
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamics of structural diffusion in phosphoric acid hydrogen-bond clusters†
Parichart Suwannakham and
Kritsana Sagarik *
*
School of Chemistry, Institute of Science, Suranaree University of Technology, Nakhon Ratchasima 30000, Thailand. E-mail: kritsana@sut.ac.th; Fax: +66 44 224635; Tel: +66 44 224635
First published on 18th April 2017
Abstract
The dynamics and mechanism of structural diffusion in H3PO4 hydrogen-bond (H-bond) clusters were studied under excess proton conditions using ab initio calculations at the RIMP2/TZVP level and Born–Oppenheimer molecular dynamics (BOMD) simulations. The outstanding feature of our theoretical approach was the use of combined vibrational and NMR spectroscopic techniques that enable local dynamical motions and their activation energies to be studied in detail. Based on the concept of presolvation, H+(H3PO4)n (n = 2–5) clusters were selected as model systems, in which the rate-determining intermediate complexes in the structural diffusion process were studied in detail. The potential energy curves for proton displacement suggested that Zundel-like complexes are preferentially formed in linear H-bond chains in low local-dielectric environments, whereas Eigen-like complexes are stabilized in high local-dielectric environments. The RIMP2/TZVP results revealed that the Eigen–Zundel–Eigen mechanism, which leads to a positive charge displacement, is a short-range process (n = 2–3), and structural diffusion is enhanced by fluctuations in the H-bond chain length and local-dielectric environment. The vibrational and 1H NMR spectra obtained from BOMD simulations confirmed the proposed mechanism and suggested that for protonated H3PO4 clusters, structural diffusion can proceed without the reorientation of H3PO4 molecules (frustrated rotation), as in the case of neat liquid H3PO4. Because the H+ species cannot be directly probed in experiments, this theoretical study suggested for the first time characteristic properties of exchanging protons in H3PO4 systems under excess proton conditions, which can be used to assist experimental investigations.
Introduction
The dynamics and mechanism of proton transfer have been topics of interest not only in fundamental physical chemistry but also in the development of proton-conducting materials, which can be effectively used in electrochemical devices such as fuel cells. Because the rate of proton transfer from anode to cathode partly determines the efficiency of a fuel cell, theoretical and experimental studies have been conducted in recent decades to develop electrolytes that possess high proton conductivity and excellent thermal, mechanical and chemical stabilities.1,2 This is because commercially available fuel cells with hydrated proton-exchange membranes such as Nafion® can only be effectively operated at temperatures lower than the boiling point of water.2Possessing high proton-solvating and self-ionization abilities, as well as the ability to form extensive hydrogen-bond (H-bond) networks in the condensed phase, phosphoric acid (H3PO4) exhibits remarkably high intrinsic proton conductivity in the pure state and in aqueous solutions1,3 and, therefore, has been used effectively as an electrolyte in distributed fuel-cell power generators.4 Due to the presence of various charged species and difficulties in directly probing proton exchange, the dynamics and mechanism of proton transfer in liquid and solid H3PO4 are complicated and not well understood;1,2,5 proton transfer in liquid H3PO4 has been suggested to occur almost exclusively through a structural diffusion process, also regarded as the protonic-chain conduction mechanism.6
Nuclear magnetic resonance (NMR) spectroscopy has been effectively used to obtain fundamental information on the molecular structures and dynamics of atoms in different local chemical environments.7 For a prototypical system such as protonated water clusters, 1H NMR experiments suggested the chemical shift of the shared proton (“naked proton”) in the Zundel complex (H5O2+) to be 21.3 ppm,8 whereas for neutral water clusters, the chemical shifts in polar and non-polar solvents were 4.6–5.5 ppm.9 In regards to H3PO4 systems, 1H and 31P pulsed gradient Hahn spin-echo (PGSE) NMR spectroscopy has been used to study mobile species in 85 wt% (14.6 M) H3PO4 solution.10 The self-diffusion coefficients (D) obtained based on the analysis of the 1H NMR peak at 9.8 ppm and the approximation that H+ and 1H diffuse at the same rate suggested that protons are transferred much faster than phosphorus-carrying species. It was concluded in ref. 10 that the activation energy obtained from the slope of the line width-temperature dependence in conventional 1H NMR experiments and that derived from a simple Arrhenius-type equation in 1H PGSE NMR experiments are not the same due to the different time and length scales probed by these two techniques, providing energies of 11 and 25 kJ mol−1, respectively.
1H and 31P pulsed field gradient (PFG)-NMR spectroscopy was used to study the dynamics of mobile species in liquid anhydrous H3PO4 over the temperature range of 290–420 K.11 The average self-diffusion coefficients (D) of all 1H atoms were measured and compared with those obtained from high resolution quasi-elastic neutron scattering (QNS)12 and conductivity measurements.6,13 It appeared that the PFG-NMR and QNS methods yielded similar proton self-diffusion coefficients, whereas that obtained from conductivity measurements is significantly smaller because the fast diffusion process was not probed in the conductivity measurements.11 Analysis of the experimental data led to the conclusion that in the glassy state, the self-diffusion coefficients obtained from 1H and 31P PFG-NMR measurements cannot be described by the simple Arrhenius equation.11
The mechanism of proton transfer in “neat” liquid H3PO4 was studied using Car–Parrinello molecular dynamics (CPMD) simulations on 54 H3PO4 molecules.14 Based on the analysis of proton coupling correlation functions, the proton transfer under neutral conditions was concluded to involve structural diffusion, which is associated with specific H-bond rearrangements in the surrounding environment. It was anticipated that to prevent the proton from returning to the original H3PO4 and to ensure that the negatively and positively charged species are fully separated, the linking H-bond chain should be broken. This can be accomplished through the rotation of an H3PO4 molecule due to “frustrated” configurations in the H-bond network. It was also concluded that in neat liquid H3PO4, strong, polarizable H-bonds produce coupled proton motion and a pronounced dielectric response in the medium, leading to the formation of extended, polarized H-bond chains.
In our previous work,15 ab initio calculations and Born–Oppenheimer molecular dynamics (BOMD) simulations were used to study the dynamics and mechanism of proton dissociation and structural diffusion in hydrated H3PO4 clusters. This theoretical study focused on the effects of the introduction of water into liquid H3PO4, which leads to an increase in the proton conductivity of the liquid.13 Because the effects of electron correlation play important roles in many-electron systems and BOMD simulations based on quantum chemical methods that include the effects of electron correlation are very CPU time consuming, the applicability and performance of second-order Møller–Plesset perturbation theory with the resolution of the identity approximation (RIMP2) were assessed in this work. Based on the concept of presolvation and the observation that the intermediate complexes are preferentially formed under excess proton conditions, H+(H3PO4)(H2O)n (n = 1–4) clusters were selected as model systems. The static results revealed that the rate-determining intermediate complex for proton dissociation is preferentially formed in low local-dielectric environments (e.g., ε = 1), whereas proton transfer from the first to the second hydration shell is driven by fluctuations in the number of water molecules in high local-dielectric environments (e.g., ε = 78). The BOMD results over the temperature range of 298–430 K validated the proposed mechanism and rate-determining process and iterated that insights into the dynamics and mechanism of structural diffusion in microhydrated H-bond systems can be obtained based on a carefully selected presolvation model.
In this work, the dynamics and mechanism of structural diffusion in H3PO4 H-bonds were studied under excess proton conditions using the RIMP2 method and BOMD simulations. Because the H+ species cannot be directly probed in experiments10 and previous theoretical studies focused on proton transfer in liquid H3PO4 under neutral conditions,14,16 to provide fundamental information that can be used as a guideline in experimental investigations, the present theoretical study focused on the dynamics of exchanging protons (H+) in the rate-determining intermediate complexes and the reorientation of the H3PO4 H-bond chain (frustrated rotation), which has been suggested to be the determining factor in the rate of proton transfer in neat liquid H3PO4.14 Based on the concept of presolvation, H+(H3PO4)n (n = 2–5) clusters were selected as model systems, for which characteristic vibrational and 1H NMR spectra of the H-bonds in the intermediate complexes were computed. The potential energy curves for proton displacement in ε = 1 and 61 (ref. 17) were constructed and used to suggest the structural diffusion mechanism. The dynamic results obtained from BOMD simulations were used to confirm the proposed mechanism and are discussed in comparison with the available theoretical and experimental data.
Computational methods
Quantum chemical calculations
To consider the effects of electron correlation, the RIMP2 method was employed with the TZVP basis set, abbreviated RIMP2/TZVP. This ab initio method has been tested and used successfully in our previous studies on protonated H-bond systems, e.g., protonated water clusters,18 H3PO4-doped imidazole (Im) systems,19 and microhydrated H3PO4 clusters.15 Because several of our previous studies involved density functional theory (DFT),19–22 to investigate the performance of the DFT method, the RIMP2/TZVP results were compared with those obtained from B3LYP/TZVP calculations; although the DFT method has been applied effectively in various H-bond systems, there have been cases in which the DFT method fails to account for the equilibrium structures and interaction energies, especially for systems dominated by dispersion interactions.23 Three basic steps that were applied successfully in our previous studies24–26 were used in this work; (1) searching for equilibrium structures of the intermediate complexes in the structural diffusion process using the Test-particle model (T-model) potentials;15,24,25 (2) refining the equilibrium structures using RIMP2/TZVP and B3LYP/TZVP geometry optimizations and; (3) performing BOMD simulations using the refined intermediate structures as the starting configurations. All of the quantum chemical calculations and BOMD simulations were performed using the TURBOMOLE 7.02 software package.27Presolvation model
The concept of presolvation,6 which was used successfully in our previous studies on liquids and aqueous solutions,15,18,19,21,28 was adopted in this work; “the local-energy fluctuations and dynamics in aqueous solutions lead to the weakening or breaking of some H-bonds in the first and second hydration shells, resulting in a reduction in the water coordination number such that the proton-accepting species possesses a hydration structure corresponding to the species into which it will be transformed to complete the structural diffusion process”.15 Because experiments have shown that H4PO4+ plays an important role in structural diffusion in liquid H3PO4 (ref. 10) and there is only a 10% probability for quasi-coherent proton transfer in a four H-bond chain,14 to apply the concept of presolvation, H+(H3PO4)n (n = 2–5) clusters were selected as model systems. This choice is justified by the conclusion that the dynamics and energetics of the structural diffusion process in protonated H-bonds can be studied reasonably well using the smallest intermediate complexes as representatives.21,22,28,29In addition, because the fluctuations of local-dielectric environment have been shown to play an important role in the formation and stabilization of the intermediate complexes,15,18,19,21,28 the polarization effects of the surrounding H3PO4 molecules were taken into account;14,30 our previous studies showed that for the intermediate complex in a low local-dielectric environment, the shape of the potential energy curve for proton displacement is characterized by a single-well potential, whereas in a high local-dielectric environment, a double-well potential dominates,15,31 which is in accordance with the localized-charge solvation (LCS) concept.32 In this work, to account for the dielectric response of the medium, a conductor-like screening model (COSMO)33 with the dielectric constant of liquid H3PO4 (ε = 61)17 was included in the static and dynamic calculations.
Static calculations
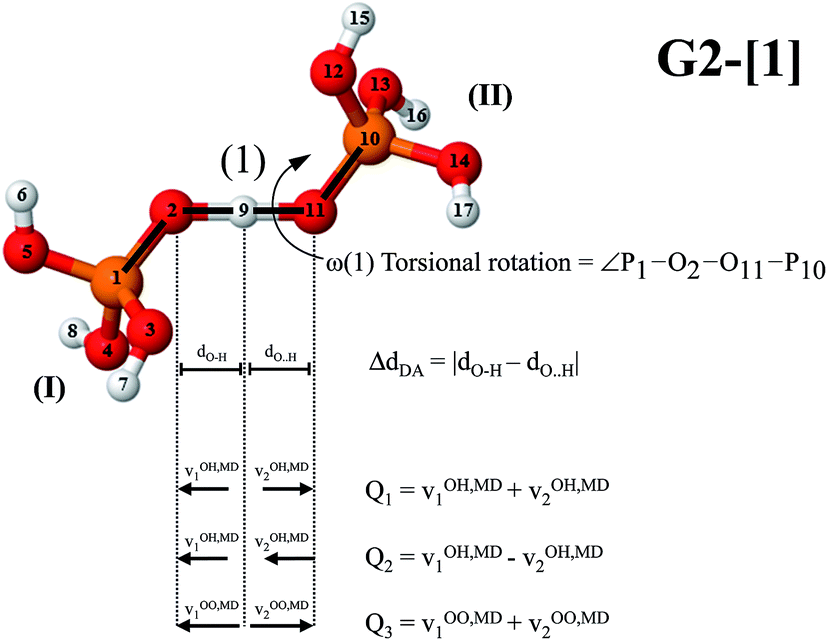 | ||
| Fig. 1 Velocity vectors (v1OH,MD, v2OH,MD, v1OO,MD and v2OO,MD) used in the calculations of vibrational spectra of O–H⋯O H-bond; Q1 = symmetric O–H stretch; Q2 = asymmetric O–H stretch and; Q3 = O–O vibration; ΔdDA = asymmetric stretching coordinate; torsional rotation of H-bond (1) (ω(1) = ∠P1–O2–O11–P10). | ||
The isotropic shielding constants of exchanging protons were calculated using the gauge-including atomic orbital (GIAO) method;34,35 the 1H NMR shielding constants at the electron correlated level 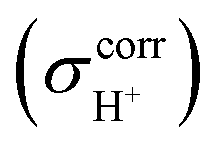 were approximated from
were approximated from
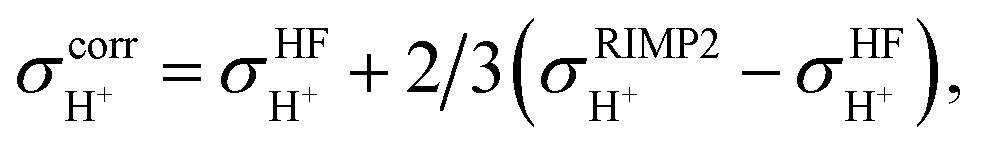 | (1) |
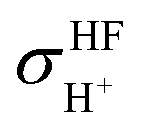 and
and 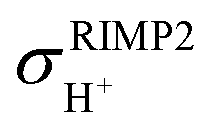 are the isotropic shielding constants computed at the Hartree–Fock (HF) and RIMP2 levels, respectively.36 The 1H NMR chemical shifts of the exchanging protons
are the isotropic shielding constants computed at the Hartree–Fock (HF) and RIMP2 levels, respectively.36 The 1H NMR chemical shifts of the exchanging protons 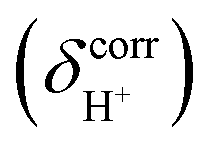 were derived from
were derived from 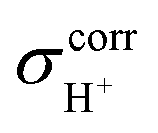 with respect to tetramethylsilane (TMS), which was 31.97 ppm at the MP2/TZVP level.37 Because the isotropic shielding constants of protons in strong, protonated H-bonds depend only on the H-bond distance and not on the neighboring solvent molecules,38 and because the effects of the continuum solvent (reaction field) on the H-bond structure are generally small,37–39 it is reasonable to discuss only the 1H NMR results in the gas phase.
with respect to tetramethylsilane (TMS), which was 31.97 ppm at the MP2/TZVP level.37 Because the isotropic shielding constants of protons in strong, protonated H-bonds depend only on the H-bond distance and not on the neighboring solvent molecules,38 and because the effects of the continuum solvent (reaction field) on the H-bond structure are generally small,37–39 it is reasonable to discuss only the 1H NMR results in the gas phase.
Dynamics calculations
In this work, the dynamics of proton exchange in ε = 1 and 61 were studied using BOMD simulations over a temperature range of 298–430 K. The time step used to solve the equations of motion was set to 1 fs. In the BOMD simulations, 2000 steps were used in the equilibration calculations and 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps were used in the property calculations. We were aware of possible statistical errors due to the restricted number of time steps used in the analysis of time-dependent properties and applied the so-called “overlapped data collection” method40 in all our dynamic calculations, e.g., the velocity autocorrelation function (VACF) and mean-square displacement (MSD). To provide extra samples without extending the simulation time, the BOMD trajectory (e.g., in Fig. S1a†) was divided into equal overlapped intervals (nstep) with equally spaced time origins (nskip). The overlapped data collection method improved the quality of the averaged dynamic properties by generating additional sets of samples for statistical analyses.40
000 steps were used in the property calculations. We were aware of possible statistical errors due to the restricted number of time steps used in the analysis of time-dependent properties and applied the so-called “overlapped data collection” method40 in all our dynamic calculations, e.g., the velocity autocorrelation function (VACF) and mean-square displacement (MSD). To provide extra samples without extending the simulation time, the BOMD trajectory (e.g., in Fig. S1a†) was divided into equal overlapped intervals (nstep) with equally spaced time origins (nskip). The overlapped data collection method improved the quality of the averaged dynamic properties by generating additional sets of samples for statistical analyses.40
Because information on the effects of temperature on the dynamics of proton exchange is necessary to calculate the activation energy,15,18,19,21,28 BOMD simulations were performed over the canonical ensemble (NVT), in which heat exchange between the model system and solvent environment was allowed using a thermostat bath. In this work, a Nosé–Hoover chain thermostat was applied to each degree of freedom in the model systems.41–44 Setting the thermostat relaxation to twenty times the time step was proven in our previous work15,19,31 to generate reasonable local-energy fluctuations in the model systems. In this work, to ensure that the energetics and dynamic properties were well represented, the energy conservation and the stability of the numerical integration, as well as drifts (δA) in the energies and velocities of the exchanging proton, were evaluated in the NVT-BOMD simulations using the method discussed in ref. 45 and the smallest intermediate complex as a representative.
In addition, the simple Arrhenius equation,
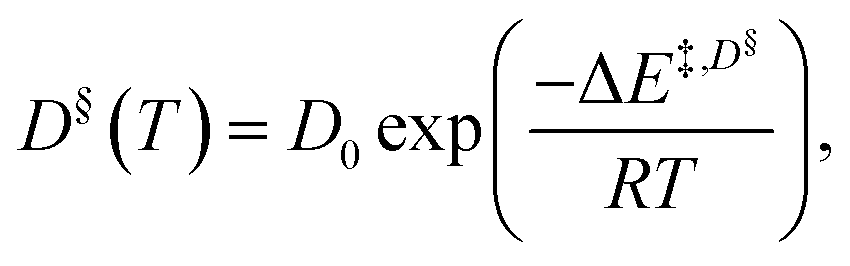 | (2) |
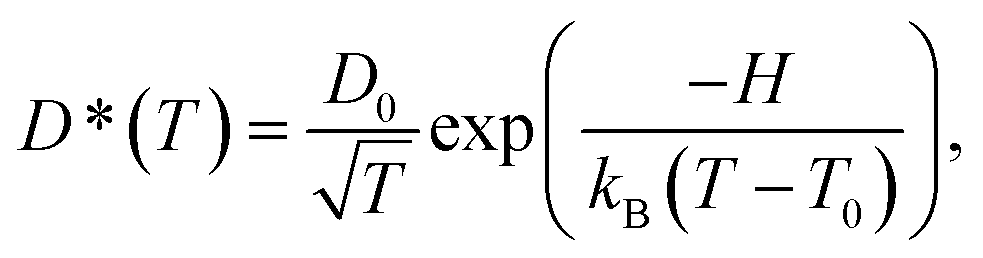 | (3) |
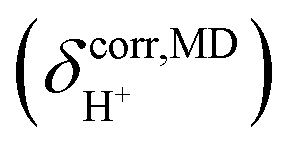 obtained from NVT-BOMD simulations over a temperature range of 298–430 K. Statistical samplings of the intermediate structures were performed every five BOMD steps, and approximately 2500 intermediate structures were used in the GIAO calculations of the instantaneous 1H NMR shielding constants
obtained from NVT-BOMD simulations over a temperature range of 298–430 K. Statistical samplings of the intermediate structures were performed every five BOMD steps, and approximately 2500 intermediate structures were used in the GIAO calculations of the instantaneous 1H NMR shielding constants 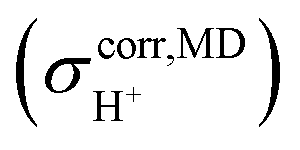 . To plot the 1H NMR spectra using the instantaneous chemical shifts,18 a histogram with a bin size of 0.05 ppm was constructed. Based on the structure of the histogram and the characteristic chemical shifts obtained from the static calculations, the centers of the peaks were assigned and used as initial estimates in a multi-peak fitting process using Lorentzian peak functions. The fitted Lorentzian peak functions were combined to generate the total 1H NMR spectra. The activation energies (ΔE‡,NMR) for the exchange between the shared-proton and close-contact structures were determined based on a 1H NMR line-width
. To plot the 1H NMR spectra using the instantaneous chemical shifts,18 a histogram with a bin size of 0.05 ppm was constructed. Based on the structure of the histogram and the characteristic chemical shifts obtained from the static calculations, the centers of the peaks were assigned and used as initial estimates in a multi-peak fitting process using Lorentzian peak functions. The fitted Lorentzian peak functions were combined to generate the total 1H NMR spectra. The activation energies (ΔE‡,NMR) for the exchange between the shared-proton and close-contact structures were determined based on a 1H NMR line-width 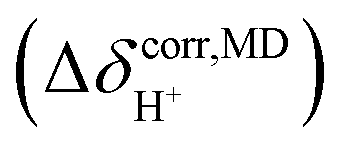 analysis;48
analysis;48 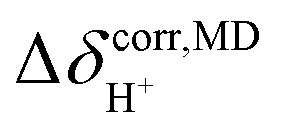 was approximated as the full width at half maximum (FWHM), and the effective transverse relaxation time
was approximated as the full width at half maximum (FWHM), and the effective transverse relaxation time  was calculated from
was calculated from 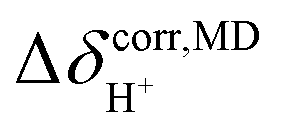 using
using 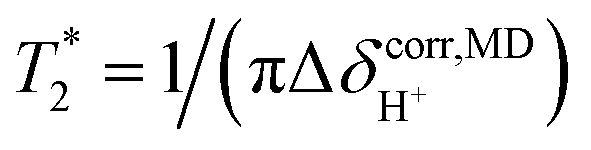 . ΔE‡,NMR was estimated from the slope of the linear relationship between
. ΔE‡,NMR was estimated from the slope of the linear relationship between 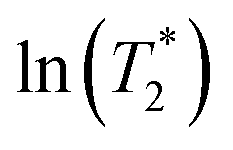 and 1000/T.48
and 1000/T.48Remarks should be made on the analysis of NMR spectra. In 1H NMR experiments, a motional correlation time (τc) with respect to the 1H NMR time scale can be used to study the dynamics of chemical exchange processes, in which either slow or fast exchange comprises the limiting cases.49 It was cited in ref. 49 “In the slow exchange limit, the NMR spectrum is a superposition of the resonances arising from each species, while in the fast exchange limit, a single peak representing an average chemical shift is observed” and “Hence, changes in the line-widths as a function of temperature can be correlated with an exchange rate Ω, which in the fast exchange limit follows the Arrhenius equation”. In H3PO4 systems, because the transverse relaxation time (T2) was reported to be much shorter than the longitudinal relaxation time (T1),11 and because the dynamics of short-life processes (e.g., oscillatory shuttling and structural diffusion motions) were of interest, we decided to discuss the energetics and dynamics of structural diffusion using the effective transverse relaxation time 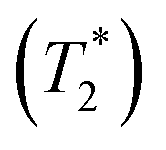 .48,49
.48,49
Results and discussion
Static results
All of the static results on H+(H3PO4)n (n = 2–5) clusters obtained from RIMP2/TZVP and B3LYP/TZVP calculations are included in Tables S1 and S2,† respectively. Correlations between the static properties obtained from these two methods are illustrated in Fig. S2.† Examples of the static results on neutral H3PO4 clusters obtained from RIMP2/TZVP calculations are listed in Table S3.† Tables S1 and S2† show that the RIMP2/TZVP and B3LYP/TZVP methods give approximately the same equilibrium structures, and excellent correlations are obtained for all of the static properties; the highest correlation coefficients (R2) are found for ΔdDA and RO–O with R2 = 0.98, whereas the lowest R2 values are for the interaction energy per H-bond (ΔEH-bond) and , R2 = 0.92 and 0.93, respectively. These R2 values lead to the conclusion that RIMP2/TZVP and B3LYP/TZVP calculations yield approximately the same results, and when computational resources are restricted, B3LYP/TZVP calculations can be applied to H3PO4 H-bond systems with confidence.
, R2 = 0.92 and 0.93, respectively. These R2 values lead to the conclusion that RIMP2/TZVP and B3LYP/TZVP calculations yield approximately the same results, and when computational resources are restricted, B3LYP/TZVP calculations can be applied to H3PO4 H-bond systems with confidence.
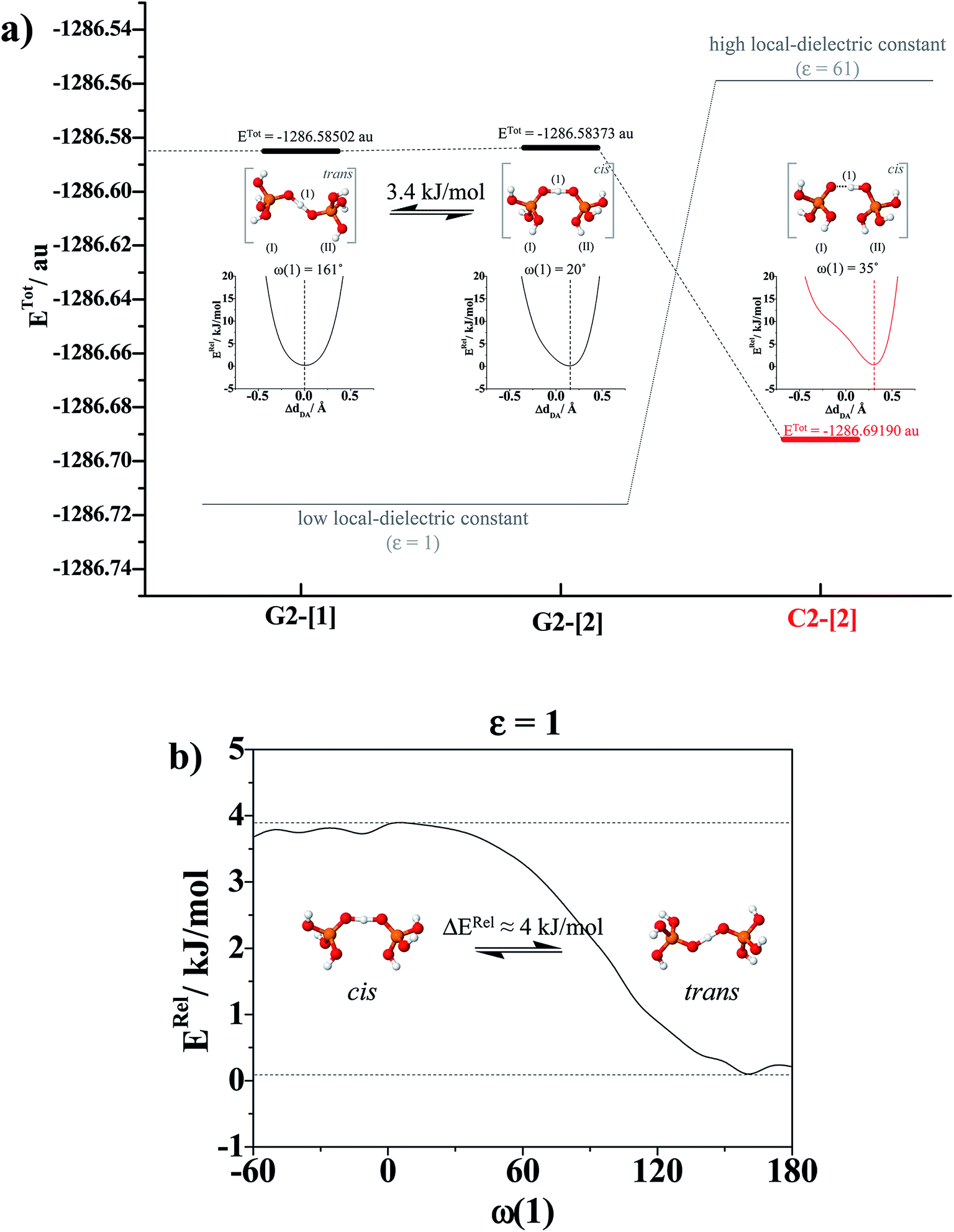
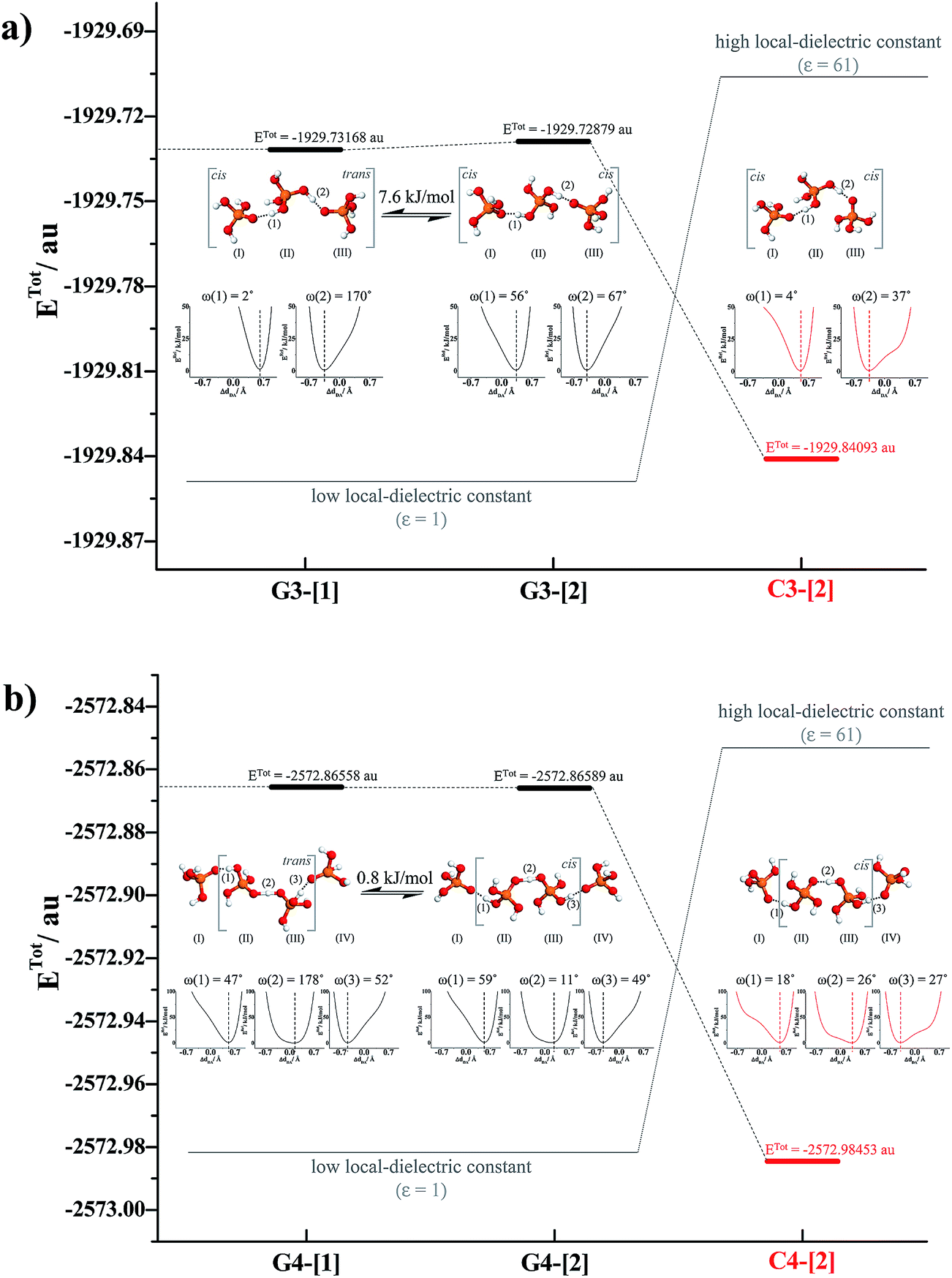
To simplify the discussion, the H-bond structures in Tables S1–S3† are labeled with a three-character code, Gn-[m] or Cn-[m], where G = H-bond structure in the gas phase, C = H-bond structure in continuum solvent, and n is the number of H3PO4 molecules. Different H-bond structures with the same number of H3PO4 molecules are distinguished by [m]. For example, according to the three-character code, G3-[1] and G3-[2] are different H-bond structures ([1] and [2], respectively) with the same number of H3PO4 molecules (n = 3) in the gas phase (G). In contrast, G2-[2] and C2-[2] are the same H-bond structure (n = 2, m = 2) in the gas phase (G) and continuum solvent (C), respectively. To discuss the direction of proton displacement, H3PO4 in the H-bond chains are labeled with Roman numerals in parentheses from (I) to (V).
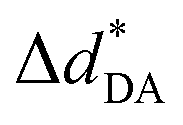 = 0.29 Å. For the neutral H3PO4 clusters (Table S3†), cyclic H-bond structures dominate with νOH and ΔdDA above the threshold values. These findings rule out the likelihood that the neutral cyclic H-bond structures are the intermediate complex in the structural diffusion process. The predominance of the cyclic H-bond structures in the neutral H3PO4 clusters is in accordance with the B3LYP/6-311G** results on (H3PO4)n (n = 2–6),16 for which the values of ΔdDA are all above the threshold value (ΔdDA ≥ 0.4). The H-bond structures that are potentially involved in the structural diffusion mechanism (n = 2–4) are included in Fig. 2. Because the static results for short and long H-bond chains are not substantially different, the smallest intermediate complexes (n = 2) in ε = 1 are discussed first.
= 0.29 Å. For the neutral H3PO4 clusters (Table S3†), cyclic H-bond structures dominate with νOH and ΔdDA above the threshold values. These findings rule out the likelihood that the neutral cyclic H-bond structures are the intermediate complex in the structural diffusion process. The predominance of the cyclic H-bond structures in the neutral H3PO4 clusters is in accordance with the B3LYP/6-311G** results on (H3PO4)n (n = 2–6),16 for which the values of ΔdDA are all above the threshold value (ΔdDA ≥ 0.4). The H-bond structures that are potentially involved in the structural diffusion mechanism (n = 2–4) are included in Fig. 2. Because the static results for short and long H-bond chains are not substantially different, the smallest intermediate complexes (n = 2) in ε = 1 are discussed first.
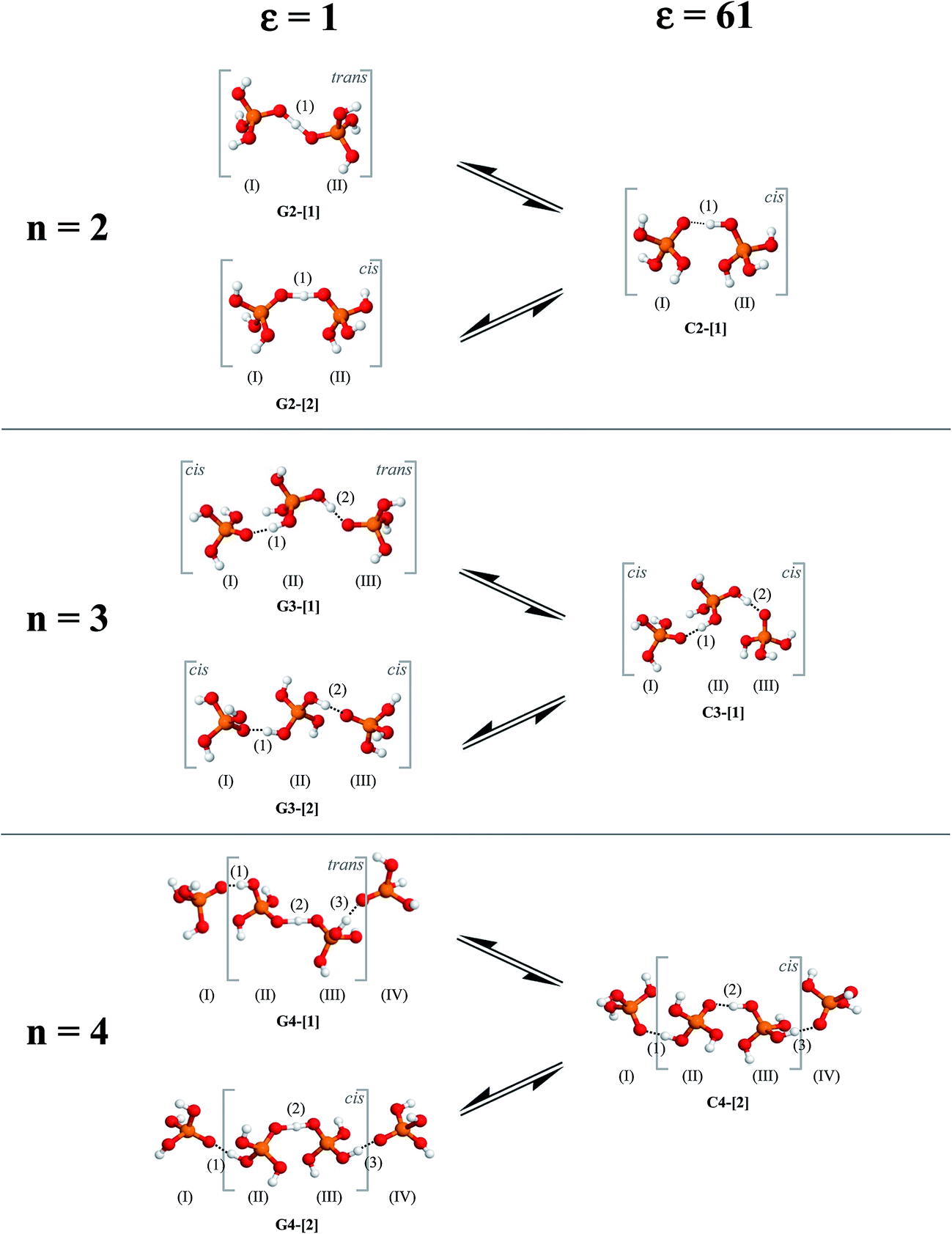 | ||
| Fig. 2 Linear H-bond structures potentially involved in proton exchange in H+(H3PO4)n (n = 2–5) clusters obtained from RIMP2/TZVP calculations in low and high local-dielectric environments, ε = 1 and 61, respectively. | ||
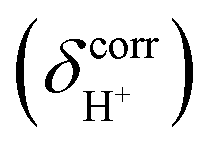 of protons in G2-[1] and G2-[2] are slightly different at
of protons in G2-[1] and G2-[2] are slightly different at 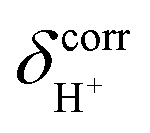 = 19.9 and 18.8 ppm, respectively. Because the potential energy curves for proton displacement in Fig. 3a show the characteristics of single-well potentials, G2-[1] and G2-[2] can be considered Zundel-like complexes. The
= 19.9 and 18.8 ppm, respectively. Because the potential energy curves for proton displacement in Fig. 3a show the characteristics of single-well potentials, G2-[1] and G2-[2] can be considered Zundel-like complexes. The 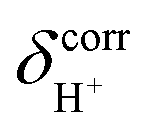 values of 19–20 ppm are regarded as 1H NMR signatures of exchanging protons in the protonated H3PO4 clusters, and are slightly smaller than that in the prototypical system (H5O2+), determined in 1H NMR experiments to be 21.3 ppm,8 and are also consistent with the
values of 19–20 ppm are regarded as 1H NMR signatures of exchanging protons in the protonated H3PO4 clusters, and are slightly smaller than that in the prototypical system (H5O2+), determined in 1H NMR experiments to be 21.3 ppm,8 and are also consistent with the 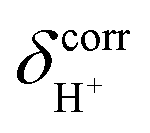 values for the hydrated H4PO4+ clusters, obtained based on the same method to be 18–20 ppm;15 the strong 1H NMR downfield shifts (14–22 ppm) are associated with the deshielded protons in short, strong H-bonds, with ΔEH-bond approximately 63–160 kJ mol−1.50
values for the hydrated H4PO4+ clusters, obtained based on the same method to be 18–20 ppm;15 the strong 1H NMR downfield shifts (14–22 ppm) are associated with the deshielded protons in short, strong H-bonds, with ΔEH-bond approximately 63–160 kJ mol−1.50
 | ||
| Fig. 3 (a) Potential energy curves of proton displacement in H+(H3PO4)2 in low and high local-dielectric environments obtained from RIMP2/TZVP calculations with a freeze-scan method. (b) Potential energy curve for the interconversion between the trans and cis conformations in low local-dielectric environment obtained from RIMP2/TZVP calculations with a relax-scan method. | ||
Because the potential energy curve in Fig. 3b suggests the energy barrier (ΔERel) for the transformation from the trans to cis conformation to be approximately 4 kJ mol−1, which is slightly higher than thermal energy fluctuations at room temperature (2.6 kJ mol−1), and because RO–O and ΔdDA do not change in the relax-scan path (approximately 2.38 and 0 Å, respectively), one can conclude that in ε = 1, rotation of the protonated H-bond (ω(1)) is feasible but does not seem to affect the energetics of structural diffusion under excess proton conditions.
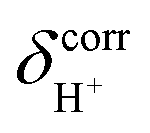 values for protons in Eigen-like complexes (close-contact structures) are 10–16 ppm, whereas those for protons in neutral cyclic H-bonds (Table S3†) are 6–12 ppm. The latter confirm that the 1H NMR peak at 9.8 ppm used in experiments10,51 is associated with protons in neutral H-bonds, which is in accordance with the observation that the undissociated H3PO4 molecules dominate in orthophosphoric acid;17 H-bonds with 1H NMR chemical shifts below 14 ppm are categorized as “moderate” or “week” H-bonds with ΔEH-bond less than 63 kJ mol−1.50
values for protons in Eigen-like complexes (close-contact structures) are 10–16 ppm, whereas those for protons in neutral cyclic H-bonds (Table S3†) are 6–12 ppm. The latter confirm that the 1H NMR peak at 9.8 ppm used in experiments10,51 is associated with protons in neutral H-bonds, which is in accordance with the observation that the undissociated H3PO4 molecules dominate in orthophosphoric acid;17 H-bonds with 1H NMR chemical shifts below 14 ppm are categorized as “moderate” or “week” H-bonds with ΔEH-bond less than 63 kJ mol−1.50
 | ||
| Fig. 4 Potential energy curves for proton displacement in low and high local-dielectric environments obtained from RIMP2/TZVP calculations with a freeze-scan method. (a) H-Bonds (1) and (2) in H+(H3PO4)3. (b) H-Bonds (1), (2) and (3) in H+(H3PO4)4. | ||
Further extension of the H-bond chains in G3-[1] and G3-[2] leads to G4-[1] and G4-[2] in Fig. 4b, respectively, in which the characteristic properties and the shape of the potential energy curves of H-bond (2) are similar to those of H-bond (1) in G2-[1] and G2-[2], implying that H-bond (2), which is formed from H3PO4 (II) and H3PO4 (III), is the precursor (Zundel-like complex) for the next transfer. Further extension of the H-bond chain at H3PO4 (IV) results in another Eigen-like complex (G5-[1]) with characteristic double-well potentials at H-bonds (2) and (3). The formation of Zundel-like and Eigen-like complexes leads to a positive charge displacement from H-bond (1) to (2), as shown in Fig. 5a, which is in accordance with the Eigen–Zundel–Eigen mechanism in protonated imidazole (Im) H-bond chains,52 in which breaking and forming H-bonds in the c crystallographic axis of a unit cell represent the most important elementary steps in the structural diffusion process.31
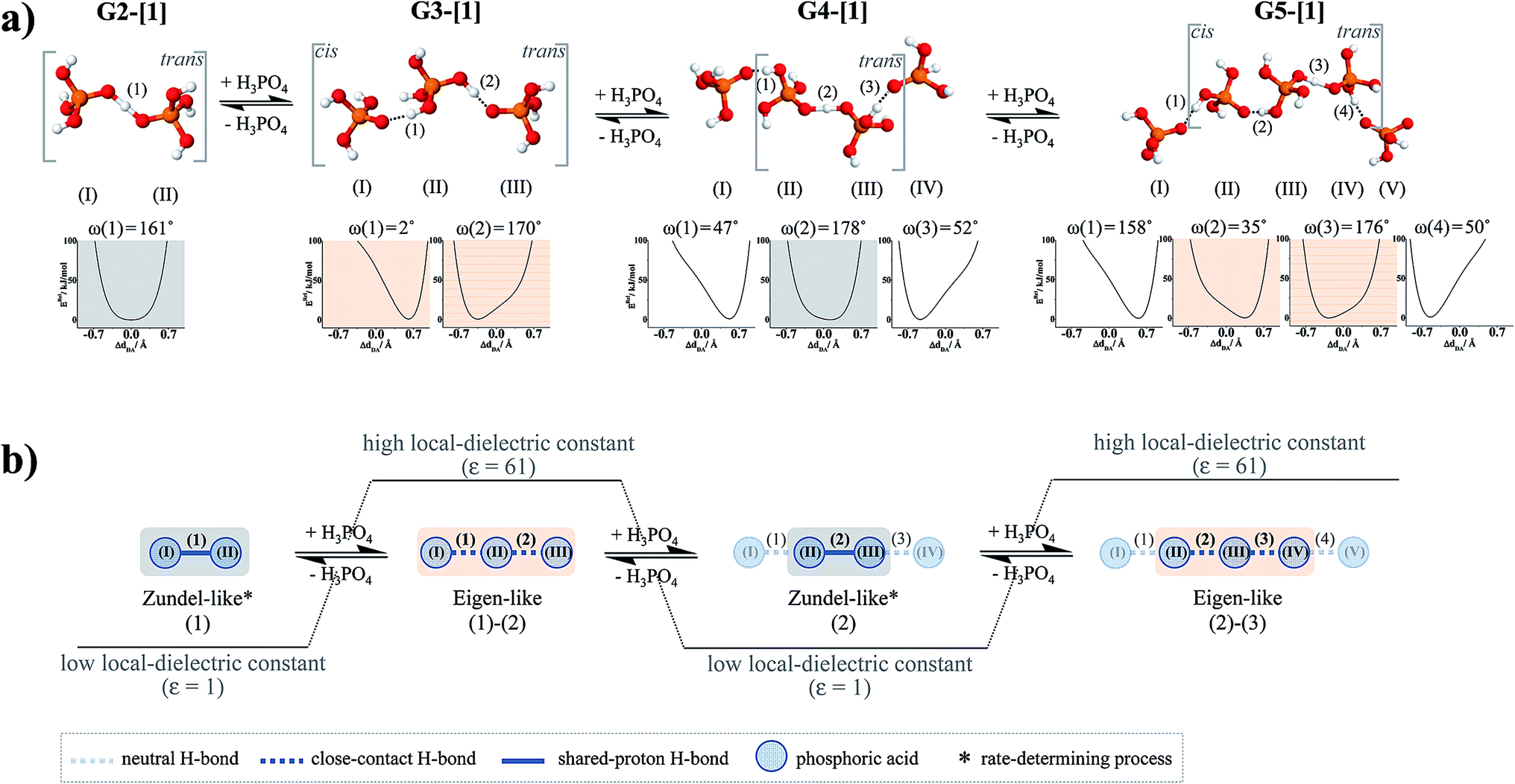 | ||
| Fig. 5 (a) Eigen–Zundel–Eigen scenario for proton exchange in H+(H3PO4)n (n = 2–5) clusters which involves fluctuations of the H-bond chain length. Zundel-like and Eigen-like complexes are shown with the potential energy curves for single-proton displacement in low local-dielectric environment, obtained from RIMP2/TZVP calculations. (b) Short-range Eigen–Zundel–Eigen mechanism for proton exchange in protonated H3PO4 H-bond chain which involves the H-bond structures with n = 2 and 3, proposed based on the NVT-BOMD results. | ||
Dynamic results
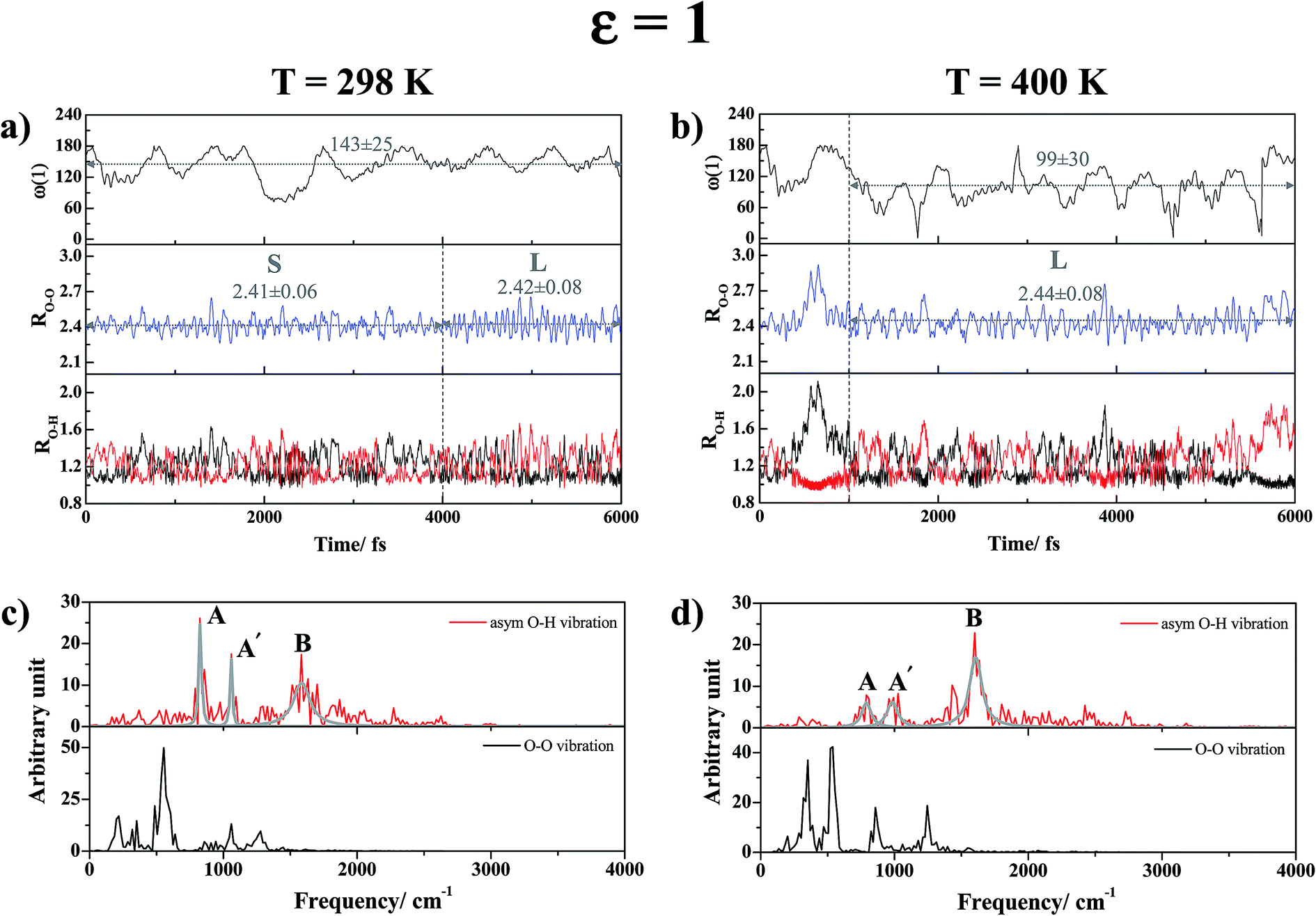 | ||
| Fig. 6 (a) and (b) Examples of proton transfer profiles and time evolutions of the dihedral angle ω(1) of H-bond (1) in H+(H3PO4)2 obtained from NVT-BOMD simulations in low local-dielectric environment. (c) and (d) Examples of vibrational spectra of H-bond (1) in H+(H3PO4)2 obtained from NVT-BOMD simulations in low local-dielectric environment. RO–O and RO–H are in Å and ω(1) in degree. L and S = large-amplitude and small-amplitude O–O vibrations, respectively; A and A′ = oscillatory shuttling peaks; B = structural diffusion peak. | ||
The vibrational spectra in Fig. 6c show two structured peaks associated with the oscillatory shuttling motions (labeled A and A′); the gray curves in all of the vibrational spectra were obtained by fittings of Lorentzian peak functions to the characteristic peaks (red curves) to differentiate the characteristic peaks from vibrational interference peaks. Peaks A and A′ are located at νAasym-OH,MD = 819 cm−1 and νA′asym-OH,MD = 1066 cm−1, whereas a broad peak associated with the motion of structural diffusion (peak B) is seen at νBasym-OH,MD = 1599 cm−1. These characteristic peaks are in accordance with the IR frequencies reviewed in ref. 50, in which the O–H stretching frequencies for complementary acid–base solutions were suggested to be 500–2000 cm−1.
Investigation of the time evolution of the dihedral angle ω(1) in Fig. 6a suggests a “librational dynamic equilibrium” in the trans conformation (ω(1) = 143 ± 25°), which results in a splitting of the oscillatory shuttling peak. Because peaks A and A′ are more structured than peak B, the oscillatory shuttling motions dominate at this temperature. The proton transfer profiles in Fig. 6b reveal that as the temperature is increased to 400 K, the large-amplitude O–O vibration (RO–O = 2.44 ± 0.08 Å) and the structural diffusion motion dominate with a strong librational motion of H-bond (1) (ω(1) = 99 ± 30°). The predominance of the structural diffusion motion is also manifested in the vibrational spectra in Fig. 6d, in which peak B is prominent at νBasym-OH,MD = 1616 cm−1, whereas peaks A and A′ are considerably smaller and slightly redshifted to νAasym-OH,MD = 791 cm−1 and νA′asym-OH,MD = 993 cm−1.
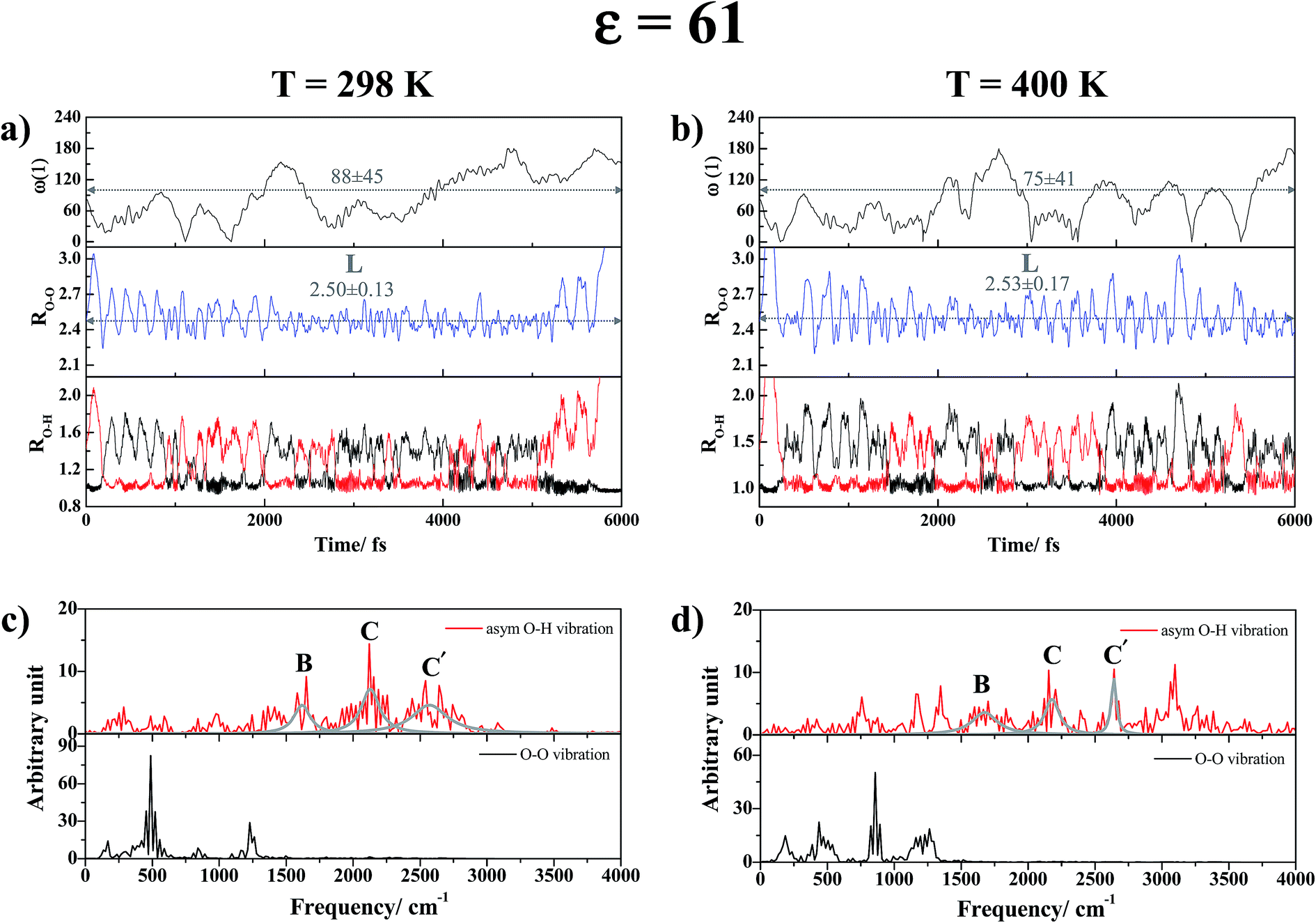 | ||
| Fig. 7 (a) and (b) Examples of proton transfer profiles and time evolutions of the dihedral angle ω(1) of H-bond (1) in H+(H3PO4)2 obtained from NVT-BOMD simulations in high local-dielectric environment. (c) and (d) Examples of vibrational spectra of H-bond (1) in H+(H3PO4)2 obtained from NVT-BOMD simulations in high local-dielectric environment. RO–O and RO–H are in Å and ω(1) in degree. L = large-amplitude O–O vibration; B = structural diffusion peak; C and C′ = characteristic peaks of proton in double-well potential. | ||
As the temperature is increased to 400 K, although the proton transfer profiles and the time evolution of the dihedral angle ω(1) in Fig. 7b show the same dynamic behaviors of the exchanging proton as those at 298 K, the number of proton exchange events is significantly decreased due to stronger thermal agitation. The effects of thermal agitation are also reflected in the vibrational spectra in Fig. 7d, in which the structural diffusion (peak B) and close-contact peaks (peaks C and C′) are blueshifted to νBasym-OH,MD = 1683 cm−1, νCasym-OH,MD = 2171 cm−1 and νC′asym-OH,MD = 2642 cm−1 with strong interference peaks over the entire spectra. The vibrational spectra are complex when the H-bond chain length is increased from n = 2 to 4 in ε = 61. Fig. S6d† shows that for C4-[1], vibrational interferences make it difficult to differentiate the characteristic peaks from the interference peaks; at 298 K, peaks B, C and C′ are observed approximately at 2018, 2459 and 2925 cm−1, respectively.
Similar vibrational analyses were applied in the study of proton transfer in solid maleimide (MI),53 in which a triclinic crystal structure including eight MI molecules and one unbounded proton was selected as the model system in CPMD simulations. Although this model system was considerably larger than our presolvation model, the envelopes of the VACF of the acidic proton showed similar exponential decay functions, with two characteristic vibrational frequency ranges, approximately from 800–1200 cm−1 and from 2900–3100 cm−1. Our analyses, which were based on the velocity vectors in Fig. 1 (the Q2 mode), suggest that the former correspond to the shuttling motion of H+ in the shared-proton structures, whereas the latter reflect the stretching vibration of O–H+ in the close-contact structures.
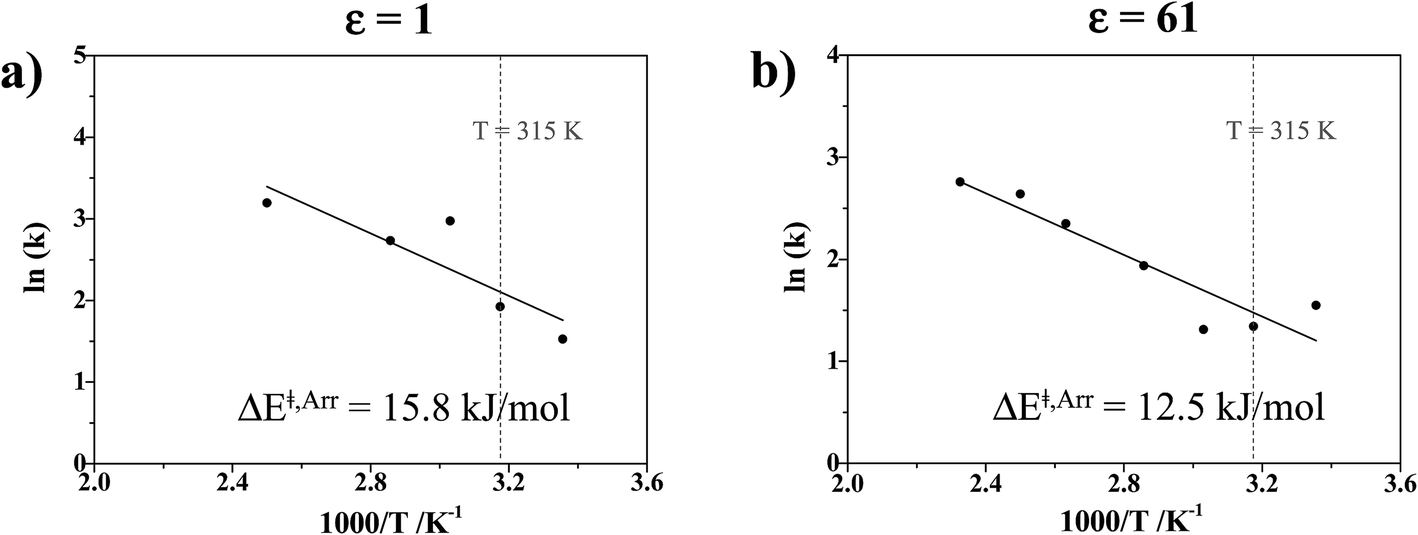 | ||
| Fig. 8 (a) and (b) Arrhenius plots obtained from NVT-BOMD simulations over the temperature range of 298–430 K in low- and high local-dielectric environments, respectively. ΔE‡,Arr = activation energy obtained from the exponential relaxation behavior of the envelope of the VACF of the O–O vibration. | ||
The activation energies for proton exchange in protonated H3PO4 clusters in ε = 1 and 61 are higher than those for proton dissociation in hydrated H4PO4+ clusters, ΔE‡,Arr = 13.1 and 9.9 kJ mol−1 in ε = 1 and 78, respectively,15 and higher than for proton exchange in the Zundel complex (H5O2+), ΔE‡,Arr = 10.2 kJ mol−1 in ε = 1.18 The trend of ΔE‡,Arr supports the results of conductivity measurements,13 in which the introduction of water into liquid H3PO4 increases proton conductivity, which in this case is by lowering the activation energies for proton exchange upon hydration.
To study the temperature dependence of the proton self-diffusion coefficients obtained from the theoretical and experimental methods, the relations between D§ and D* and 1000/T are illustrated in Fig. 9. It appears that reasonable agreement between the theoretical and experimental values (D§ and D*) is observed above the melting temperature, whereas at lower temperatures (298–350 K), the proton self-diffusion coefficients obtained from the 1H PFG-NMR experiments suggest a slower proton diffusion rate. The discrepancy between the theoretical and experimental values at low temperatures occurs because the 1H PFG-NMR measurements yield the average proton self-diffusion coefficients (D*) of all 1H in the system,11 including protons that are not directly involved in structural diffusion, whereas D§ values are obtained from the MSD plots that are constructed based only on the displacement of the exchanging proton (H+).
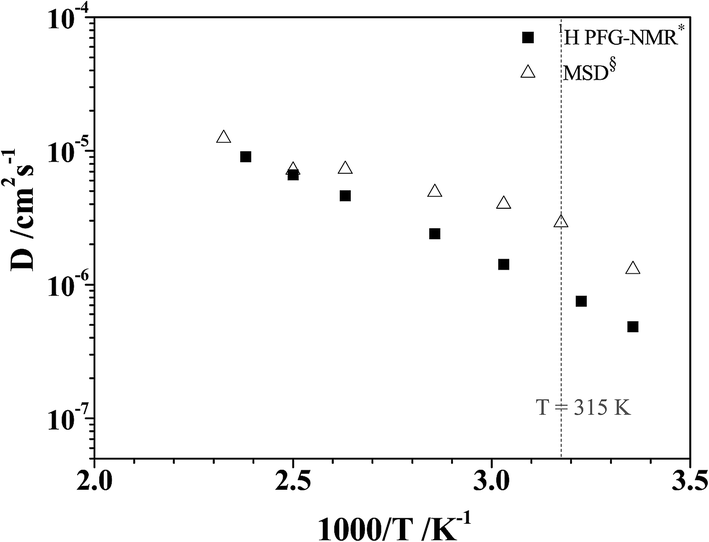 | ||
| Fig. 9 Plots of proton self-diffusion coefficients (D§) in low local-dielectric environment as a function of 1000/T. D* = values obtained from 1H PFG-NMR experiment in ref. 11; D§ = values obtained from the MSD plots. | ||
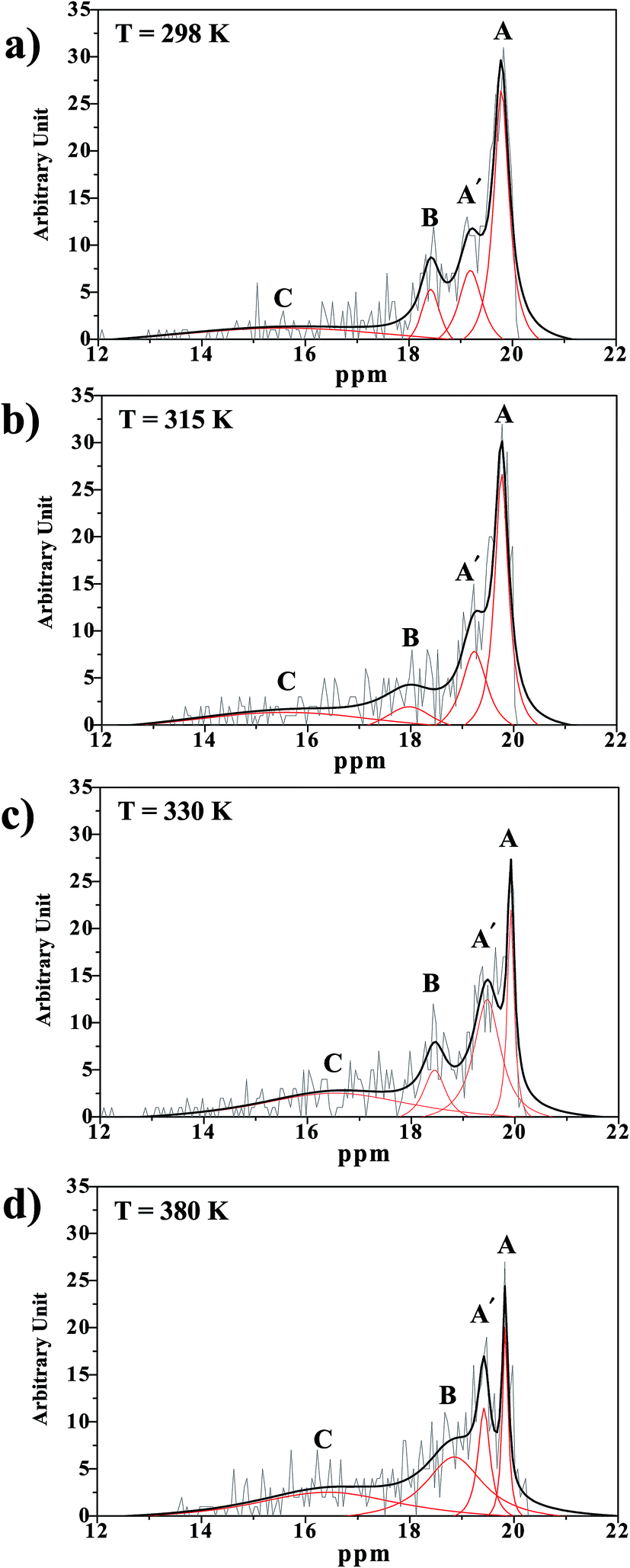 | ||
| Fig. 10 (a)–(d) 1H NMR chemical shift spectra of the exchanging proton in H-bond (1) in H+(H3PO4)2 obtained from NVT-BOMD simulations. A and A′ = oscillatory shuttling peaks; B = structural diffusion peak; C = close-contact peak. | ||
In Fig. 10a, it appears that at 298 K, peak A is outstanding at 19.8 ppm, and peak A′ appears as a shoulder at 19.2 ppm, whereas peak B is centered at 18.4 ppm. Integrations of peaks A and A′ yield the populations of the two oscillatory shuttling modes in arbitrary units of approximately 16 and 8, respectively (2:1). Investigation of the structures of peaks A and A′ over a temperature range of 298–430 K reveals the temperature dependence of oscillatory shuttling, as well as the motional narrowing of these two modes (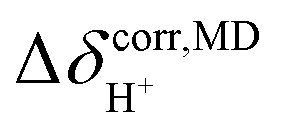 in Table S6†); although all of the characteristic peaks are located at approximately the same frequency, the shapes and relative intensities are temperature dependent. For example, at 380 K, peak A′ is more well-defined and separated from peak A, indicating a stronger librational motion in the rate-determining Zundel-like structure at elevated temperatures. Integrations of these two peaks yield a ratio for these two oscillatory shuttling modes of approximately 0.8:1; this result is in accordance with the proton transfer profiles and the vibrational analyses, in which the likelihood of oscillatory shuttling is decreased at elevated temperatures. The plots of
in Table S6†); although all of the characteristic peaks are located at approximately the same frequency, the shapes and relative intensities are temperature dependent. For example, at 380 K, peak A′ is more well-defined and separated from peak A, indicating a stronger librational motion in the rate-determining Zundel-like structure at elevated temperatures. Integrations of these two peaks yield a ratio for these two oscillatory shuttling modes of approximately 0.8:1; this result is in accordance with the proton transfer profiles and the vibrational analyses, in which the likelihood of oscillatory shuttling is decreased at elevated temperatures. The plots of 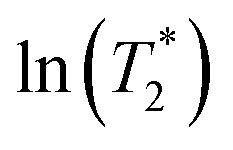 and 1000/T in Fig. 11 suggest that the activation energies (ΔE‡,NMR) for proton exchange in the oscillatory shuttling modes (A and A′) are 13.8 and 8.6 kJ mol−1, respectively. The values of ΔE‡,NMR are consistent with ΔE‡,D§ and ΔE‡,Arr, which are obtained based on MSD and VACF, respectively.
and 1000/T in Fig. 11 suggest that the activation energies (ΔE‡,NMR) for proton exchange in the oscillatory shuttling modes (A and A′) are 13.8 and 8.6 kJ mol−1, respectively. The values of ΔE‡,NMR are consistent with ΔE‡,D§ and ΔE‡,Arr, which are obtained based on MSD and VACF, respectively.
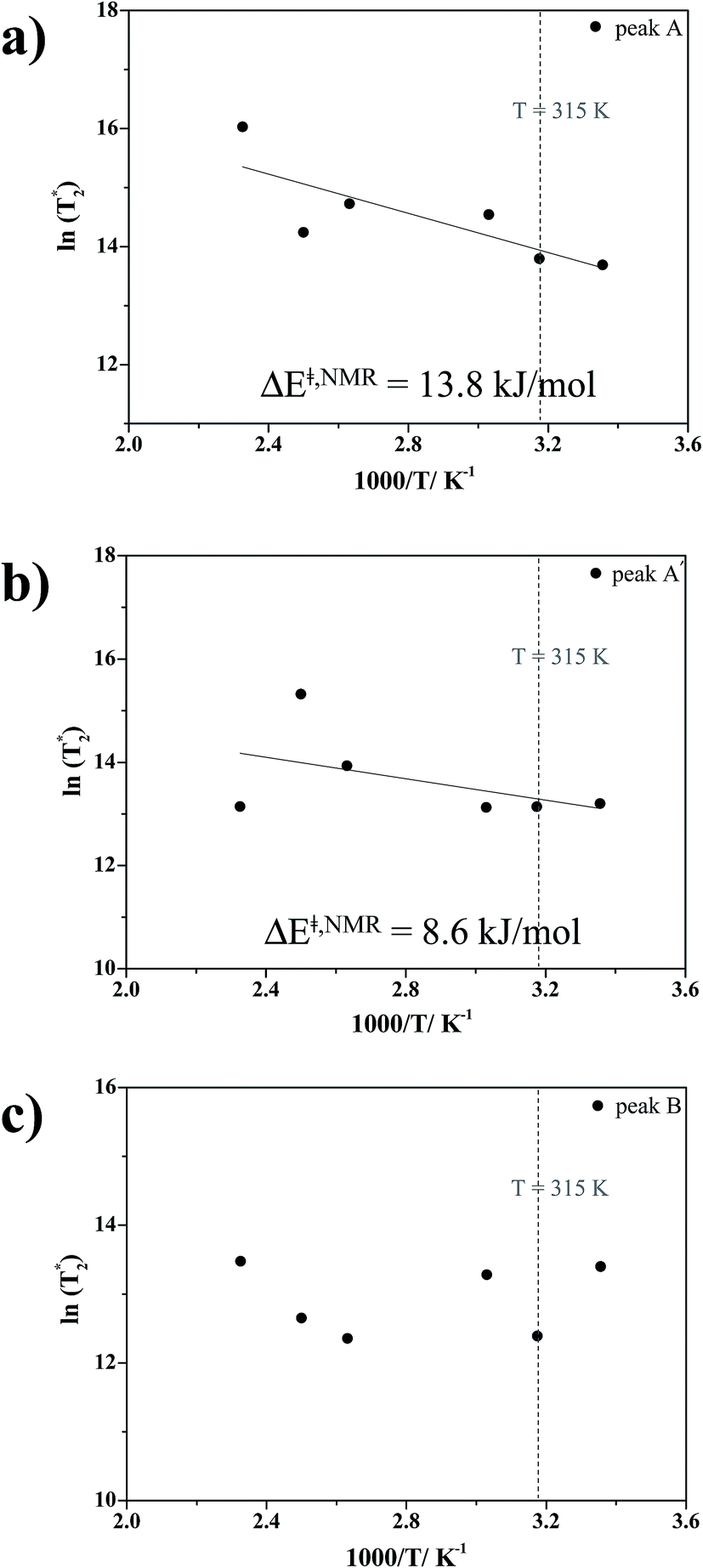 | ||
Fig. 11 Plots of the natural log of effective transverse relaxation time 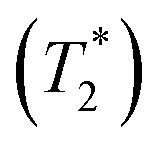 as a function of 1000/T. ΔE‡,NMR = activation energy obtained from 1H NMR line-width analysis. (a) and (b) For the oscillatory shuttling motions (peaks A and A′, respectively). (c) For the structural diffusion motion (peak B). as a function of 1000/T. ΔE‡,NMR = activation energy obtained from 1H NMR line-width analysis. (a) and (b) For the oscillatory shuttling motions (peaks A and A′, respectively). (c) For the structural diffusion motion (peak B). | ||
Remarks should be made on the 1H NMR results discussed in this work and those obtained from 1H PGSE NMR measurements.51 To understand the ion conduction mechanisms in H3PO4 fuel cells, 1H, 2H and 31P NMR experiments were conducted to measure diffusion coefficients and activation energies of mobile species in water-free H3PO4 solutions. Based on the analysis of the 1H NMR peaks at 10.6 and 12.4 ppm (protons in H-bonds in 105 and 116 wt% H3PO4 solutions, respectively) and the approximation that the diffusion rate of 1H and H+ are the same, the activation energies for diffusion obtained from the analysis of these two peaks are 32.4 and 36.1 kJ mol−1, respectively, whereas the activation energies for the correlation times (τc) estimated from the 1H NMR spin-lattice relaxation (T1) measurements at high temperatures are 21.2 and 21.5 kJ mol−1, respectively. The discrepancies between the experimental activation energies are due to the different methods used in the NMR analyses10 and the fact that H3PO4 solutions are complicated due to the presence of various mobile species that cannot be easily separated (e.g., H3PO4, H6P2O7 and H8P3O10). Whereas the discrepancies between the theoretical and experimental values are due to the approximation that the diffusion rates of 1H and H+ are the same and the fact that the H+ species cannot be directly probed in 1H NMR experiments. Because the experimental activation energies are systematically higher, they are anticipated to include the dynamics of slower processes (e.g., mobile 1H species) other than the dynamics of the H+ species in the structural diffusion mechanism.
Conclusions
The dynamics and mechanism of structural diffusion in the H3PO4 H-bond clusters were studied under excess proton conditions using quantum chemical methods at the RIMP2/TZVP level and NVT-BOMD simulations over a temperature range of 298–430 K. This theoretical study focused on characteristic properties of exchanging protons and on local dynamics that could determine the rate of structural diffusion, as well as on the likelihood that the reorientation of H3PO4 molecules plays an important role in the structural diffusion process.To accomplish our designated goals, based on the concept of presolvation, H+(H3PO4)n (n = 2–5) clusters were selected as model systems, for which structures, energetics and spectroscopic properties of potential intermediate complexes in low and high local-dielectric environments were examined. RIMP2/TZVP geometry optimizations suggested that only the linear H-bond structures are involved in the structural diffusion process and that intermediate complexes with shared-proton structures (Zundel-like complexes) are preferentially formed in low local-dielectric environments, whereas close-contact structures (Eigen-like complexes) with stronger O–H+ covalent character are stabilized in high local-dielectric environments. The static results also suggested that the positive charge displacement along the H3PO4 H-bond chain involves a short-range process, and the fluctuations of the H-bond chain length and local-dielectric environment enhance structural diffusion by transforming the Zundel-like complex (n = 2) to an Eigen-like complex (n = 3), which can be regarded as an Eigen–Zundel–Eigen mechanism. In addition, the potential energy curve for the rotation of the H-bond in the smallest Zundel-like complex revealed that the interconversion between the trans and cis conformations is feasible but not necessarily involved in the structural diffusion process.
Analyses of the vibrational and 1H NMR spectra obtained from BOMD simulations validated the proposed short-range Eigen–Zundel–Eigen mechanism by showing that the oscillatory shuttling, which is the rate-determining process, is dominant in the smallest Zundel-like complex in low local-dielectric environments. The dynamic results also led to the conclusion that for the protonated H3PO4 clusters, in which the Grotthuss mechanism dominates, structural diffusion can proceed without the frustrated rotation, as in the case of neat liquid H3PO4. Based on our analyses, the proton self-diffusion coefficients and activation energies obtained from 1H NMR experiments are anticipated to include the dynamics of slower processes (e.g., mobile 1H species) other than the diffusion of the H+ species. Therefore, the present theoretical study suggested for the first time the energetics, dynamics and spectroscopic properties of the H+ species in H3PO4 H-bond systems under excess proton conditions, which can be used as guidelines for further experimental investigation.
Acknowledgements
The authors would like to acknowledge the financial support provided by the Royal Golden Jubilee (RGJ) Ph.D. Program of Thailand Research Fund (TRF) and Suranaree University of Technology (SUT) (Grant No. PHD/0085/2552) to Prof. Kritsana Sagarik and Parichart Suwannakham and the Advanced Research Scholarship (Grant No. BRG-5580011) to Prof. Kritsana Sagarik. The high-performance computer facilities provided by the following organizations are gratefully acknowledged: Schools of Mathematics and School of Chemistry, SUT; National e-Science project of the National Electronics and Computer Technology Centre (NECTEC), the National Science and Technology Development Agency (NSTDA). This work was supported in part by the Higher Education Research Promotion and National Research University (HERP-NRU) Project of the Office of the Higher Education Commission (OHEC), Thailand.References
- M. F. H. Schuster and W. Meyer, Annu. Rev. Mater. Res., 2003, 33, 233–261 CrossRef CAS.
- K. Sundmacher, Ind. Eng. Chem. Res., 2010, 49, 10159 CrossRef CAS.
- M. Schuster, K. D. Kreuer, H. Steininger and J. Maier, Solid State Ionics, 2008, 179, 523–528 CrossRef CAS.
- S. J. Peighambardoust, S. Rowshanzamir and M. Amjadi, Int. J. Hydrogen Energy, 2010, 35, 9349–9384 CrossRef CAS.
- H. Pu, W. H. Meyer and G. Wegner, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 663 CrossRef CAS.
- N. N. Greenwood and A. Thompson, J. Chem. Soc., 1959, 3485 RSC.
- J. A. Pople, W. G. Schneider and H. J. Bernstein, High Resolution Nuclear Magnetic Resonance, McGraw-Hill, New York, 1959 Search PubMed.
- H.-H. Limbach, P. M. Tolstoy, N. Pérez-Hernández, J. Guo, I. G. Shenderovich and G. S. Denisov, Isr. J. Chem., 2009, 49, 199–216 CrossRef CAS.
- M. N. Nakahara and C. Wakai, Chem. Lett., 1992, 21, 809–812 CrossRef.
- S. H. Chung, S. Bajue and S. G. Greenbaum, J. Chem. Phys., 2000, 112, 8515 CrossRef CAS.
- T. Dippel, K. D. Kreuer, J. C. Lassègues and D. Rodriguez, Solid State Ionics, 1993, 61, 41–46 CrossRef CAS.
- J. C. Lassègues and D. Cavagnat, Phys. B, 1992, 180–181, 645 CrossRef.
- R. A. Munson and M. E. Lazarus, J. Phys. Chem., 1967, 71, 3245–3248 CrossRef CAS.
- L. Vilčiauskas, M. E. Tuckerman, G. Bester, S. J. Paddison and K. D. Kreuer, Nat. Chem., 2012, 4, 461 CrossRef PubMed.
- P. Suwannakham, S. Chaiwongwattana and K. Sagarik, Int. J. Quantum Chem., 2015, 115, 486–501 CrossRef CAS.
- L. Vilciauskas, S. J. Paddison and K.-D. Kreuer, J. Phys. Chem. A, 2009, 113, 9193–9201 CrossRef CAS PubMed.
- R. A. Munson, J. Chem. Phys., 1964, 40, 2044 CrossRef CAS.
- C. Lao-ngam, M. Phonyiem, S. Chaiwongwattana, Y. Kawazoe and K. Sagarik, Chem. Phys., 2013, 420, 50–61 CrossRef CAS.
- J. Thisuwan and K. Sagarik, RSC Adv., 2014, 4, 61992–62008 RSC.
- C. Lao-ngam, P. Asawakun, S. Wannarat and K. Sagarik, Phys. Chem. Chem. Phys., 2011, 13, 4562–4575 RSC.
- S. Chaiwongwattana, M. Phonyiem, V. Vchirawongkwin, S. Prueksaaroon and K. Sagarik, J. Comput. Chem., 2012, 33, 175–188 CrossRef CAS PubMed.
- K. Sagarik, S. Chaiwongwattana, V. Vchirawongkwin and S. Prueksaaroon, Phys. Chem. Chem. Phys., 2010, 12, 918 RSC.
- K. Kwac, C. Lee, Y. Jung and J. Han, J. Chem. Phys., 2006, 125, 244508 CrossRef PubMed.
- H. J. Boehm and R. Ahlrichs, J. Chem. Phys., 1982, 77, 2028 CrossRef CAS.
- H. J. Boehm and R. Ahlrichs, Mol. Phys., 1985, 55, 1159 CrossRef CAS.
- K. P. Sagarik and R. Ahlrichs, J. Chem. Phys., 1987, 86, 5117 CrossRef CAS.
- Turbomole Tutorial V7.0, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2015, Turbomole GmbH, since 2007, available from http://www.turbomole.com.
- M. Phonyiem, S. Chaiwongwattana, C. Lao-ngam and K. Sagarik, Phys. Chem. Chem. Phys., 2011, 13, 10923 RSC.
- K. Sagarik, M. Phonyiem, C. Lao-ngam and S. Chaiwongwattana, Phys. Chem. Chem. Phys., 2008, 10, 2098 RSC.
- R. Vuilleumier and D. Borgis, Nat. Chem., 2012, 4, 432–433 CrossRef CAS PubMed.
- W. Bua-ngern, S. Chaiwongwattana, P. Suwannakham and K. Sagarik, RSC Adv., 2016, 6, 99391–99403 RSC.
- B. Koeppe, J. Guo, P. M. Tolstoy, G. S. Denisov and H.-H. Limbach, J. Am. Chem. Soc., 2013, 135, 7553–7566 CrossRef CAS PubMed.
- A. Klamt and G. Schuuermann, J. Chem. Soc., Perkin Trans. 2, 1993, 799 RSC.
- R. Ditchfield, Mol. Phys., 1974, 27, 789 CrossRef CAS.
- R. Ditchfield, J. Chem. Phys., 1976, 65, 3123 CrossRef CAS.
- D. B. Chesnut, Chem. Phys. Lett., 1995, 246, 235 CrossRef CAS.
- S. Taubert, H. Konschin and D. Sundholm, Phys. Chem. Chem. Phys., 2005, 7, 2561–2569 RSC.
- D. B. Chesnut and B. E. Rusiloski, J. Mol. Struct.: THEOCHEM, 1994, 314, 19–30 CrossRef.
- D. B. Chesnut, J. Phys. Chem. A, 2005, 109, 11962 CrossRef CAS PubMed.
- D. C. Rapaport, The Art of Molecular Dynamics Simulation, Cambridge University Press, London, 1995 Search PubMed.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695 CrossRef.
- S. Nosé, J. Chem. Phys., 1984, 81, 511 CrossRef.
- S. Nosé, Prog. Theor. Phys. Supp., 1991, 103, 1 CrossRef.
- S. Nosé and M. L. Klein, Mol. Phys., 1983, 50, 1055 CrossRef.
- R. L. Davidchack, J. Comput. Phys., 2010, 229, 9323–9346 CrossRef CAS.
- P. Bopp, Chem. Phys., 1986, 106, 205 CrossRef CAS.
- J. M. Haile, Molecular Dynamics Simulations, John Wiley & Sons Ltd, New York, 1997 Search PubMed.
- Y. J. Lee, B. Bingöl, T. Murakhtina, D. Sebastiani, W. H. Meyer, G. Wegner and H. W. Spiess, J. Phys. Chem. B, 2007, 111, 9711–9721 CrossRef CAS PubMed.
- G. Brunklaus, S. Schauff, D. Markova, M. Klapper, K. Müllen and H. W. Spiess, J. Phys. Chem. B, 2009, 113, 6583–7040 CrossRef PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- Y. Aihara, A. Sonai, M. Hattori and K. Hayamizu, J. Phys. Chem. B, 2006, 110, 24999–25006 CrossRef CAS PubMed.
- A. Li, Z. Cao, Y. Li, T. Yan and P. Shen, J. Phys. Chem. B, 2012, 116, 12793–12800 CrossRef CAS PubMed.
- X. Li, L. Yan and B. Yue, RSC Adv., 2015, 5, 80220–80227 RSC.
- R. He, Q. Li, G. Xiao and N. J. Bjerrum, J. Membr. Sci., 2003, 226, 169–184 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01829k |
| This journal is © The Royal Society of Chemistry 2017 |