
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible and near-infrared electrochromic thiophene–diketopyrrolopyrrole polymers†
Xiuhong Chenab,
Wenqiang Qiao*a and
Zhi Yuan Wang *ac
*ac
aState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: wqqiao@ciac.ac.cn, wayne_wang@carleton.ca
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cDepartment of Chemistry, Carleton University, 1125 Colonel By Drive, Ottawa, Ontario, Canada K1S 5B6
First published on 8th March 2017
Abstract
A series of low bandgap polymers containing thiophene and diketopyrrolopyrrole (DPP) units were synthesized and their electrochromic (EC) properties were investigated. It was found that the ratio (Rda) of thiophene to DPP units correlates to the EC properties, in particular optical contrast (ΔT) and coloration efficiency (CE) in the visible and near-infrared spectral regions (e.g., 1310 nm). At the wavelength of 1310 nm, the CE of polymer P2 (Rda = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and ΔT of polymer P4 (Rda = 4:1) reached the high values of 1383 cm2 C−1 and 81.5%, respectively. Polymer P3 (Rda = 3.5:1) had a relatively short switching time of 1.24 s from the doped state to neutral state. Colors of polymer films changed reversibly between green or blue (neutral state) and grey (doped state) upon the redox cycling.
:1) and ΔT of polymer P4 (Rda = 4:1) reached the high values of 1383 cm2 C−1 and 81.5%, respectively. Polymer P3 (Rda = 3.5:1) had a relatively short switching time of 1.24 s from the doped state to neutral state. Colors of polymer films changed reversibly between green or blue (neutral state) and grey (doped state) upon the redox cycling.
Introduction
Many π-conjugated polymers are known to be electrochromic and have been explored for use in various EC devices,1,2 owing to their unique redox and optoelectronic properties, multiple colorations, rapid switching, high coloration efficiency, and good solution processability.3–6 To date, blue-, green-, magenta-, red-colored and black EC polymers have been reported.7–10 In addition, conjugated polymers that are able to switch reversibly between colored and transparent states in both visible and near-infrared (NIR) regions by electrochemical means are reported.11,12 Visible and NIR EC polymers have potential applications in display, camouflage materials, optical communications and thermal control such as in buildings.For EC applications, a wide variety of homopolymers (e.g., poly(3,4-ethylenedioxythiophene),13 polythiophene14 and polypyrrole15) and copolymers16,17 have been developed. Furthermore, donor–acceptor (D–A) type polymers are versatile candidates due to an ease of band-gap tuning or color selection by changing the D and A units. One of the common acceptor units is diketopyrrolopyrrole (DPP), which has been incorporated into D–A polymers for potential EC applications.9 The Reynolds group has studied extensively the EC properties of various conjugated polymers and D–A polymers by changing the structures in the polymer mainchain and sidechain in recent years.18 For example, by introducing the pendent electron-donating units and increasing the steric hindrance in the backbone they obtained a range of yellow or orange polymers with the improved redox stability, higher optical contrast (ΔT > 65%) and fast switching.19 By lowering steric strain of the polymer backbone, delocalization in the charged state was relaxed, which led to a high optical contrast (ΔT > 50%) for the black-to-transmissive EC polymers.20 More recent studies revealed the influence of side chains on the EC properties.21,22
The objective of this work intends to study the effect of the D/A ratio in D–A polymers with structurally simple thiophene donor and DPP acceptor units on the EC properties, such as optical contrast, coloration efficiency and redox switching stability, and attempt to correlate these properties with the energy level and charge transfer in polymer.23 Thus, we synthesized a series of thiophene–DPP polymers P1–P4 with the different thiophene content or ratio of thiophene to DPP units (Rda).
Experimental section
Materials
All the reagents and indium tin oxide (ITO)-coated glass substrates were purchased from commercial sources. Solvents were purified by distillation before use. ITO glass substrates were cleaned by sonication in alkaline and rinsed with deionized water, acetone and isopropyl alcohol in sequence prior to use.Characterization
1H and 13C NMR spectra were recorded in CDCl3 on Bruker Acance 300 (300 M) with TMS as an internal standard. Thermogravimetric analysis (TGA) was conducted on a PerkinElmer Pyris Diamond TG instrument from 20 to 800 °C at a heating rate of 10 °C min−1 under continuous nitrogen flow. Gel permeation chromatography (GPC) was performed on a Waters 2414 system with chloroform as eluent and polystyrene as standard at room temperature. Element analysis was recorded on Elementar Vario EL cube analyzer. Cyclic voltammetry (CV) was carried out on a CHI660b electrochemical workstation under argon atmosphere at room temperature with a scan rate of 50 mV s−1 in an acetonitrile (ACN) solution of n-Bu4NPF6 (0.1 M). A Pt disk (2 mm diameter) as the working electrode, Pt wire as the counter electrode and Ag/Ag+ electrode as the reference electrode were used. The redox potential was calibrated by ferrocene as the internal standard. The UV-Vis-NIR absorption spectra were recorded on Shimadzu UV-3700 spectrophotometers. Spectroelectrochemical experiment was carried out in a transparent electrochemical cell containing an ITO glass coated with polymer film as working electrode.Synthesis of monomers and polymers
3,6-Bis(5-bromo-2-thienyl)-2,5-bis(2-hexyldecyl)-2,5-dihydropyrrolo[3,4-c]-pyrrole-1,4-dione (monomer A), 3,6-bis[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thiophen-2-yl]-2,5-bis(2-hexyldecyl)-2,5-dihydropyrrolo[3,4-c]-pyrrole-1,4-dione (monomer B), 2,5-bis(trimethylstannyl)thiophene (monomer C) and 5,5′-bis(trimethylstannyl)-2,2′-bithiophene (monomer D) were synthesized according to the literature procedures.24,25Polymer P1
Monomer A (181 mg, 0.20 mmol), monomer B (201 mg, 0.20 mmol), 1 M K3PO4 aqueous solution (1 mL), one drop of Aliquat 336 and 5 mL of toluene were added to a 50 mL Schlenk tube. The solution was degassed with argon for 40 min. Then Pd2(dba)3 (8 mg) and P(o-tol)3 (30 mg) were added to the solution, followed by degassing and charging with argon several times. The reaction mixture was stirred at 100 °C for 72 h. After cooling down to 70 °C, 10 mL of aqueous solution of sodium diethyldithiocarbamate trihydrate was added to the mixture and the mixture was stirred for 12 hours to remove the residual catalyst. The solution was then poured into 300 mL of water and extracted with chloroform. The combined organic phase was washed with deionized water and dried over anhydrous Na2SO4 for 4 h. After removal of most of the solvent, the residue (∼6 mL) was poured into 300 mL of methanol with stirring. The resulting solid was collected by filtration and placed in a Soxhlet extractor. After washing with acetone and hexane and extracting with chloroform in sequence, the extracted chloroform solution was concentrated and the polymer was precipitated in methanol and collected by filtration. After drying under reduced pressure at 40 °C, polymer P1 was obtained as black solid (225 mg, 70% yield). 1H NMR (400 MHz, CDCl3, 25 °C), 9.24 (br), 7.10 (br), 3.88–4.25 (br), 1.17–1.41 (br), 0.79–0.97 (br). Anal. calcd C, 74.17; H, 9.86; N, 3.60; S, 8.25. Found C, 73.99; H, 9.76; N, 3.52; S, 8.53. GPC Mn = 18.7 kDa, Mw = 41.2 kDa; PDI = 2.21.Polymer P2
P2 was synthesized by the same procedure as for P1 without using aqueous K3PO4 solution and Aliquat 336. Monomers used: monomer A (181 mg, 0.20 mmol) and monomer C (85.0 mg, 0.20 mmol). P2 was obtained as black solid (148 mg, 86% yield). 1H NMR (400 MHz, CDCl3, 25 °C), 8.92 (br), 6.99 (br), 4.03 (br), 0.85–1.55 (br). Anal. calcd C, 72.67; H, 9.15; N, 3.26; S, 11.19. Found C, 72.84; H, 9.30; N, 3.13; S, 11.01. GPC Mn = 17.6 kDa, Mw = 37.9 kDa; PDI = 2.15.Polymer P3
P3 was synthesized by the same procedure as for P2. Monomers used: monomer A (181 mg, 0.20 mmol), monomer C (42.5 mg, 0.10 mmol) and monomer D (47.0 mg, 0.10 mmol). P3 as a black solid was obtained (146 mg, 84% yield). 1H NMR (400 MHz, CDCl3, 25 °C), 8.96 (br), 7.00 (br), 4.02 (br), 0.67–1.94 (br). Anal. calcd C, 71.59; H, 8.67; N, 3.21; S, 12.86. Found C, 71.37; H, 8.46; N, 2.97; S, 13.37. GPC Mn = 50.7 kDa, Mw = 112.3 kDa; PDI = 2.22.Polymer P4
P4 was synthesized by the same procedure as for P2. Monomers used: monomer A (181 mg, 0.20 mmol) and monomer D (94.0 mg, 0.20 mmol). P4 as black solid was obtained (146 mg, 80% yield). 1H NMR (400 MHz, CDCl3, 25 °C), 8.98 (br), 7.00 (br), 4.02 (br), 0.84–1.93 (br). Anal. calcd C, 70.85; H, 8.59; N, 3.06; S, 14.01. Fould C, 70.96; H, 8.32; N, 2.97; S, 13.95. GPC Mn = 37.1 kDa, Mw = 80.4 kDa; PDI = 2.17.Results and discussion
Synthesis
Polymers P1–P4 were synthesized by the Suzuki or Stille cross-coupling reaction of the four monomers in different ratios (Scheme 1). The Rda values were designed as 2:1, 3:1, 3.5:1 and 4:1 for polymers P1, P2, P3 and P4, respectively (Table 1). The molecular weights of P1–P4 were in the range of 18–50 kDa (Mn) with the polydispersity indices (PDI) of 2.15–2.22 as measured by GPC (Fig. S1†). The actual ratios of thiophene–DPP in polymers correlate well to the Rda values as confirmed by the elemental analysis. From thermogravimetric analysis, the onset temperatures (Td) for 5% weight loss in nitrogen of all the polymers were around 400 °C. Polymers P1–P4 had good solubility in common organic solvents, such as chloroform and chlorobenzene and could be cast into thin films.
 | ||
| Scheme 1 Synthetic routs to polymers P1–P4. | ||
Optical and electrochemical property
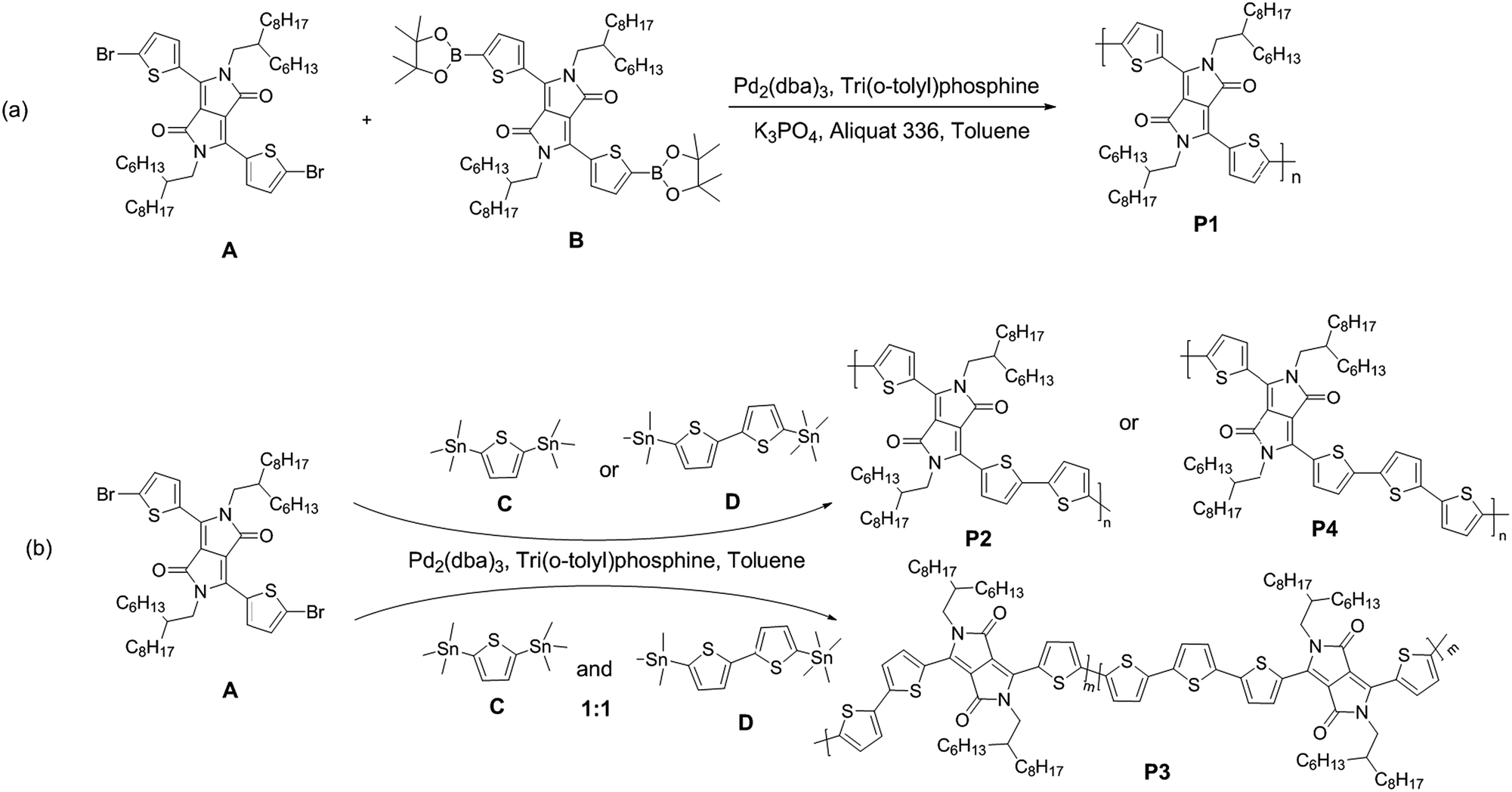
The UV-Vis-NIR absorption spectra of the polymers in chloroform and as thin films spin-coated on quartz are shown in Fig. 1. In solution, polymer P1 is green in color and has dual-band π–π* absorption at 369–445 nm. It also has a low-energy absorption with a maximum at 912 nm and a shoulder at 826 nm, assigned to the D–A charge transfer between donor and acceptor unit. As the thiophene content increases, the D–A charge transfer becomes less effective, which leads to a blue shift in maximal absorptions. Thus, the absorption spectra of P2–P4 exhibit the maximal peaks at 815 nm, 768 nm and 767 nm, respectively. Compared with the spectra of polymers in solution, the absorption of the polymer films is red shifted and broadened. The optical bandgap (Eoptg) is estimated from the onset absorption of polymer films. P1 has the lowest Eoptg of 1.20 eV. The onset absorptions of P3 and P4 are bathochromic with reference to P2 and thus have lower optical bandgaps. The colors of polymers P2–P4 in solution and as solid film are basically same and appear to be slightly darker in green than P1.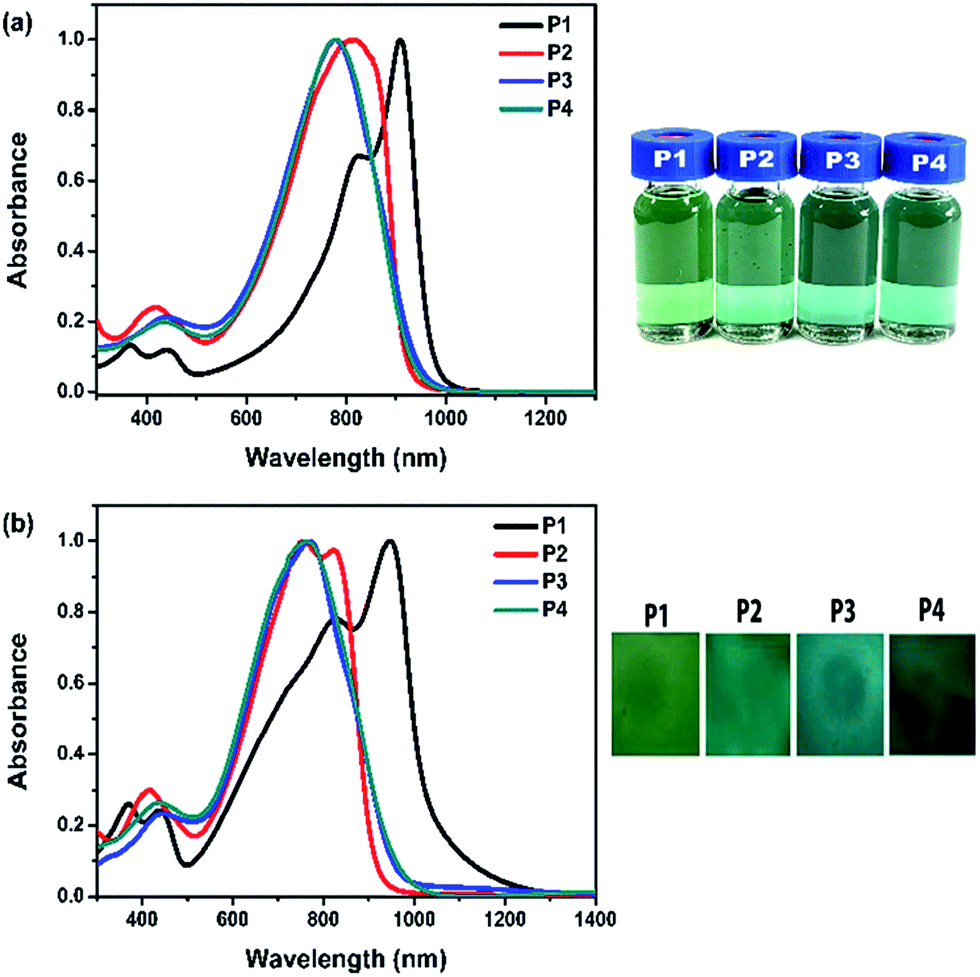 | ||
| Fig. 1 Absorption spectra of polymers P1–P4 (a) in chloroform and (b) as films on quartz. The corresponding photographs are shown on the right. | ||
| Polymer | Rda | Mw (kDa) | Mn (kDa) | PDI | Td (°C) |
|---|---|---|---|---|---|
| P1 | 2:1 |
41.2 | 18.7 | 2.21 | 401 |
| P2 | 3:1 |
37.9 | 17.6 | 2.15 | 399 |
| P3 | 3.5:1 |
112.3 | 50.7 | 2.22 | 402 |
| P4 | 4:1 |
80.4 | 37.1 | 2.17 | 403 |
Cyclic voltammetry (CV) was done to determine the redox potentials and energy levels of the polymers. Polymers P1–P4 show a great tendency towards p-doping, as indicated by the strong oxidation peaks at 0.95 V, 0.86 V, 0.74 V and 0.73 V, respectively (Fig. S2†). The high Rda values or high optical bandgaps relate to a decrease in the first oxidation potential of the polymers. The anodic peaks appear to be more prominent than the cathodic ones. The reason for such dissymmetry is not fully understood, although it was proposed that the neutral and oxidized polymers would have different conformations and relaxation, thus resulting in different redox behaviors.26 P1 and P2 had similar quasi-reversible redox behaviors from −1.2 to −1.5 V, which can be assigned to the reduction of DPP units and the formation of anionic radicals.
From the onset oxidation potential (Eonset,ox), the HOMO energy level is calculated according to the equation EHOMO = −(Eonset,ox − Eferrocene,ox) eV − 4.8 eV. The HOMO energy level of the polymer increases with the increase of Rda value or thiophene content. The lowest unoccupied molecular orbital (LUMO) energy level is calculated according to the equation ELUMO = EHOMO + Eoptg eV and increases in steps as the acceptor content decreases (Table 2).
| Polymer | λmax (nm) | λonset (nm) | Eoptg (eV) | Potential (V) | HOMO (eV) | LUMO (eV) | ||
|---|---|---|---|---|---|---|---|---|
| Vis | NIR | Eox | Eonset,ox | |||||
| P1 | 444 | 946 | 1032 | 1.20 | 0.95 | 0.69 | −5.43 | −4.23 |
| P2 | 420 | 825 | 913 | 1.36 | 0.86 | 0.74 | −5.48 | −4.12 |
| P3 | 446 | 761 | 963 | 1.29 | 0.74 | 0.61 | −5.35 | −4.06 |
| P4 | 441 | 768 | 954 | 1.30 | 0.73 | 0.58 | −5.32 | −4.02 |
Spectroelectrochemistry
The optical changes upon doping were performed in a [BMIM]PF4 (1-butyl-3-methylimidazolium tetrafluoroborate)-ACN solution (the volume ratio of [BMIM]PF4 to ACN was 3:1) under the progressive potential from 0 to 1.2 V. Polymer films were spin-coated on ITO glass substrates. The spectroelectrochemical changes of P1 are presented in Fig. 2. The spectral changes of P2–P4 are illustrated in Fig. S4.† These results indicate the formation of positively charge carriers upon electrochemical oxidation. Upon the progressive oxidation, new absorption bands appear in the NIR region (>1000 nm), due to the formation of polarons and bipolarions (low-energy charge carriers). The polaron peaks reach a maximum under applied potential of 1.0 V and 1.2 V for polymers P1 and P2–P4, respectively. As for P2–P4, the polaronic absorption bands are blue-shifted and become more intense (λmax 1240–1260 nm) compared with P1 (λmax 1300 nm). At the same time, the absorptions at 761–946 nm are diminished upon oxidation due to the interruption of charge transfer between donor and acceptor units. In particular, when P1 is completely oxidized, the absorption in the visible region becomes a minimum and a highly transmissive state (60–70% transmission in the visible region) is achieved (Fig. 2b). The color changes from green to grey (Fig. 2c). Similarly, P2–P4 change the colors to grey at the oxidation states (Fig. S4†).
 | ||
| Fig. 2 (a) The absorption spectra of P1 film under different voltages; (b) transmittance and absorption (inset) of P1 film at the neutral and oxidation states; (c) photographs of P1 film at the neutral state and oxidation state. | ||
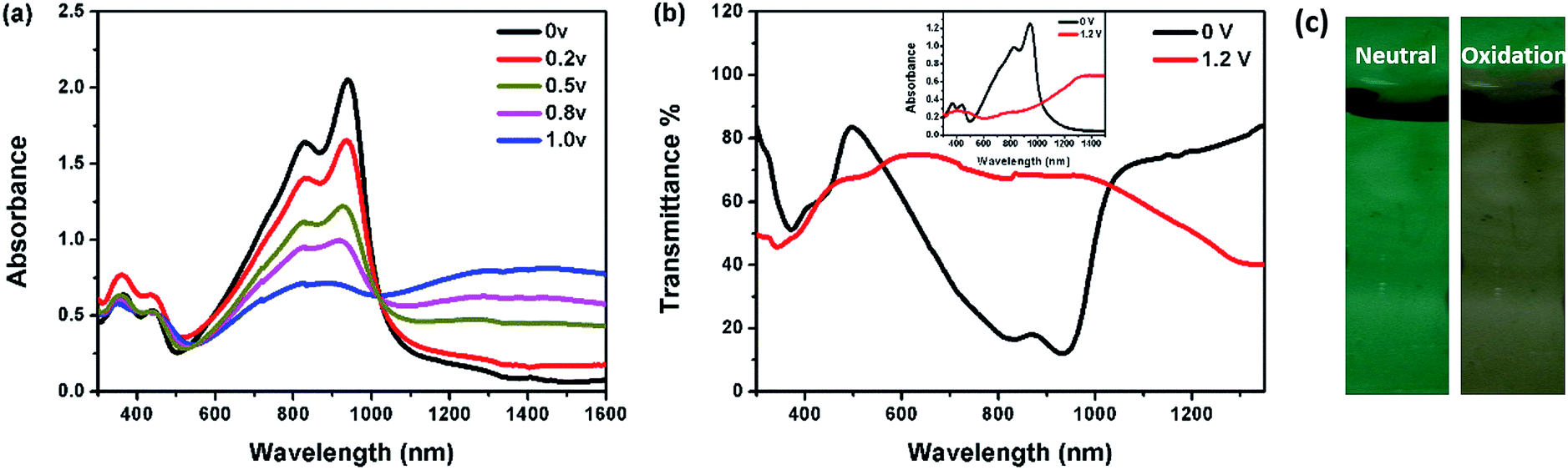
At both neutral and oxidation states, P1 is more transparent in the visible region than the other three (Fig. 3). The transmittance of P2–P4 in the visible region decreases with the increase of the Rda values or more thiophene content. While in the NIR region, P4 is the most transparent at the neutral state and becomes the least at the oxidation state, thus representing a high optical contrast in the NIR region.
 | ||
| Fig. 3 Transmittance of polymer films at (a) neutral states and (b) oxidation states. | ||
Electrochromic switching
The EC switching was studied by probing the transmittance changes under a square-wave potential step in the [BMIM]PF4–ACN solution. The polymers were spin-coated on ITO glass substrates with an active area of 2.7 cm2. The voltage was switched between 0 V and 1.2 V at an interval of 20 s. The transmittance was monitored as a function of time at a given wavelength. Optical contrast (ΔT) and response time (t) were of 946 nm for P1, 825 nm for P2, 761 nm for P3 and 768 nm for P4. (b) Electrochromic switching of P1 film at 1310 nm measured from the spectra of transmittance versus time (Fig. 4a). The inject/eject charge per area (Q) was determined by in situ square-wave potential and calculated from the integration of current density and time. The coloration efficiency (CE) is defined as the optical density change at a specific wavelength per unit of charge inject/eject, which is calculated according to the equation CE = log[Tb/Tc]/Q, where Tb is transmittance at the bleached (oxidation) state and Tc is transmittance at the colored (neutral) state. CE represents the power requirement of an electrochromic material. All the data were collected at the wavelengths of the maximal absorption and at 1310 nm for each polymer, based on 95% of the full switch (Table 3). | ||
| Fig. 4 (a) Electrochromic switching of polymer films at 1310 nm (solid line) and at maximal absorption (dash line) and (c) at 946 nm for different cycles. | ||
| Polymer | P1 | P2 | P3 | P4 | ||||
|---|---|---|---|---|---|---|---|---|
| λ (nm) | 1310 | 946 | 1310 | 825 | 1310 | 761 | 1310 | 768 |
| ΔT (%) | 37.3 | 13.2 | 66.9 | 23.8 | 42.2 | 39.3 | 81.5 | 41.3 |
| tb (s) | 5.36 | 11.02 | 6.61 | 5.33 | 8.11 | 3.12 | 4.51 | 7.75 |
| tc (s) | 12.82 | 4.36 | 5.61 | 5.14 | 1.24 | 1.85 | 2.02 | 3.37 |
| Q (mC cm−2) | 0.66 | 0.75 | 0.47 | 0.69 | 5.62 | 4.97 | 2.63 | 3.62 |
| CE (cm2 C−1) | 543 | 348 | 1383 | 424 | 92 | 243 | 328 | 261 |
It is interesting to note that optical contrasts at the maximal absorption are lower than those at 1310 nm for all the polymers. With the increase of Rda values, ΔT of P1–P4 increases from 13.2% to 41.3% at the maximal absorption peaks. It may be ascribed to an ease of charge transfer along the polymer backbone containing more thiophene units. Similarly, ΔT increases significantly from 37.3% to 81.5% at 1310 nm as the Rda values increase, except for P3 due to a possible restriction of polaronic charge transfer.
These polymers are able to switch from the colored states to bleached states within 3–11 s and vice versa within 1–13 s. The coloring time tc decreases noticeably for those having more thiophene units in polymer. Especially for P3, the coloring time is 1.24 s at 1310 nm and 1.85 s at 761 nm, which are far more quickly than the others. The coloration efficiencies of all the polymers are greater than 200 cm2 C−1 at λmax which are a bit larger than the typical CE of PEDOT (183 cm2 C−1).27 The CE value of P2 at 1310 nm is the largest among all, up to 1383 cm2 C−1.
The switching stability was preliminarily tested under ambient conditions in air by subjecting polymer films to 500 cycles of redox switching (Fig. 4b, c and S5†). There was a slight drop in optical contrast after 500 cycles at 1310 nm. Under inert atmosphere and optimized conditions, these polymers are expected to have an excellent switching stability and suitable for EC applications.
Conclusions
A series of low bandgap D–A polymers containing thiophene and DPP units are electrochromic in the visible and NIR spectral region. Their EC properties, in particular optical contrast, response time and coloration efficiency are found to depend on the ratio of thiophene to DPP in polymer. Among all, polymer P2 (Rda = 3:1) has the highest CE of 1383 cm2 C−1, P3 (Rda = 3.5:1) has the fastest coloring speed of 1.24 s and P4 (Rda = 4:1) gives the highest optical contrast of 85% at 1310 nm. These polymers show high contrast in the NIR region and good switching stability, thus making them a good candidate for use in NIR electrochromic applications.
Acknowledgements
This work was supported by National Natural Science Foundation of China (21474102 and 21474105) and the Natural Science and Engineering Research Council of Canada.References
- E. M. Sanford, M. E. Tori, T. M. Smeltzer, C. K. Beaudoin, B. H. Bowser, M. E. Anderson and K. L. Brown, Electrochemistry, 2015, 83, 1061 CrossRef CAS.
- A. L. Dyer, R. H. Bulloch, Y. Zhou, B. Kippelen, J. R. Reynolds and F. Zhang, Adv. Mater., 2014, 26, 4895 CrossRef CAS PubMed.
- J. Z. Low, W. T. Neo, Q. Ye, W. J. Ong, I. H. K. Wong, T. T. Lin and J. Xu, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1287 CrossRef CAS.
- Y. Fu, X. Cheng, J. Zhao, T. Kong, C. Cui and X. Zhang, Polym. J., 2012, 44, 1048 CrossRef CAS.
- W. T. Neo, Q. Ye, S. J. Chua and J. Xu, J. Mater. Chem. C, 2016, 4, 7364 RSC.
- M. Sassi, M. M. Salamone, R. Ruffo, G. E. Patriarca, C. M. Mari, G. A. Pagani, U. Posset and L. Beverina, Adv. Funct. Mater., 2016, 26, 5240 CrossRef CAS.
- W. T. Neo, C. M. Cho, Z. Shi, S. J. Chua and J. Xu, J. Mater. Chem. C, 2016, 4, 28 RSC.
- I. Welterlich, J. M. Neudörfl and B. Tieke, Polym. Chem., 2015, 6, 1005 RSC.
- M. Gora, W. Krzywiec, J. Mieczkowski, E. C. Rodrigues Maia, G. Louarn, M. Zagorska and A. Pron, Electrochim. Acta, 2014, 144, 211 CrossRef CAS.
- A. R. Rabindranath, Y. Zhu, K. Zhang and B. Tieke, Polymer, 2009, 50, 1637 CrossRef CAS.
- M. Akbayrak and A. M. Önal, Polym. Chem., 2016, 7, 6110 RSC.
- M. İçli, M. Pamuk, F. Algi, A. M. Önal and A. Cihaner, Chem. Mater., 2010, 22, 4034 CrossRef.
- C. G. Cutler, M. Bouguettaya, T. Kang and J. R. Reynolds, Macromolecules, 2008, 38, 3068 CrossRef.
- M. R. Andersson, O. Thomas, W. Mammo, M. Svensson, M. Theander and O. Inganas, J. Mater. Chem., 1999, 9, 1933 RSC.
- M. Ak, B. Gacal, B. Kiskan, Y. Yagci and L. Toppare, Polymer, 2008, 49, 2202 CrossRef CAS.
- I. Pochorovski and F. Diederich, Acc. Chem. Res., 2014, 47, 2096 CrossRef CAS PubMed.
- H. J. Byker, Electrochim. Acta, 2001, 46, 2015 CrossRef CAS.
- P. M. Beaujuge, S. V. Vasilyeva, D. Y. Liu, S. Ellinger, T. D. McCarley and J. R. Reynolds, Chem. Mater., 2012, 24, 255 CrossRef CAS.
- K. Cao, D. E. Shen, A. M. Österholm, J. A. Kerszulis and J. R. Reynolds, Macromolecules, 2016, 49, 8498 CrossRef CAS.
- J. A. Kerszulis, R. H. Bulloch, N. B. Teran, R. M. W. Wolfe and J. R. Reynolds, Macromolecules, 2016, 49, 6350 CrossRef CAS.
- C. Lin, T. Endo, M. Takase, M. Iyoda and T. Nishinaga, J. Am. Chem. Soc., 2011, 133, 11339 CrossRef CAS PubMed.
- Ö. Çelikbilek, M. İçliÖzkut, F. Algi, A. M. Önal and A. Cihaner, Org. Electron., 2012, 13, 206 CrossRef.
- P. C. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268 CrossRef CAS PubMed.
- J. S. Ha, K. H. Kim and D. H. Choi, J. Am. Chem. Soc., 2011, 133, 10364 CrossRef CAS PubMed.
- Y. Li, B. Sun, P. Sonar and S. P. Singh, Org. Electron., 2012, 13, 1606 CrossRef CAS.
- G. Tourillon and F. Garnier, J. Electroanal. Chem., 1984, 161, 51 CrossRef CAS.
- C. L. Gaupp, D. M. Welsh, R. D. Rauh and J. R. Reynolds, Chem. Mater., 2002, 14, 3964 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CV and TGA traces of all the polymers and spectroelectrochemical data of P2, P3 and P4. See DOI: 10.1039/c7ra01828b |
| This journal is © The Royal Society of Chemistry 2017 |