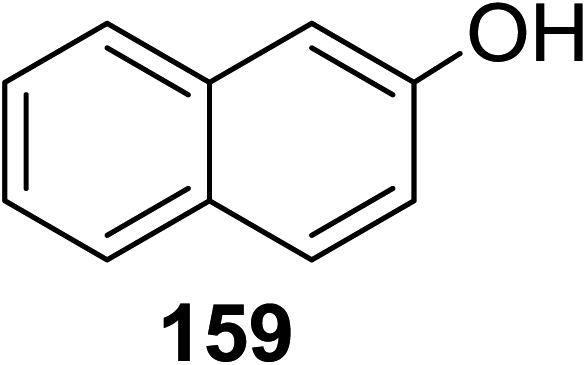 |
0 | Et2O | 35 | 7 | 8 | Trace |
| 2 | 159 | 12 | Et2O | 35 | 7 | 79 (54:46) |
20 |
| 3 | 159 | 27 | MeCN | 50 | 2 | 56 (23:77) |
37 |
| 4 | 159 | 27 | i-Pr2O | 50 | 7 | 68 (54:46) |
24 |
| 5 | 159 | 12 | Et2O | 35 | 20 | 72 (61:39) |
27 |
| 6 | 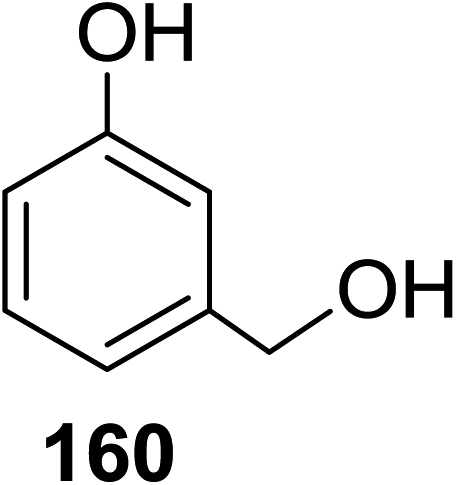 |
12 | Et2O | 35 | 20 | 67 (49:51) |
22 |
| 7 | 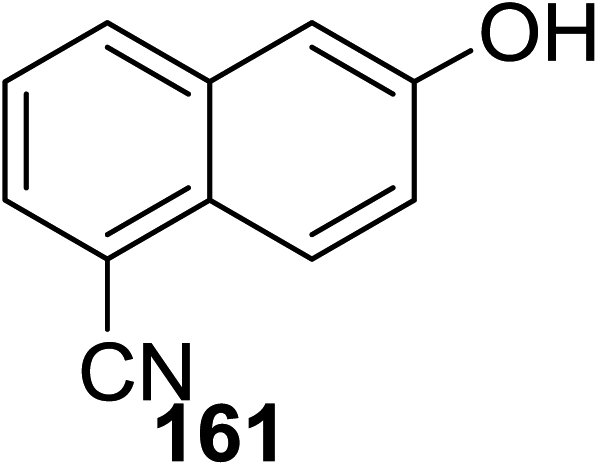 |
12 | Et2O | 35 | 20 | 52 (61:39) |
22 |
| 8 | 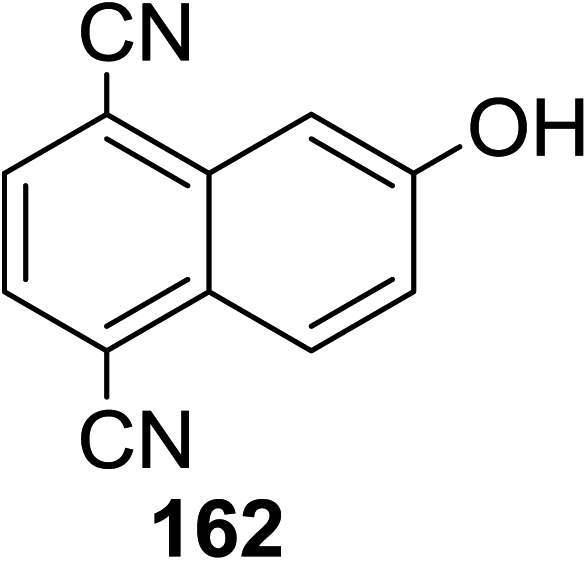 |
12 | Et2O | 35 | 20 | 75 (59:41) |
23 |
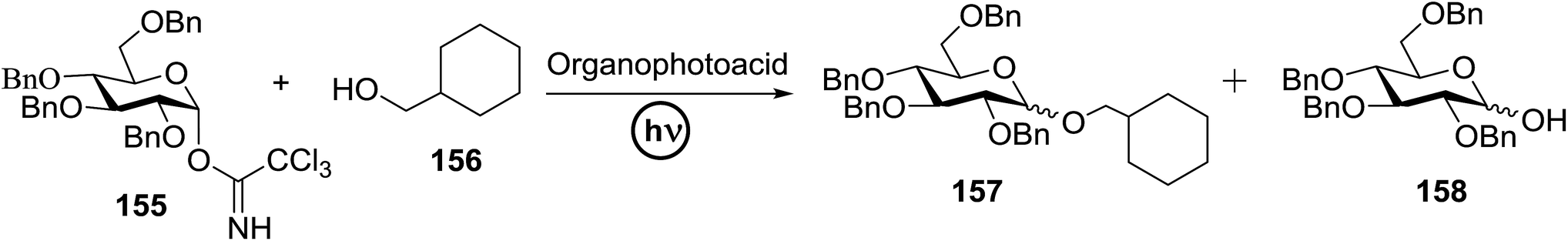
The universality of the glycosylation method using several alcohols and different types of glycosyl donors is summarized in Table 23. Glycosylation of different alcohol acceptors as well as 156 with 155 using 159 under photoirradiation conditions progressed smoothly to form their respective glycosides 163–170 in good yields (Fig. 20).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Organophoto acid | Time (h) | Yield 157 (α:β) |
Entry | Organophoto acid | Time (h) | Yield 157 (α:β) |
| 1 | 159 | 20 | 72 (59:41) |
8 | 159 | 20 | 76 (59:41) |
| 2 | 159 | 20 | 83 (48:52) |
9 | 162 | 20 | 71 (48:42) |
| 3 | 159 | 20 | 81 (53:47) |
10 | 162 | 20 | 81 (54:46) |
| 4 | 162 | 20 | 75 (59:41) |
11 | 159 | 2 | 60 (49:51) |
| 5 | 162 | 20 | 79 (47:53) |
12 | 159 | 4 | 83 (48:52) |
| 6 | 162 | 20 | 72 (52:48) |
13 | 162 | 2 | 44 (50:50) |
| 7 | 159 | 20 | 74 (51:49) |
14 | 162 | 4 | 80 (53:47) |
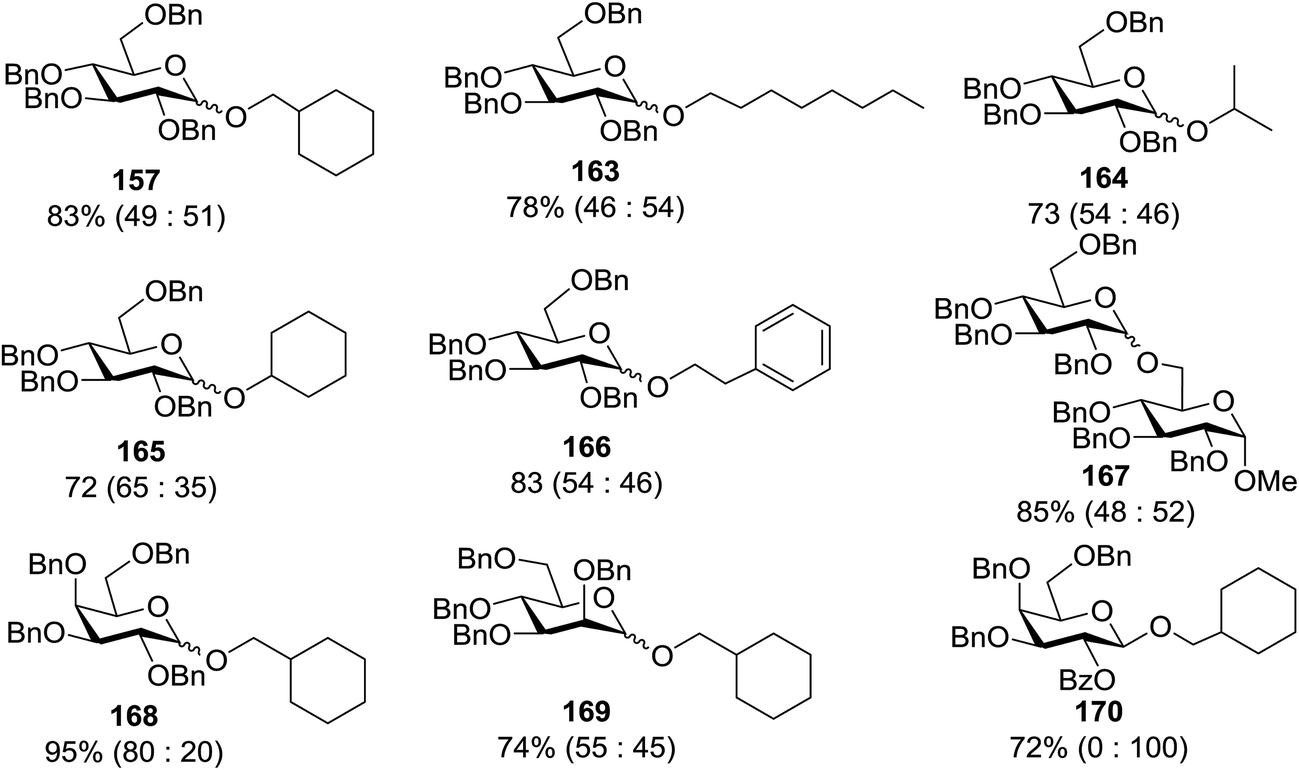 | ||
| Fig. 20 Substrate scope of the glycosylation reaction using organophotoacid. | ||
The above glycosylation profile using organophotoacids under photoirradiation is described by the mechanism given in Fig. 21, which indicates that organophotoacid 159 was in the excited state, and its acidity significantly increased under photoirradiation. This increased acidity activated the glycosyl donor 155, as shown in Fig. 21. Furthermore, it has been observed that 159 in the excited state exhibited low nucleophilicity and the naphthol glycoside 171 was not detected at all.
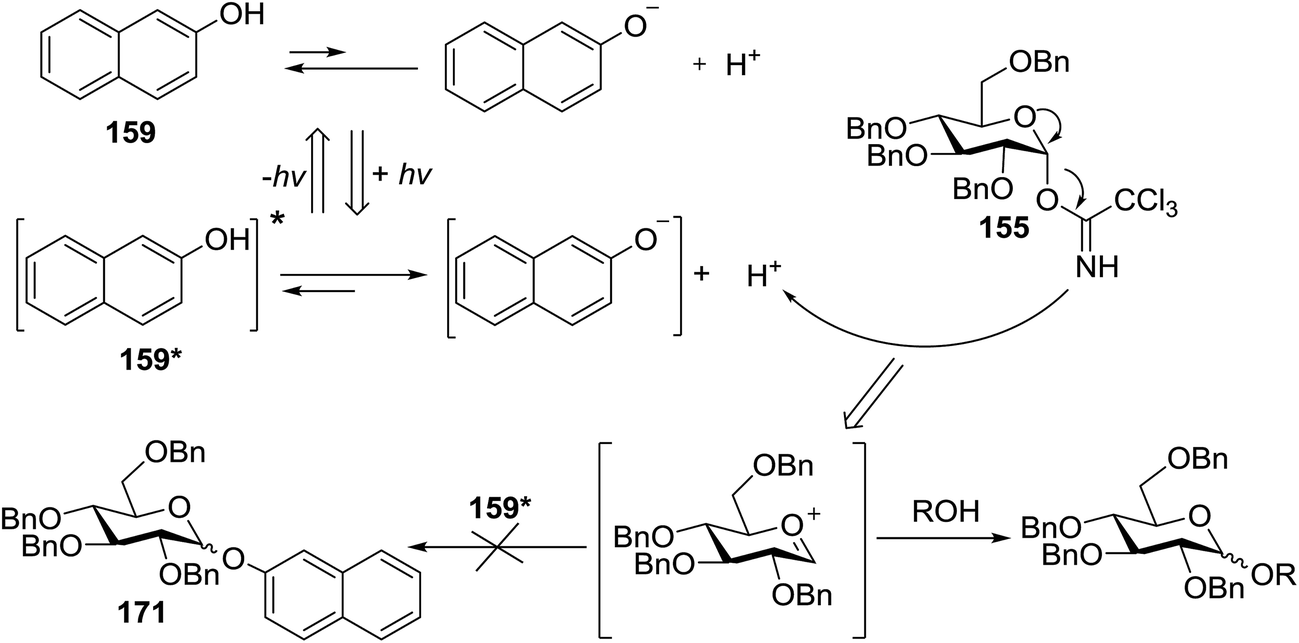 | ||
| Fig. 21 Glycosylation reaction profile using organophotoacid 159 under photoirradiation. | ||
Organophotoacids exhibit a unique property where in the photoexcited state their acidity increases. Therefore, organophotoacids can be used to control glycosylation by light switching and neutralization is not required for switching the reaction. However, α/β-stereoselectivity cannot be controlled in photoinduced glycosylation using naphthol derivatives as organophotoacids. In 2016, Kimura and coworkers84 examined aryl thioureas as organophotoacids to overcome these limitations. Photoinduced glycosylation of alcohols with α-glucosyl trichloroacetimidates was explored under long wavelength UV, using aryl urea and thiourea as organophotoacids. Ultraviolet irradiation glycosylations progressed energetically to provide the respective glycosides in considerable yields. In addition, high β-stereoselectivity was attained using higher concentrations of thiourea as the organophotoacid, whereas high α-stereoselectivity was achieved under low concentration (Scheme 12). Aryl thioureas have been used as hydrogen bond-donating organocatalysts for many asymmetric syntheses and in glycosylation reactions as triplet sensitizers. The nitrogen anion undergoes electron delocalization (resonance effect) over the sulfur atom and the aromatic rings for its stabilisation, and the acidity of aryl thioureas increases somewhat upon photoirradiation. Glucosyl trichloroacetimidate was predicted to be activated by the aryl thioureas in their excited state, which requires a pKa value less than 5 for activation at room temperature (Fig. 22).85
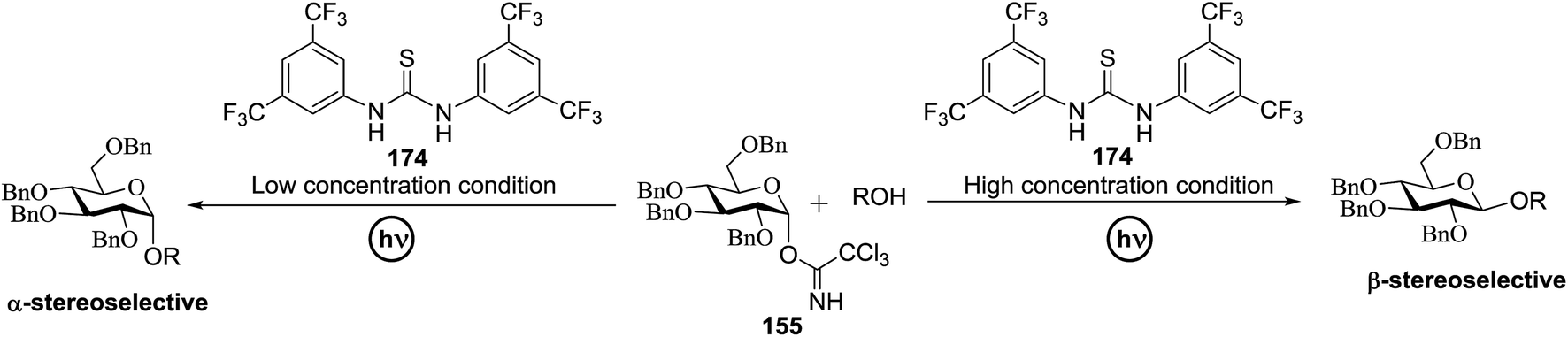 | ||
| Scheme 12 Stereocontrolled photoinduced glycosylation of alcohols with α-glucosyl trichloroacetimidates using aryl thiourea as an organophotoacid. | ||
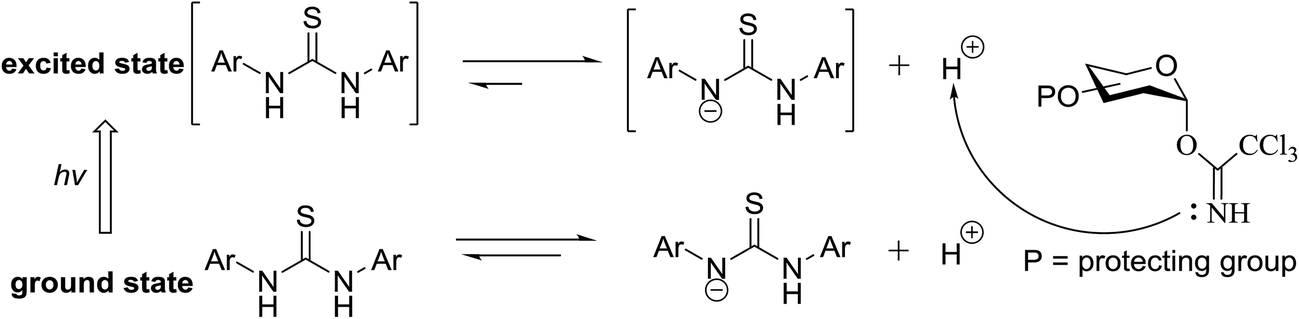 | ||
| Fig. 22 Photoinduced glycosylation using an aryl thiourea. | ||
The glucosyl trichloroacetimidate 155, donor aryl urea 173, and thioureas 172 and 174 as organophotoacids were investigated by Toshima and co-workers. The glycosylation reaction of 155 with alcohol 156 (2.0 equiv.) using aryl urea 173 and thioureas 172 and 174 (0.3 equiv.) in the presence of powdered 5 Å molecular sieves (MS) in Et2O was examined.
The results, which are outlined in Table 24, indicated that the use of aryl urea or thioureas 172–174 resulted in the analogous glycoside 175 in low to high yield (Table 24, entries 1–3). It was confirmed that in the absence of organophotoacid and photoirradiation the reaction proceeded and 155 was recovered (Table 24, entry 4 and 5). These results clearly concluded that aryl thiourea 174 as an organophotoacid improves glycosylation. Further the effect of the substituent was examined. It was found that the two trifluoromethyl electron-withdrawing groups on the benzene ring are responsible for increasing the acidity of the aryl thiourea (Table 24, entry 1 vs. 3). Significantly, β-stereoselectivity was achieved even when Et2O was used as the solvent, which generally induces α-stereoselectivity.86 Further results revealed that β-stereoselectivity was enhanced with an increase in the concentration of 155 in Et2O (Table 24, entries 6 and 7).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Organophotoacids | Concn of 1 (M) | hν | 175 (α/β) | Yield | |
| 155 | 176 | |||||
| 1 |  |
0.5 | + | 19 (33:67) |
54 | 0 |
| 2 | 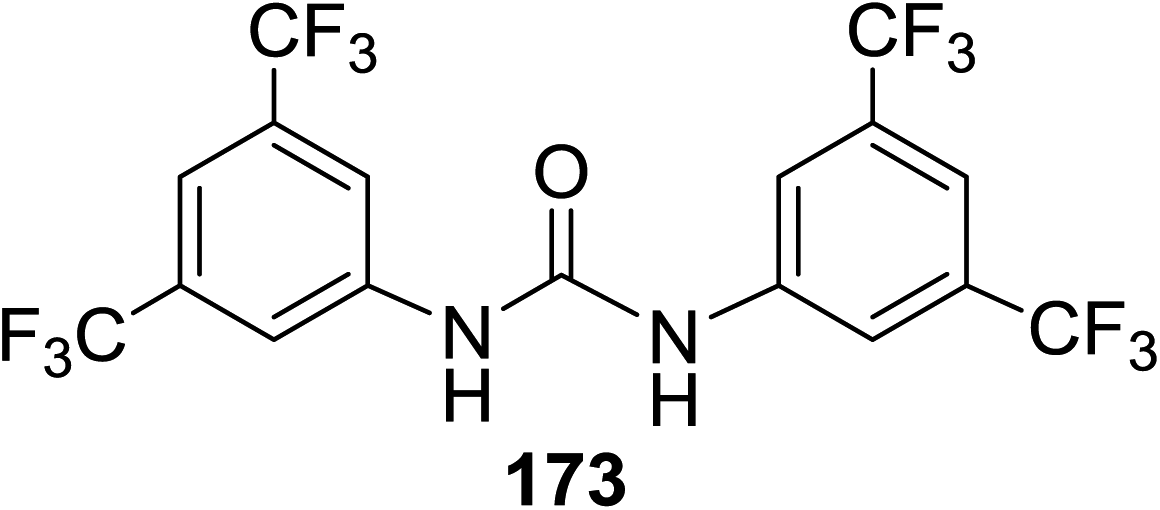 |
0.5 | + | 62 (33:68) |
30 | 0 |
| 3 | 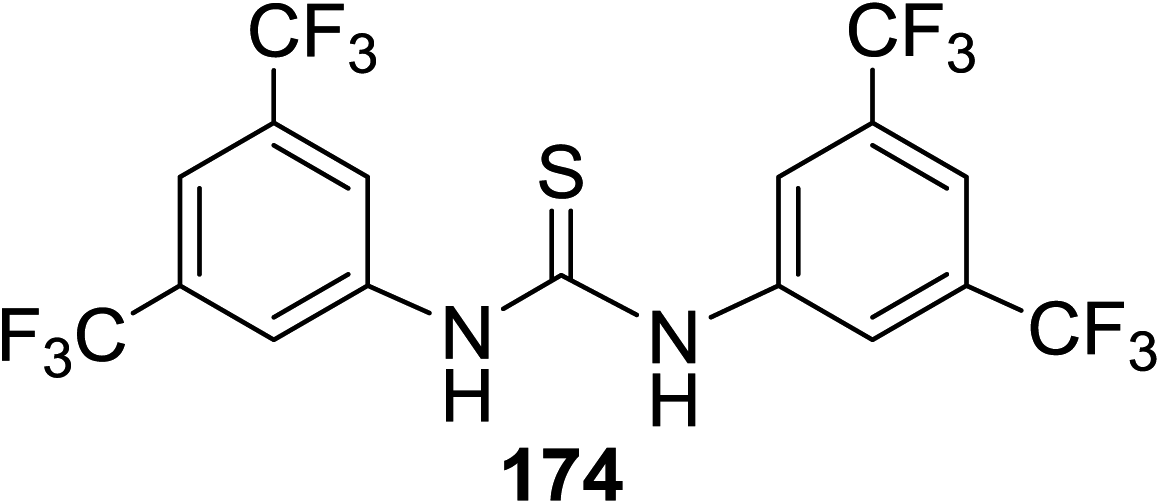 |
0.5 | + | 91 (35:65) |
0 | 0 |
| 4 | 174 | 0.5 | − | 0 | 84 | 9 |
| 5 | 174 | 0.5 | + | 0 | 92 | 0 |
| 6 | 174 | 0.8 | + | 87 (30:70) |
0 | 0 |
| 7 | 174 | 1.0 | + | 85 (24:76) |
0 | 5 |
The optimal conditions showed that glycosylation involving 155 and 156 using a higher equivalent of thiourea 174 progressed with greater β-stereoselectivity. 0.1–0.5 equivalent of 174 was used under photoirradiation and it was found that 0.3 equivalent of 174 was sufficient, which resulted in high reproducibility (Table 25). The solvent effect was also optimised using THF and MeCN in addition to Et2O, which showed that MeCN provided the best yield with β-stereoselectivity (Table 25, entries 6 and 7).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Solv (M) | MS 5 Å | 174 (equiv.) | Yield (%) (α/β) | Entry | Solv (M) | MS 5 Å | 174 (equiv.) | Yield (%) (α/β) |
| 1 | Et2O (1.0) | + | 0.5 | 31 (22:78) |
10 | Et2O (0.1) | + | 0.3 | 84 (64:36) |
| 2 | Et2O (1.0) | + | 0.1 | 82 (26:74) |
11 | Et2O (0.01) | + | 0.3 | 83 (81:19) |
| 3 | Et2O (1.0) | + | 0.3 | 85 (24:76) |
12 | Et2O (0.01) | + | 0.3 | 78 (80:20) |
| 4 | Et2O (1.0) | + | 0.5 | 87 (25:75) |
13 | Et2O (0.01) | + | 0.1 | 79 (82:18) |
| 5 | Et2O (2.0) | − | 0.3 | 85 (16:64) |
14 | THF (2.0) | − | 0.5 | 82 (19:81) |
| 6 | Et2O (0.005) | + | 0.3 | 73 (83:17) |
15 | THF (0.01) | − | 1.0 | 93 (08:92) |
| 7 | Et2O (0.01) | + | 0.3 | 81 (83:17) |
16 | MeCN (2.0) | + | 0.3 | 86 (81:19) |
| 8 | Et2O (0.025) | + | 0.3 | 88 (77:23) |
17 | MeCN (0.01) | + | 0.3 | 51 (41:59) |
| 9 | Et2O (0.05) | + | 0.3 | 89 (67:33) |
|||||
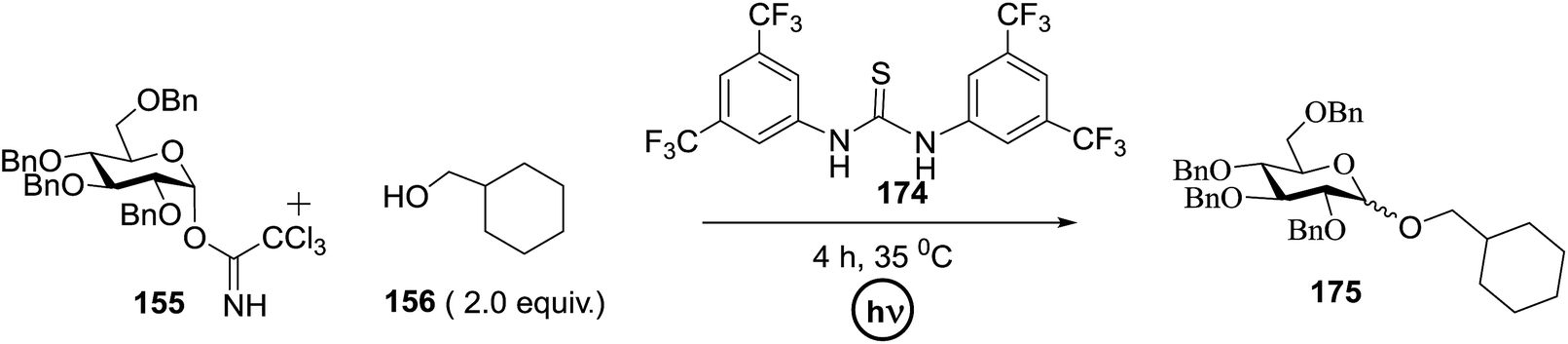
However, when glycosylation was performed at a lower equivalent (0.005 to 0.1 M, Table 25, entries 6–10) the results revealed that the highest α-stereoselectivity occurred in 0.01 M Et2O. More experiments were conducted to standardize the reaction conditions using 0.1–1.0 equivalent of 174 (Table 25, entries 11–13) and THF and MeCN as solvents (Table 25, entries 14–15) at the low concentration of 0.01 M.
The reason for the α/β stereoselectivity could be explained by the mechanism given in Scheme 13. The higher β-stereoselectivity is due to the SN2 reaction mechanism of glycosylation due to the use of high concentrations of aryl thiourea (174) (Scheme 13, path a). On the other hand, the high α-stereoselectivity is found to be due to the involvement of the SN1 reaction mechanism of glycosylation via the oxionium cation intermediate under a low concentration of aryl thiourea (174) (Scheme 13, path b).
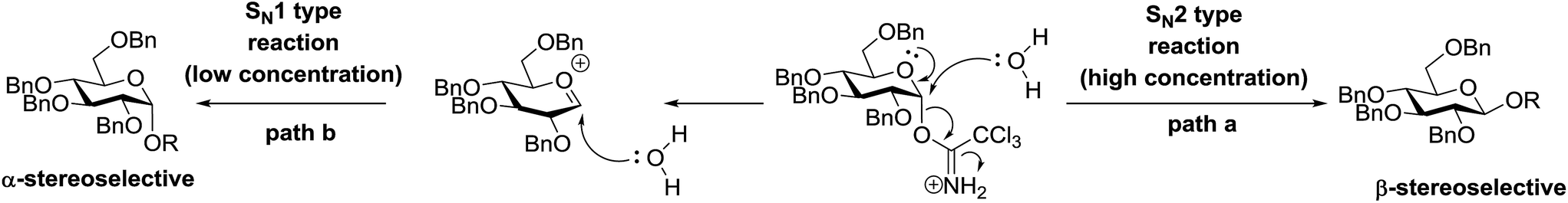 | ||
| Scheme 13 Proposed mechanism of stereocontrolled photoinduced glycosylation using aryl thiourea 174. | ||
Next, the scope and limitations of the glycosylation method are summarized in Table 26 using the different alcohols. The photoinduced glycosylation of alcohols using thiourea 174 in Et2O in the presence of 5 Å MS at low concentrations progressed smoothly to form the respective glycosides 178a–h in good to high yield with α-stereoselectivity (Table 26, entries 1–8).
|
||||
|---|---|---|---|---|
| Entry | ROH | Product | Yield (%) (α/β) | |
| Et2O | MeCN | |||
| 1 |  |
 |
84 (81:19) |
90 (9:91) |
| 2 |  |
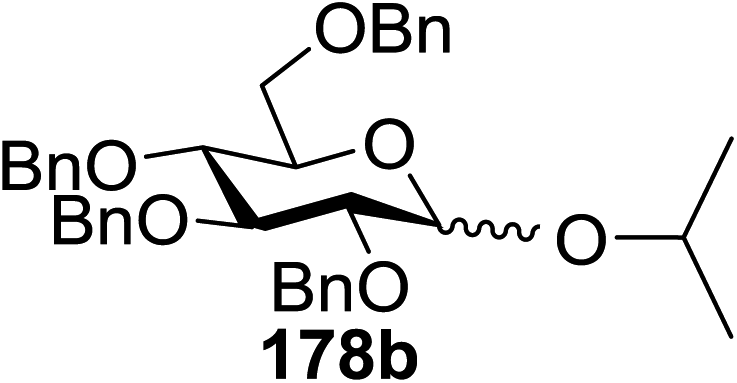 |
90 (85:15) |
87 (9:91) |
| 3 |  |
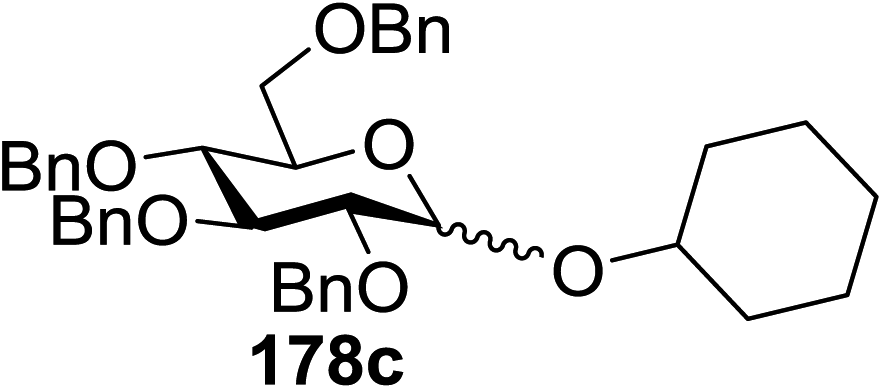 |
85 (84:16) |
87 (12:88) |
| 4 |  |
 |
83 (83:17) |
88 (16:84) |
| 5 |  |
 |
78 (81:19) |
89 (11:89) |
| 6 |  |
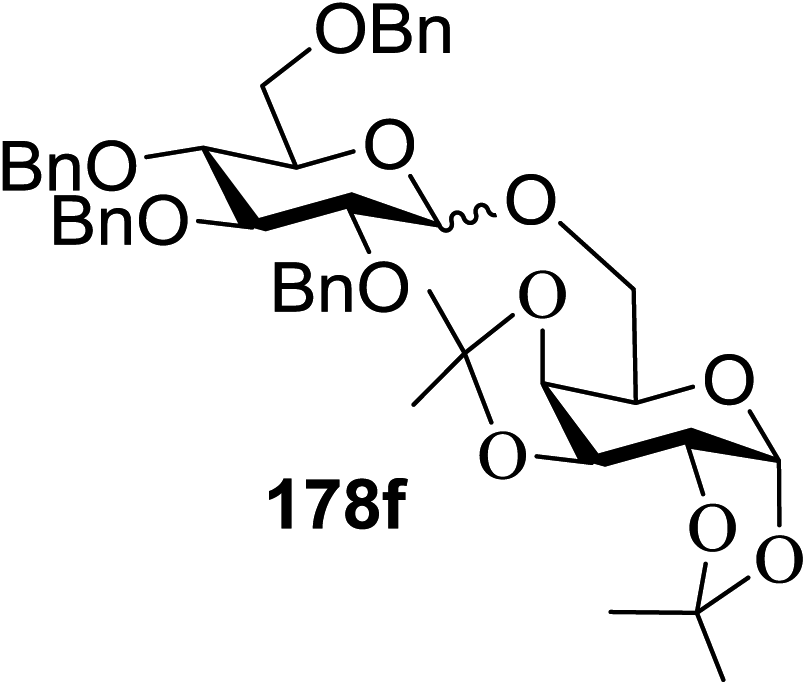 |
75 (81:19) |
85 (12:88) |
| 7 | 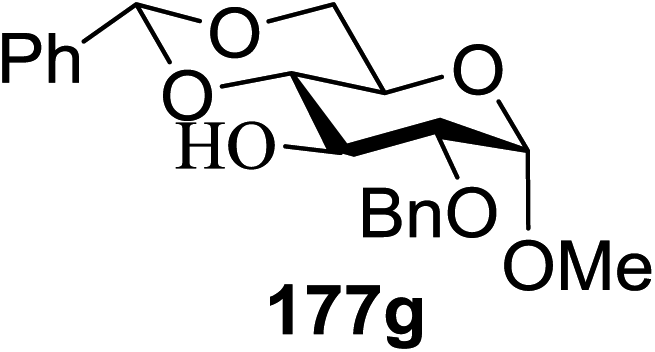 |
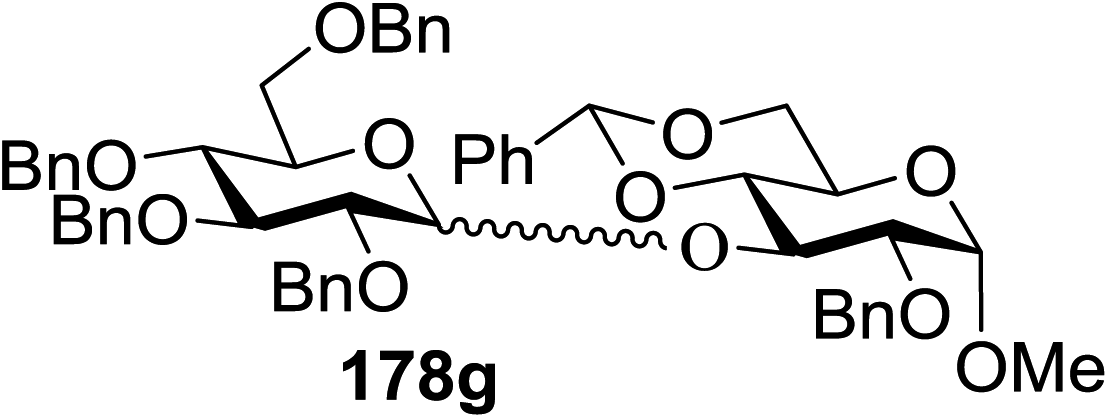 |
68 (79:21) |
83 (28:72) |
| 8 | 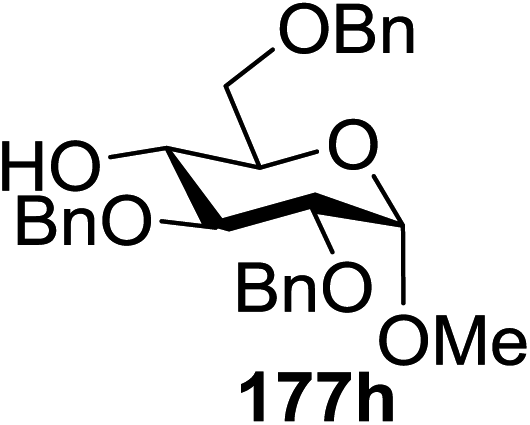 |
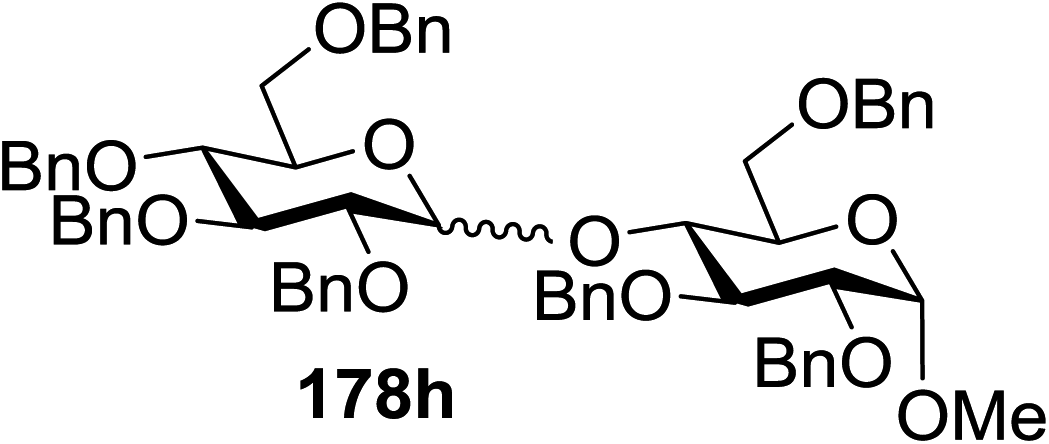 |
56 (75:25) |
84 (24:76) |
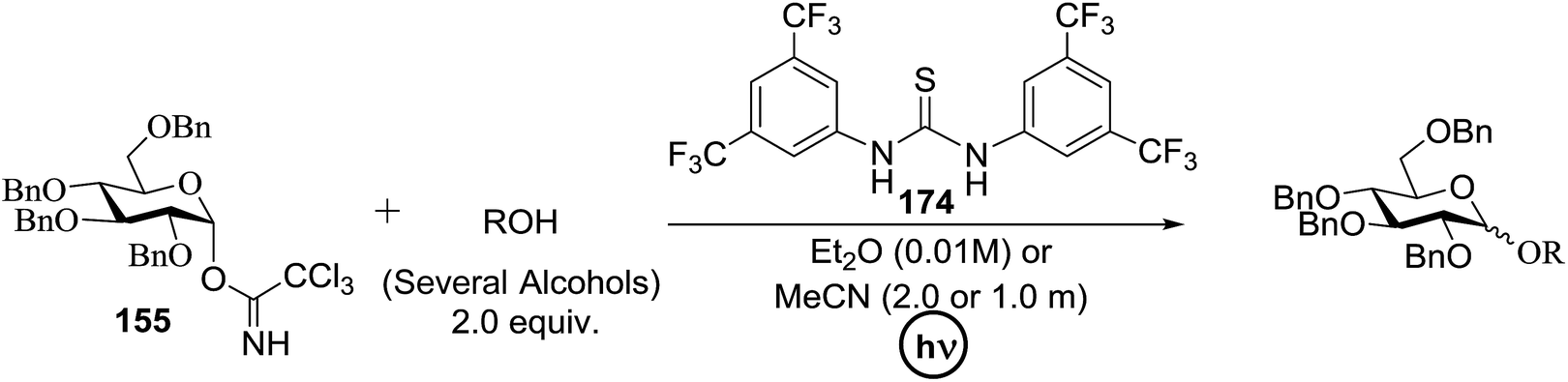
4.2. S-Linked glycoconjugate synthesis via photoinduced thiol–ene coupling (TEC)
The great potential and selectivity of the thiol–ene radical reaction have been verified by the photoinduced coupling of anomeric sugar thiols with sugar alkenes to give 1,6-linked S-disaccharides in good to excellent yields (76–92%) and excellent diastereoselectivities. Another approach includes the glycosylation of thio sugar acceptors with activated glycosyl donors and Michael additions of sugar thiolates to α,β-unsaturated systems. The centenary old thiol–ene coupling (TEC)87 was extended via a radical mechanism88 to accord an anti-Markovnikov sulfide adduct in very great yield. Fiore M. and coworkers89 conceptualized the photoinduced thiol–ene coupling as a click ligation tool for thiodisaccharide synthesis. The exemplary reaction was conducted at room temperature, without deoxygenation and under irradiation from a UV-visible lamp (λmax 420 nm, 40 W) in the presence of 2,2-dimethoxy-2-phenylacetophenone (DPAP) as the photoinitiator (Table 27). Assemblies of thiodisaccharides by this ligation tool justify its synthetic potential as well as contribute new and universal elements to these important carbohydrates mimics.
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | 179:180 |
PI (%) | Solvent | Time (min) | λmax (nm) | 181 (%) | Entry | 179:180 |
PI (%) | Solvent | Time (min) | λmax (nm) | 181 (%) |
| 1 | 1.1:1 |
15 | MeOH | 960 | 420 | 14 | 9 | 1.3:1 |
50 | MeOH | 30 | 365 | 78 |
| 2 | 1.5:1 |
10 | MeOH | 480 | 420 | 20 | 10 | 1.3:1 |
50 | MeOH | 15 | 365 | 81 |
| 3 | 3.0:1 |
15 | MeOH | 960 | 420 | 42 | 11 | 1.3:1 |
50 | MeOH | 60 | 365 | 83 |
| 4 | 1.0:1 |
50 | MeOH | 60 | 365 | 46 | 12 | 1.2:1 |
10 | MeOH | 15 | 365 | 75 |
| 5 | 1.2:1 |
50 | MeOH | 60 | 365 | 79 | 13 | 1.2:1 |
10 | CH2Cl2 | 15 | 365 | 80 |
| 6 | 1.3:1 |
50 | MeOH | 60 | 365 | 91 | 14 | 1.2:1 |
10 | PhCH3 | 15 | 365 | 81 |
| 7 | 3.0:1 |
50 | MeOH | 60 | 365 | 79 | 15 | 1.2:1 |
10 | — | 15 | 365 | 89 |
| 8 | 1.3:1 |
25 | MeOH | 60 | 365 | 61 | 16 | 1.3:1 |
50 | MeOH | 540 | Sunlight | 77 |
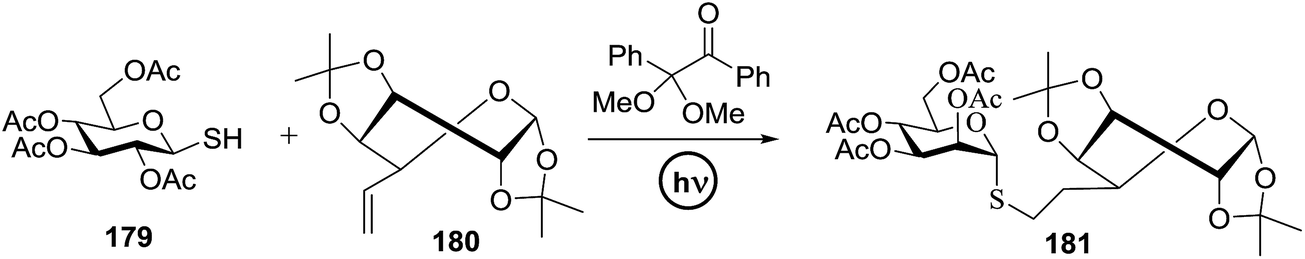
Due to the uncommon characteristics of TEC, the linking process of TEC in polymer and bioorganic chemistry has been reported in a recent paper, which introduces high efficiency, total atom economy, orthogonality to a wide range of reagents, and affinity with water and oxygen. In recent times, TEC has been discussed as an exemplary case of the click process developed by Sharpless and co-workers.90 There has been considerable use of this reaction in polymer chemistry; however, only a few recent applications in bioorganic chemistry have been reported.
The Table 27 displays the optimization conditions of the reaction. Entries 1–7 were carried out in MeOH with different thiol/ene ratios which resulted in 181 in low to good isolated yields. Similar results were attained by reducing the amount of photoinitiator DPAP from 50 to 10 mol% and the irradiation time from 60 to 15 min. Finally, it was established that the reaction could be achieved in aprotic solvents such as CH2Cl2 and toluene (entries 13 and 14) with the same efficiency as in MeOH. In addition, reactions were performed under solvent-free conditions (entry 15) or sun light irradiation (entry 16).
After determining the suitable reaction conditions, the substrate scope of TEC for S-disaccharide synthesis was explored. Sugar thiols and alkenes all having acetyl or isopropylidene protective groups were studied and the results of their photoinitiated reactions are summarized in Table 28. Coupling of the peracetylated glucosyl thiol 179 with hex-5-enopyranosides 185c and pent-4-enofuranoside 187 proceeded rapidly to give the corresponding S-disaccharides 188–190 in high isolated yields and diastereoselectivities (entries 1–3). The efficiency of the coupling reaction was affected by the anomeric configuration of the sugar thiol, which was tested by screening the reactions of the peracetylated α- and β-mannosyl derivatives 182 and 183 with alkenes 180 and 186 (entries 4–7). Adequate and stereoselective reactions were realised by coupling 2-acetamido-2-deoxyglucosyl thiol 184 with alkenes 180, 185 and 186 (entries 8–10), which contribute to the evidence of TEC compatibility with carbohydrate fragments containing the N-acetylamino group.
|
||||
|---|---|---|---|---|
| Entry | Thiol | Alkene | Product | Yield and d.r. |
| 1 | 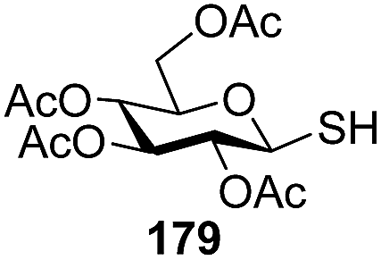 |
 |
 |
82%, 75:25 |
| 2 | 179 |  |
 |
89%, >99:1 |
| 3 | 179 |  |
 |
92%, >99:1 |
| 4 |  |
 |
 |
76% |
| 5 | 182 | 186 |  |
89%, >99:1 |
| 6 |  |
180 |  |
80% |
| 7 | 183 | 186 | 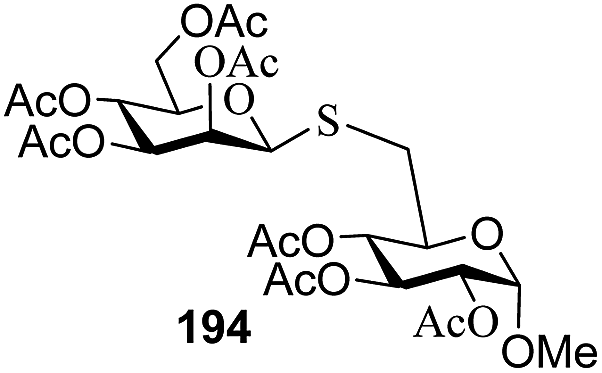 |
85%, 97:3 |
| 8 | 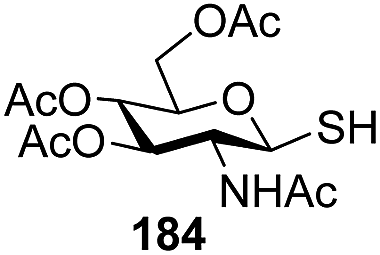 |
180 | 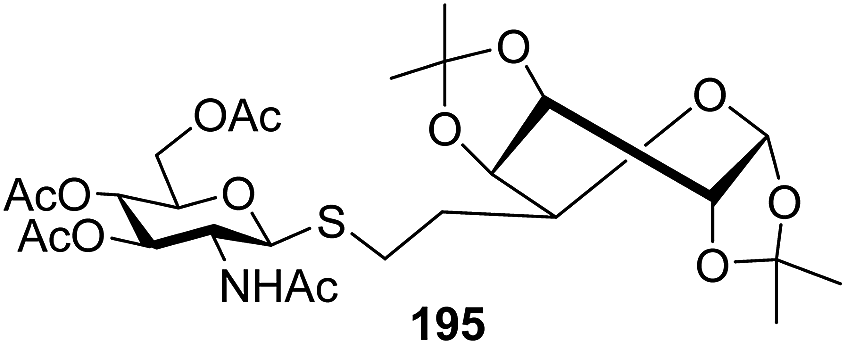 |
77% |
| 9 | 184 | 185 | 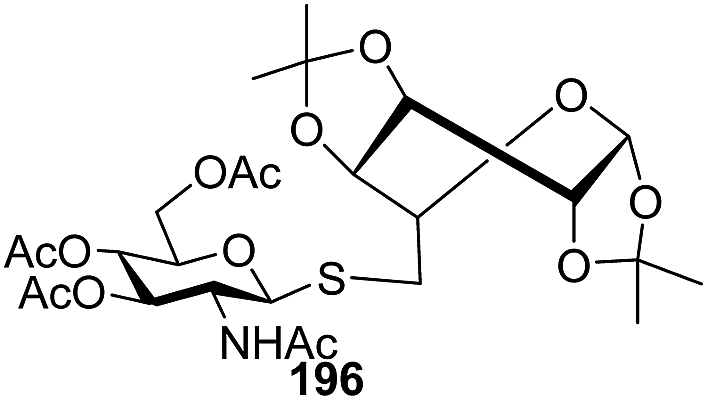 |
83%, 66:34 |
| 10 | 184 | 186 |  |
84%, 94:6 |
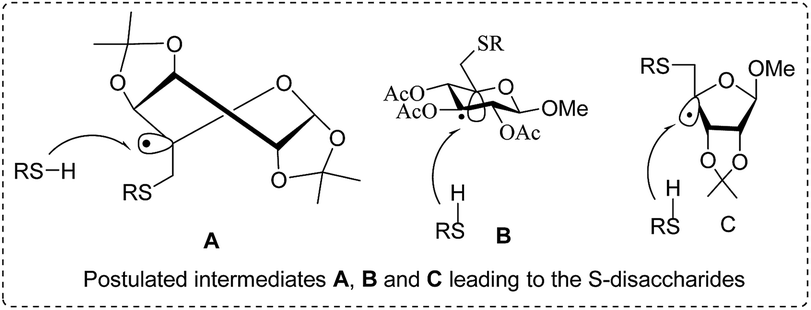 |
||||
The reason for the formation of the stereoisomer is given in Table 28, which is explained on the basis of the chain-reaction-type mechanism involving the formation of thio alkyl radical intermediates A, B, and C. These all have trans arrangements of their substituents on the C4–C5 bond. These intermediates present the less hindered face to a molecule of thiol from which they abstract an H˙ radical in the final and irreversible locking step.
For the development of the thiol–ene strategy, Dondoni and co-workers91 in 2009 reported a new ligation phenomenon for peptide and protein glycosylation with the application of photoinduced thiol–ene coupling. The method was based on the photoinduced coupling of an alkenyl C-glycoside with a peptide or protein containing a free sulfhydryl group, which occurred via a radical mechanism in an anti-Markovnikov regioselective manner (Fig. 23). The vital role of this ligation tool in carbohydrate and protein chemistry has been developed, which has significant applications such as the synthesis of heteroglycoclusters, vaccines, polymers and S-disaccharides.
 | ||
| Fig. 23 General mechanism of thiol–ene coupling (TEC). | ||
The coupling of peracetylated allyl α-C-galactoside (198) and N-Fmoc cysteine (199) resulted in the C-linked galactosyl amino acid 200 (Table 29). Waldmann and co-workers92 described the induced coupling of cysteine 199 with different alkenes successfully. However, Dondoni and co-workers carried out the reaction of 198 and 199 in refluxing dichloroethane (DCE) and in the presence of AIBN as the radical initiator. To optimize the reaction conditions, a number of experiments in different reaction conditions were performed. Under photochemical conditions, the reaction of 198 with excess 199 (3 equiv.) occurred readily at room temperature by irradiation at λmax 365 nm and using 10 mol% of 2,2-dimethoxy-2-phenyl-acetophenone (DPAP) as the sensitizer. Almost total conversion of 198 was observed and the galactosyl cysteine 200 was isolated in a very good yield (Table 29, entry 4).
|
|||||
|---|---|---|---|---|---|
| Entry | Conditions | 198/199 | Solvent (c [M]) | Time [h] | Yield (%) |
| 1 | Δ | 3:1 |
DCE (0.25) | 14 | 12 |
| 2 | Δ | 1:1 |
DCE (0.25) | 14 | — |
| 3 | Δ | 1:3 |
DCE (0.25) | 14 | 15 |
| 4 | hν | 1:3 |
DMF (0.5) | 1 | 78 |
| 5 | hν | 1:1 |
DMF (0.5) | 1 | 40 |
| 6 | hν | 3:1 |
DMF (0.5) | 1 | 21 |
| 7 | hν | 1:3 |
DMF (0.05) | 1 | 52 |
| 8 | hν | 1:3 |
DCM (0.05) | 1 | 48 |
| 9 | hν | 1:3 |
DMF (0.5) | 1 | 56 |
From the optimization reaction conditions, further exploration of this scheme involved the easy isolation of the galactosyl amino ester 205 in excellent yield (92%, Table 30). The glycosylation of cysteine containing tripeptide glutathione 204 (γ-L-Glu-L-Cys-Gly, GSH) with the allyl α-C-galactoside 198 was carried out in DMF/H2O which provided the coupling product 208 in 81% isolated yield (Table 30, entry 4).
|
|||
|---|---|---|---|
| Entry | Alkene | Thiol | Sulfide |
| 1 | 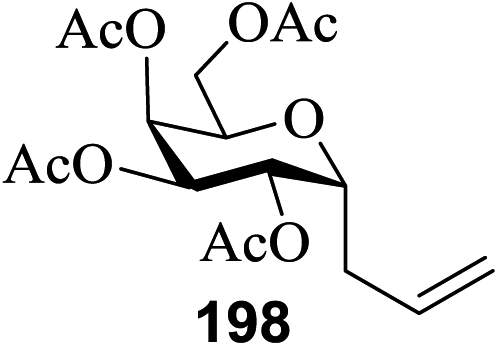 |
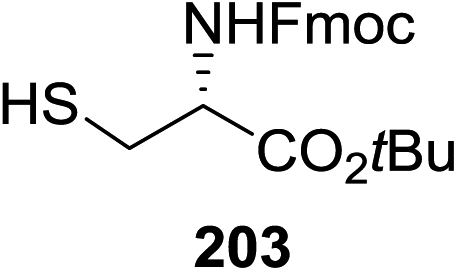 |
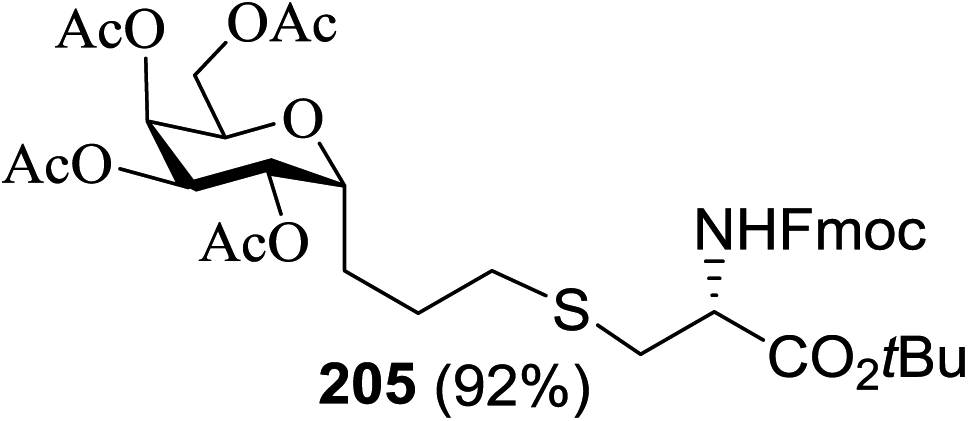 |
| 2 | 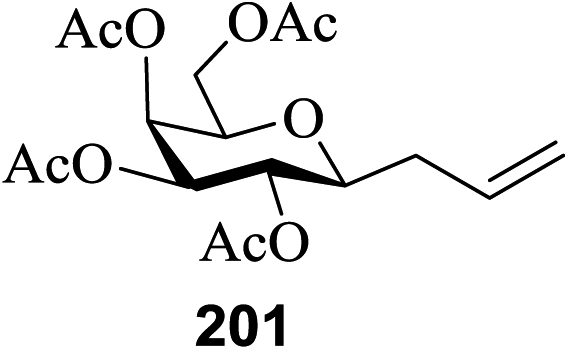 |
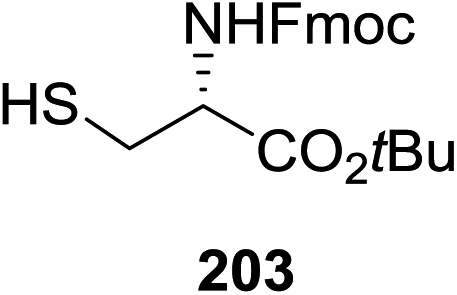 |
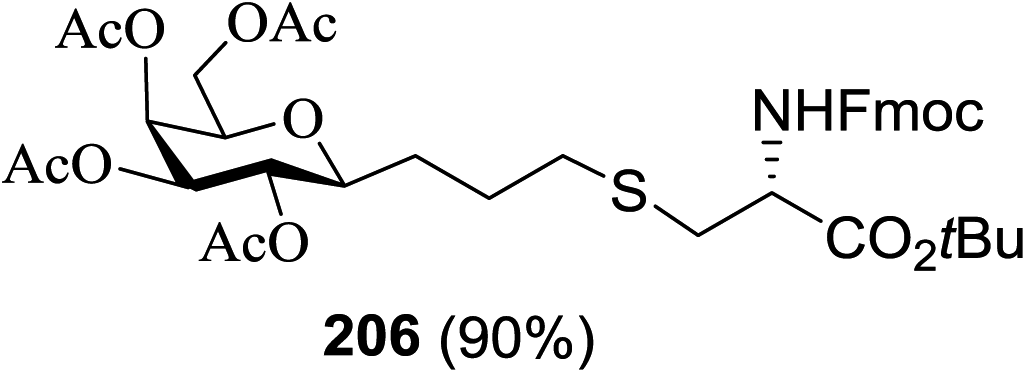 |
| 3 | 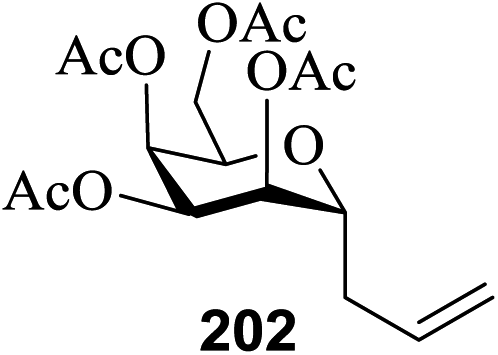 |
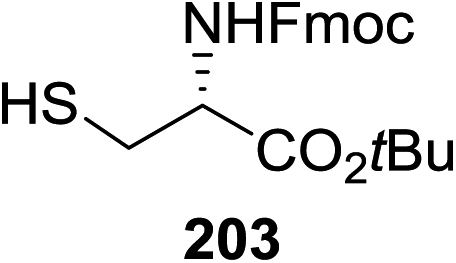 |
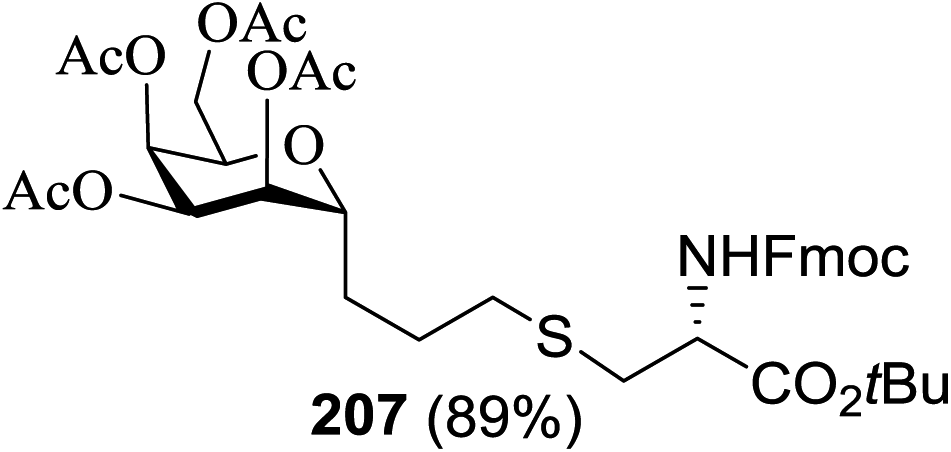 |
| 4 | 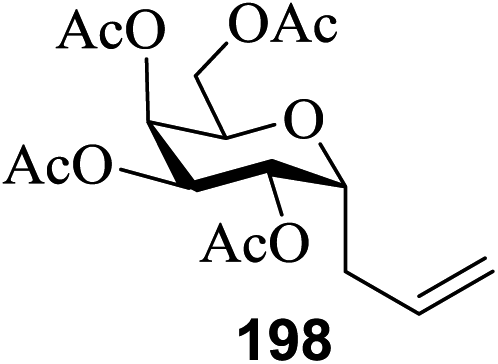 |
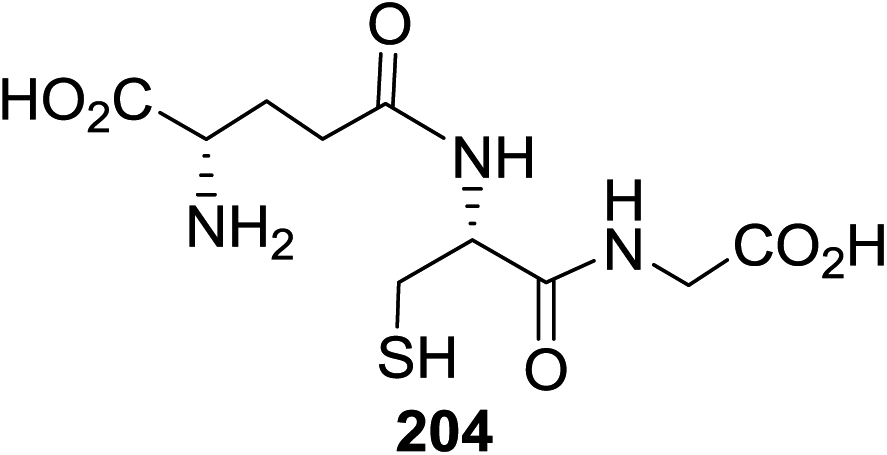 |
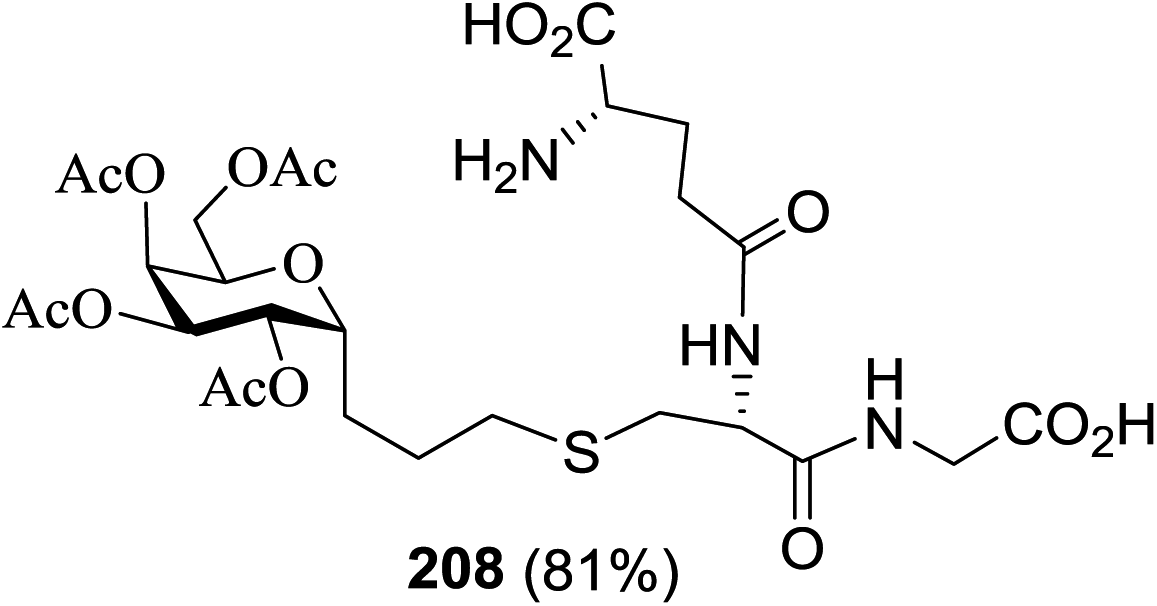 |
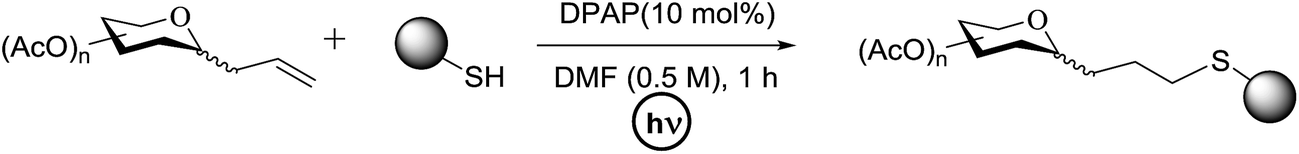
The usefulness of this photoinduced reaction was demonstrated via synthesis of nonapeptide TALNCNDSL (209) using the allyl α-C-galactoside 198 with a single cysteine residue (Scheme 14). Hence, the reaction condition standardized the use of aqueous solutions at physiological pH diluted with salts for protein stabilization. The coupling reaction conditions of 198 with 209 were optimized by adding a DPAP (80 mol%) containing solution of 198 (30 equiv.) in DMSO (0.12 M, 4% final volume content) to a 0.16 mm solution of 209 in 20 mM phosphate buffer (pH 7.4) and applying irradiation at λmax 365 nm for 1 h under argon. It was reported that the reactions performed at higher nonapeptide concentrations resulted in a partial loss of selectivity toward the cysteine residue. This novel strategy of protein modification may offer new opportunities in biotechnology and proteomics.
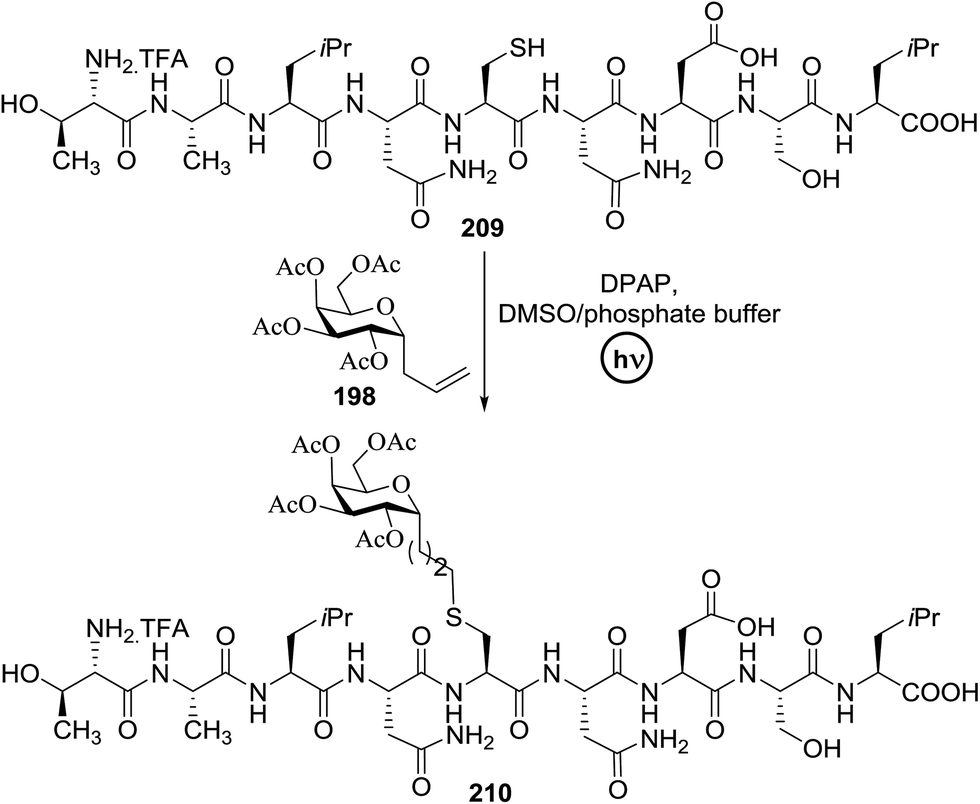 | ||
| Scheme 14 Preparation of glycopeptide 210. | ||
Davis and co-workers93 described the development of a convergent approach for the synthesis of a novel class of S-linked glycoconjugate proteins using the site-specific ligation strategy of 1-glycosyl thiols to proteins (Fig. 24). One of the most elegant examples of thiol–ene ligation for protein modification was reported in 2009, which involves the “tag-modify” approach for protein glycosylation.94 This process was investigated for a variety of protected and unprotected thiosugars including a disaccharide and on a wide range of different proteins to explore the compatibility of variables such as steric bulk, configuration and stereochemistry.
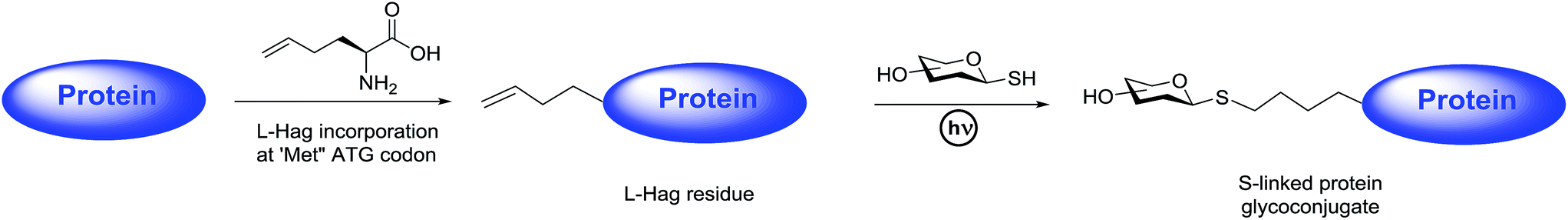 | ||
| Fig. 24 Synthesis of S-linked glycoproteins by thiol–ene ligation. | ||
The free-radical reaction of glycosyl thiyls was investigated and optimized initially for representative amino acid model systems containing Hag (Table 31). A wide range of 1-glycosyl thiols, in both the protected and unprotected forms, as starting materials were used, in which good regioselectivity (>98% e-thiolation) and retention of the anomeric configuration in the product were observed. The use of extended reaction times or excess thiol (3–10 equiv.) allowed complete conversion.
| Entry | Glycosyl thiol | Olefin | Solvent | % yield | Entry | Glycosyl thiol | Olefin | Solvent | % yield |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 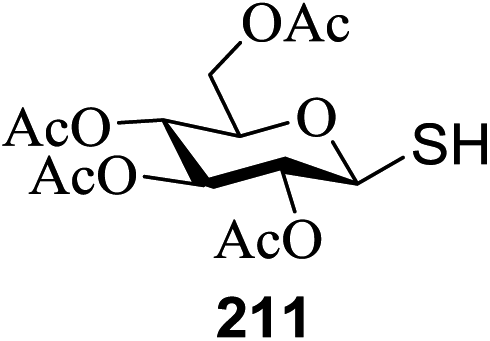 |
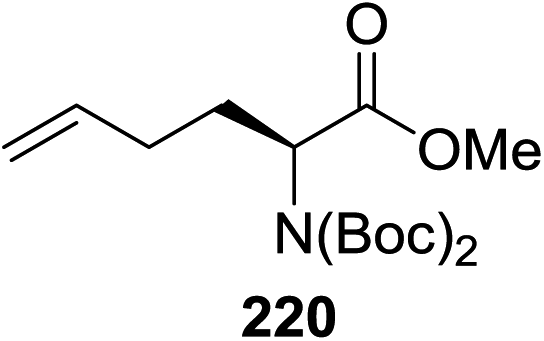 |
H2O/MeOH 1:1 |
79 | 6 | 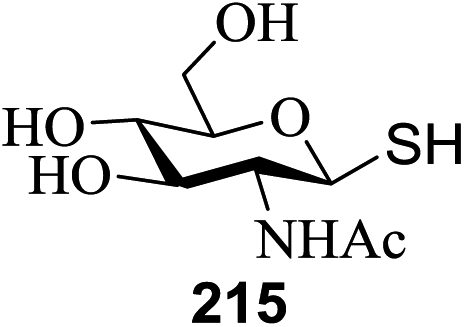 |
221 | pH 4 acetate | 63 |
| 2 | 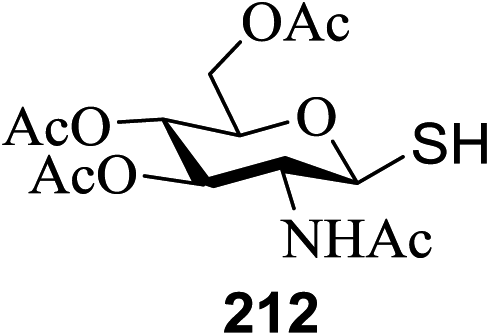 |
220 | H2O/MeOH 1:1 |
71 | 7 | 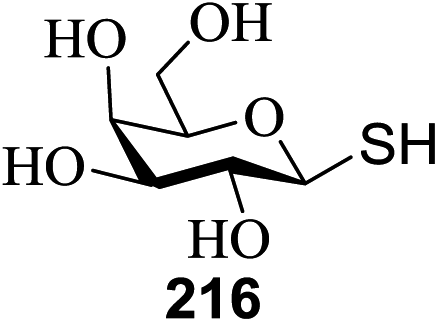 |
221 | pH 4 acetate | 70 |
| 3 | 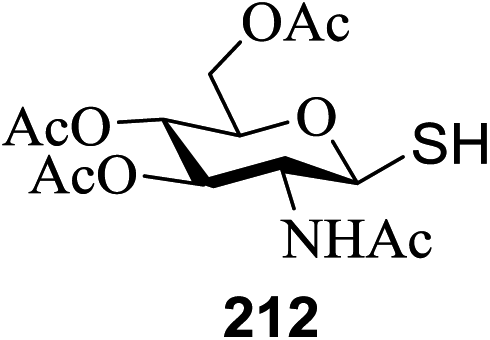 |
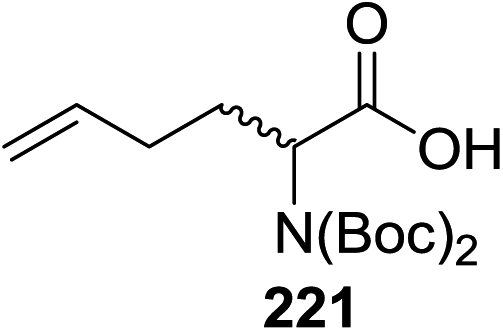 |
H2O | 81 | 8 | 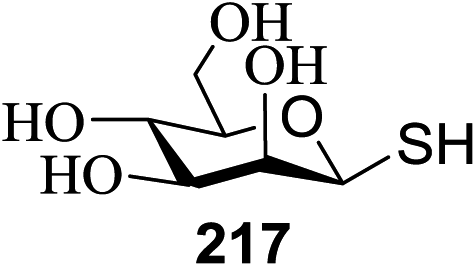 |
221 | pH 4 acetate | 28 |
| 4 | 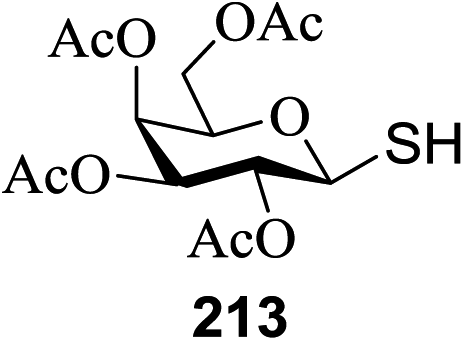 |
221 | H2O/MeOH 1:1 |
57 | 9 | 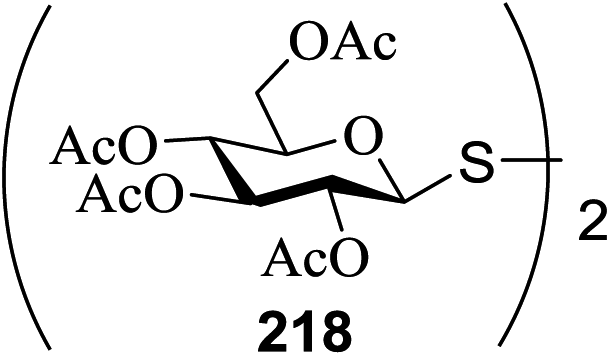 |
221 | pH 4 acetate | 0 |
| 5 | 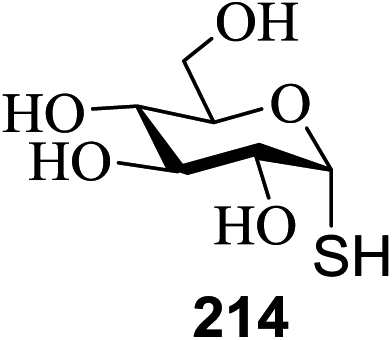 |
221 | pH 4 acetate | 42 | 10 | 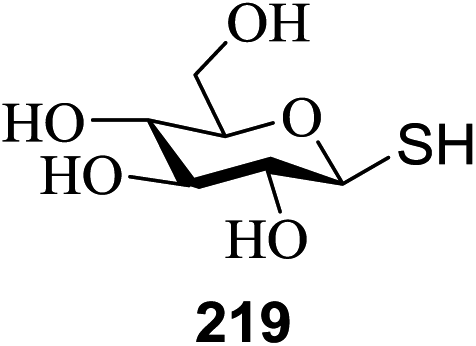 |
221 | pH 4 acetate | 91 |
For the optimization of the protein scaffold, SsβG-Hag43 was used as a substrate in reactions with β-GlcSH, which exposed the rational conditions compared to that discussed for amino acid systems. The fastest, complete transformations (>95%) were seen at pH < 7 with the use of photoinitiation. A relatively high concentration of thiol (up to 100 mm) was important for rapid reaction and hydride abstraction. Reaction conditions were developed for the site selective glycoconjugation to the single olefinic side-chain in SsβG-Hag43 which was expanded to other glycans and proteins. It was shown that glycoconjugations at pH 6 were equally successful, yielding >95% product, although the reaction was slightly slower. No degradation was seen with any of the proteins at pH 4. The yields of the reactions with different thiols are presented in Table 28; for example, β-GlcNAcSH (Table 32, entry 2) was slightly slower than β-GlcSH (entry 1). The use of SsβG-Met43, as a negative control substrate that does not contain Hag, showed no coupling (entry 3), which confirms the chemoselectivity of the reaction for the olefinic “tag” and the lack of reaction with other amino acids in SsβG. The glycoconjugation was further extended to the self-assembled multimeric, virus-like particle Qβ-WT (Table 32, entry 6).95
|
||||
|---|---|---|---|---|
| Entry | Glycosyl thiol | Olefinic protein | Product | Yield% |
| 1 | 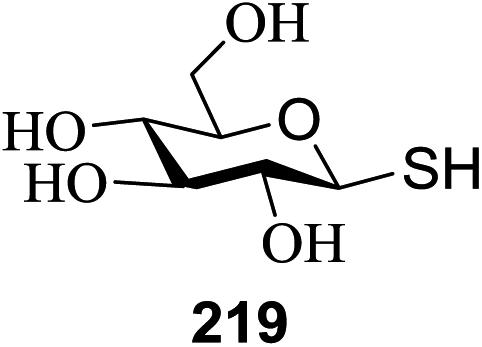 |
 |
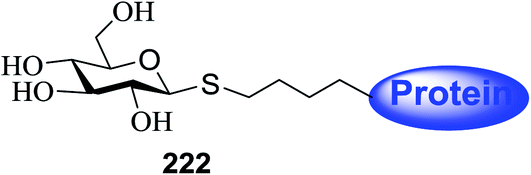 |
>95 |
| 2 | 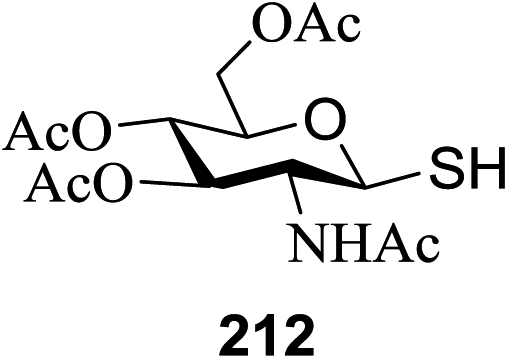 |
 |
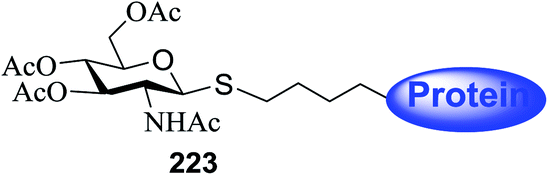 |
>95 |
| 3 | 219 |  |
 |
0 |
| 4 | 219 |  |
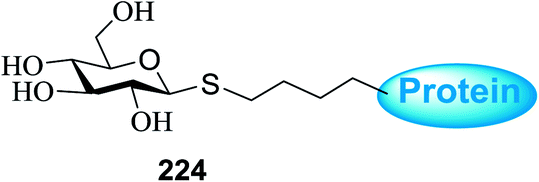 |
>95 |
| 5 | 219 | 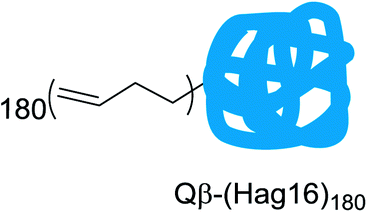 |
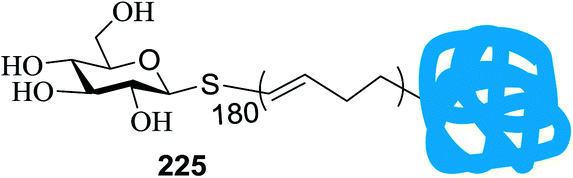 |
>95 |
| 6 | 219 |  |
— | 0 |
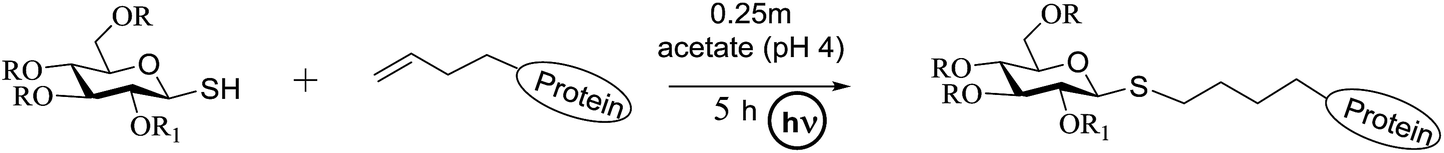
Among the various methods for S-glycosyl amino acid and peptide synthesis, the coupling of an activated sugar derivative with a protected cysteine residue is the most widely explored. Dondoni and co-workers96 reported the same type of approach to S-glycosyl amino acids based on the photoinduced hydrothiolation of alkenyl glycines by glycosyl thiols (Scheme 15). This photoinduced thiol–ene coupling (TEC) reaction proceeds via a radical mechanism and allows the addition of thiols to non-activated alkenes according to the anti-Markovnikov rule. The main characteristic of photoinduced TEC is that it involves irradiation close to visible light and orthogonality to a broad range of functional groups.
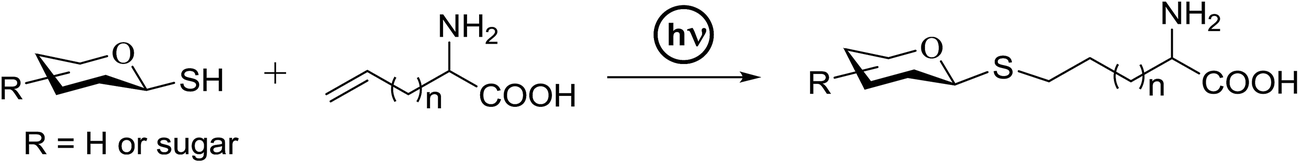 | ||
| Scheme 15 General strategy for S-glycosyl amino acid synthesis via photoinduced TEC. | ||
Due to its efficiency and specificity, TEC has been further explored for the glycosylation of natural cysteine containing peptides such as glutathione (GSH) and the protein bovine serum albumin. To perform this glycosyl, thiols 226–228 and allyl or vinyl glycines 229 or 230, respectively, were used as substrates (Table 33).88,95
| Entry | Thiol | Alkene | Product (yield%) |
|---|---|---|---|
| 1 | 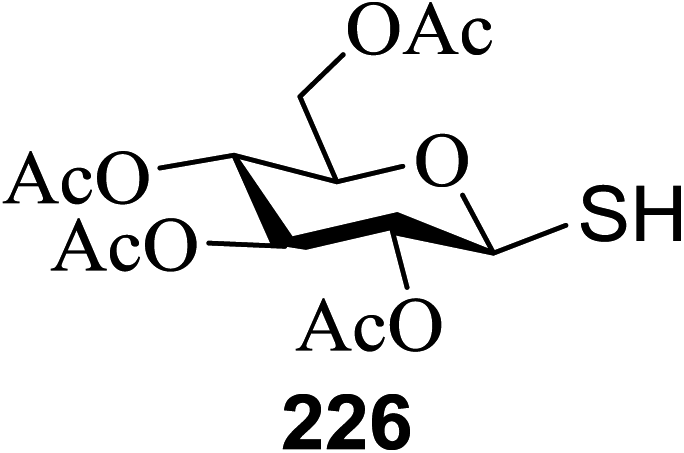 |
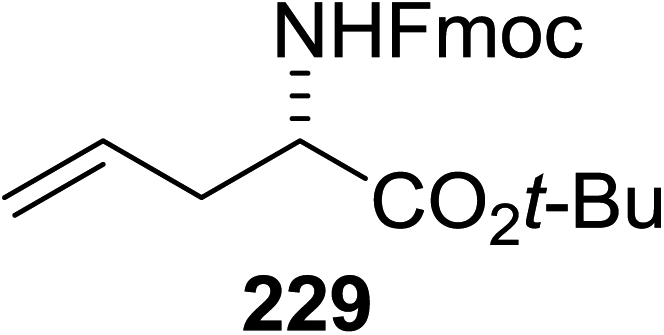 |
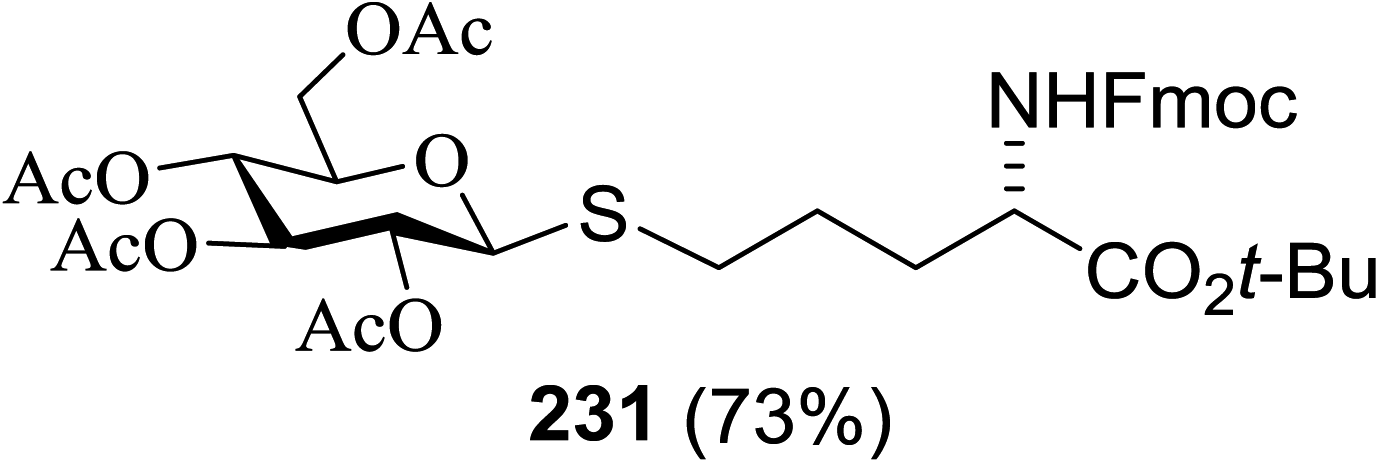 |
| 2 | 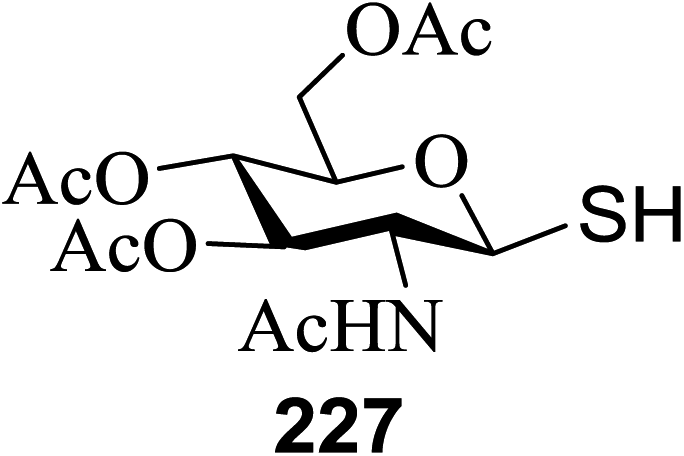 |
229 | 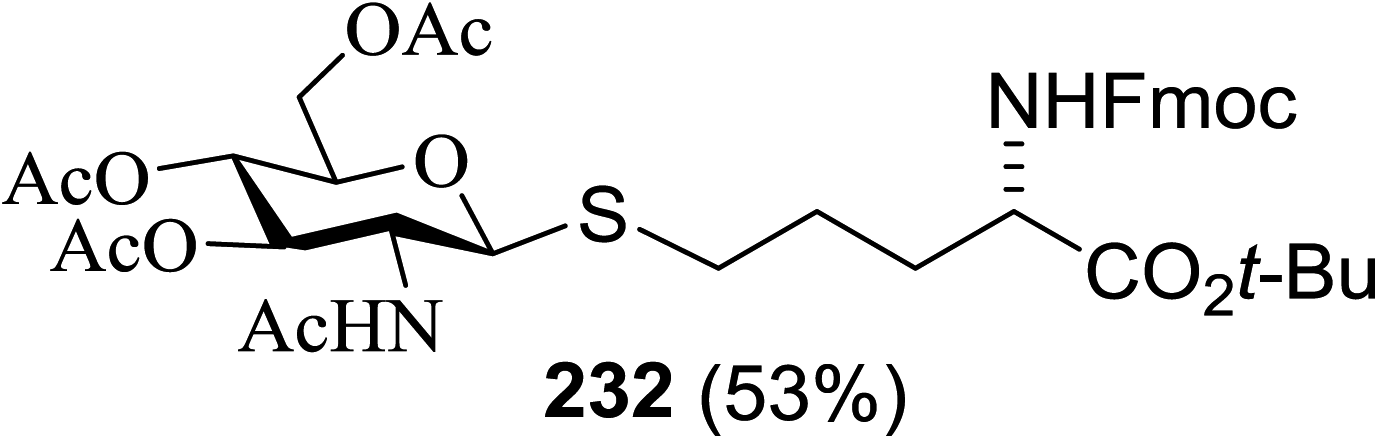 |
| 3 | 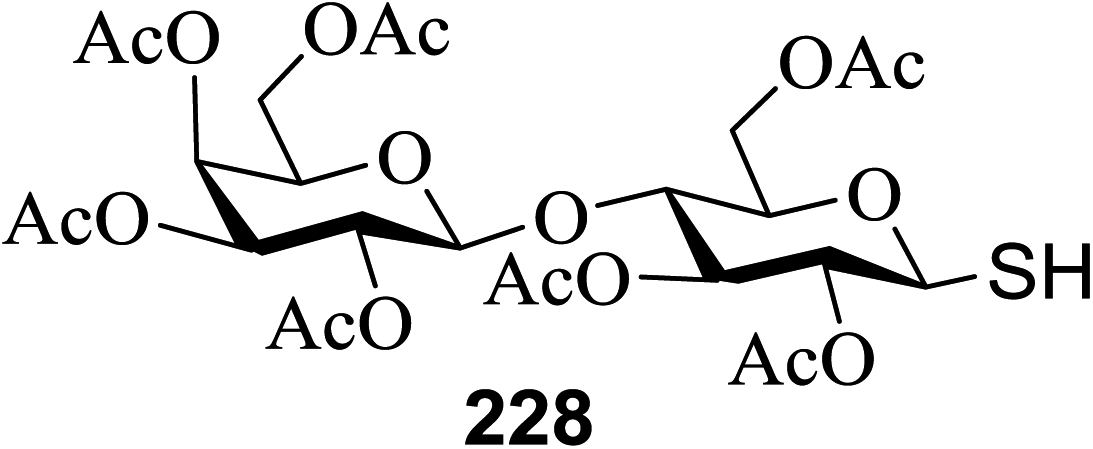 |
229 | 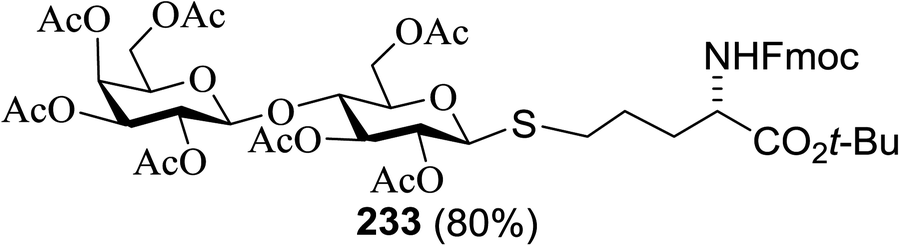 |
| 4 | 226 | 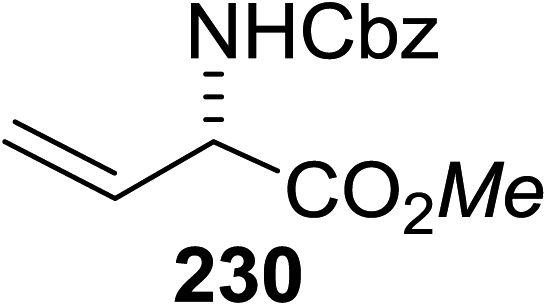 |
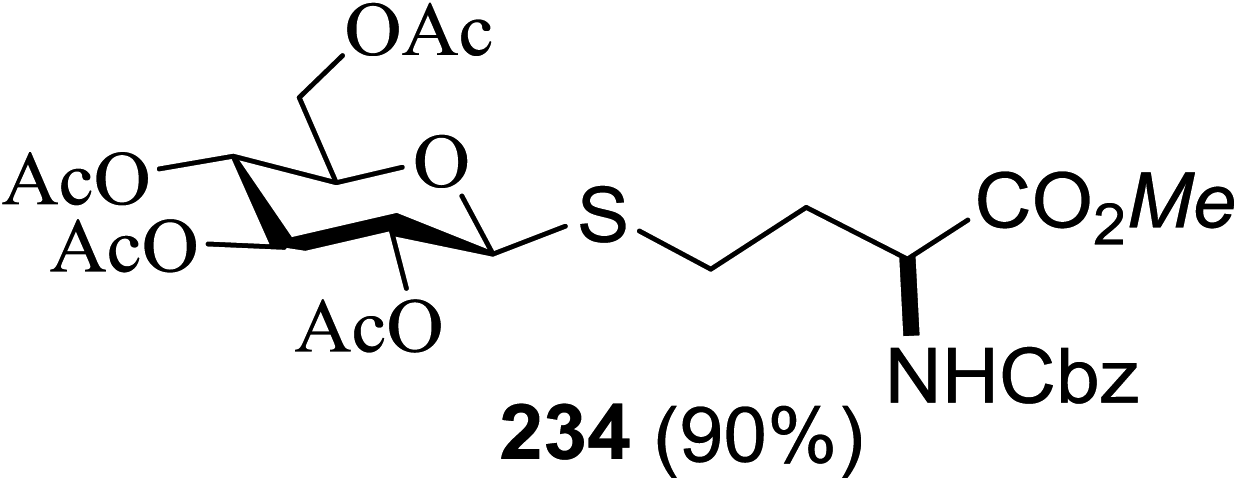 |
| 5 | 227 | 230 | 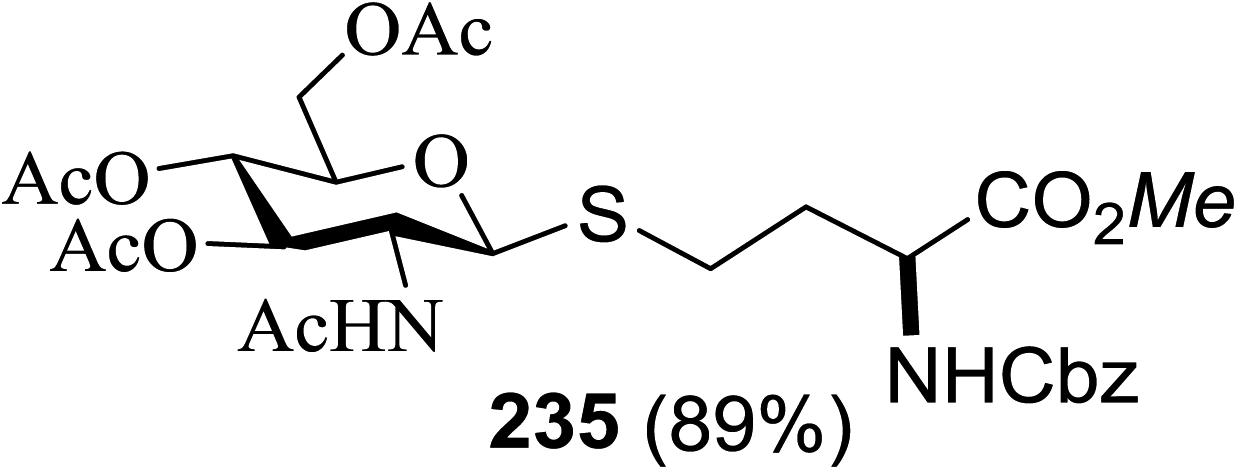 |
| 6 | 228 | 230 | 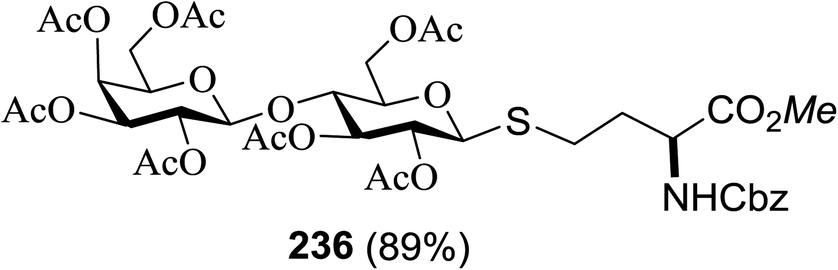 |
Different types of protective groups in the glycinyl motifs were employed in order to obtain S-glycosyl amino acids suitable for peptide synthesis. Optimized conditions were established, in which TEC was conveniently explored for S-disaccharide synthesis, peptide–protein glycosylation and calixarene-glycoclustering. The photo reactions were performed using a slight excess of thiols 226–228 (1.2 equiv.) with alkenes 229 or 230 by irradiation at λmax 365 nm in the presence of 2,2-dimethoxy-2-phenylacetophenone (DPAP) as the photoinitiator (0.1 equiv.). The yields of the isolated S-glycosyl amino acids 231–236 are summarized in Table 33, which were used to develop a TEC-based postsynthetic approach to S-glycopeptides.
In a second instance, the development of a TEC-based postsynthetic approach for the formation of S-glycopeptides was explored. In a model study, a homoallyl chain in L-cysteine was reacted with bromo alkene in the presence of Et3N and DPAP. Encouraged by this model, the approach was further extended to the S-glycosylation of glutathione 237 (Scheme 16).
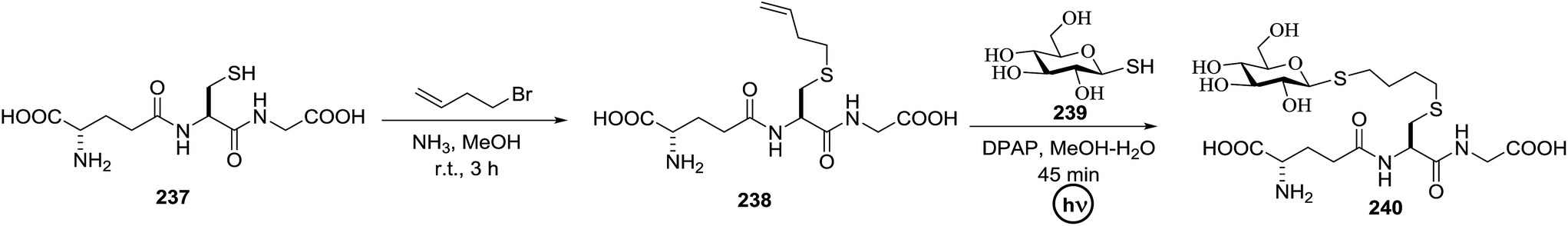 | ||
| Scheme 16 Synthesis of S-glycopeptides via photoinduced click thiol–ene coupling. | ||
The synthesis of a new family of 1-deoxy S-disaccharides was established by Staderini and co-workers97 via the free radical hydrothiolation of glycals by sugar thiols. Several chemical syntheses of S-disaccharides have been developed. The free-radical thiol–ene coupling (TEC) between alkenyl sugars and sugar thiols was reported for the first time by the same group in 2009. Investigation of the exemplary reaction between 3,4,6-tri-O-acetyl-D-glucal 241a and peracetylated 1-thio-β-D-glucopyranose 242a (Scheme 17) was conducted under standard conditions for S-disaccharide synthesis via TEC. Optimal conditions were established involving irradiation at a wavelength close to visible light (λmax 365 nm) in the presence of a catalytic amount of 2,2-dimethoxy-2-phenylacetophenone (DPAP) as a radical initiator, using 6 equiv. of 242a and 20:1 EtOH–CH2Cl2 as the solvent.
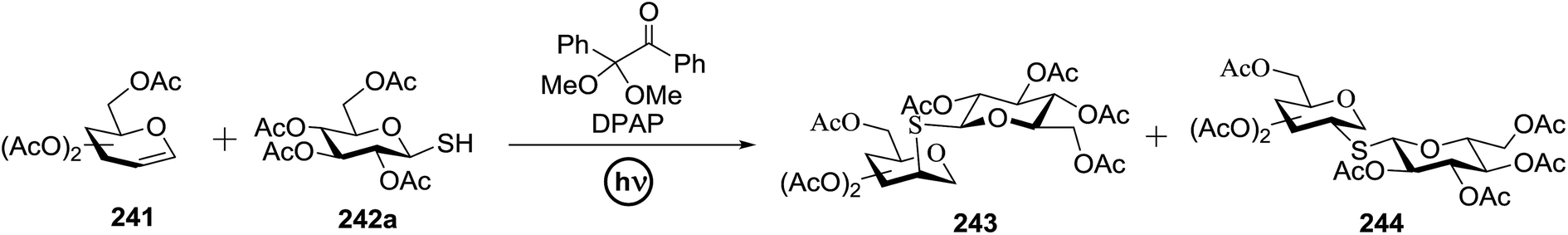 | ||
| Scheme 17 Synthesis of S-disaccharides via the hydrothiolation of glycals. | ||
Under the optimized conditions the glucal 241a was completely consumed (100% conversion) to give a mixture of products 243a and 244a in 80% isolated overall yield and 57:43 ratio. The reaction suffered from a lack of stereoselectivity. Similarly, the addition of the sugar thiol 242a to the D-galactal triacetate 241b under the optimized conditions gave a mixture of diastereomeric sulfides 243b and 244b in good yield. The same stereoselectivity was investigated in the reaction of 242a with D-glucal triacetate 241d to afford the diastereoisomer 243d despite the presence of the axial OAc group at C4 (Table 34).
| Entry | Glycal | Thiol | Products | Yield (%) | Product ratio | |
|---|---|---|---|---|---|---|
| 1 |  |
242a | 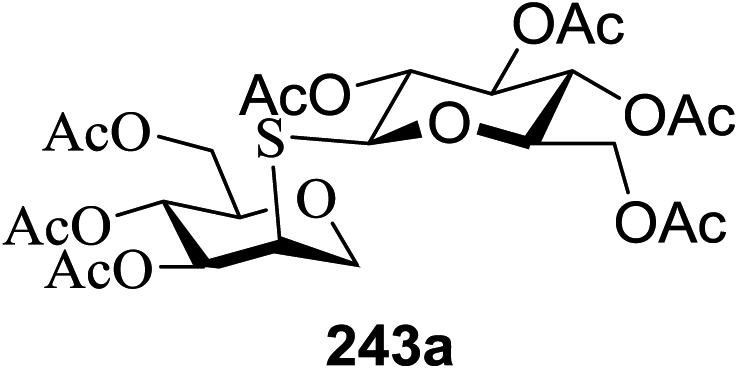 |
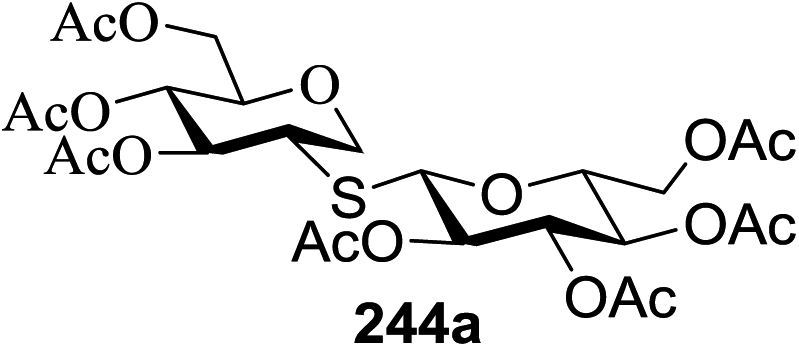 |
80 | 57:43 |
| 2 | 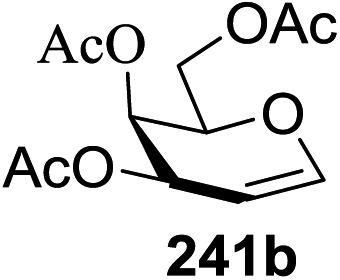 |
242a | 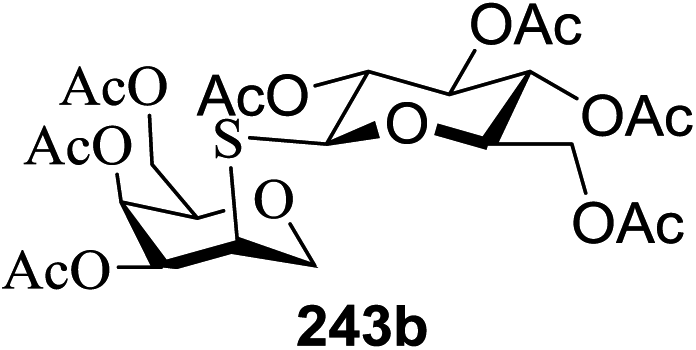 |
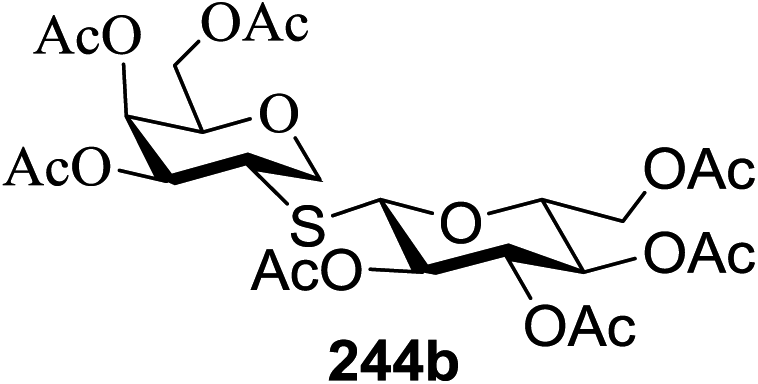 |
59 | 37:63 |
| 3 | 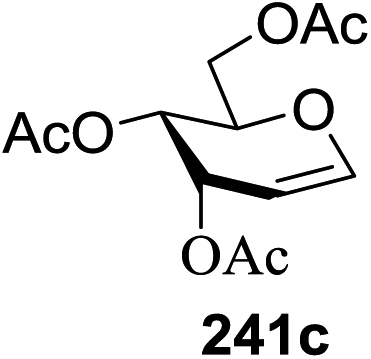 |
242a | 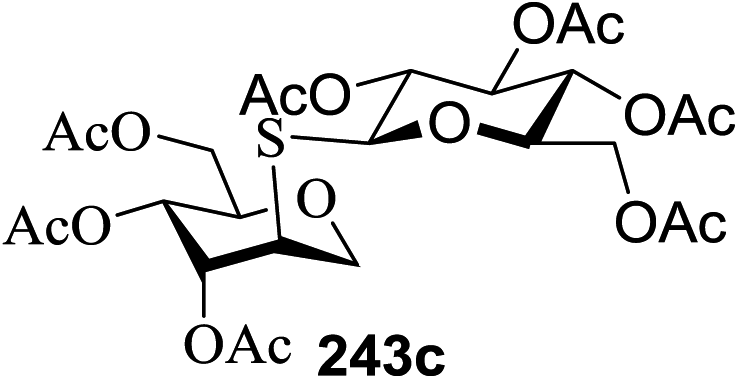 |
38 | 100:0 |
|
| 4 | 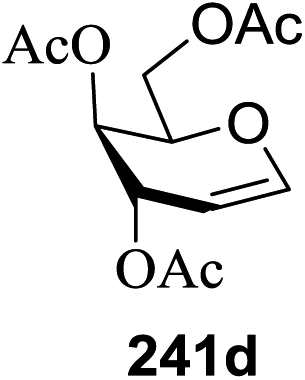 |
242a | 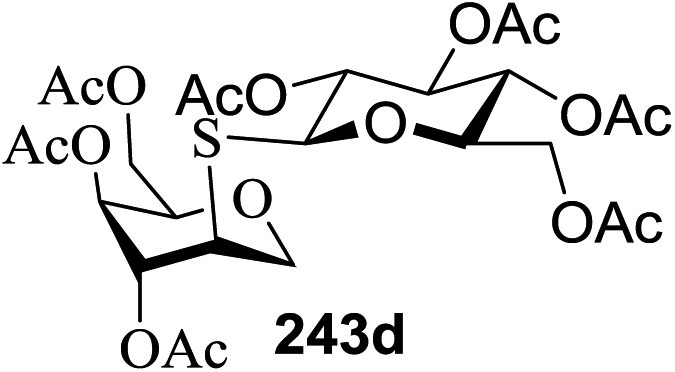 |
20 | 100:0 |
|
| 5 | 241a | 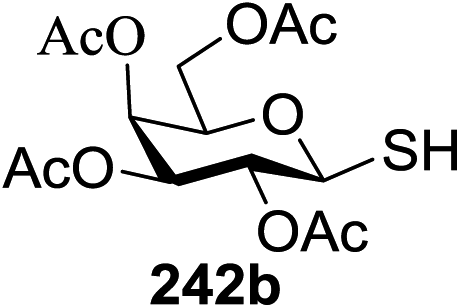 |
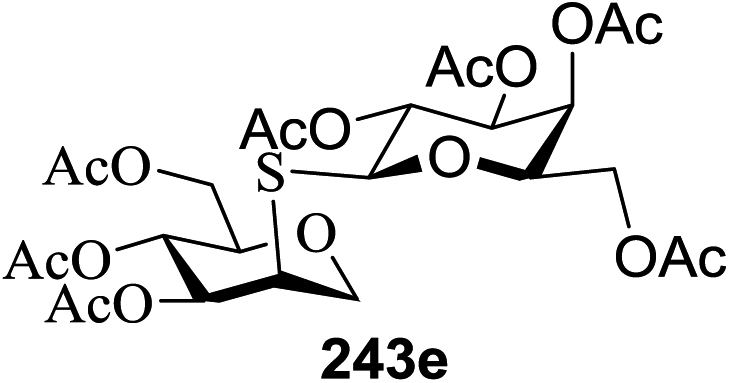 |
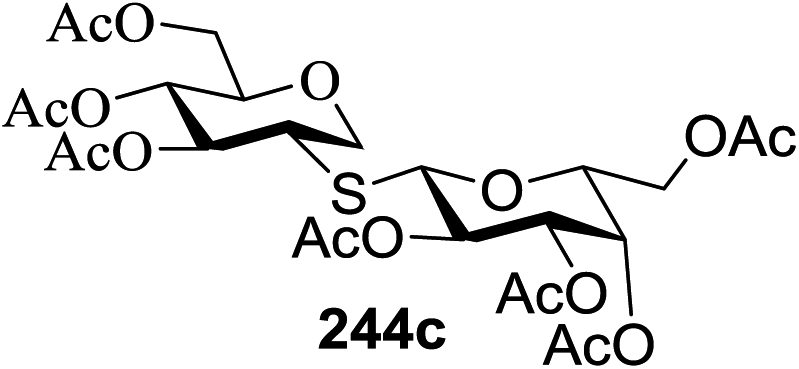 |
100 | 54:46 |
| 6 | 241a | 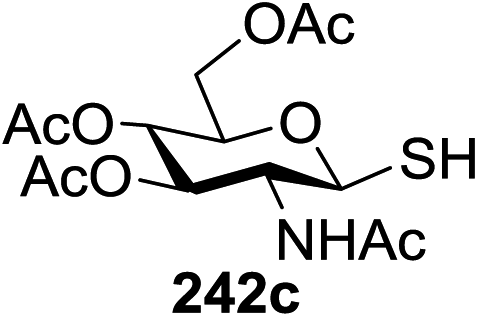 |
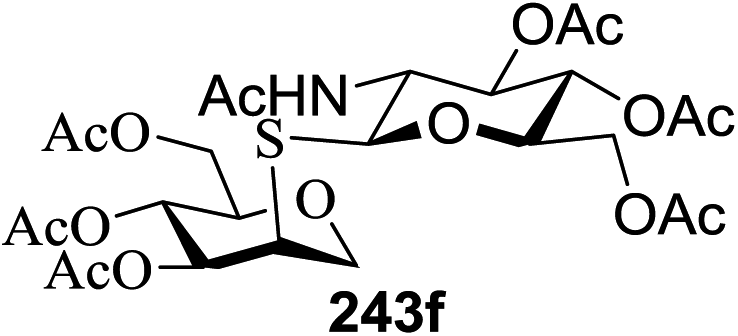 |
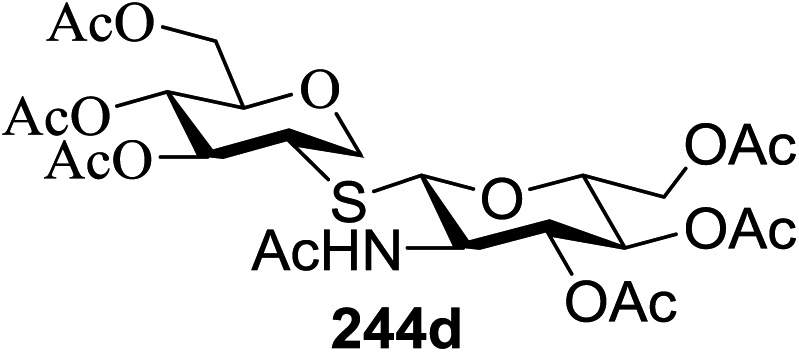 |
82 | 74:26 |
| 7 | 241b | 242b | 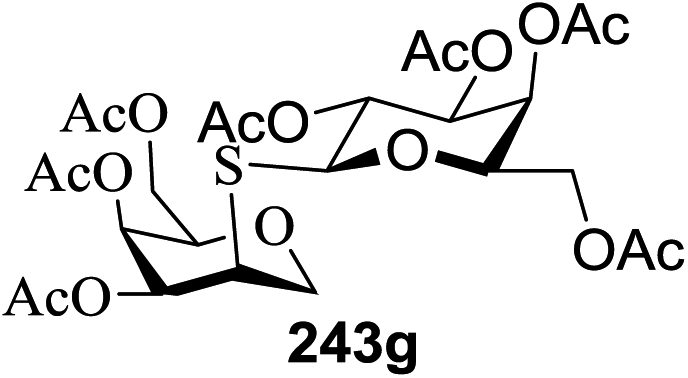 |
 |
100 | 38:62 |
| 8 | 241b | 242c |  |
 |
100 | 50:50 |
For further exploration of the substrate scope, the reaction was conducted with the addition of two more thiols 242b and 242c to glucal 241a and galactal 241b (Table 34). Moreover the presence of the NHAc group may serve to establish the stability of this important functionality under the conditions of TEC. The photoinduced hydrothiolation of 241a and 241b by thiols 242b and 242c under the optimized conditions occurred with essentially the transformation of the glycals to give the respective pairs of diastereoisomers 243e–244c, 243f–244d, 243g–244e and 243h–244f in very high isolated yields.
Schlaad and co-workers promoted the synthesis of heterofunctional glycopolypeptides via in situ glycosylation and polymerization. There have been many efforts involving the polymerization of glycosylated amino acid N-carboxy anhydrides (glyco-NCAs)98 as well as the post-polymerization functionalization of ready-made polypeptides bearing applicable functional groups in the side chains.99 All of these paths require many steps and difficult purification protocols and are usually laborious and time-consuming procedures. Krannig and co-workers100 developed an approach for the synthesis of glycopolypeptides via in situ glycosylation and subsequent polymerization of 2-aminopent-4-enoic acid (allylglycine, AGly) NCA, which links the radical thiol–ene photochemistry with nucleophilic ring-opening polymerization (ROP) (Scheme 18). The synthesized glycopolypeptides consist of a prearranged sugar and vinyl groups, which were vulnerable to additional functional groups, e.g. sugar, carboxylic acid, amine and thiol, through a second reaction. Glycosylation of AGly NCA (→glyco-NCA 247 (DL) and 247 (L)) was predicted to take place through the radical addition of 1-thio-β-D-glucopyranose-2,3,4,6-tetraacetate (AcGlcSH, 246) to the exocyclic vinyl group, as defined in Scheme 18.
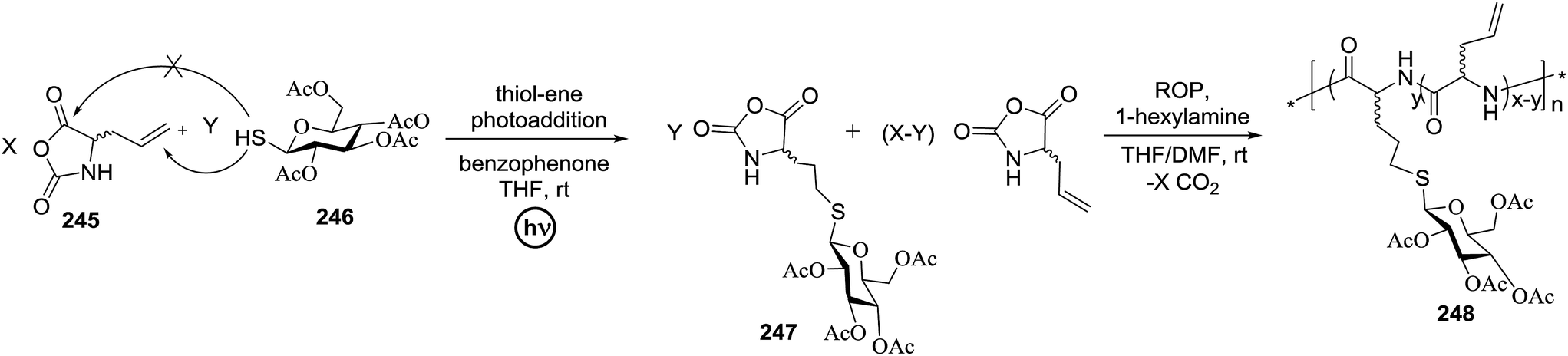 | ||
| Scheme 18 One-pot partial glycosylation and copolymerization of AGly NCA. | ||
However, the thiol may also behave as a nucleophile and attach to the heterocyclic ring (at C5), conclusively beginning the polymerization of the NCA. This one-pot path allows the synthesis of well known glycopolypeptides with good control over composition, chain length and dispersity. The AGly units in the partially glycosylated copolypeptides were vulnerable to post-polymerization conversion via thiol–ene photochemistry (Scheme 19). For example, P1 (248) was reacted with three different ω-functional thiols, i.e. 247-mercaptopropionic acid, N-acetylcysteamine and thioacetic acid, to insert carboxyl, amine and thiol functionalities respectively. Glycopolypeptides having more complex sugars or oligosaccharides can be synthesized using this approach.
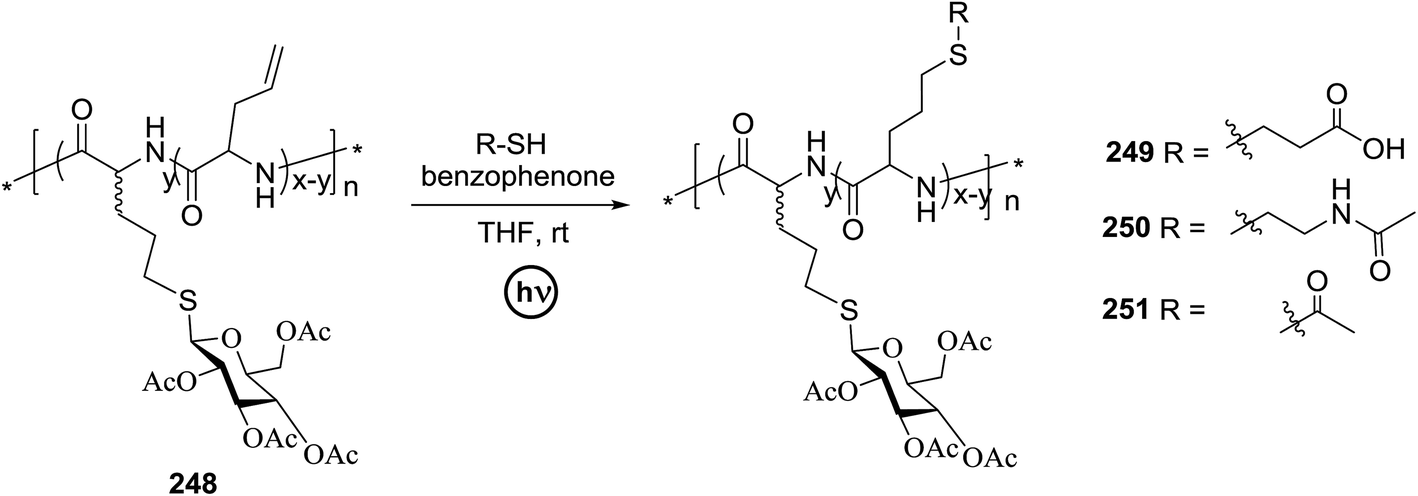 | ||
| Scheme 19 Modification of the partially glycosylated copolypeptide P1 with different thiols. | ||
4.3. S-Linked glycoconjugate synthesis via photoinduced thiol–yne coupling (TYC)
In recent years synthetic glycopeptides and glycoproteins involving unnatural linkages between the carbohydrate and aglycone moieties have been reported. Conte and co-workers101 reported the radical-mediated hydrothiolation of terminal alkynes. This is a metal-free ligation process102 for the assembly of biomolecules under mild and neutral reaction conditions. The distinct formation of powerful sulfide bridges is an interesting target due to the suitable features of the C–S bond.A discovery dating back to the mid-1900s103 reported the application of a sister thiol–ene reaction, that is the thiol–yne process, which serves to introduce two thiol fragments across a carbon–carbon triple bond. Fig. 25 illustrates the reaction steps, in which two thiol fragments are introduced across a carbon–carbon triple bond via a multi step mechanism. The first step involves the anti-Markovnikov-like addition of a thiyl radical to the alkyne bond to yield an intermediate vinyl thioether which undergoes a second, faster, thiyl radical addition, a formal thiol–ene reaction, leading to a dithioether with the exclusive 1,2-addition mode.
 | ||
| Fig. 25 General strategic approach for the introduction of two thiol fragments across a carbon–carbon triple bond via a multistep mechanism. | ||
The exemplary reaction of protected cysteine 252 with a slight excess of propargyl bromide (1.2 equiv.) and triethylamine (2 equiv.) in CH2Cl2 at room temperature, as shown in Scheme 20, afforded the S-propargyl derivative 253 in 86% yield. Then, the photoinduced reaction of 253 with the peracetylated glucosyl thiol 254 was investigated, which was carried out in methanol as the solvent under irradiation at λmax 365 nm in the presence (10 mol%) of 2,2-dimethoxy-2 phenylacetophenone (DPAP) as the sensitizer. The optimal conditions involved the use of excess thiol 254 (4 equiv.) with respect to alkyne 253.
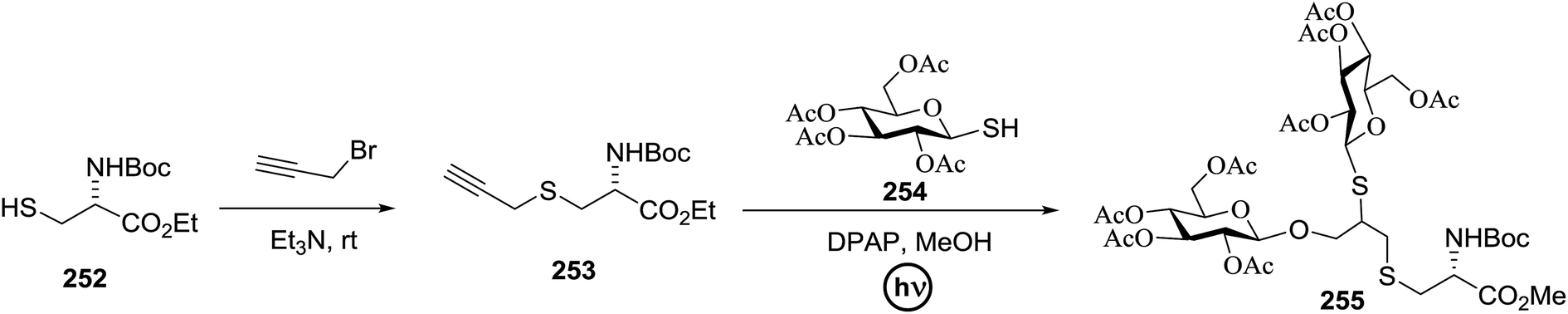 | ||
| Scheme 20 Photoinduced addition of glycosyl thiols to alkynyl peptides. | ||
By using the natural tripeptide Glu-Cys-Glc (glutathione, GSH 256) and the synthetic tetrapeptide Arg-Gly-Asp-Cys (RGDC 257) as substrates, the glycosylation of small peptides via this approach defined was investigated. A one-pot two-step sequence containing peptide alkynylation and thiol–yne coupling was proposed to bypass the intermediate isolation. Particularly, the use of the unprotected peptides GSH 256 and RGDC 257 allowed the alkynylation which was performed under very mild conditions. After this photoinduced double hydrothiolation was carried out using 4 equivalent of the unprotected glucosyl thiol 260 under the conditions defined above except that the solvent was a 1:1 H2O/MeOH mixture. It was observed that the thiol–yne reactions took place with successfully with conversions >95% into the corresponding S-glucosyl peptides 263, 264, 265, 266, 267 and 268 (Table 35). The scope of the one-pot two-step glycosylation strategy was expanded by using higher peptides such as the synthetic cysteine-containing octapeptides 258 and 259 (Table 35).
|
||||
|---|---|---|---|---|
| Entry | Peptide | Sugar thiol | Product | Yield (%) conversion (%) |
| 1 |  |
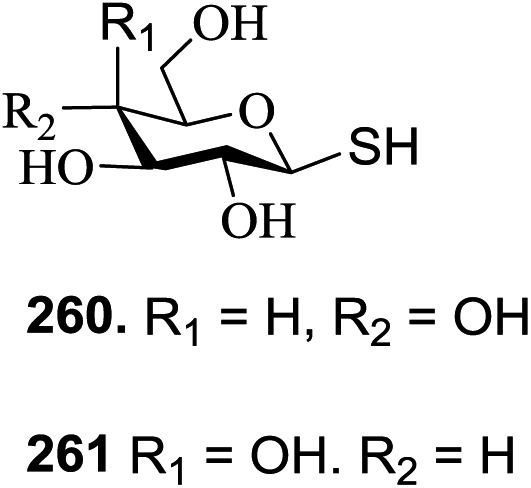 |
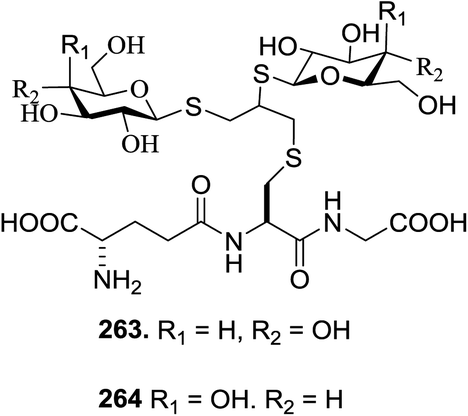 |
77 (>95) |
| 74 (>95) | ||||
| 2 | 256 | 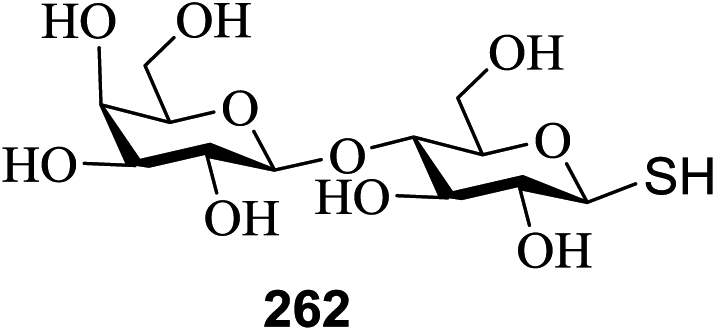 |
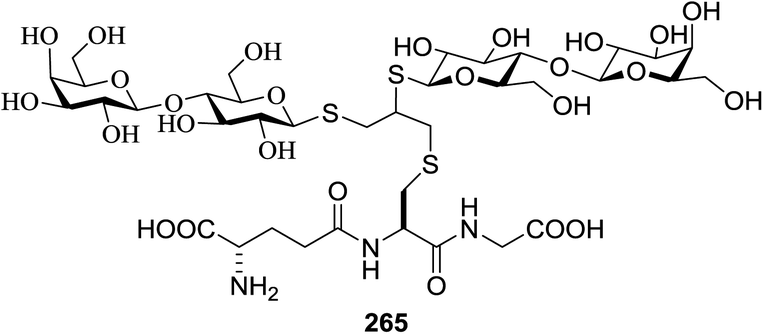 |
57 (>95) |
| 3 |  |
 |
 |
36 (95) |
| 27 (>95) | ||||
| 4 | 257 | 262 |  |
55 (>95) |
| 5 | H-Asp-Leu-Tyr-Cys-Tyr-Glu-Gln-Leu-NH2 (258) | 260 | 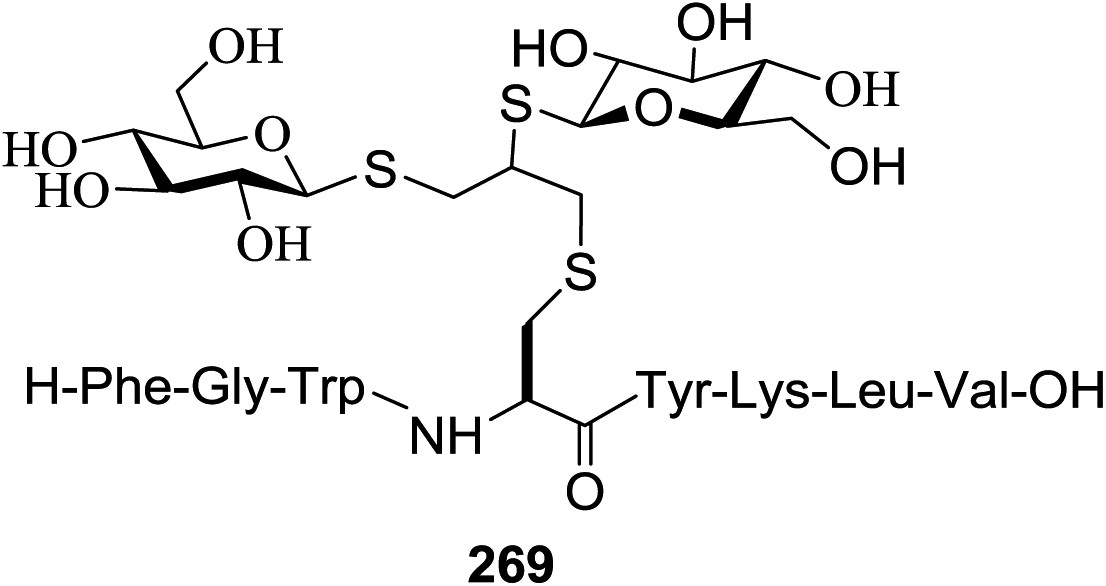 |
(>95) |
| 6 | H-Phe-Gly-Trp-Cys-Tyr![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Lys-Leu-Val-OH (259) Lys-Leu-Val-OH (259) |
260 | 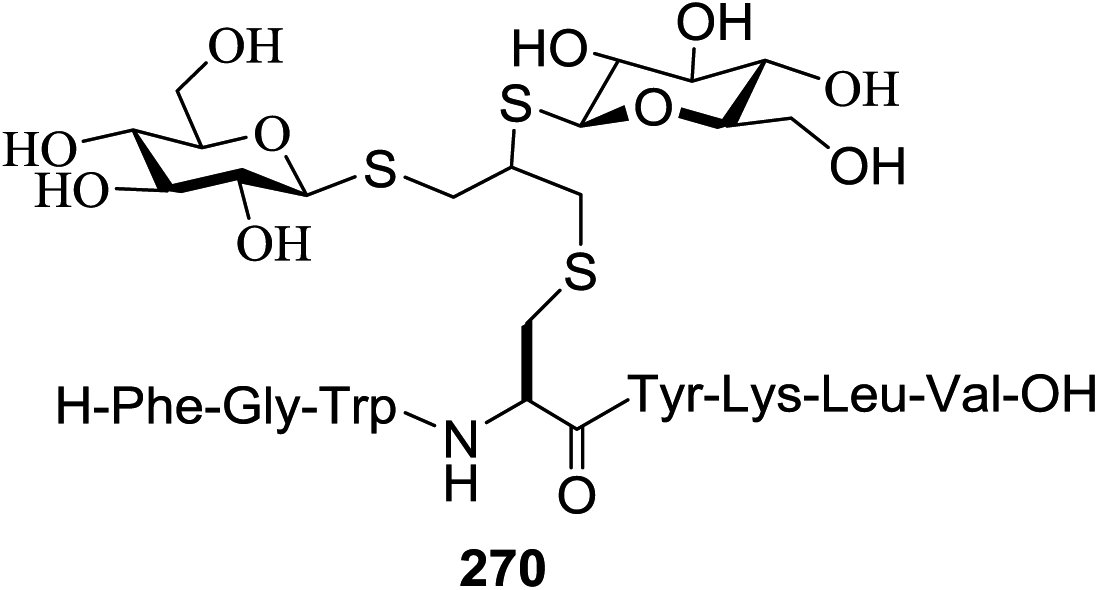 |
(>95) |
By utilizing the above procedure and conditions for the propargylation of GSH 256 and RGDC 257 followed by thiol–yne coupling with thiol 260, these peptides were converted into the corresponding S-glycosides 269 and 270. The same procedure was adopted for the preparation of the regioisomer of 272 with use of a reversed order hydrothiolation, i.e. biotinylation first and then glycosylation (Scheme 21). Since biotin is an important biological vitamin B complex104 which is typically conjugated to peptides and proteins via primary amines (e.g., lysines) this method established a different path for the selective biotin labeling of cysteine-containing compounds.
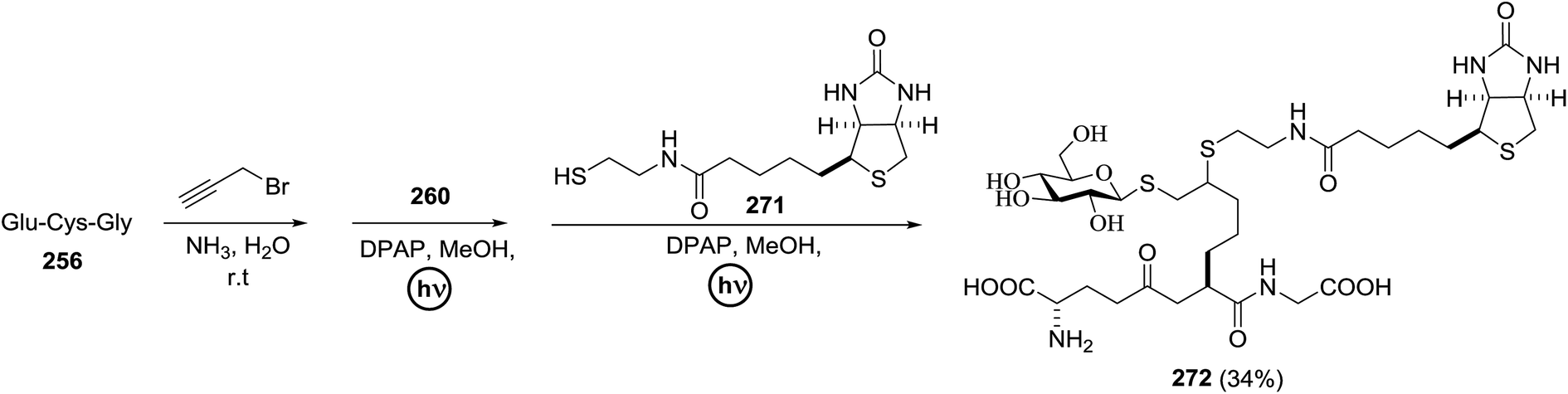 | ||
| Scheme 21 Preparation of the regioisomer 272 with use of a reversed order hydrothiolation. | ||
A sugar displaying an aryl acetylene moiety gave rise to click-reaction involving the glycosylation of the tripeptide glutathione in its native form. This reaction was successfully applied under physiological conditions to a cysteine-containing nonapeptide with marked advantages over the similar type thiol–ene coupling (TEC) derivatization. A different study on the thiol–yne coupling (TYC) reaction was carried out by Massi and co-workers using an aliphatic (1-octyne) and an aromatic alkyne (phenylacetylene) and two alkane thiols (methyl thioglycolate and N-acetyl-L-cysteine methyl ester, Scheme 22).105
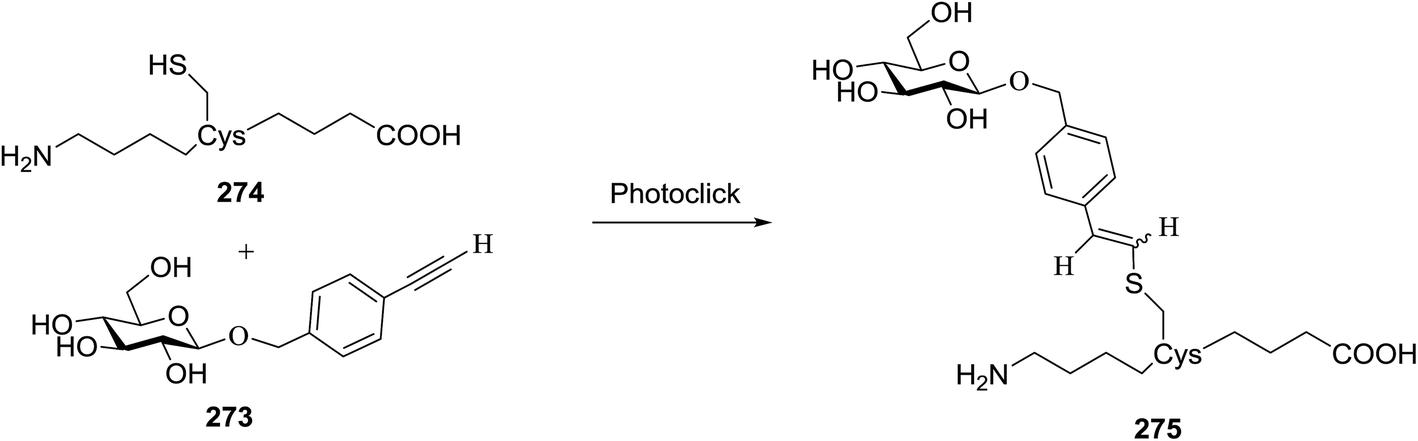 | ||
| Scheme 22 Photoinduced glycosylation of cysteine-containing peptides. | ||
The main disadvantage of TEC is that an excess of both reagents is required in order to obtain complete conversions due to the reversibility of the attack of sulfur-centered radicals to C–C double bonds. In that case, additional work-up is essential to get rid of the extra alkene/thiol, thus the main advantage of click-reactions is lost. In principle, an expected way to overcome this drawback could be to carry out the reaction in the presence of alkynes instead of alkenes, that is, to perform thiol–yne couplings (TYCs).
The usefulness of this radical reaction in glycopeptides chemistry has been recently verified by the synthesis of doubly glycosylated peptides via a two-step strategy, in which S-propargylation of a cysteine involving a peptide first occurs followed by photoinduced coupling with an excess of glycosyl thiol. Coupling between the peracetylated O-propargyl β-glycoside 276 with a single cysteine residue 277 was initially performed to determine the optimal conditions for the selective mono hydrothiolation procedure of more complex substrates (Table 36). The formation of the expected bis-adduct 279 seemed to be highly inhibited by steric factors. The ‘sticky’ nature of 277 resulted in agglomeration and hence excluded intimate contact between the reactants (entry 3). On the other hand, when an equimolar amount of 276 and 277 was dissolved in toluene, the subsequent addition of H2O (0.01 M) resulted in the production of organic droplets which disappeared under vigorous magnetic stirring.
|
||||
|---|---|---|---|---|
| Entry | 276/277 | Solvent (c [M]) | Time (h) | Yield 278/279 [%] |
| 1 | 1:1 |
DMF (0.5) | 1 | 25/— |
| 2 | 3:1 |
DMF (0.5) | 1 | 88/— |
| 3 | 1:1 |
H2O (0.5) | 2 | —/— |
| 4 | 1:1 |
H2O–PhMe (0.01) | 2 | 6/81 |
| 5 | 1:2 |
H2O–PhMe (0.01) | 2 | —/85 |
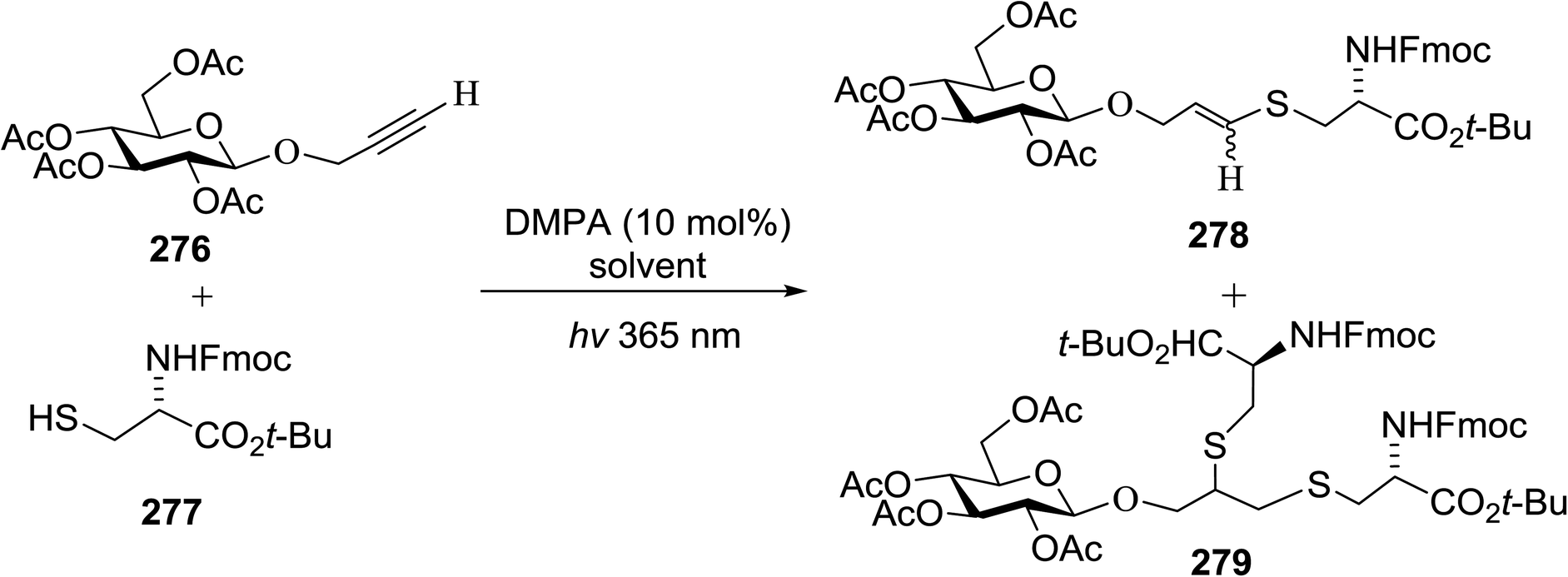
Irradiation of that mixture for 2 h afforded the bis-glycosylated cysteine derivative 279 as the main product (81%, d.r. ∼ 1:1) along with a small amount of the mono adduct 278 (6%; entry 4). The selective formation of the bis-adduct 279 was finally attained by simply using 2 equivalents of cysteine 277 under the same conditions (entry 5).
The glycosyl alkyne 280 and the natural tripeptide glutathione (γ-LGlu-L-Cys-Gly, GSH) in its native form were treated with the relevant substrates to test the efficiency of this path (Scheme 23) by forming the glycoconjugate 282.
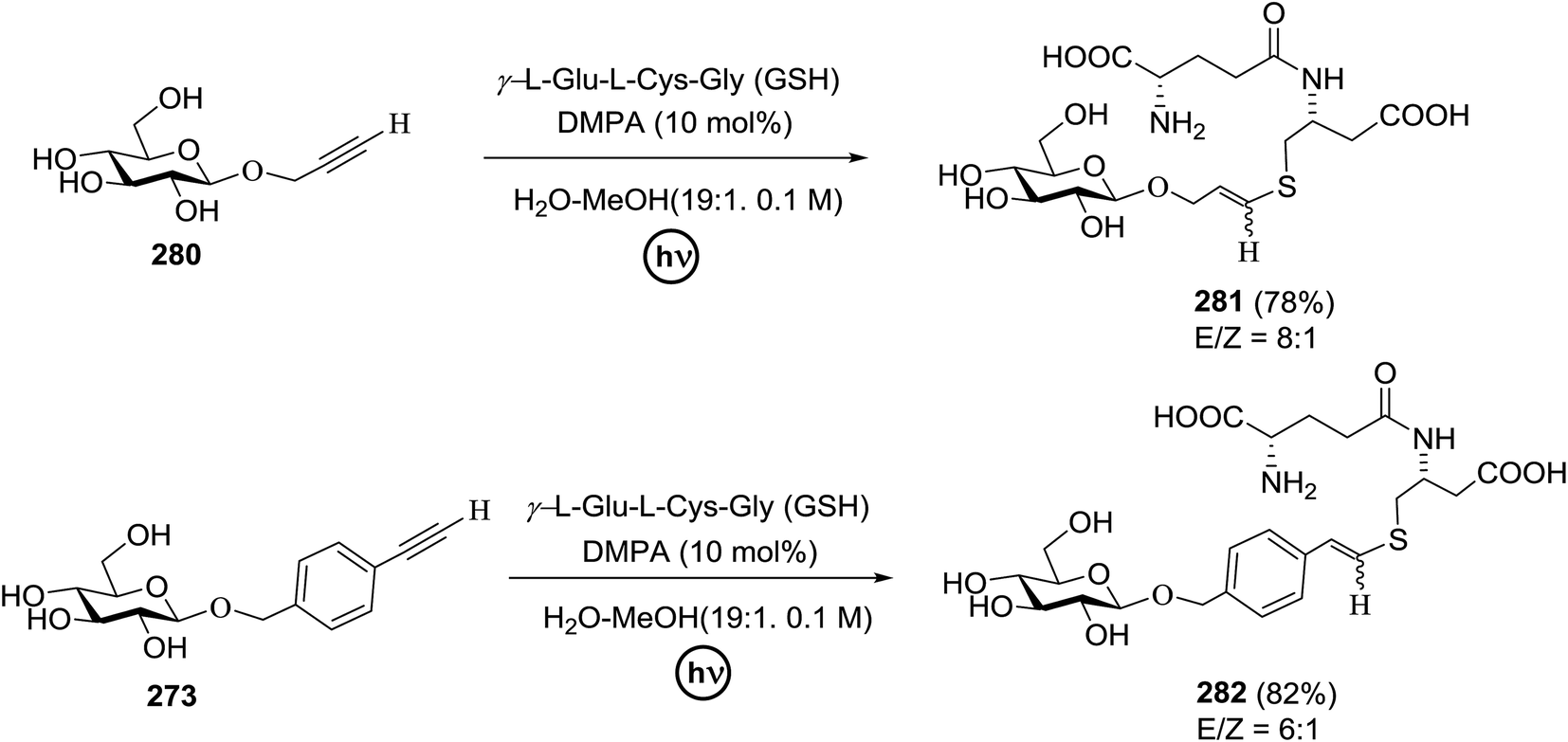 | ||
| Scheme 23 Optimized conditions for the complete glycosylation of glutathione by sugar alkynes. | ||
The exploration of the reaction profile for both the mono- and double-hydrothiolation of sugar alkyne 276 was conducted using the two different cysteine derivatives 277 and 283 (Scheme 24). The vinyl thioether intermediate 278 was first obtained by the TYC of alkyne 276 with cysteine 277 and then subjected to photoinduced TEC with cysteine 283 to give the target bis-adduct 284 (d.r. ∼ 1:1) in 66% overall yield (Scheme 24).
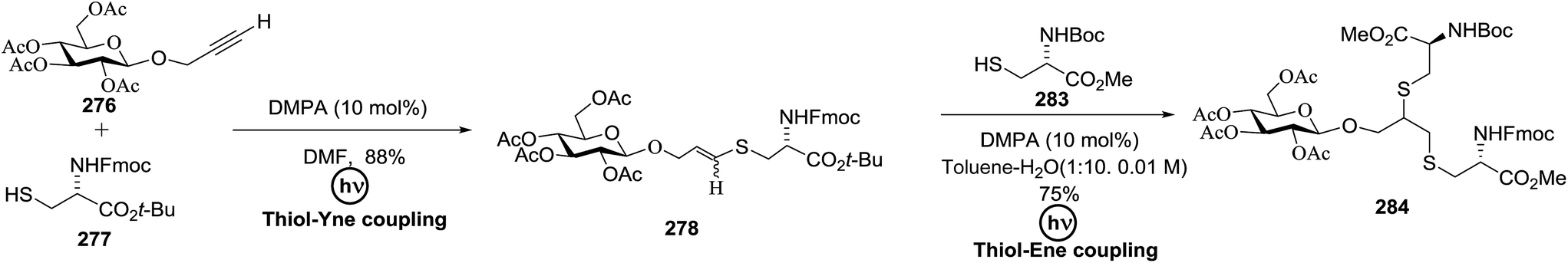 | ||
| Scheme 24 Synthesis of orthogonally protected bis-armed cysteine. | ||
4.4. Photoinduced hydrothiolation of endocyclic double bond enoses
Free-radical addition of thiols to alkenes, which is called thiol–ene coupling or thiol–ene click reactions, has already been perfectly applied for the formation of thioglycosidic linkages. Free-radical hydrothiolation of the endocyclic double bond of enoses was reported by Borbas and co-workers.106 The reaction of 2-acetoxy-D-glucal with a wide range of thiols, including amino acids, peptide, glycosyl thiols, and sugars with primary or secondary thiol functional groups, gave S-linked α-glucoconjugates and S-disaccharides with full regio- and stereoselectivity (Scheme 25). The successful synthesis of glycodendrimers, S-linked protein glycoconjugates107 and thiodisaccharides through the addition of glycosyl 1-thiols to terminal double bonds via the light-mediated radical mechanism has been reported.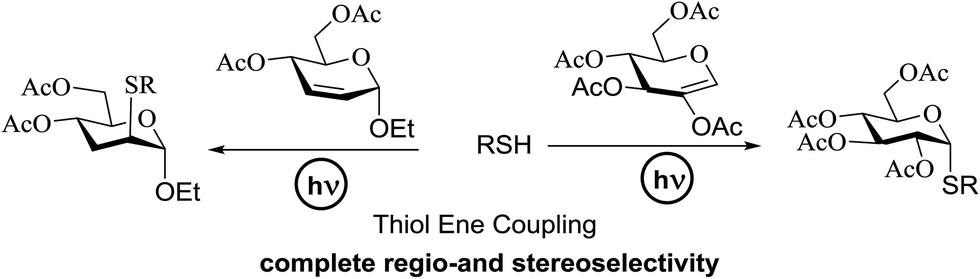 | ||
| Scheme 25 Preparation of S-linked glycoconjugates by thiol–ene coupling reaction of enoses. | ||
The addition of ethanethiol to 2-acetoxy-3,4,6-tri-O-acetyl-D-glucal 285a in toluene was examined and standardised initially. Irradiation at λmax 365 nm in the presence of 2,2-dimethoxy-2-phenylacetophenone (DPAP) as a cleavage-type photoinitiator (Fig. 26) was chosen. The reaction resulted in the exclusive formation of the ethyl 2,3,4,6-tetra-O-acetyl-α-thioglucoside 286a. They examined the function of the easily used enoses such as glycals, 2-acetoxy glycals and Ferrier glycals (2,3-unsaturated glycosides). The hydrothiolation of 2-acetoxy glycals 285b–d with ethane thiol under the standardised conditions resulted in complete regio- and stereoselectivity and high yield in all cases, affording the analogous 1,2-cis α-thioglycosides 286b–d, respectively. The reactions of the galactal derivative 285b and the allal 285c indicated that the orientation of the 3- and 4-O-acetyl moieties did not affect the stereochemical outcome of the hydrothiolation. However, hydrothiolation of the pentose-derived 2-acetoxy-glycal 287a under these conditions resulted in a stereoisomeric mixture of the 1,2-cis and 1,2-trans α-thioglycosides 288a and 289a in a ratio of 2:1. Similarly, the reaction between ethane thiol and 287b led to the formation of both the 1,2-cis and 1,2-trans α-thioglycosides 288b and 289b, respectively (Table 37).
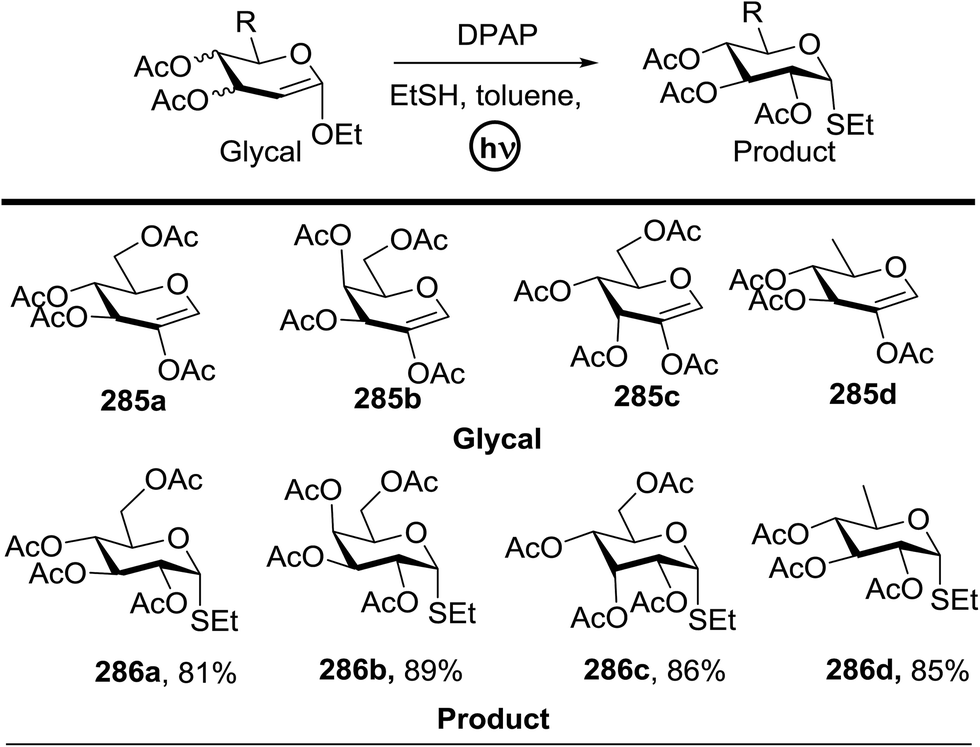 | ||
| Fig. 26 Free-radical addition of ethanethiol to 2-acetoxy glycals. | ||
|
|
|---|---|
| Glycal | Products (yield) |
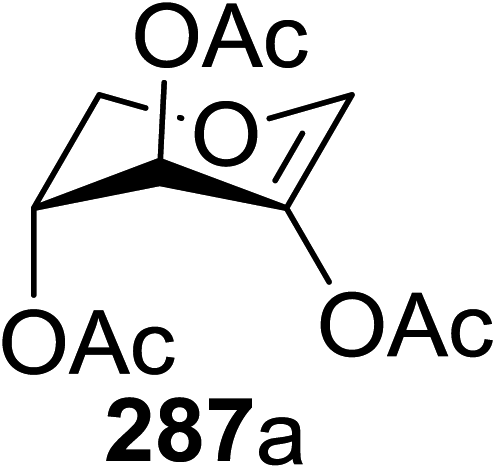 |
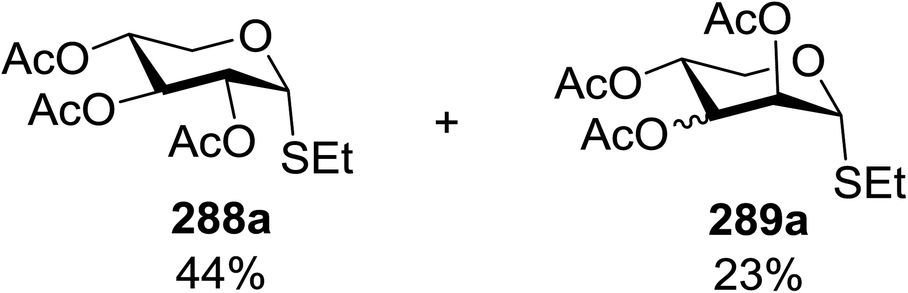 |
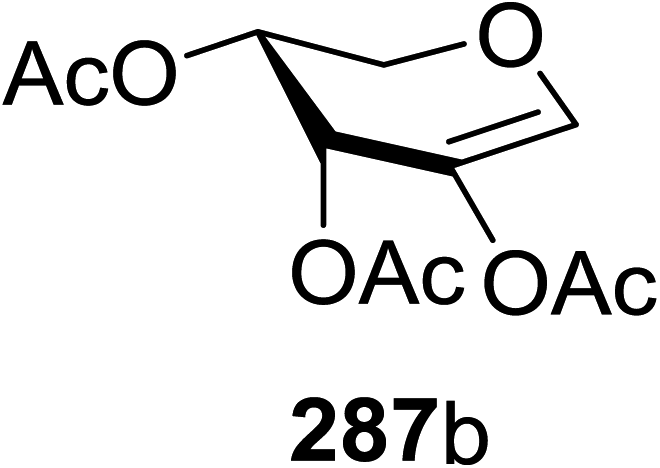 |
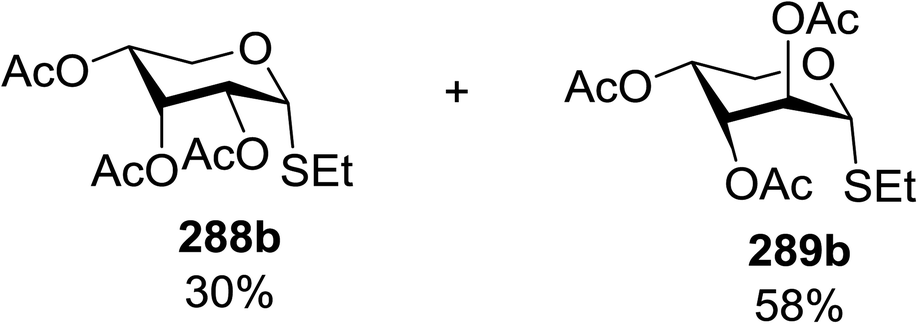 |
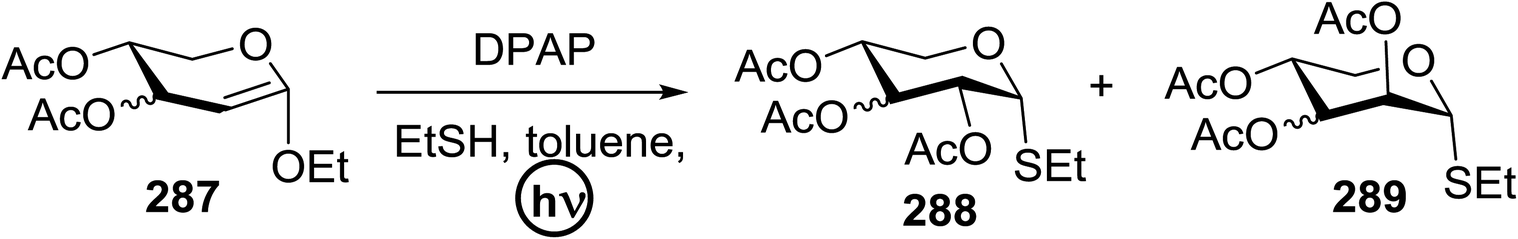
These reactions undoubtedly demonstrated that a bulky C-6 group (either acetoxymethyl or methyl) anchored on the β-side of the sugar ring could afford the stereoselective formation of the 1,2-cis C-2-radical intermediates (I) upon addition of the thiyl radical (Fig. 27). In the absence of a bulky substituent, the 1,2-cis radical (IIcis), in which the 2-acetoxygroup is equatorial and the 1,2-trans radical (IItrans), in which the same group is axial, can both can be formed and the ratio of their formation depends on the stereochemistry of the adjacent C-3 substituent.
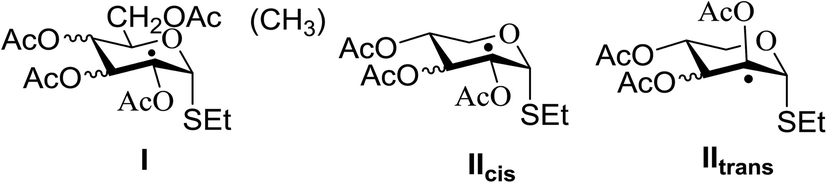 | ||
| Fig. 27 Radical intermediates formed by the addition of ethanethiyl to hexose- (I) and pentose-derived (II) 2-acetoxy glycals. | ||
The scope of this approach for the synthesis of biorelevant glycoconjugates is presented in Table 38. All the reactions progressed with immense selectivity in favour of the α-thioglucosides, and the solvent plays a key role in the reaction efficiency. N-acetyl-L-cysteine (290) reacted readily with the endocyclic double bond of 285a in toluene–MeOH and gave 298 in good yield. However, low conversion of the reactants was observed when 285a was reacted with captopril 291, which is an angiotensin-converting enzyme (ACE) inhibitor. Changing the solvent to MeOH enhanced the conversion significantly and afforded thioglucoside 299 in excellent yield. For S-disaccharide synthesis, sugar thiols including acetyl-protected glycosyl thiols (294 and 295) and di-O-isopropylidenated sugars with primary (296) or secondary thiol functions (297) were tested as thiol partners.
| Entry | Thiol | Product | Yield% | Entry | Thiol | Product | Yield% |
|---|---|---|---|---|---|---|---|
| 1 | 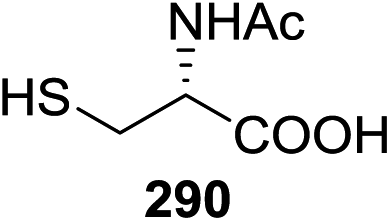 |
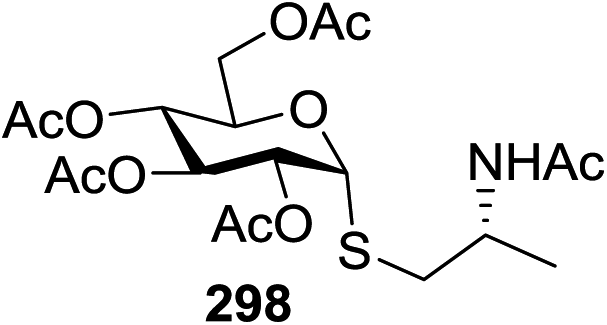 |
61 | 5 | 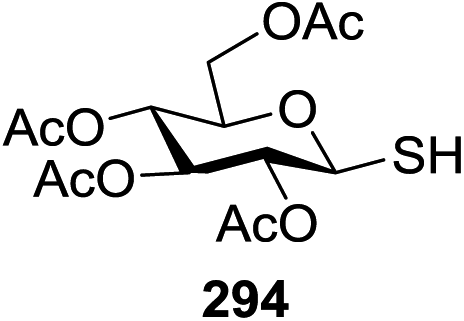 |
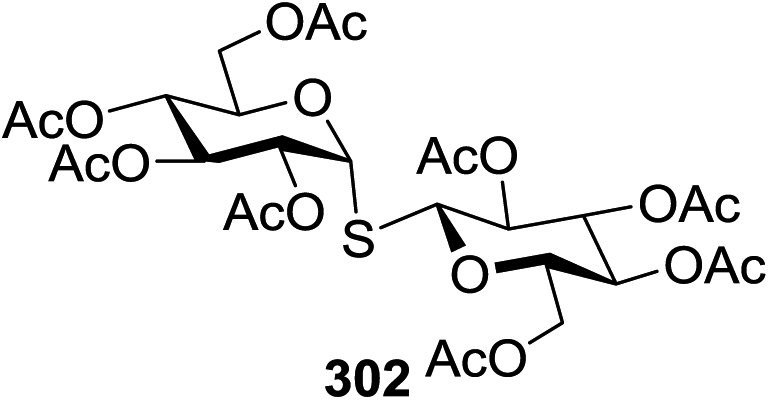 |
87 |
| 2 | 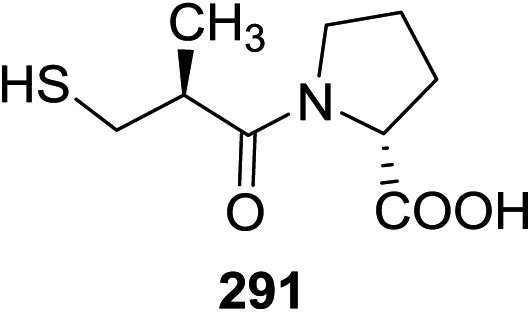 |
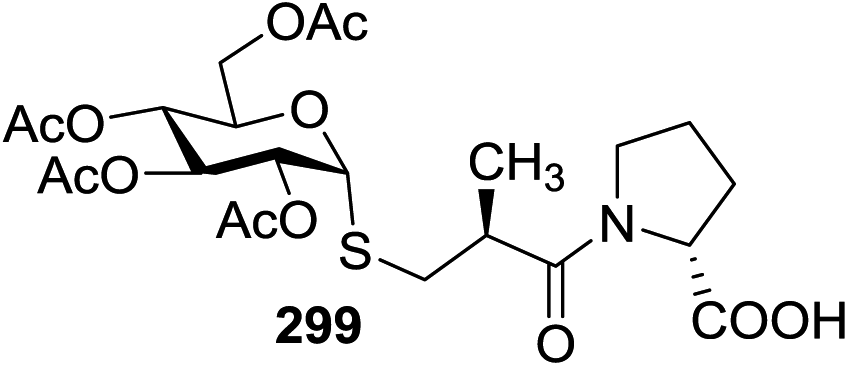 |
88 | 6 | 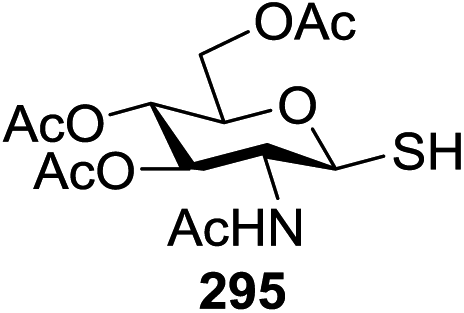 |
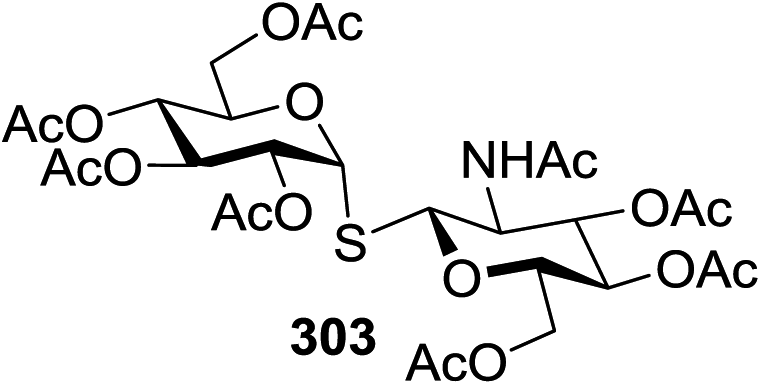 |
55 |
| 3 |  |
 |
51 | 7 |  |
 |
53 |
| 4 | 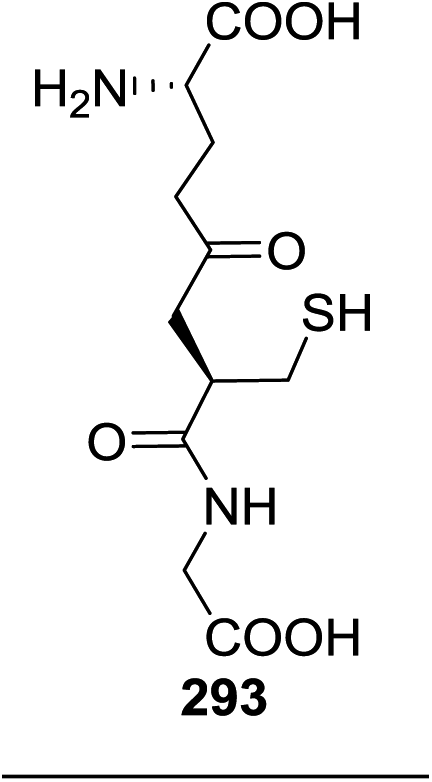 |
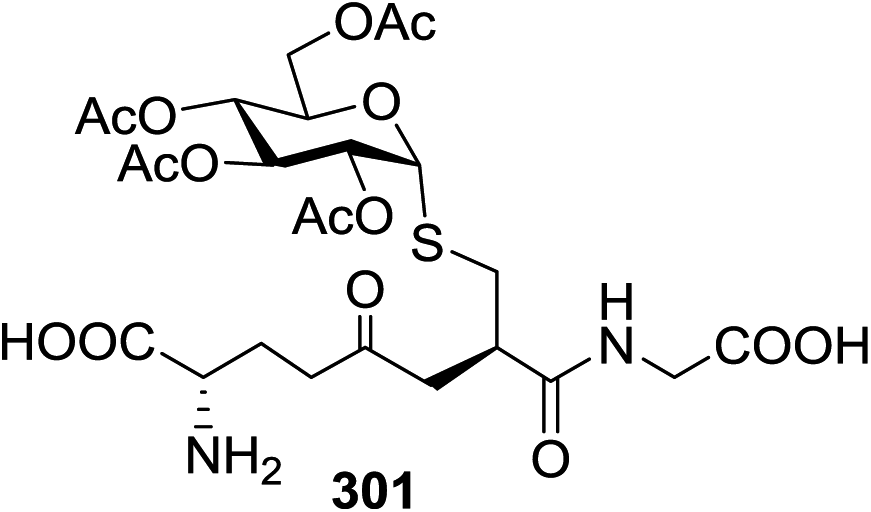 |
60 | 8 | 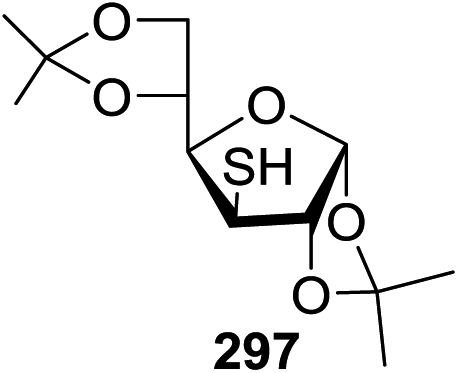 |
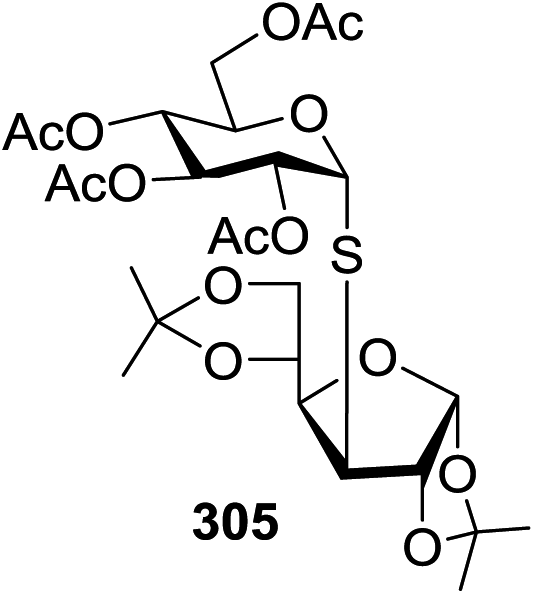 |
69 |
For the synthesis of S-disaccharide compounds, 306 was subjected coupling with a range of glycosyl thiols (294, 295, 307 and 308) in toluene or toluene–MeOH. Selective addition occurred in all cases with the thiyl radical adding axially to C-2 of the enose, thus resulting in the 3-deoxy thiodisaccharides 309–312, respectively (Table 39).
| Entry | Enose | Thiols | Product | Yields (%) |
|---|---|---|---|---|
| 1 | 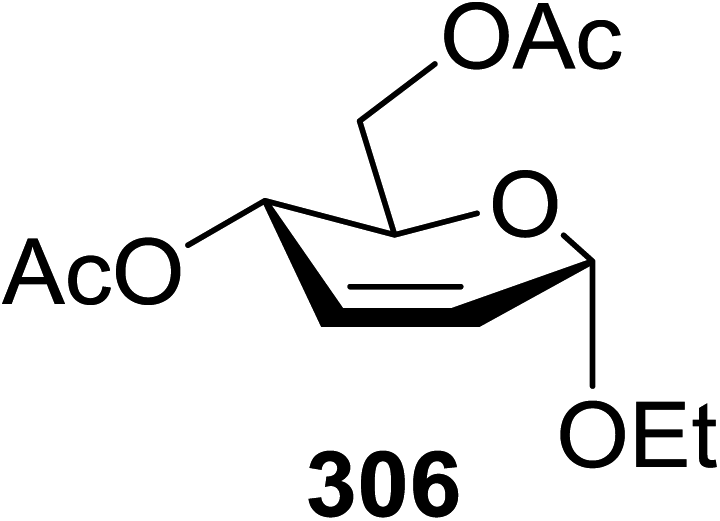 |
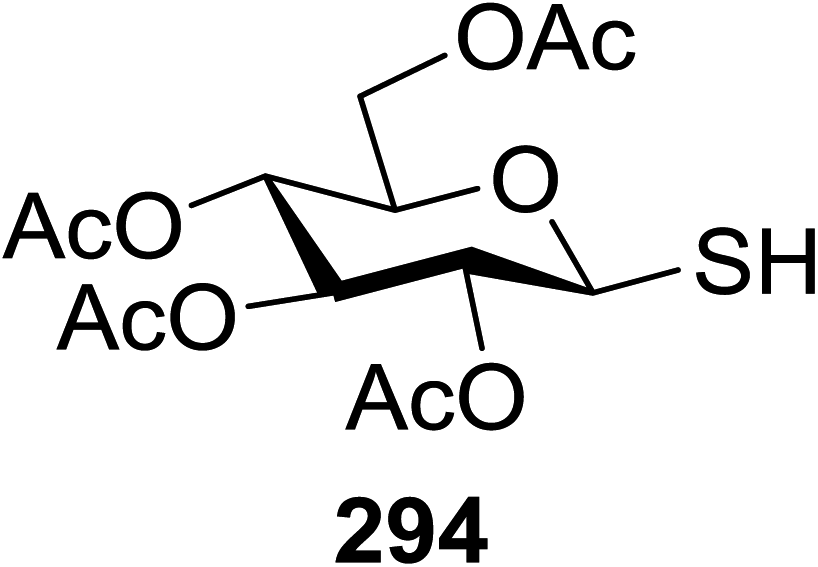 |
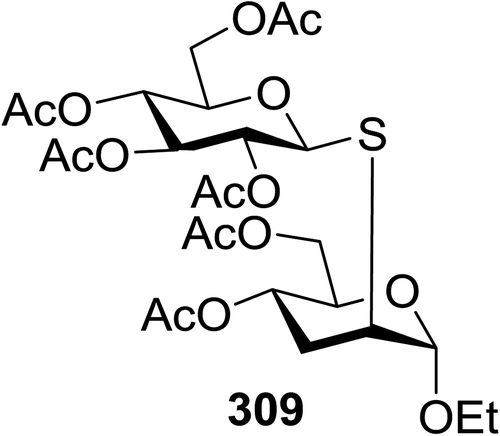 |
71 |
| 2 | 306 | 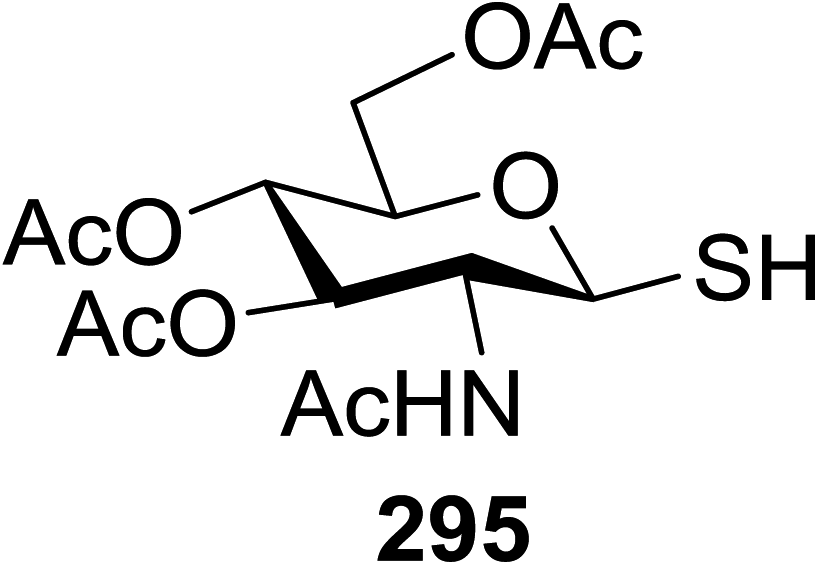 |
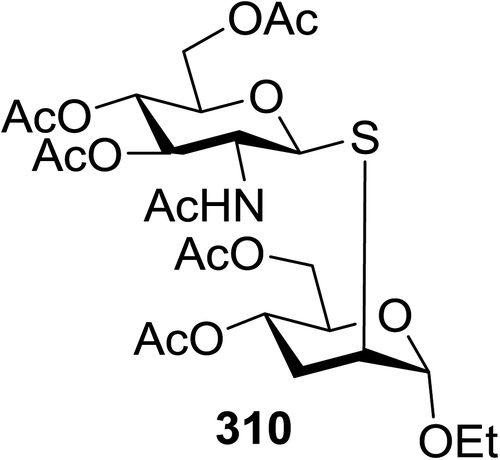 |
63 |
| 3 | 306 | 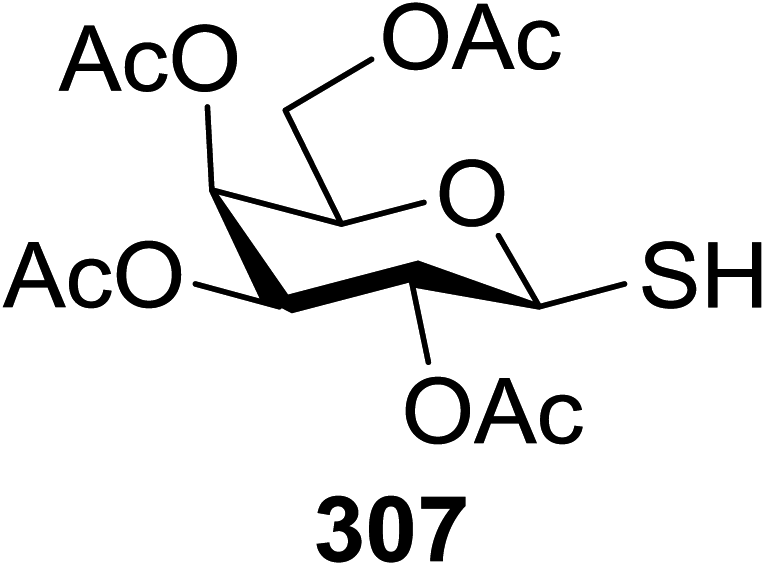 |
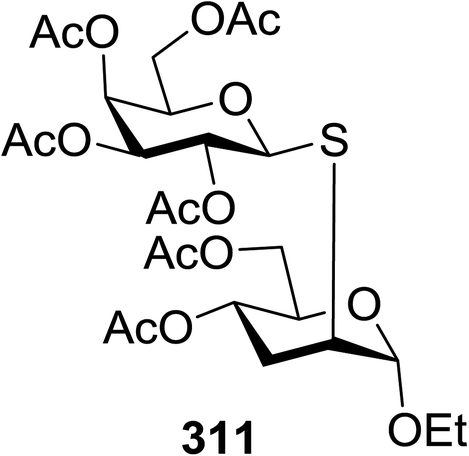 |
72 |
| 4 | 306 | 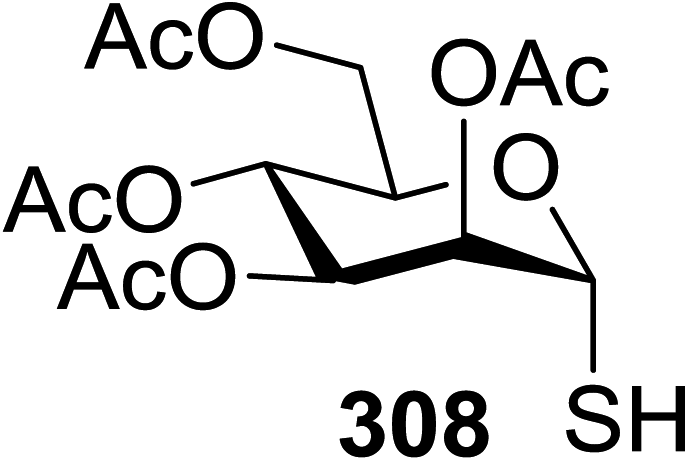 |
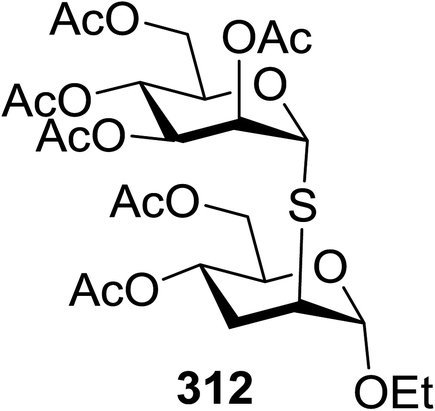 |
78 |
5. Photoinduced energy transfer
Another type of photocatalytic activation mechanism involves the sensitization of organic substrates through energy transfer. The mechanism for the activation of substrates in this method is the Dexter electron exchange mechanism (Fig. 28). Due to the outcome of the electron exchange process there is no net redox chemistry. Thus, electrochemical potentials are not useful predictors of reactivity for these reactions. The relative triplet state energies for both photocatalyst and substrate are the most important elements for the feasibility of energy transfer. This process can be summarized as simultaneous photoinduced electron transfer both to and from the excited state of the photocatalyst. This results in the generation of an electronically excited substrate molecule and concomitant relaxation of the photocatalyst to its electronic ground state.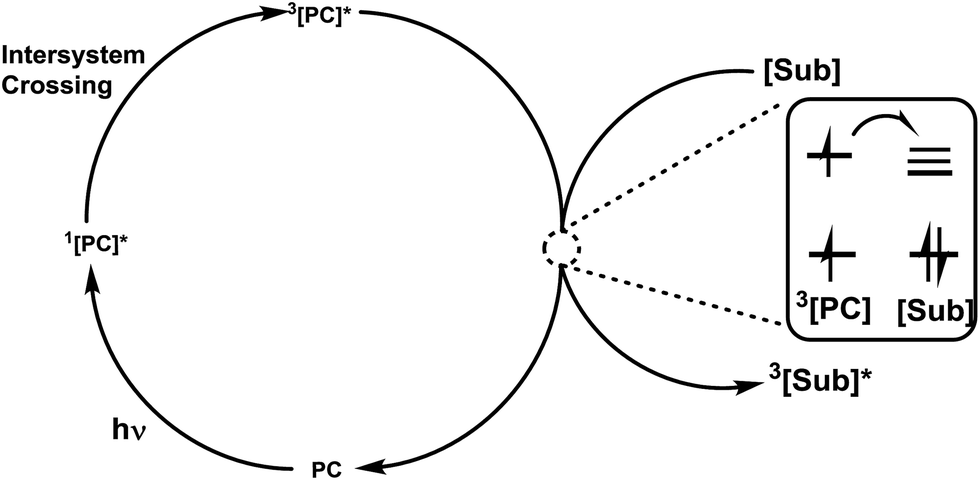 | ||
| Fig. 28 Mechanism for triplet energy transfer. | ||
Photosensitization by energy transfer provides access to the special reactivity of electronically excited molecules, which has considerable advantages over direct photoexcitation of the substrate. First, direct irradiation of ground-state, closed-shell organic molecules leads to the formation of excited singlets, which never undergo efficient intersystem crossing to long-lived triplet states and thus relax quickly to the ground state before useful bimolecular reactions can occur. On the other hand, triplet sensitizers generally undergo rapid intersystem crossing and provide a more efficient route for the production of triplet-state organic compounds. Additionally, the direct photoexcitation of most organic compounds requires relatively high-energy UV light which can be incompatible with common organic functional groups.
6. Application in chemical biology and materials chemistry
The carbohydrate motif greatly affects protein folding, immunogenicity and stability toward proteases as well as governs biological properties and activities. Hence, the expansion of methods for peptide and protein glycosylation via efficient and site-specific ligation tools is the focus of attention in the fields of biotechnology and proteomics. Synthetic glycopeptides and glycoproteins enclosing unnatural linkages between carbohydrate and aglycone moieties have been reported in recent years. Methods for the synthesis of glycopeptides utilizing diverse peptide-ligation protocols based on chemoselective reactions have also been reported in recent years.6.1. THP ring C1–C10 fragment of madeirolide A synthesis
Lee and co-workers108 developed a concise synthetic route for madeirolide A, which features an iridium-catalyzed visible light induced radical cyclization for the construction of the THP ring and palladium-catalyzed glycosylation for the formation of the α-cineruloside linkage. The synthesis of a fully elaborated C1–C10 fragment of madeirolide A (315) was performed via the photoinduced glycosylation approach based on a series of stereospecific processes, which exhibited nanomolar cytotoxicity against tumor cell lines. Their study resulted in a stereoselective and robust synthetic pathway to a fully adorned C1–C10 fragment of madeirolide A. Starting from a readily available aldol product, total synthesis contributes the key intermediate through a series of diastereoselective processes which introduce Claisen rearrangement, iodolactonization and radical cyclization to establish all of the stereocenters along the C1–C10 chain with high stereocontrol (Fig. 29). The 2,6-cis-tetrahydropyran and α-cinerulose glycosidic linkage was established with complete stereospecificity by taking advantage of the iridium catalyzed reductive cyclization and palladium-catalyzed glycosylation, which demonstrates the efficiency of visible light photoredox catalysis.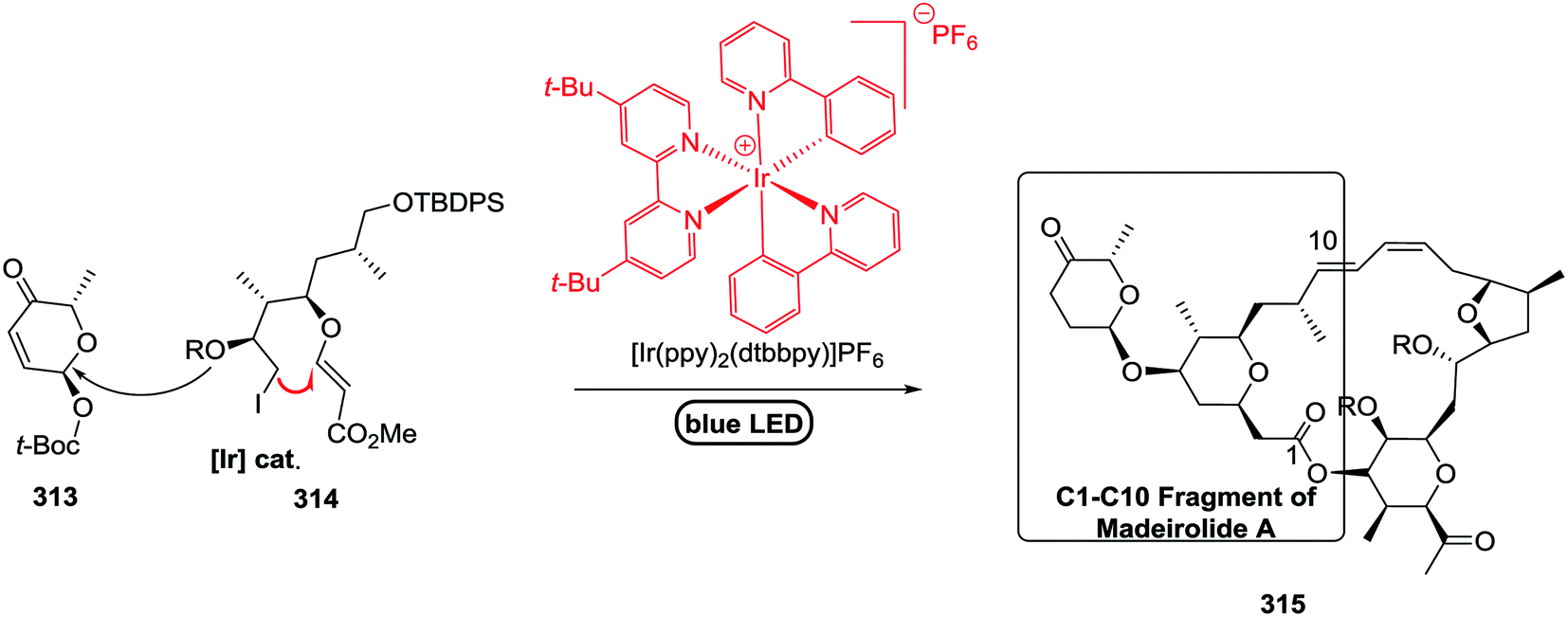 | ||
| Fig. 29 Construction of the THP ring via iridium-catalyzed visible light induced radical cyclization. | ||
6.2. Vaccines based on MUC1 glycopeptides
The use of vaccines based on MUC1 glycopeptides is an encouraging approach to treat cancer. Corzana and co-workers109 provided an improved way to synthesize these desirable compounds via a photoinduced method. However, only a slight improvement of the yield was achieved by this as compared to the thermal method. The reaction was comprised of the thiol–ene coupling of alkenyl-α-amino acids with a three-methylene bridge between S-α-GalNAc, and amino acid moieties were incorporated following the hydrothiolation protocol, which was investigated again by both thermally and photochemically (Scheme 26). Several sulfa-Tn antigens incorporated in MUC1 sequences possess a variable linker between the carbohydrate (GalNAc) and the peptide backbone.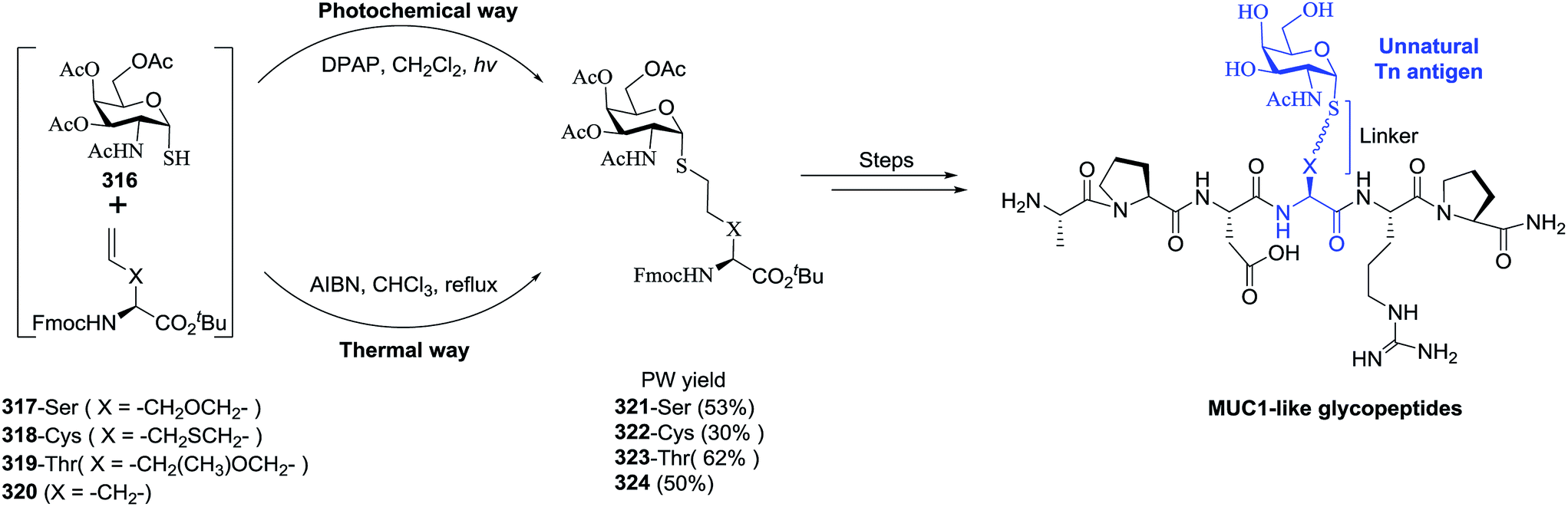 | ||
| Scheme 26 Synthesis of S-glycosyl amino acid building blocks via hydrothiolation. | ||
6.3. Glycodendrimer ligation
The use of free-radical thiol–ene coupling (TEC) for the introduction of carbohydrate, poly(ethylene glycol), and peptide fragments (Fig. 30) at the periphery of an alkene functional dendrimer was reported by Dondoni and co-workers.110 Four different sugar thiols including glucose, mannose, lactose and sialic acid, two PEGylated thiols, and the natural tripeptide glutathione were treated with a fourth generation alkene functional dendrimer[G4]-ene48 (Fig. 31) under irradiation at λmax 365 nm.111 This reaction is assigned as a click process due to its high chemoselectivity and regioselectivity as well as complete atom economy. These molecules are applied as globular multivalent tools in view of the participation of neuraminic acid in a variety of biomolecular recognition processes such as in viral adhesion of cells.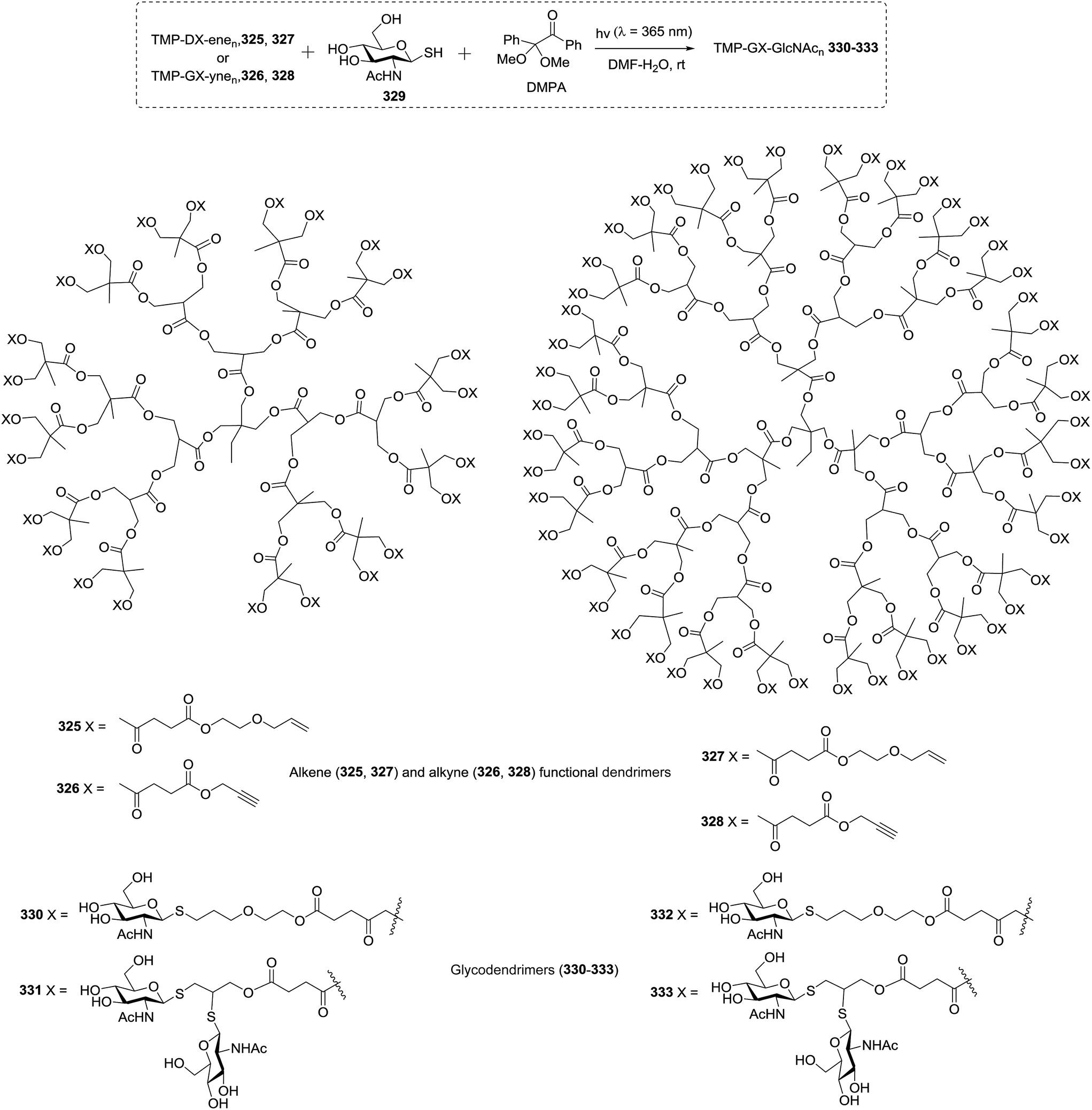 | ||
| Fig. 30 Thiol–ene and thiol–yne based synthesis of glycodendrimers. | ||
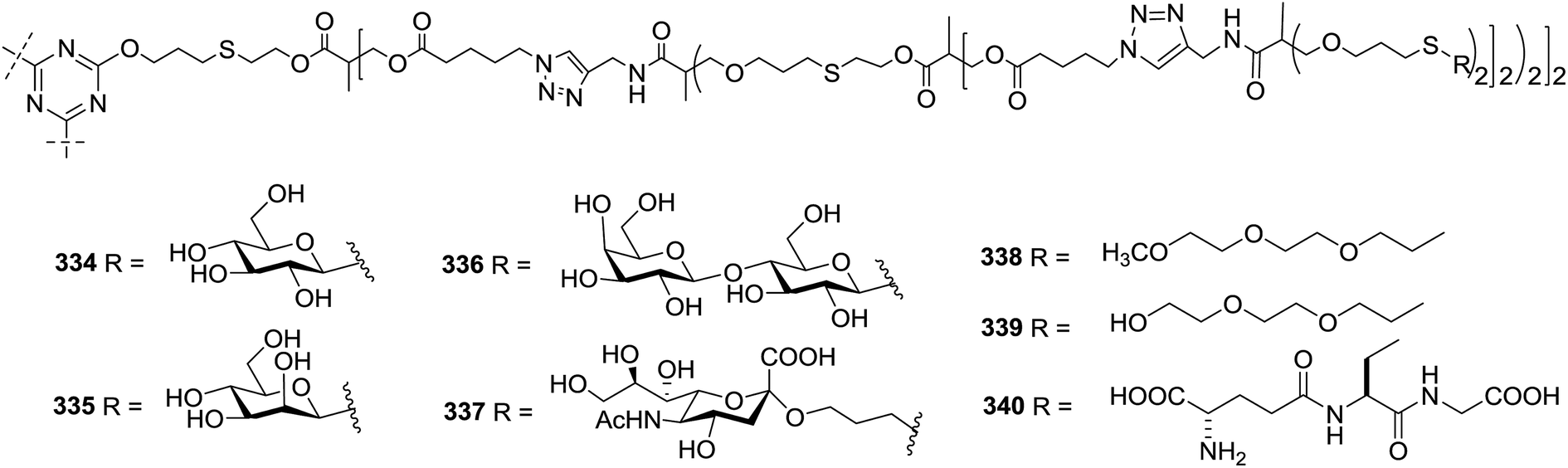 | ||
| Fig. 31 Functionalized dendrimers 334–340 with carbohydrate, PEG and peptide fragments. | ||
Glycodendrimers have been used to study biological processes occurring on cell surfaces, such as bacterial adhesion, and they have been synthesized by use of carbohydrate derivatives as branching elements with the aid of reductive amination or peptide chemistry. They were designed as mimetics of cell surface oligosaccharides of the high-mannose type, which are part of the glycocalyx. Many biological processes occurring on cell surfaces are dependent on interactions with the glycocalyx. One of these processes is the adhesion of bacteria to their host cells, which might trigger inflammation, apoptosis, or peptic ulcer, or might initiate other disease states of a cell. Stoddart and co-workers reported the synthesis of two glycodendrimers via the TEC of 6-O-allyl-glycosides with anomeric thiodisaccharides, followed by reductive amination to grow the dendrimers. Carbohydrate-based dendrons were synthesized by combining these two approaches, i.e. the photoaddition and reductive amination methodologies (Scheme 27).112 Trisaccharide monomers were used in the synthesis of di- and tetravalent lactoside dendrons. These reactions are chemoselective and work well in polar solvents such as methanol and water.
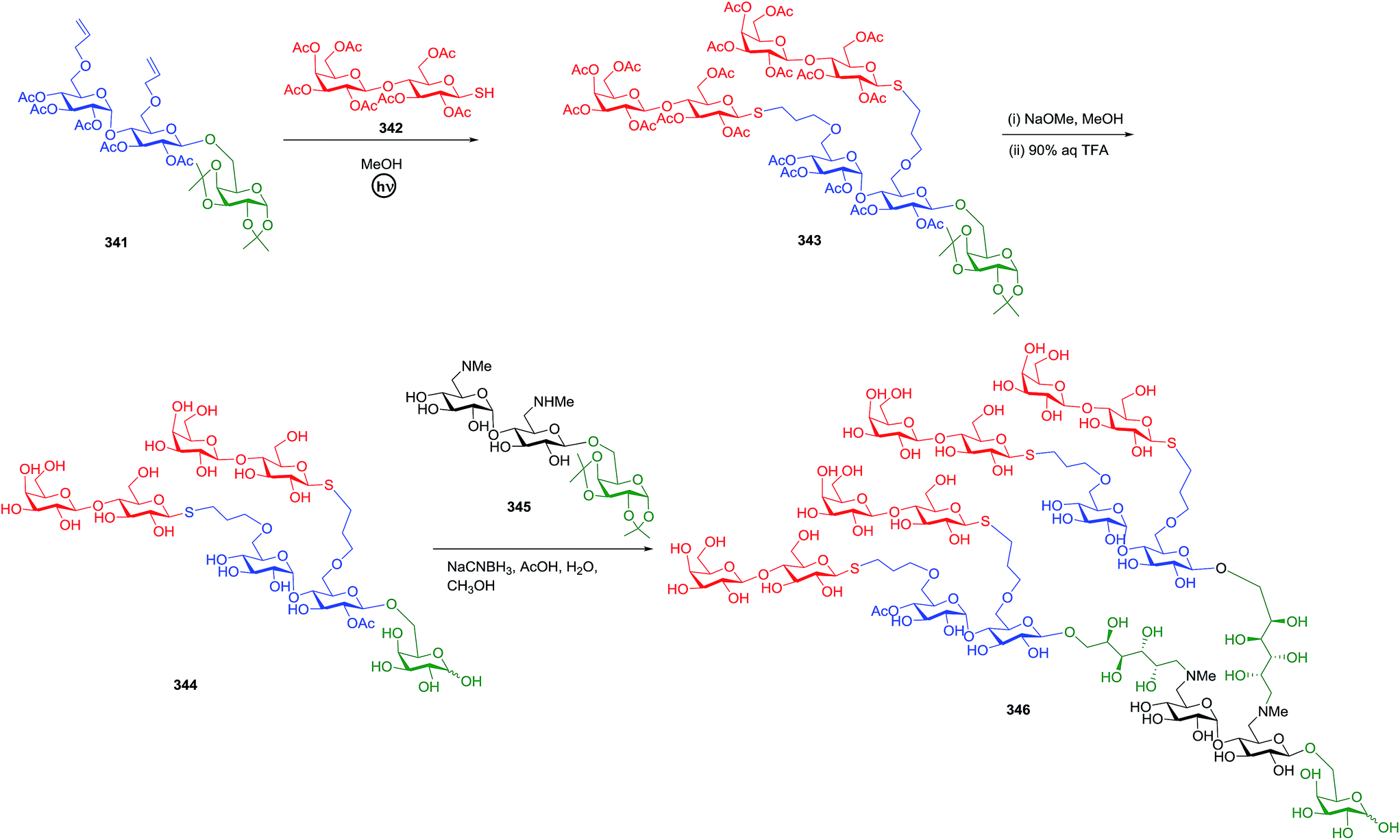 | ||
| Scheme 27 Synthesis of lactoside glycodendrons using photoaddition and reductive amination. | ||
6.4. Functionalization of glycoclusters
In 2000, Fulton D. A. and Stoddart J. F.113 synthesized carbohydrate clusters based on cyclodextrin via photoaddition reactions. The main step in their synthetic methodology was the attachment of carbohydrate residues to the cyclodextrin torus, which resulted in moderate-good yields (42–70%) by the photoaddition of thiol groups. Cyclodextrins (CDs) (Fig. 32) are naturally occurring cyclic oligosaccharides that exhibit a high demand for guest inclusion, which has already been incorporated in drug formulations. It has been found that CD-based glycoclusters could prove more useful for receptor-mediated drug delivery than that based on calixarenes. The synthesis of homogeneous cyclodextrin-based carbohydrate clusters, each substituted with β-D-thioglucosyl or D-thiolactosyl (Scheme 28) residues on either (a) the primary face, (b) the secondary face, (Scheme 29) or (c) both the primary and the secondary faces of their cyclodextrin was illustrated.114 | ||
| Fig. 32 Graphical representation of the synthesis of CD-based carbohydrate cluster compounds. | ||
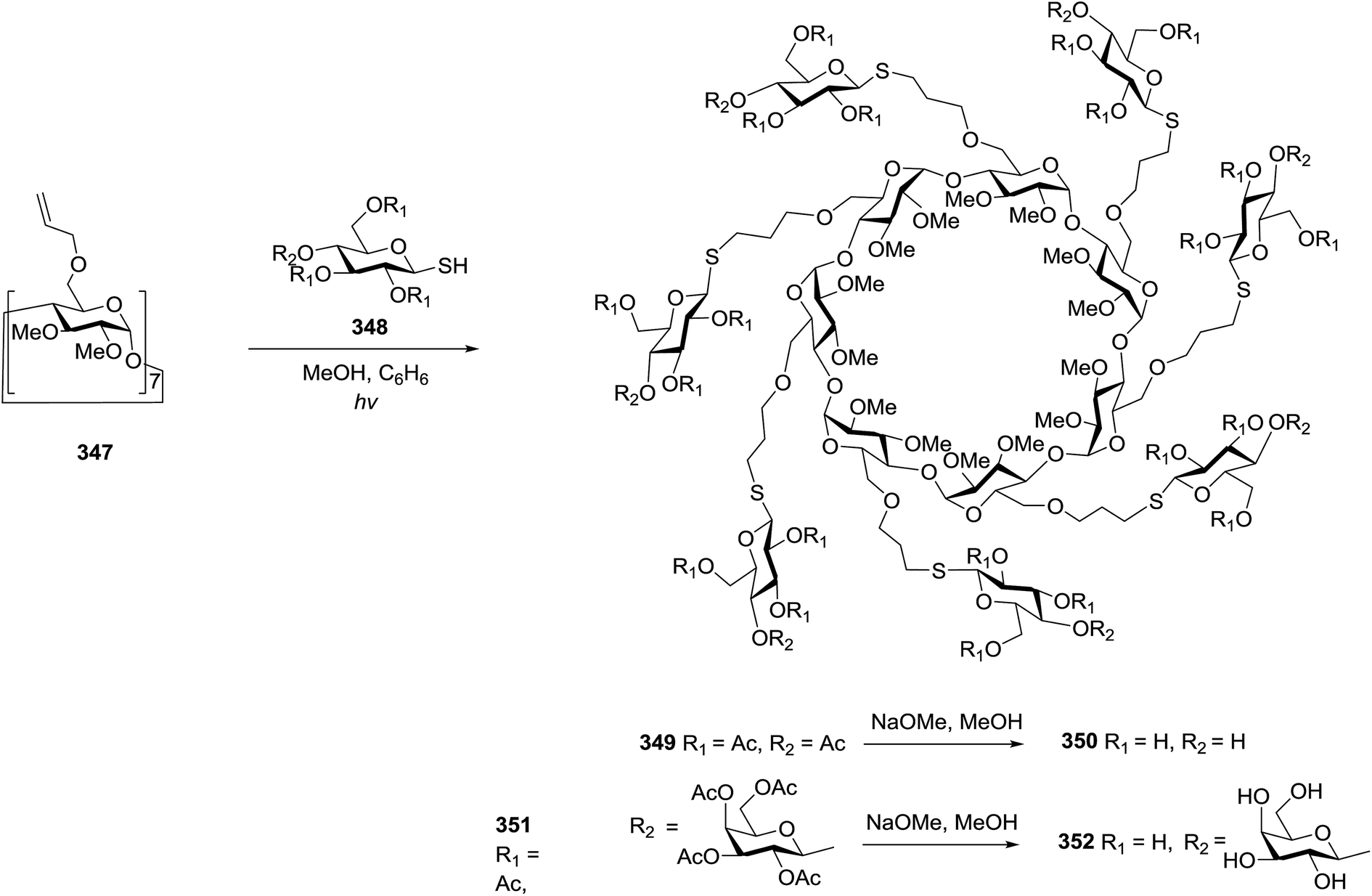 | ||
| Scheme 28 Synthesis of primary face-substituted clusters. | ||
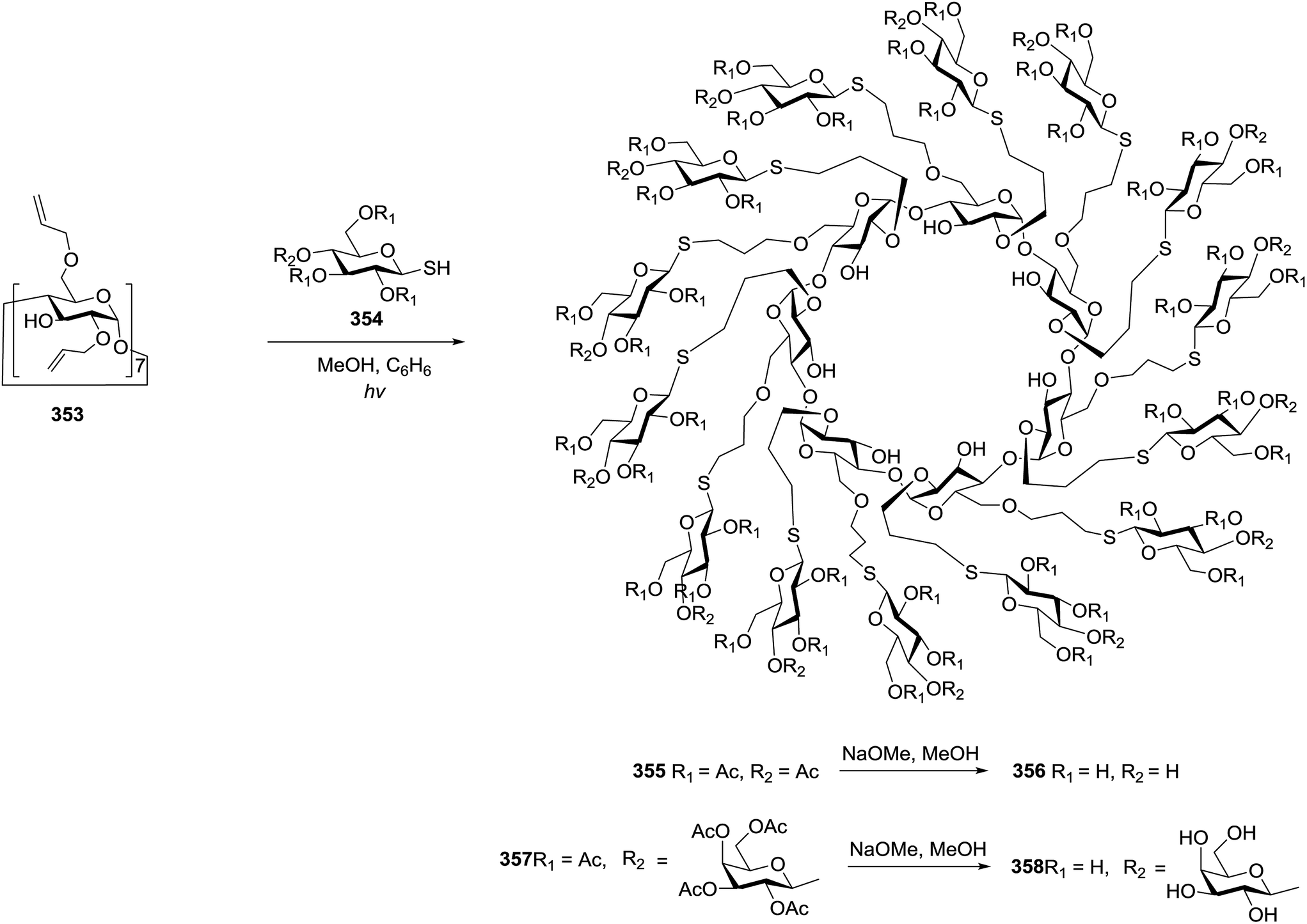 | ||
| Scheme 29 Synthesis of clusters substituted on both faces. | ||
Further modifications of CDs were reported by the Ravoo group in 2013,115 where TEC was employed to functionalize CDs with a broad range of thiols and hydrophilic and fluorinated moieties were incorporated at one, two or three sites on the each sugar unit. Cyclodextrins allylated at the 2-OH, 3-OH and/or 6-OH positions and the use of alkane thiols substituted with hydroxy, carboxylic acid, ester, protected amine and tetraethylene glycol groups resulted in a broad variety of functionalized cyclodextrins (Scheme 30). The introduction of both hydrophilic and fluorinated substituents on the same cyclodextrin is of interest in the synthesis of highly water-soluble fluorinated cyclodextrins.
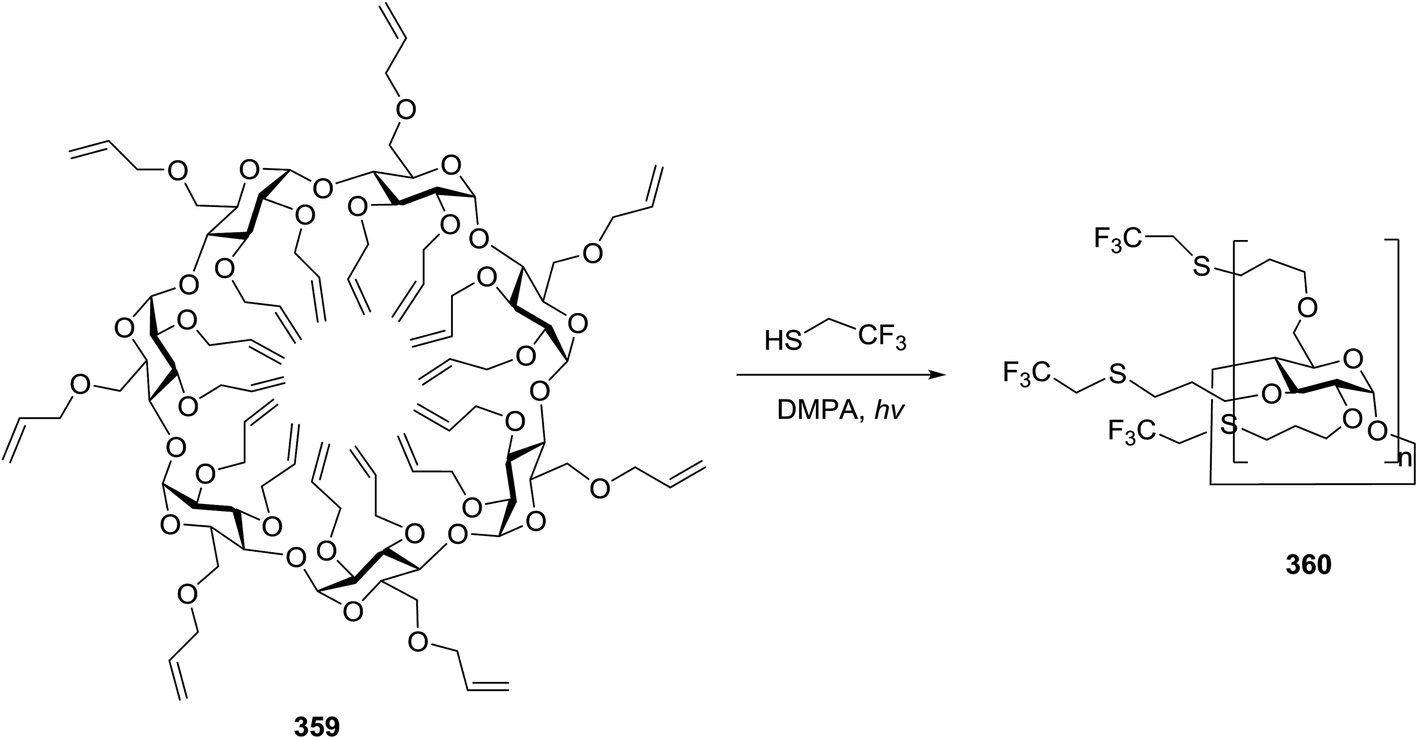 | ||
| Scheme 30 Functionalization of cyclodextrins via the photochemical thiol–ene ligation strategy. | ||
Dondoni and co-workers116 reported single and dual glycoside clustering around calix[4]arene scaffolds via photoinduced multiple thiol–ene coupling and azide–alkyne cycloaddition (Fig. 33). They synthesized compounds in which the carbohydrate fragments were implanted in the macrocycle through a carbon chain formed via TEC and CuAAC of calix[4]arene aldehydes and sugar phosphoranes. They described the dual clustering at the upper and lower rim of a calix[4]arene with two different sugars (galactose and glucose) via sequential copper(I)-catalyzed azide–alkyne cycloaddition and photoinduced thiol–ene coupling.
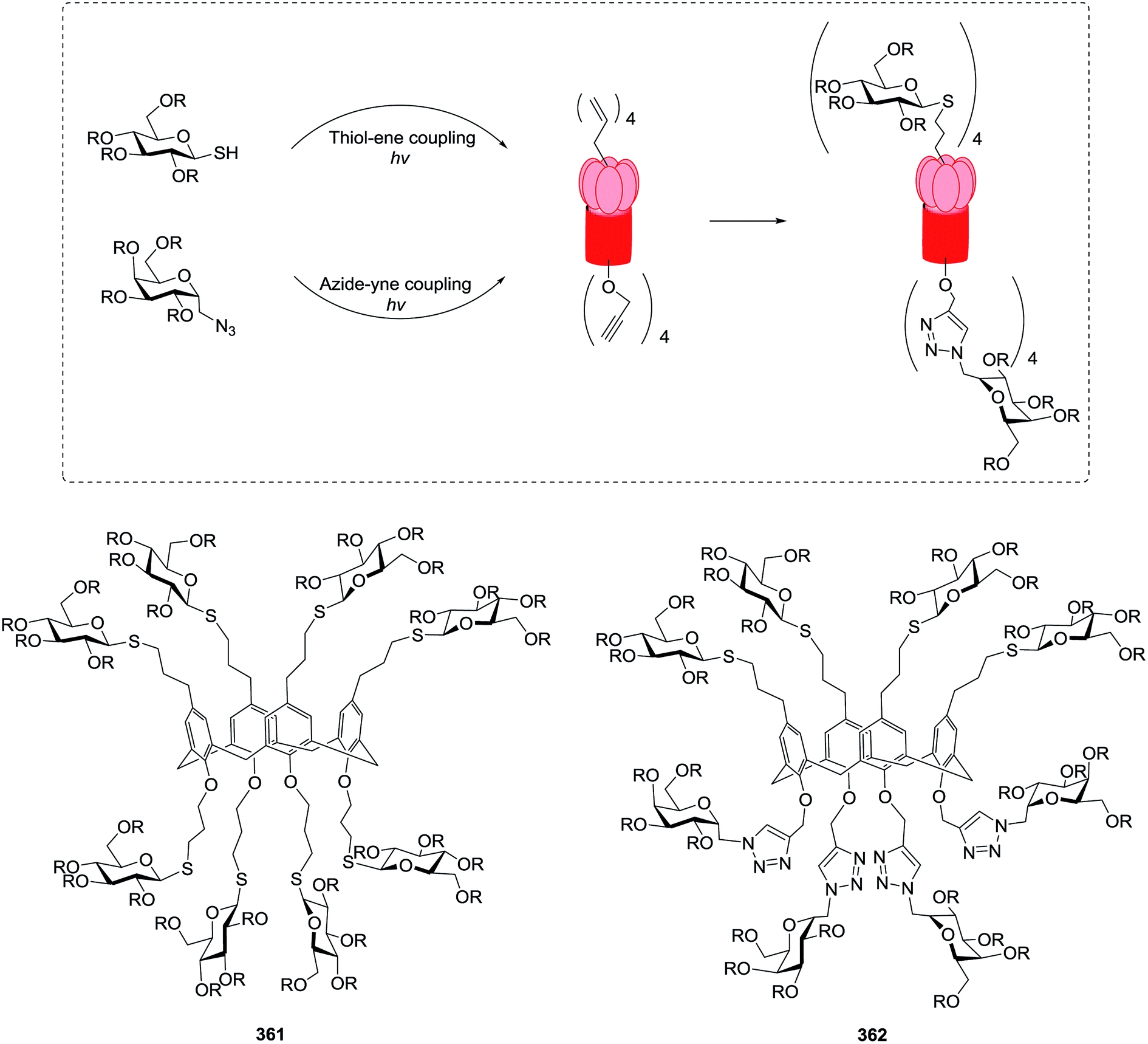 | ||
| Fig. 33 Glycoside clustering around calix[4]arene via click thiol–ene and azide–alkyne coupling. | ||
The research on similar types of motifs was further extended by Fessner and coworkers who synthesized a hybrid nanocluster possessing cubic symmetry.117 Chiara and coworkers discovered a versatile approach for the synthesis of highly functionalized cube-octameric polyhedral oligosilsesquioxane frameworks through click assembly.118 A new type of glycocluster based on polyhedral oligosilsesquioxanes (POSS) was efficiently prepared by Lee in 2004 (Fig. 34). The synthesis utilized unprotected mannoside and lactoside and exploited a diverse approach for the thiol-radical addition reaction. The diversity of this approach was exposed by functionalization of mannosides and lactosides with different-length spacers.
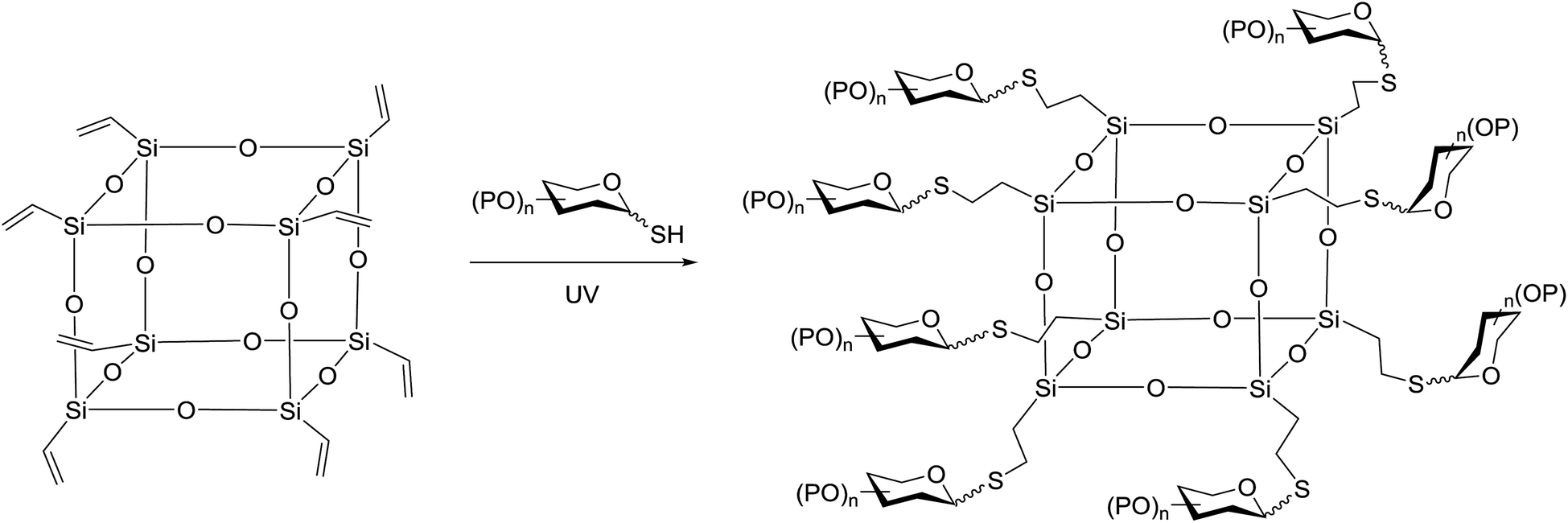 | ||
| Fig. 34 Synthesis of glycosylated POSS derivatives using thiol–ene. | ||
Multivalent octasilsesquioxane glyco- and peptido-conjugates were synthesized by Dondoni to explore the versatility of clustering compounds using the photoinduced free-radical thiol–ene coupling (TEC).
Octasilsesquioxane glyco-conjugates were obtained by coupling C-glycosyl propyl thiols and cysteine having peptides with the known octavinyl octasilsesquioxane and protein-conjugates were obtained by reacting glycosyl thiols with a new octasilsesquioxane derivative (Scheme 31).119
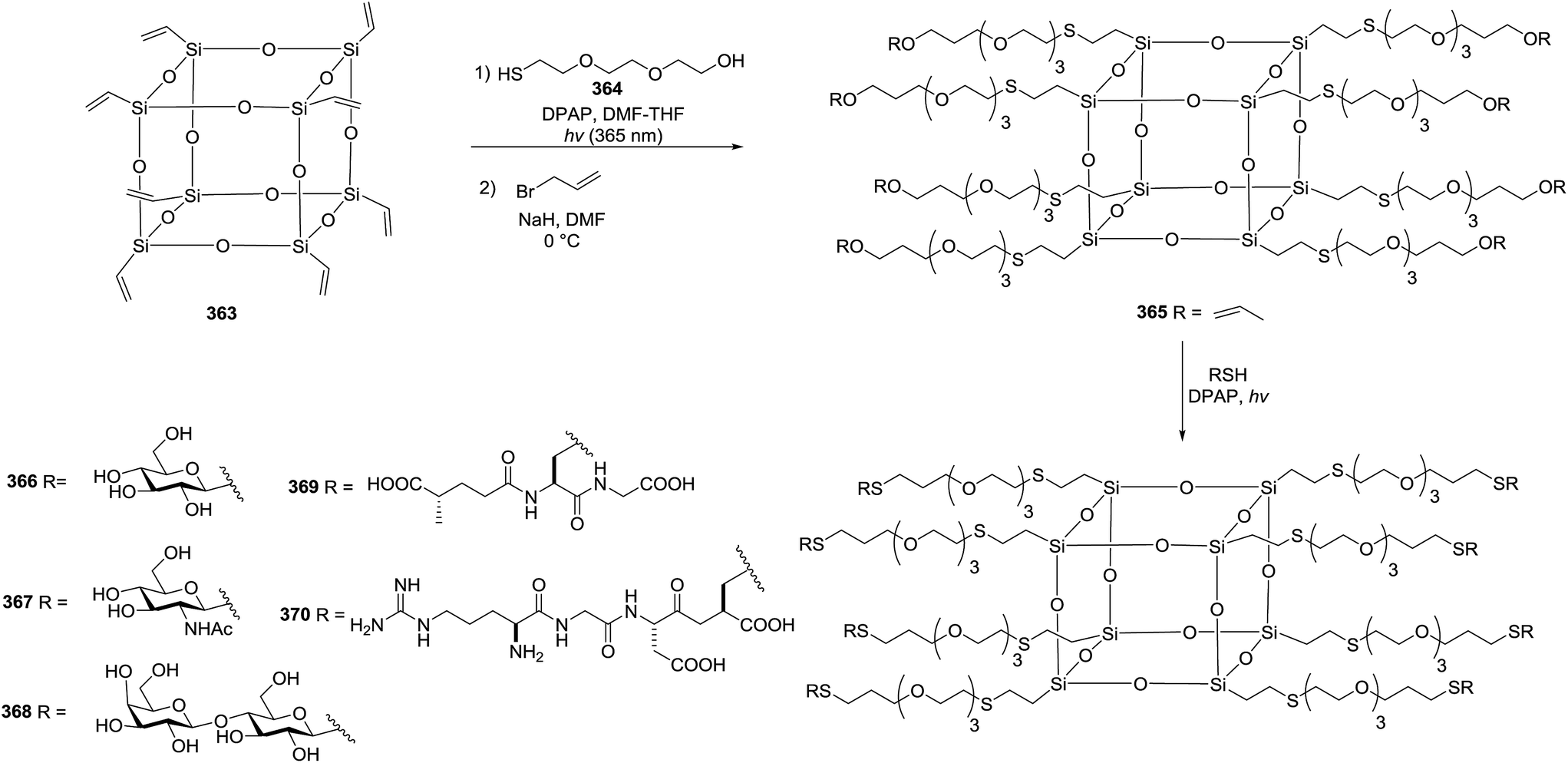 | ||
| Scheme 31 Synthesis of glyco- and peptido-conjugates from PEGylated POSS. | ||
6.5. Glycopeptide and glycoprotein synthesis
TEC as a strategy for the bioconjugation of carbohydrates with peptides and proteins has been employed in the search for novel vaccines against cancer. Sven Wittrock, Torsten Becker and Horst Kunz120 explored synthetic vaccines of tumor-associated glycopeptide antigens by thioether linkage to bovine serum albumin. The conjugates of synthetic glycopeptides with carrier proteins constitute the most versatile forms of synthetic vaccines (Scheme 32).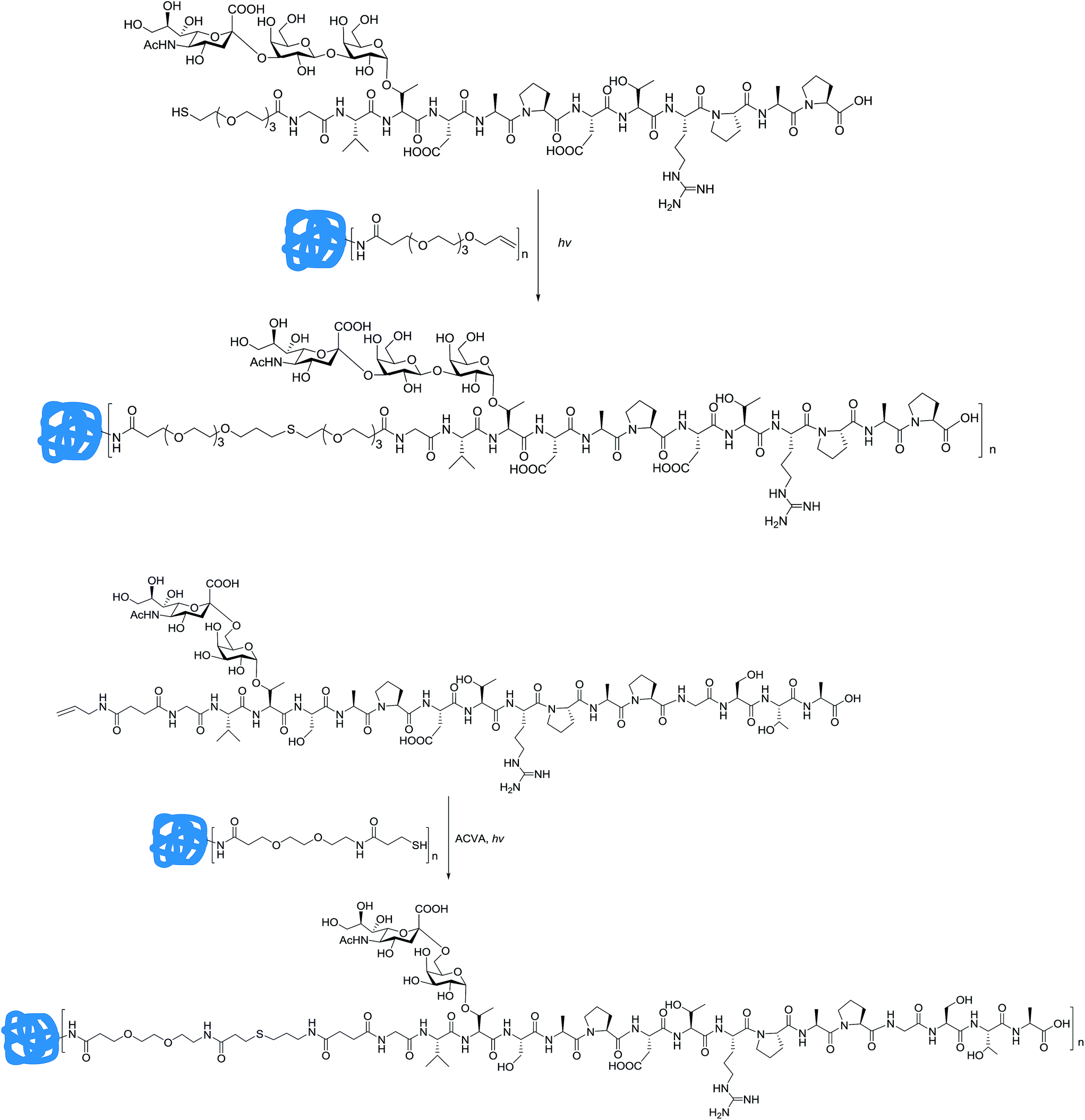 | ||
| Scheme 32 Thiol–ene ligation strategy with glycopeptides for the functionalisation of proteins by irradiation. | ||
Dondoni et al.121 reported the utilization of photoinduced glycosylation for the modification of peptides and proteins, which allows glycoconjugation and fluorescent labeling. Further development involves the sequential dual-ligation approach for the direct modification of bovine serum albumin via the free-radical hydrothiolation of alkynes (thiol–yne coupling, TYC) to unite two thiol fragments across the carbon–carbon triple bond to give a dithioether derivative with exclusive 1,2-addition. These results have significance not only as a flexible strategy for attaching two modifications at a single site in proteins, but also for the unexpected side-reactions of reagents (such as cycloalkynes) used in other protein coupling reactions (Scheme 33).
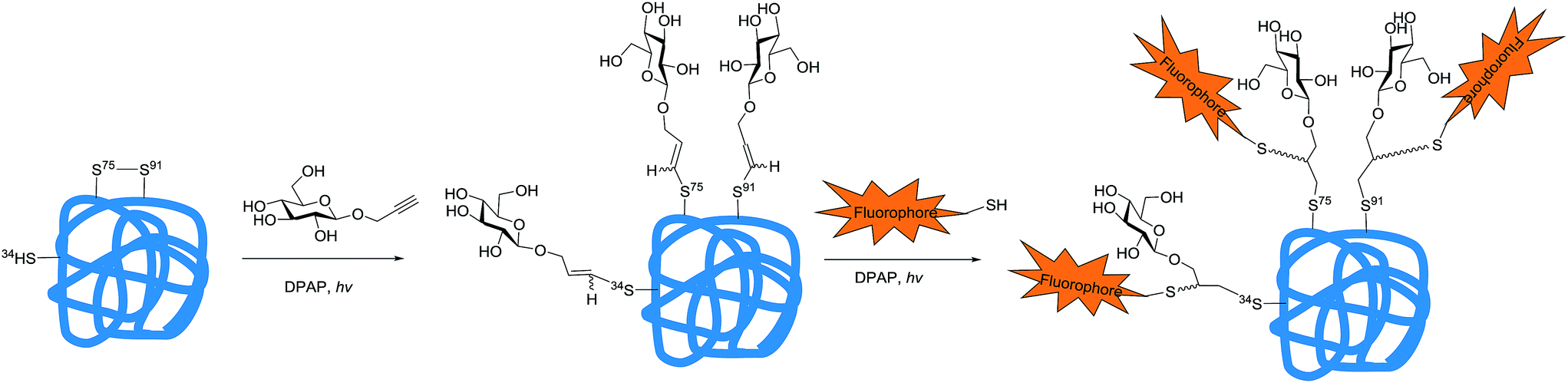 | ||
| Scheme 33 Bovine serum albumin ligation with carbohydrate using thiol–yne coupling. | ||
6.6. Polymerisation
Surface chemistry offers many applications for TEC and TYC ligation reactions, and the utility of these ligations to polymerization has been explored. Garrell and co-workers122 reported that a variety of either thiols or alkenes were conjugated to an allyltrialkoxysilane or a thiotrialkoxysilane via TEC to furnish active surface coating agents. Debuigne and co-workers123 prepared glycosurfactants via esterification at the C-6 position of mannose with mercaptopropanoic acid through TEC/TYC. Deviating from the standard solution phase applications of TEC in carbohydrate chemistry, Seeberger, Hartmann and coworkers124 applied thiol–ene ligation chemistry to solid phase poly(amidoamine) synthesis using a continuous flow photoreactor. For the preparation of sequence-defined carbohydrate-functionalised PAAs, the use of photochemical thiol–ene coupling (TEC) was an alternative tool to the established azide–alkyne cycloaddition reaction. In their study, two synthetic routes to carbohydrate-functionalised polymers were developed; one employed an olefin-bearing building block unit and the other employed a post-TEC carbohydrate-bearing monomer. In the former, the polymerisation step is carried out before the TEC step and in the latter the carbohydrate functionalised peptide is polymerised (Scheme 34). Schubert and co-workers125 employed TEC in polymer chemistry by using glycosyl thiols as the thiyl radical donor. The glycosylated polymers were found to be bio-responsive.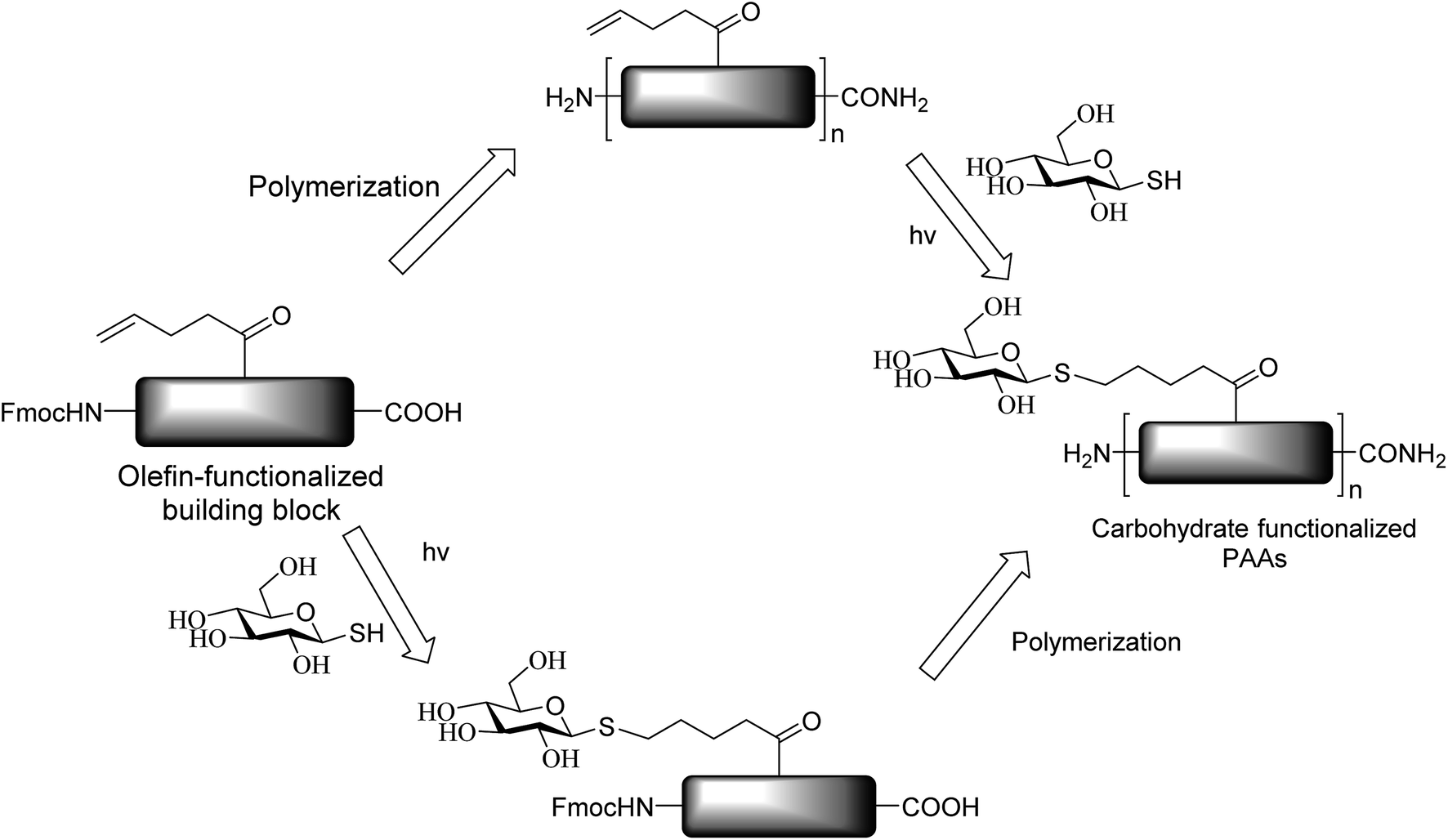 | ||
| Scheme 34 Synthesis of polymerized building blocks via the thiol–ene addition of thioglycoside with amidoamine. | ||
7. Summary and outlook
Widely useful glycosylation approaches are hugely fascinating for the synthesis of oligosaccharides, vaccines, thiosugars and glycoconjugates. The main targets of glycosylation are to diminish the extent of waste resulting from undesired reactivity, broaden the scope of the reaction to reactants with a broad range of protecting groups, high yields and increasing the chemoselectivity of donors. The use of photoinduced glycosylation protocols attains impressive selectivity in the construction of glycosidic bonds, which are is longer confined to neighboring group participation or the anomeric effect for directing orientation at the newly formed anomeric linkage.The impact of photoinduced glycosylation strategies in photochemistry might be uniquely rationalized as a consequence of the ways in which photocatalysis differs from other modes of catalysis. The capacity to absorb and convert the energy of a photon into useful chemical feature does not require energetically bonding interactions with the substrate. Thus, photocatalysts often do not require potentially reactive binding sites and they have a broad range of cocatalysts that are compatible with photocatalysis. As photochemistry becomes an increasingly common tool for synthesis, the advancement of new approaches will extend it use as powerful strategy for the photochemical construction of complex organic carbohydrate molecules.
Thiyl-radical mediated ligation reactions will play a vital role in advancing the future of glycoscience and in the advancement of novel therapeutics and materials. Moreover, photoinduced synthesis relies upon the regio- and stereoselective glycosylation of chemically derived substrates which results in an opportunity to advance the parallel and combinatorial syntheses of natural or non-natural carbohydrates of biological importance.
The broadness and scope of the applications discussed in this review indicate that glycosylation reactions mediated via light are highly compatible with carbohydrates and that these processes have significant relevance to the field of glycoscience and beyond. The substrates extend this highly functional ligation process to thiosugar synthesis, glycoconjugation, protein modification, glycodendrimer synthesis and the synthesis of functional materials.
Development in the synthesis of complex carbohydrates will also facilitate the expansion of new protocols for the synthesis of homogeneous glycoproteins, glycoclusters, glycodendrimers and glycovaccines.
The diverse range of applications of photoinduced glycosylation was highlighted for the preparation of glycosylated substrates and materials. We anticipate that this review on photoinduced glycosylation strategies will help researchers to further broaden the field of carbohydrate chemistry in the green aspects and open new doors for the utilisation of photoinduced glycosylation for a variety of applications in research.
Conflict of interest
The authors declare no competing financial interest.Acknowledgements
Rekha Sangwan highly acknowledges Council of Scientific and Industrial Research (CSIR) for Junior Research Fellowship. Author gratefully acknowledges financial support by Department of Science and Technology (DST), India (SB/ST-127/2012). This is CDRI communication No 9483.References
- (a) R. Owens and J. Nettleship, Functional and Structural Proteomics of Glycoproteins, Springer, Dordrecht, New York, 2011 Search PubMed; (b) A. Varki, Glycobiology, 1993, 3, 97–130 CrossRef CAS PubMed; (c) H. J. Allen and E. C. Kisailus, Glycoconjugates: Composition, Structure, and Function, Dekker, New York, 1992 Search PubMed; (d) R. A. Dwek, Chem. Rev., 1996, 96, 683–720 CrossRef CAS PubMed.
- M. Sznaidman, in Bioorganic Chemistry: Carbohydrates, ed. S. M. Hecht, Oxford University Press, New York, 1999, pp. 1–56 Search PubMed.
- (a) K. J. Doores, D. P. Gamblin and B. G. Davis, Chem.–Eur. J., 2006, 12, 656–665 CrossRef CAS PubMed; (b) S. Danishefsky and J. R. Allen, Angew. Chem., 2000, 112, 882–912 (Angew. Chem., Int. Ed., 2000, 39, 836–863) CrossRef; (c) S.-i. Hakomori and Y. Zhang, Chem. Biol., 1997, 4, 97–104 CrossRef CAS PubMed; (d) G.-J. Boons and Z. Guo, Carbohydrate-Based Vaccines and Immunotherapies, Wiley, Hoboken, NJ, 2009 Search PubMed; (e) B. Kuberan and R. J. Lindhardt, Curr. Org. Chem., 2000, 4, 653–677 CrossRef CAS.
- D. P. Galonic and D. Y. Gin, Nature, 2007, 446, 1000–1007 CrossRef CAS PubMed.
- (a) K. Toshima and K. Tatsuta, Chem. Rev., 1993, 93, 1503–1531 CrossRef CAS; (b) P. H. Seeberger and W.-C. Haase, Chem. Rev., 2000, 100, 4349–4393 CrossRef CAS PubMed; (c) J. D. C. Code, R. E. J. N. Litjens, L. J. van den Bos, H. S. Overkleeft and G. A. van der Marel, Chem. Soc. Rev., 2005, 34, 769–782 RSC; (d) P. H. Seeberger and D. B. Werz, Nature, 2007, 446, 1046–1051 CrossRef CAS PubMed; (e) P. H. Seeberger, Chem. Soc. Rev., 2008, 37, 19–28 RSC; (f) X. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900–1934 CrossRef CAS PubMed; (g) T. J. Boltje, T. Buskas and G.-J. Boons, Nat. Chem., 2009, 1, 611–622 CrossRef CAS PubMed; (h) C.-H. Hsu, S.-C. Hung, C.-Y. Wu and C.-H. Wong, Angew. Chem., Int. Ed., 2011, 50, 11872–11923 CrossRef CAS PubMed; (i) T. Nokami, K. Saito and J. Yoshida, Carbohydr. Res., 2012, 363, 1–6 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- (a) L. Furst, J. M. R. Narayanam and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2011, 50, 9655–9659 CrossRef CAS PubMed; (b) M. J. Schnermann and L. E. Overman, Angew. Chem., Int. Ed., 2012, 51, 9576–9580 CrossRef CAS PubMed; (c) D. S. Muller, N. L. Untiedt, A. P. Dieskau, G. L. Lackner and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 660–663 CrossRef CAS PubMed.
- (a) D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224–228 CrossRef CAS PubMed; (b) D. A. DiRocco, K. Dykstra, S. Krska, P. Vachal, D. V. Conway and M. Tudge, Angew. Chem., Int. Ed., 2014, 53, 4802–4806 CrossRef CAS PubMed.
- (a) B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed; (b) B. P. Fors, J. E. Poelma, M. S. Menyo, M. J. Robb, D. M. Spokoyny, J. W. Kramer, J. H. Waite and C. J. Hawker, J. Am. Chem. Soc., 2013, 135, 14106–14109 CrossRef CAS PubMed.
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillian, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- M. A. Ischay, Z. Lu and T. P. Yoon, J. Am. Chem. Soc., 2010, 132, 8572–8574 CrossRef CAS PubMed.
- (a) T. Furuta, K. Takeuchi and M. Iwamura, Chem. Commun., 1996, 157–158 RSC; (b) I. Cumpstey and D. Crich, J. Carbohydr. Chem., 2011, 30, 469–485 CrossRef CAS; (c) G. W. Griffin, N. C. Bandara, M. A. Clarke, W.-S. Tsang, P. J. Garegg, S. Oscarson and B. A. Silwanis, Heterocycles, 1990, 30, 939–947 CrossRef CAS.
- X. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900–1934 CrossRef CAS PubMed.
- A. Studer, Chem. Soc. Rev., 2004, 33, 267–273 RSC.
- A. Studer, Chem.–Eur. J., 2001, 7, 1159–1164 CrossRef CAS.
- G. J. Kavarnos and N. J. Turro, Chem. Rev., 1986, 86, 401–449 CrossRef CAS.
- J. C. Scaiano, J. Photochem., 1973, 2, 81–118 CrossRef CAS.
- M. D. Tzirakis, I. N. Lykakis and M. Orfanopoulos, Chem. Soc. Rev., 2009, 38, 2609–2621 RSC.
- N. J. Turro, Pure Appl. Chem., 1977, 49, 405–429 CrossRef CAS.
- D. A. Nicewicz and D. W. C. MacMillian, Science, 2008, 322, 73–77 CrossRef PubMed.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed.
- M. A. Ischay and T. P. Yoon, Eur. J. Org. Chem., 2012, 3359–3372 CrossRef CAS.
- K. Mizuno and Y. Otsugi, Topics in current chemistry, Berlin-Heidelberg, 1994, vol. 169 Search PubMed.
- (a) A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef CAS PubMed; (b) K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS; (c) A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS; (d) V. Balzani, G. Bergamini, F. Marchioni and P. Ceroni, Coord. Chem. Rev., 2006, 250, 1254–1266 CrossRef CAS; (e) M. Z. Hoffman, F. Bolletta, L. Moggi and G. L. Hug, J. Phys. Chem. Ref. Data, 1989, 18, 219–543 CrossRef CAS.
- G.-W. Griffin and N. C. Bandara, Heterocycles, 1990, 30, 201–203 CrossRef.
- M. Nakanishi, D. Takahashi and K. Toshima, Org. Biomol. Chem., 2013, 11, 5079–5082 CAS.
- K. Sakurai and D. Kahne, Tetrahedron Lett., 2010, 51, 3724–3727 CrossRef CAS PubMed.
- W. D. Hitz, P. J. Card and K. G. Ripp, J. Biol. Chem., 1986, 261, 11986–11991 CAS.
- H. Tanaka, A. Yoshizawa and T. Takahashi, Angew. Chem., Int. Ed., 2007, 46, 2505–2507 CrossRef CAS PubMed.
- K. Toshima and K. Tatsuta, Chem. Rev., 1993, 93, 1503–1531 CrossRef CAS.
- L. Mathew and S. Sankararaman, J. Org. Chem., 1993, 58, 7576–7577 CrossRef CAS.
- E. Kaji, T. Nishino, K. Ishige, Y. Ohya and Y. Shirai, Tetrahedron Lett., 2010, 51, 1570–1573 CrossRef CAS.
- W. J. Wever, M. A. Cinelli and A. A. Bowers, Org. Lett., 2013, 15, 30–33 CrossRef CAS PubMed.
- (a) J. M. Zen, S. L. Liou, A. S. Kumar and M. S. Hsia, Angew. Chem., Int. Ed., 2003, 42, 577–579 CrossRef CAS PubMed; (b) K. Okada, K. Okubo, N. Morita and M. Oda, Tetrahedron Lett., 1992, 33, 7377–7380 CrossRef CAS.
- M. L. Spell, K. Deveaux, C. G. Bresnahan, B. L. Bernard, W. Sheffield, R. Kumar and J. R. Ragains, Angew. Chem., Int. Ed., 2016, 55, 6515–6519 CrossRef CAS PubMed.
- N. Romero and D. A. Nicewicz, J. Am. Chem. Soc., 2014, 136, 17024–17035 CrossRef CAS PubMed.
- (a) J. Inanaga, Y. Yokoyama and T. Hanamoto, Tetrahedron Lett., 1993, 34, 2791–2794 CrossRef CAS; (b) J. Dinkelaar, A. R. De Jong, R. van Meer, M. Somers, G. Lodder, H. S. Overkleeft, J. D. C. Codee and G. A. van der Marel, J. Org. Chem., 2009, 74, 4982–4991 CrossRef CAS PubMed.
- Y. Yasu, T. Koike and M. Akita, Angew. Chem., 2012, 124, 9705–9709 (Angew. Chem., Int. Ed., 2012, 51, 9567–9571) CrossRef.
- S. Mattia, E. Arceo, I. D. Jurberg, C. Cassani and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 6120–6123 CrossRef PubMed.
- R.-Z. Mao, D.-C. Xiong, F. Guo, Q. Li, J. Duanb and X.-S. Ye, Org. Chem. Front., 2016, 3, 737–743 RSC.
- A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950–8958 CrossRef CAS PubMed.
- T. Koike and M. Akita, Top. Catal., 2014, 57, 967–974 CrossRef CAS.
- N. J. Straathof, B. J. Tegelbeckers, V. Hessel, X. Wang and T. Noel, Chem. Sci., 2014, 5, 4768–4773 RSC.
- T. Umemoto and S. Ishihara, J. Am. Chem. Soc., 1993, 115, 2151–2156 CrossRef.
- J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2014, 115, 650–682 CrossRef PubMed.
- I. Kieltsch, P. Eisenberger and A. Togni, Angew. Chem., Int. Ed., 2007, 46, 754–757 CrossRef CAS PubMed.
- P. Eisenberger, S. Gischig and A. Togni, Chem.–Eur. J., 2006, 12, 2579–2586 CrossRef CAS PubMed.
- T. Billard, B. R. Langlois, S. Large, D. Anker, N. Roidot and P. Roure, J. Org. Chem., 1996, 61, 7545–7550 CrossRef CAS PubMed.
- T. Billard, N. Roques and B. R. Langlois, Tetrahedron Lett., 2000, 41, 3069–3148 CrossRef CAS.
- T. Billard, N. Roques and B. R. Langlois, J. Org. Chem., 1999, 64, 3813–3820 CrossRef CAS.
- Y. Yu, D.-C. Xiong, R.-Z. Mao and X.-S. Ye, J. Org. Chem., 2016, 81, 7134–7138 CrossRef CAS PubMed.
- R. R. Schmidt and E. Rucker, Tetrahedron Lett., 1980, 21, 1421–1424 CrossRef CAS.
- T. J. Martin and R. R. Schmidt, Tetrahedron Lett., 1992, 33, 6123–6126 CrossRef CAS.
- Y. Zhao, Y.-P. Lu and L. Zhu, J. Carbohydr. Chem., 2008, 27, 113–119 CrossRef CAS.
- P.-J. Garegg, L. Maron and C.-G. Swahn, Acta Chem. Scand., 1972, 26, 3895–3901 CrossRef CAS.
- R.-Z. Mao, F. Guo, D.-C. Xiong, Q. Li, J. Duan and X.-S. Ye, Org. Lett., 2015, 17, 5606–5609 CrossRef CAS PubMed.
- R. S. Andrews, J. J. Becker and M. R. Gagne, Angew. Chem., Int. Ed., 2010, 49, 7274–7276 CrossRef CAS PubMed.
- J. Dupuis, B. Giese, D. Ruegge, H. Fishcer, H. G. Korth and R. Sustmann, Angew. Chem., 1984, 96, 887–888 CrossRef CAS.
- J.-P. Cheng, Y. Lu, X.-Q. Zhu, Y. Sun, F. Bi and J. He, J. Org. Chem., 2000, 65, 3853–3857 CrossRef CAS PubMed.
- R. S. Andrews, J. J. Becker and M. R. Gagne, Org. Lett., 2011, 13, 2406–2409 CrossRef CAS PubMed.
- R. S. Andrews, J. J. Becker and M. R. Gagne, Angew. Chem., Int. Ed., 2012, 51, 4140–4143 CrossRef CAS PubMed.
- (a) T. Ghaffar and A. W. Parkins, Tetrahedron Lett., 1995, 36, 8657–8660 CrossRef CAS; (b) T. Ghaffar and A. W. Parkins, J. Mol. Catal. A: Chem., 2000, 160, 249–261 CrossRef CAS.
- A. Cordova, S.-i. Watanabe, F. Tanaka, W. Notz and C. F. Barbas, J. Am. Chem. Soc., 2002, 124, 1866–1867 CrossRef CAS PubMed.
- T. Furuta, K. Takeuchi and M. Iwamura, Chem. Commun., 1996, 157–158 RSC.
- I. Cumpstey and D. Crich, J. Carbohydr. Chem., 2011, 30, 469–485 CrossRef CAS.
- V. S. Kumar and P. E. Floreancig, J. Am. Chem. Soc., 2001, 123, 3842–3843 CrossRef CAS PubMed.
- V. S. Kumar, D. L. Aubele and P. E. Floreancig, Org. Lett., 2001, 3, 4123–4125 CrossRef CAS PubMed.
- S. Mehta and B. M. Pinto, Tetrahedron Lett., 1991, 32, 4435–4438 CrossRef CAS.
- M. Spell, X. Wang, A. E. Wahba, E. Conner and J. Ragains, Carbohydr. Res., 2013, 369, 42–47 CrossRef CAS PubMed.
- A. J. Wain, I. Streeter, M. Thompson, N. Fietkau, L. Drouin, A. J. Fairbanks and R. G. Compton, J. Phys. Chem. B, 2006, 110, 2681–2691 CrossRef CAS PubMed.
- G. J. Kavarnos and N. J. Turro, Chem. Rev., 1986, 86, 401–449 CrossRef CAS.
- K. Tsuchii, M. Doi, T. Hirao and A. Ogawa, Angew. Chem., Int. Ed., 2003, 42, 3490–3493 CrossRef CAS PubMed.
- M. Tingoli, M. Tiecco, L. Testaferri and A. Temperini, J. Chem. Soc., Chem. Commun., 1994, 1883–1884 RSC.
- (a) F. E. McDonald and K. S. Reddy, Angew. Chem., Int. Ed., 2001, 40, 3653–3655 (Angew. Chem., 2001, 113, 3765–3767) CrossRef CAS; (b) K. N. Baryal, S. Adhikari and J. Zhu, J. Org. Chem., 2013, 78, 12469–12476 CrossRef CAS PubMed.
- H. Wang, J. Tao, X. Cai, W. Chen, Y. Zhao, Y. Xu, W. Yao, J. Zeng and Q. Wan, Chem.–Eur. J., 2014, 20, 17319–17323 CrossRef CAS PubMed.
- (a) V. Kren and T. Rezanka, FEMS Microbiol. Rev., 2008, 32, 858–889 CrossRef CAS PubMed; (b) J. M. Langenhan, B. R. Griffith and J. S. Thorson, J. Nat. Prod., 2005, 68, 1696–1711 CrossRef CAS PubMed.
- J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed.
- D. Liu, S. Sarrafour, W. Guo, B. Goulart and C. S. Bennett, J. Carbohydr. Chem., 2014, 33, 423–434 CrossRef CAS.
- F. Rasool, A. H. Bhat, N. Hussain and D. Mukherjee, ChemistrySelect, 2016, 1, 6553–6557 CrossRef.
- (a) J. Li, J. Zhang, H. Tan and D. Z. Wang, Org. Lett., 2015, 17, 2522–2525 CrossRef CAS PubMed; (b) W. Yang, S. Yang, P. Li and L. Wang, Chem. Commun., 2015, 51, 7520–7523 RSC; (c) S. Devari, M. A. Rizvi and B. A. Shah, Tetrahedron Lett., 2016, 57, 3294–3297 CrossRef CAS.
- Y. Nishikubo, S. Kanzaki, S. Matsumura and K. Toshima, Tetrahedron Lett., 2006, 47, 8125–8128 CrossRef CAS.
- R. Iwata, K. Uda, D. Takahashi and K. Toshima, Chem. Commun., 2014, 50, 10695–10698 RSC.
- T. Kimura, T. Eto, D. Takahashi and K. Toshima, Org. Lett., 2016, 18, 3190–3193 CrossRef CAS PubMed.
- Y. Geng, A. Kumar, H. M. Faidallah, H. A. Albar, I. A. Mhkalid and R. R. Schmidt, Angew. Chem., Int. Ed., 2013, 52, 10089–10092 CrossRef CAS PubMed.
- A. V. Demchenko, Synlett, 2003, 1225–1240 CrossRef CAS.
- T. Posner, Ber. Dtsch. Chem. Ges., 1905, 38, 646–657 CrossRef.
- K. Griesbaum, Angew. Chem., Int. Ed., 1970, 9, 273–287 CrossRef CAS.
- M. Fiore, A. Marra and A. Dondoni, J. Org. Chem., 2009, 74, 4422–4425 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- A. Dondoni, A. Massi, P. Nanni and A. Roda, Chem.–Eur. J., 2009, 15, 11444–11449 CrossRef CAS PubMed.
- G. Triola, L. Brunsveld and H. Waldmann, J. Org. Chem., 2008, 73, 3646–3649 CrossRef CAS PubMed.
- N. Floyd, B. Vijayakrishnan, J. R. Koeppe and B. G. Davis, Angew. Chem., Int. Ed., 2009, 48, 7798–7802 (Angew. Chem., 2009, 121, 7938–7942) CrossRef CAS PubMed.
- B. G. Davis, Pure Appl. Chem., 2009, 81, 285–298 CrossRef CAS.
- T. M. Kozlovska, I. Cielens, D. Dreilinna, A. Dislers, V. Baumanis, V. Ose and P. Pumpens, Gene, 1993, 137, 133–137 CrossRef CAS PubMed.
- M. Fiore, M. Lo Conte, S. Pacifico, A. Marra and A. Dondoni, Tetrahedron Lett., 2011, 52, 444–447 CrossRef CAS.
- S. Staderini, A. Chambery, A. Marra and A. Dondoni, Tetrahedron Lett., 2012, 53, 702–704 CrossRef CAS.
- K. Aoi, K. Tsutsumiuchi and M. Okada, Macromolecules, 1994, 27, 875–877 CrossRef CAS.
- (a) N. Franz and H.-A. Klok, Macromol. Chem. Phys., 2010, 809–820 CrossRef CAS; (b) R. Mildner and H. Menzel, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3925–3931 CrossRef CAS.
- (a) K.-S. Krannig, A. Doriti and H. Schlaad, Macromolecules, 2014, 47, 2536–2539 CrossRef CAS; (b) J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2010, 132, 15068–15071 CrossRef CAS PubMed; (c) J. R. Kramer and T. J. Deming, Biomacromolecules, 2012, 13, 1719–1723 CrossRef CAS PubMed.
- M. L. Conte, S. Pacifico, A. Chambery, A. Marra and A. Dondoni, J. Org. Chem., 2010, 75, 4644–4647 CrossRef PubMed.
- A. Dondoni, Angew. Chem., Int. Ed., 2008, 47, 8995–8997 CrossRef CAS PubMed.
- H. Bader, L. C. Cross, I. Heilbron and E. R. H. Jones, J. Chem. Soc., 1949, 619–623 RSC.
- (a) A. Chapman-Smith and J. E. Cronan, Trends Biochem. Sci., 1999, 24, 359–363 CrossRef CAS PubMed; (b) D. S. Y. Yeo, R. Srinivasan, G. Y. J. Chen and S. Q. Yao, Chem.–Eur. J., 2004, 10, 4664–4672 CrossRef CAS PubMed.
- M. Minozzi, A. Monesi, D. Nanni, P. Spagnolo, N. Marchetti and A. Massi, J. Org. Chem., 2011, 76, 450–459 CrossRef CAS PubMed.
- L. Lazar, M. Csavas, M. Herczeg, P. Herczegh and A. Borbas, Org. Lett., 2012, 14, 4650–4653 CrossRef CAS PubMed.
- N. Floyd, B. Vijayakrishnan, J. R. Koeppe and B. G. Davis, Angew. Chem., Int. Ed., 2009, 48, 7798–7802 CrossRef CAS PubMed.
- S. Hwang, I. Baek and C. Lee, Org. Lett., 2016, 18, 2154–2157 CrossRef CAS PubMed.
- V. R. Ocariz, I. Companon, C. Aydillo, J. Castro-Lopez, J. J. Barbero, R. H. Guerrero, A. Avenoza, M. M. Zurbano, J. M. Peregrina, J. H. Busto and F. Corzana, J. Org. Chem., 2016, 81, 5929–5941 CrossRef PubMed.
- M. Ghirardello, K. Oberg, S. Staderini, O. Renaudet, N. Berthet, P. Dumy, Y. Hed, A. Marra, M. Malkoch and A. Dondoni, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2422–2433 CrossRef CAS.
- M. L. Conte, M. J. Robb, Y. Hed, A. Marra, M. Malkoch, C. J. Hawker and A. Dondoni, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4468–4475 CrossRef CAS PubMed.
- A. Nelson and J. F. Stoddart, Carbohydr. Res., 2004, 339, 2069–2075 CrossRef CAS PubMed.
- D. A. Fulton and J. F. Stoddart, J. Org. Chem., 2001, 66, 8309–8319 CrossRef CAS PubMed.
- D. A. Fulton and J. F. Stoddart, Org. Lett., 2000, 2, 1113–1116 CrossRef CAS PubMed.
- M. M. Becker, Z. Zeng and B. J. Ravoo, Eur. J. Org. Chem., 2013, 6831–6843 CrossRef CAS.
- M. Fiore, A. Chambery, A. Marra and A. Dondoni, Org. Biomol. Chem., 2009, 7, 3910–3913 CAS.
- D. Heyl, E. Rikowski, R. C. Hoffmann, J. J. Schneider and W.-D. Fessner, Chem.–Eur. J., 2010, 16, 554–5548 Search PubMed.
- B. Trastoy, M. E. Perez-Ojeda, R. Sastre and J. L. Chiara, Chem.–Eur. J., 2010, 16, 3833–3841 CrossRef CAS PubMed.
- M. L. Conte, S. Staderini, A. Chambery, N. Berthet, P. Dumy, O. Renaudet, A. Marra and A. Dondoni, Org. Biomol. Chem., 2012, 10, 3269–3277 Search PubMed.
- S. Wittrock, T. Becker and H. Kunz, Angew. Chem., Int. Ed., 2007, 46, 5226–5230 CrossRef CAS PubMed.
- M. L. Conte, S. Staderini, A. Marra, M. Sanchez-Navarro, B. G. Davis and A. Dondoni, Chem. Commun., 2011, 47, 11086–11088 RSC.
- A. K. Tucker-Schwartz, R. A. Farrell and R. L. Garrell, J. Am. Chem. Soc., 2011, 133, 11026–11029 CrossRef CAS PubMed.
- C. Boyere, G. Broze, C. Blecker, C. Jerome and A. Debuigne, Carbohydr. Res., 2013, 380, 29–36 CrossRef CAS PubMed.
- F. Wojcik, A. G. O'Brien, S. Goetze, P. H. Seeberger and L. Hartmann, Chem.–Eur. J., 2013, 19, 3090–3098 CrossRef CAS PubMed.
- C. von der Ehe, J. A. Czaplewska, M. Gottschaldt and U. S. Schubert, Eur. Polym. J., 2013, 49, 2660–2669 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |