
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-selective biotransformation of ursane triterpenes by Streptomyces griseus ATCC 13273†
Shao-Hua Xua,
Chao Zhanga,
Wei-Wei Wangb,
Bo-Yang Yu *b and
Jian Zhang*a
*b and
Jian Zhang*a
aState Key Laboratory of Natural Medicines, China Pharmaceutical University, 24# Tong Jia Xiang Street, Nanjing, Jiangsu 210009, China. E-mail: 1020071849@cpu.edu.cn; Fax: +86-25-86185158; Tel: +86-25-86185157
bJiangsu Key Laboratory of TCM Evaluation and Translational Research, China Pharmaceutical University, 639# Long Mian Avenue, Nanjing, Jiangsu 211198, China. E-mail: boyangyu59@163.com; Fax: +86-25-83313080; Tel: +86-25-83271321
First published on 11th April 2017
Abstract
The oxidization of unactivated C–H bonds of pentacyclic triterpenes (PTs) is of great interest for the structural modification of PTs. Herein, we discovered the unique capability of Streptomyces griseus ATCC 13273 to catalyze the site-selective oxidation of the C-30 methyl group to the carboxyl group and hydroxylation of the C-24 methyl group over a range of ursane triterpenes, including ursolic acid (1), 3-oxo ursolic acid (2), and corosolic acid (3). It is noteworthy that while using asiatic acid (4) and madasiatic acid (5), which bear one hydroxyl group on C-23 as substrates, the hydroxylation on C-24 was blocked. As a result, eight new compounds (1a–3a, 5a, 1b–3b and 5b) of the metabolites were isolated and their structures were elucidated based on 1D and 2D NMR and HR-MS data. In addition, the cytotoxicity of substrates and transformed products was preliminarily evaluated by an MTT assay.
Introduction
Pentacyclic triterpenes (PTs) are bioactive natural products, which display a remarkable spectrum of biological activities, such as anti-tumor, anti-HIV, anti-microbial, anti-diabetic, and anti-inflammatory activities.1–4 The substitution of hydroxyl or carbonyl groups to the methyl or methenyl carbons, respectively, of the skeleton greatly enrich the structural diversity2,5,6 and is also essential for the biological activities and structural modification of PTs.7–9 Herein, the research on the structural modification of PTs was focused on the functionalization of the unactivated C–H bonds on the skeleton; it could also enrich the structural diversities and generate more effective and/or less toxic derivatives.10–12 However, selective C–H activation remains a challenge for synthetic chemists.13–15 In the chemical modification of natural small molecules including PTs, regioselective oxidation of CH3 or CH2 is one of the most difficult steps, and the present chemical processes always involve the use of heavy metals and tedious steps.16,17 With the versatile enzyme systems of microbes, biotransformation makes the C–H oxidation modification of small molecules including PTs facile and green in a single step.18,19 In fact, when using microorganisms as a biocatalyst, the cheap and readily available compounds in nature can be directionally converted to high added-value natural compounds or new natural product-like compounds.20–22In our previous studies, some novel microbial transformations of pentacyclic triterpenoids such as asymmetric ketone hydroxylation23 and methyl hydroxylation24 have been reported individually. Streptomyces griseus ATCC 13273 has been proven as a potent strain that can achieve oxygenation of unactivated C–H bonds of the olean-type pentacyclic triterpenes.24 In order to investigate the regioselective oxidizing capability of S. griseus ATCC 13273 to ursane triterpenes and expand the structural diversities of PTs, five ursane triterpenes, ursolic acid (1), 3-oxo ursolic acid (2), corosolic acid (3), asiatic acid (4) and madasiatic acid (5), were used as the substrates in this study, resulting in eight new metabolites and one known metabolite. This was the first time the biotransformation of ursane triterpenes by S. griseus ATCC 13273 was screened. Moreover, the influence of the hydroxyl substitutions on the A and B rings on the reactions was also analyzed as the five ursane-type substrates bear different numbers of hydroxyl groups on the A and B ring skeleton: 3-oxo ursolic acid (2, with no hydroxyl group), ursolic acid (1, with one hydroxyl group at C-3), corosolic acid (3, with two hydroxyl groups at C-3 and C-2), asiatic acid (4, with three hydroxyl groups at C-3, C-2 and C-23) and madasiatic acid (5, with four hydroxyl groups at C-3, C-2, C-23 and C-6). In addition, the cytotoxicity of the substrates and metabolites against HepG2 and MCF-7 cells was evaluated by an MTT assay.
Results and discussion
Identification of transformed products
Ursolic acid (1) (150 mg) was added into a 2 day-old culture of S. griseus ATCC 13273, and two additional polar metabolites, compounds 1a (48.6 mg, yield 30.4%) and 1b (52.1 mg, yield 31.6%), were isolated (Fig. 1).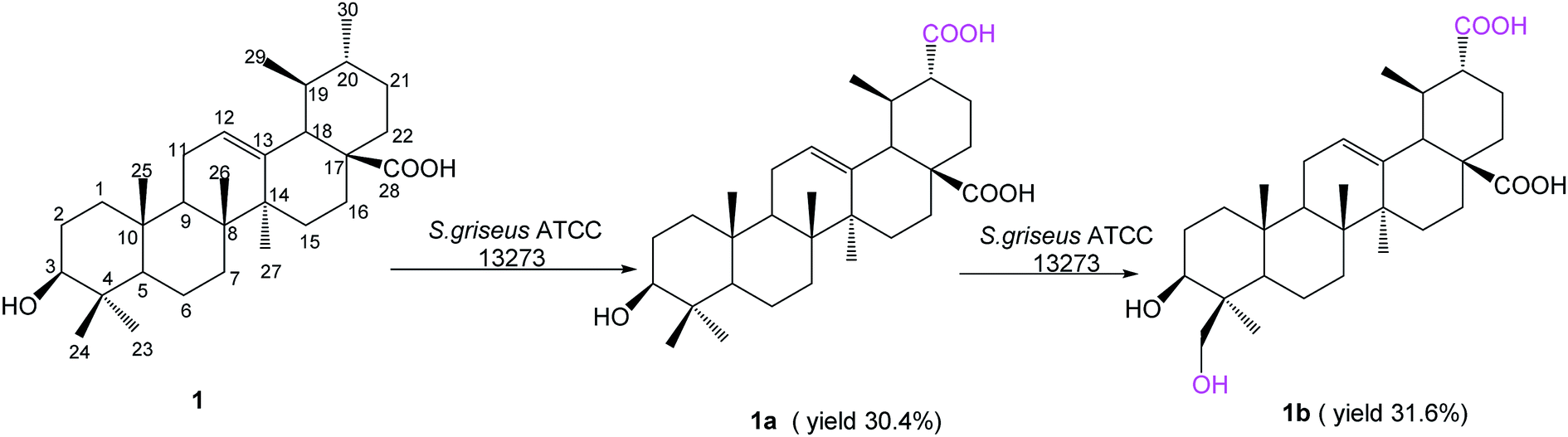 | ||
| Fig. 1 Biotransformation of ursolic acid (1) by S. griseus ATCC 13273. | ||
Compound 1a was isolated as a colorless powder, and the HR-ESI-MS of compound 1a showed an [M − H]− ion at m/z 485.3260 (calcd for C30H45O5, 485.3272), indicating a 30 amu mass increase compared to that of compound 1. The 1H NMR spectrum of compound 1a displayed six characteristic methyl groups at δH 0.94 (3H, s), 1.05 (3H, s), 1.09 (3H, s), 1.26 (3H, s), 1.31 (3H, d) and 1.32 (3H, s), with the absence of one splitting methyl signal in the substrate. The 13C NMR spectrum exhibited a new signal at δC 179.02 ppm, and only six characteristic methyl signals appeared at δC 29.25, 24.48, 19.43, 17.90, 17.04 and 16.14 ppm. This evidence suggested that one methyl group might be oxidized to carboxyl group. Namely, the resonance of C-30 (a methyl group at δC 21.41 in ursolic acid) disappeared, and the carbon signal at δC 17.52 ppm attributed to C-29 shifted down-field 1.91 ppm to δC 19.43 ppm in 1a, indicating that the oxidization was on C-30. In addition, the three-bond correlation between δH 1.31 (d, 3H, H-29) ppm and δC 53.10 ppm (C-20) was also observed in the HMBC spectrum. The relative stereochemistry of the carboxyl group was established as equatorial based on NOESY experiments. The NOESY spectrum of 1a showed NOE enhancements between δH 1.31 (d, 3H, H-29) and δH 2.80 (d, 1H, J = 11.4 Hz, β-H-18) ppm. This further evidenced that the methyl group of C-30 was oxidized to a carboxyl group in the biotransformation. Thus, compound 1a was elucidated as 3β-hydroxy-urs-12-en-28,30-dioic acid, which is a new compound.
Compound 1b was isolated as a colorless powder, and the HR-ESI-MS of compound 1b showed an [M − H]− ion at m/z 501.3222 (calcd for C30H45O6, 501.3222), which indicated an increment of 46 amu as compared to compound 1. The new signal at δC 179.02 ppm in the 13C NMR spectrum could be attributed to C-30 for the same 2D NMR evidence of 1a. The 1H NMR spectrum of 1b showed two new proton signals at δH 4.49 (d, 1H, J = 11.0 Hz) and 3.67 (d, 1H, J = 11.0 Hz) ppm, whereas the DEPT spectrum showed the presence of a new CH2 signal at δC 65.04 ppm, with the disappearance of one methyl group's carbon signals, confirming that 1b was a monohydroxylated metabolite of 1a. In the HSQC experiment, the new proton signals showed correlations with the new carbon signal at δC 65.04 ppm, and in the HMBC experiment, the new CH2 signals showed long-range (two- and three-bond) 1H–13C correlations with C-3 and C-4, confirming C-23 or C-24 as the site of hydroxylation. Furthermore, the relative stereochemistry of the hydroxyl group was established as axial based on NOESY data (Fig. 2). The NOESY spectrum of 1b showed NOE enhancements between the new proton signals at δH 4.49 (d, 1H, J = 11.0 Hz) and 3.67 (d, 1H, J = 11.0 Hz) ppm with δH 0.88 ppm (s, 3H, H-25), indicating that the hydroxyl group was substituted at C-24. Based on all the evidence, metabolite 1b was characterized as 3β,24-dihydroxy-urs-12-en-28,30-dioic acid, which is a new compound.
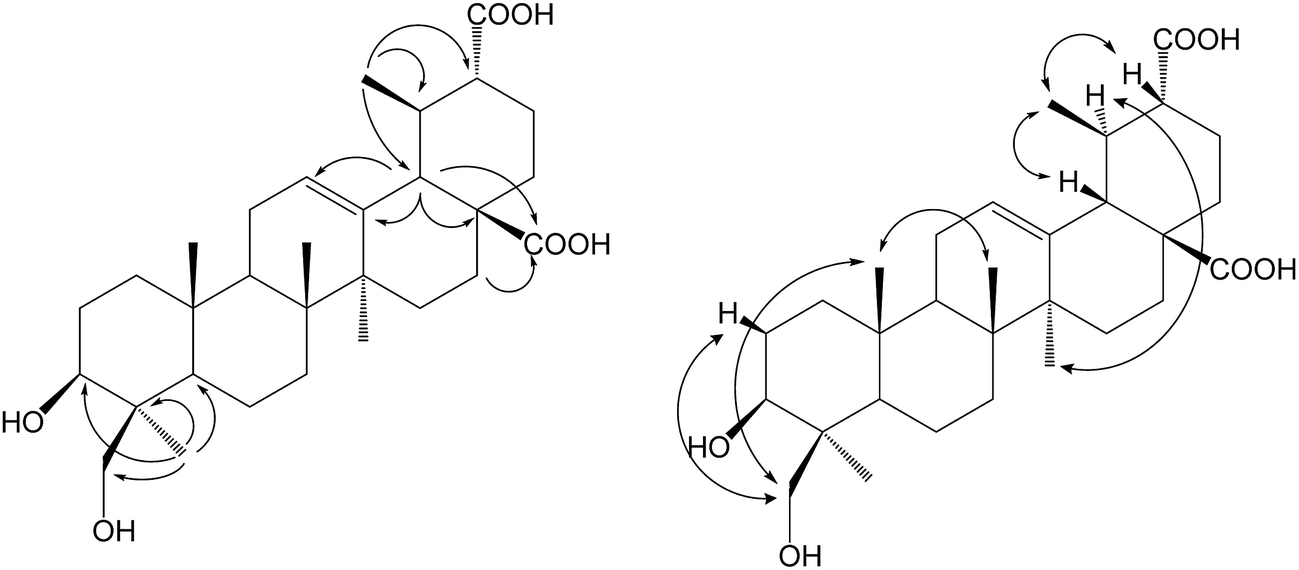 | ||
| Fig. 2 Key HMBC and NOESY correlations of compound 2b. | ||
S. griseus ATCC 13273 was found to be a potent strain based on the regioselective unactivated C–H bond oxidation of PTs, as it conducted the same oxidation reactions to ursolic acid (1) as well as oleanolic acid24 on the same reaction sites (the methyl group on C-4 and C-20).
To understand the conversion process of ursolic acid (1) by S. griseus ATCC 13273, we next conducted a time-course analysis of the biotransformation process of 1 (ESI Fig. S21†). Then, we subjected 1a as the substrate, which also resulted in the product of 1b. Therefore, in the biotransformation process, 1a was the initial product of 1, and 1b was the further oxidized product via 1a.
To further explore the biotransformation capability of S. griseus ATCC 13273 to ursane triterpenes and investigate the influence of the hydroxyl substitutions on A and B rings in the reactions, another four ursane-type substrates, 3-oxo ursolic acid (2), corosolic acid (3), asiatic acid (4) and madasiatic acid (5), were screened.
Biotransformation of 3-oxo ursolic acid (2, 150 mg) with S. griseus ATCC 13273 also resulted in two additional polar metabolites 2a (38.5 mg, yield 24.1%) and 2b (75.9 mg, yield 45.9%) (Fig. 3A). Two additional polar metabolites, 3a (46.2 mg, yield 29.0%) and 3b (29.2 mg, yield 15.9%) (Fig. 3B), were isolated from the biotransformation of corosolic acid (3, 150 mg).
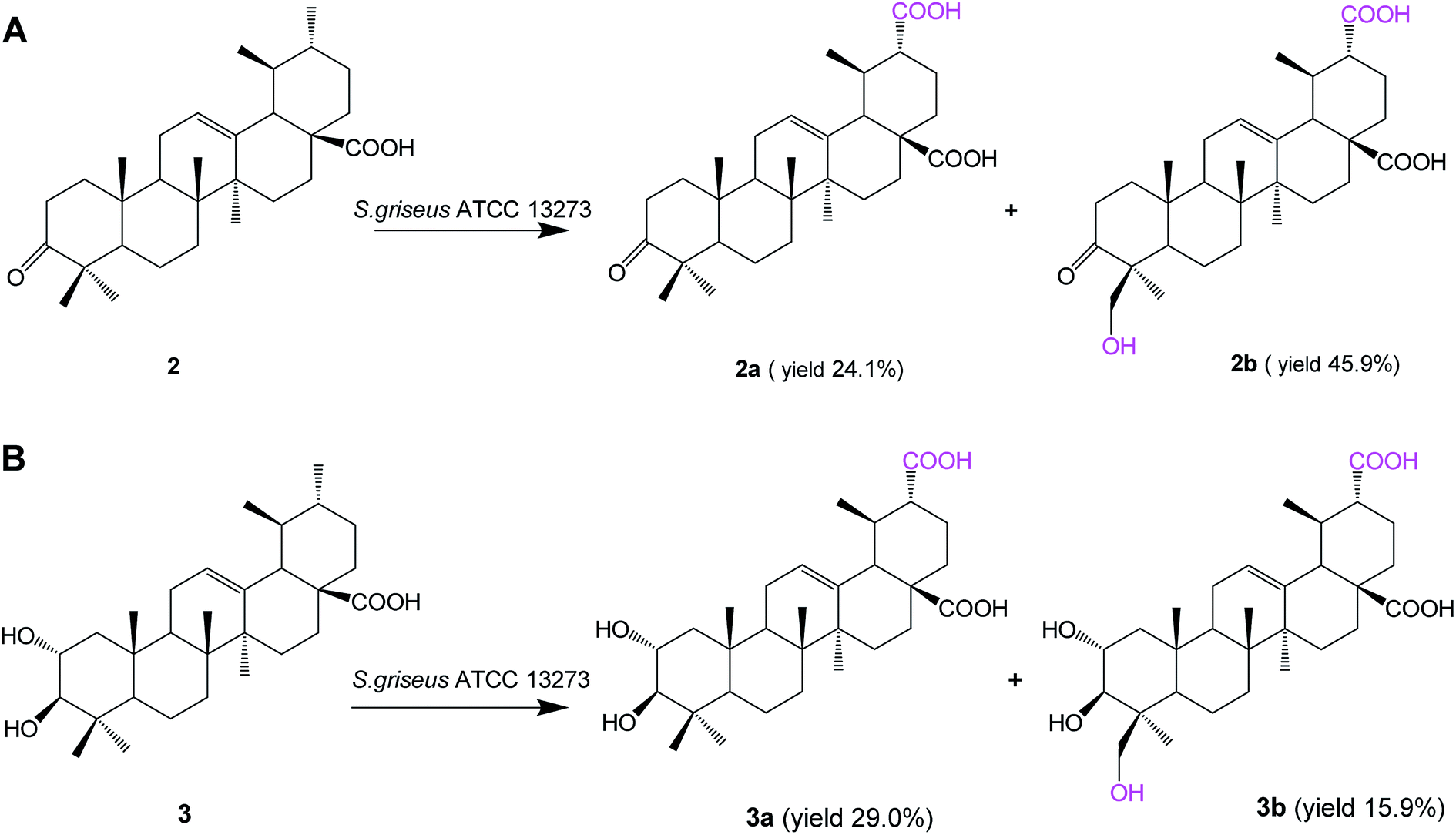 | ||
| Fig. 3 Biotransformation of 3-oxo ursolic acid (2) and corosolic acid (3) by S. griseus ATCC 13273. | ||
Based on the HR-ESI-MS, 1D and 2D NMR data and the reaction types on ursolic acid, compound 2a was identified as 3-oxo-urs-12-en-28, 30-dioic acid; compound 2b was identified as 24-hydroxy-3-oxo-urs-12-en-28,30-dioic acid;24 compound 3a was identified as 2α,3β-dihydroxy-urs-12-ene-28,30-dioic acid, and compound 3b was identified as 2α,3β,24-trihydroxy-urs-12-ene-28,30-dioic acid (the structure elucidation can be seen in the ESI†).
Biotransformation of asiatic acid (4, 100 mg) with S. griseus ATCC 13273 resulted in only one additional polar metabolite, 4a (52.1 mg, yield 49.1%) (Fig. 4).
 | ||
| Fig. 4 Biotransformation of asiatic acid (4) by S. griseus ATCC 13273. | ||
Compound 4a was obtained as a white amorphous powder. The HR-ESI-MS of compound 4a showed an [M − H]− ion at m/z 517.3164 (calcd for C30H45O7, 517.3171), which indicated an increment of 30 amu as compared to compound 4. Then, 4a was characterized as 2α,3β,23-trihydroxy-urs-12-ene-28,30-dioic acid due to its NMR spectral data being identical to those reported in the literature.25
Biotransformation of madasiatic acid (5, 100 mg) with S. griseus ATCC 13273 resulted in two additional polar metabolites, 5a (12.5 mg, yield 12.1%) and 5b (33.4 mg, yield 31.5%) (Fig. 5).
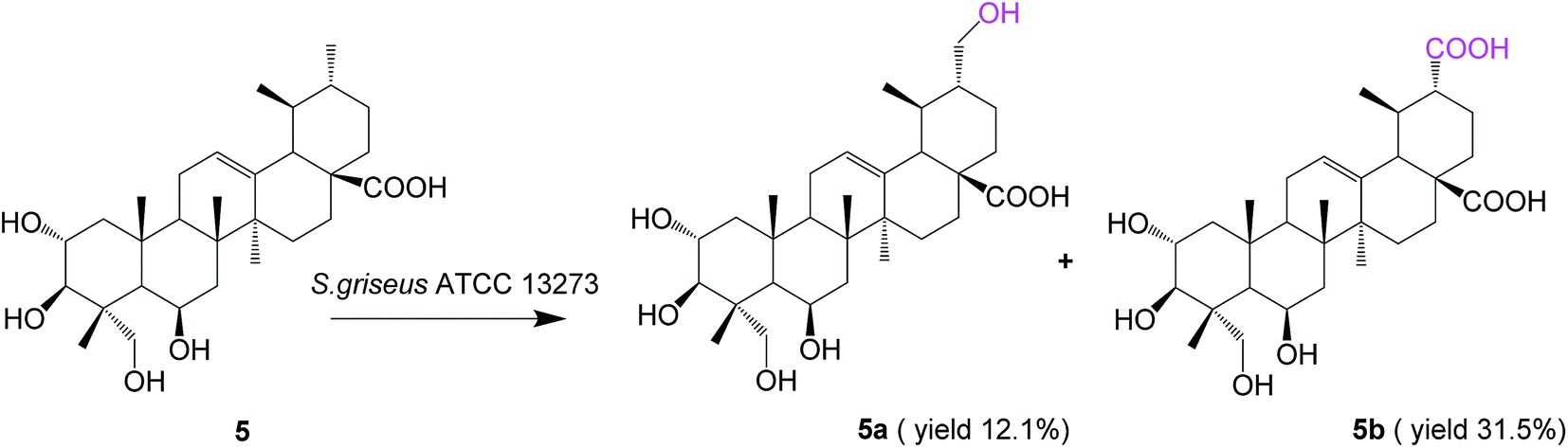 | ||
| Fig. 5 Biotransformation of madasiatic acid (5) by S. griseus ATCC 13273. | ||
The HR-ESI-MS of compound 5a showed an [M − H]− ion at m/z 519.3319 (calcd for C30H47O7, 519.3327), indicating a 16 amu mass increase compared to that of compound 5. The 1H NMR spectrum of compound 5a displayed five characteristic methyl groups in the absence of one splitting methyl signal in the substrate. The DEPT spectrum exhibited a new methylene carbon signal at δC 65.66 ppm, confirming C-30 or C-29 as the site of hydroxylation. The hydroxylation at C-30 was authenticated by the paramagnetic shifts of the neighboring carbons C-20 (from δC 39.99 ppm of the substrate to δC 47.97 ppm), whereas the signals of C-19 and C-21 shifted upfield by 5.52 and 5.41 ppm, respectively. The HMBC spectrum exhibited correlations of H-30 with C-19 and C-21, as well as H-29 with C-18, C-19 and C-20, which further confirmed the structure of the compound 5a as 2α,3β,6β,23,30-pentahydroxy-urs-12-ene-28-oic acid.
The molecular formula of 5b was established as C30H46O8 based on HR-ESI-MS, in which a quasimolecular ion was detected at m/z 533.3113 [M − H]− (calcd for C30H45O8, 533.3120), indicating a 30 amu mass increase compared to that of compound 5. According to a similar analysis of the NMR spectrum with 3a and 4a, we judged that the C-30 methyl group of 5 was oxidized to the corresponding carboxyl group. Compound 5b was identified as 2α,3β,6β,23-tetrahydroxy-urs-12-ene-28,30-dioic acid, and it is reported for the first time.
It was encouraging to find that S. griseus ATCC 13273 could catalyze highly efficient regioselective methyl oxidation at C-30 for all of these ursane triterpenes, regardless of the variation of hydroxyl substitutions on A and B rings. The hydroxylation of methyl at C-24 would be shielded when the substrates (compound 4 and 5) bear the vicinal hydroxymethyl group at C-23. Moreover, the methyl group at C-30 of compounds 1–4 could be deeply oxidized to a carboxyl group rapidly, and no potential intermediates were observed in the HPLC analysis, but to the substrate 5, bearing one hydroxyl substitution at C-6, the hydroxylated intermediate (compound 5a) was isolated. Therefore, we speculated that the hydroxyl substitution at C-6 or excessive hydroxyl substitutions on A and B rings may slow the progress of further oxidation of methyl at C-30. This reaction would also provide a new active-site on the E rings for further structural modification of PTs. In general, the facile biocatalytic oxidation of the unactivated C–H bonds by whole cells illustrates the advantages and properties of microbial transformations very well in a reaction that is difficult to achieve using synthetic chemical methods.26,27 Developing green and highly regioselective catalytic methods for the oxidation of unactivated sp3 C–H bonds will have widespread applications in synthetic chemistry.
Cytotoxic activities of the compounds
All compounds were tested for in vitro cytotoxicity. Ursolic acid (1), corosolic acid (3) and asiatic acid (4) showed an inhibitory effect against HepG-2 human hepatoma cancer cells and MCF-7 human breast cancer cells (see Table 1). None of the metabolites showed significant inhibitory activities against these cancer cell lines, which may be due to the toxicity attenuated effect of microbial metabolism on exogenous substrates.| Compounds | Inhibition rate (%) 72 h | |||||
|---|---|---|---|---|---|---|
| HepG-2 | MCF-7 | |||||
| 50 μM | 10 μM | 1 μM | 50 μM | 10 μM | 1 μM | |
| 1 | 60.1 | 5.4 | −7.2 | 78.1 | −32.6 | 5.7 |
| 3 | 94.6 | −7.5 | −4.8 | 83.9 | 6.7 | 5.6 |
| 4 | 91.8 | −3.3 | −0.9 | 70.5 | 23.1 | 5.2 |
Experimental
General procedures
NMR spectra were recorded on a Bruker AV-500 spectrometer in C5D5N solution with TMS as the internal standard, and chemical shifts were expressed in δ (parts per million). HR-ESI-MS experiments were performed on an Agilent 6210 ESI-TOF spectrometer. Preparative HPLC was carried out on a SHIMADZU system with an Ultimate® LP-C18 column (10 × 250 mm). The high performance liquid chromatography (HPLC) analysis was performed on an Agilent 1260 system with an Ultimate® LP-C18 column (4.6 × 200 mm) eluent: acetonitrile/water, flow rate: 1.0 mL min−1, detector: Alltech 3300 ELSD. All of the solvents used for extraction and isolation were of analytical grade. TLC was performed on precoated silica gel GF254 plates. Separation and purification were carried out by column chromatography on silica gel (200–300 mesh). Silica gel was purchased from Qingdao Marine Chemical Group Co., PR China.Substrates and microorganism
Ursolic acid (1) and corosolic acid (3) were purchased from Spring&Autumn Pharmacy Co., Ltd., Nanjing China. 3-Oxo ursolic acid (2) was prepared by the oxidation of 1 with Jones reagent and purified by crystallization in methanol. Asiatic acid (4) and madasiatic acid (5) were isolated from Centella asiatica in our lab. Their structures were characterized by 1H NMR, 13C NMR, and MS spectra (see Table S1†).S. griseus ATCC 13273 was obtained by courtesy of Prof. John P. N. Rosazza of the University of Iowa, USA.
Analytical and preparative scale biotransformation, isolation, and identification of biotransformation products
Cultures were grown by a two-stage procedure in 50 mL of soybean meal glucose medium held in 250 mL culture flasks. The soybean meal glucose medium contained (in g L−1) 20 glucose, 5 yeast extract, 5 soybean meal, 5 NaCl, and 5 K2HPO4 in distilled water and was adjusted to pH 7.0 with 6 N HCl before being autoclaved at 121 °C for 15 min. Cultures were incubated with shaking at 180 rpm at 28 °C. A 10% inoculum derived from 48 h-old stage I cultures was used to initiate stage II cultures, which were incubated for 24 h before receiving 10 mg of substrates in 1 mL of DMSO, and incubations were conducted as before. Substrate controls consisted of sterile medium and substrates incubated under the same conditions but without microorganisms. Cultures were incubated for 3–5 days and extracted with equal volumes of EtOAc. The organic phase was concentrated and spotted on a silica gel TLC plate, which was developed by chloroform/methanol (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). The results were visualized by spraying with H2SO4 (10% in ethanol) and heating at 120 °C for 1–2 min.
:1, v/v). The results were visualized by spraying with H2SO4 (10% in ethanol) and heating at 120 °C for 1–2 min.
Using 24 h-old stage II cultures, the substrate (1–3, 150 mg; 4 and 5, 100 mg) was distributed evenly among thirty 125 mL culture flasks. Substrates containing cultures were incubated for 3–5 days and then extracted with equal volumes of EtOAc three times. The organic solvent layer was evaporated to dryness. The crude extracts of substrates 1–5 were subjected to silica gel column chromatography eluted with a solvent system of chloroform/methanol (99:1 to 90:10) to afford the fractions 1–5, respectively. Fraction 1 was purified by preparative HPLC with acetonitrile:H2O = 45:55, 14 mL min−1. Metabolites 1a (48.6 mg) and 1b (52.1 mg) were isolated. Fraction 2 was purified by preparative HPLC with acetonitrile:H2O = 55:45, 14 mL min−1. Metabolites 2a (38.5 mg) and 2b (75.9 mg) were isolated. Fraction 3 was purified by preparative HPLC with acetonitrile:H2O = 42:58, 14 mL min−1. Metabolites 3a (46.2 mg) and 3b (29.2 mg) were isolated. Fraction 4 was purified again by silica gel column chromatography eluted with a solvent system of chloroform/methanol (93:7 to 90:10) to afford 4a (52.1 mg). Fraction 5 was purified by preparative HPLC with acetonitrile:H2O = 40:60, 14 mL min−1. Metabolites 5a (12.5 mg) and 5b (33.4 mg) were isolated. The structures were identified based on their HR-MS and NMR.
Time course for the biotransformation of ursolic acid (1)
Using 24 h-old stage II cultures, 10 mg of substrate 1 in 1 mL of DMSO was added in each 250 mL culture flask, and incubations were conducted as before. Substrates containing cultures were incubated for 0, 14, 24, 36, 48 and 72 h, and extracted with equal volumes of EtOAc. The compound contents in the extract were measured by HPLC-ELSD analysis.Cytotoxicity assay
Two cancer cell lines, HepG2 and MCF-7, were purchased from Peking Union Medical College (PUMC) Cell Bank (Beijing, China). All cells were maintained in DMEM medium supplemented with 10% fetal bovine serum. The culture was maintained at 37 °C, 5% CO2, and grown in 96-well microtiter plates for the assay. All media were supplemented with 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. The cytotoxic activity in vitro was measured using the MTT assay. The MTT solution (10.0 mL per well) was added in culture media after cells were treated with various concentrations of compounds for 72 h, and cells were incubated for a further 4 h at 37 °C. The purple formazan crystals were dissolved in 100 mL DMSO. After 10 min, the plates were read on an automated microplate spectrophotometer (Bio-Tek Instruments, Winooski, VT) at 570 nm and 650 nm. Assays were performed in triplicate on three independent experiments. In all of these experiments, three replicate wells were used to determine each point.Conclusions
In conclusion, S. griseus ATCC 13273 showed highly efficient site-selective methyl oxidizing capacity (especially at C-30) over all five substrates of ursane triterpenes, resulting in eight additional unreported metabolites. Moreover, the site-selective oxidation of PTs obtained is particularly valuable for the fact that it provided the new active-site with the ability to access a greater number and larger variety of further derivatives. The wild microbe with such a specific site-selectivity and high conversion rate to multiple substrates is even more remarkable and uncommon. The whole genome of S. griseus 13273 in our lab has been sequenced. Future efforts will focus on the identification, cloning and heterologous expression of cytochrome P450 genes for the discovery of new biocatalysts for the specific oxidation of unactivated C–H bonds.Acknowledgements
This study was supported by the National Nature Science Foundation of China (NSFC No. 21302052) and the “Program for New Century Excellent Talents in University” awarded to Prof. Jian Zhang. We also acknowledge the ‘111 Project’ from the Ministry of Education of China, and the Fundamental Research Funds for Central Universities(JKZ2011017), and the scientific and innovation research of college graduate in Jiangsu province (CXLX11_0788).References
- F. J. Reyeszurita, M. Medinao'Donnell, R. M. Ferrermartín, E. E. Rufinopalomares, S. Martinfonseca, F. Rivas, A. Martinez, A. Garciagranados, A. Pérezjiménez and L. Garcíasalguero, RSC Adv., 2016, 6, 93590–93601 RSC.
- K. Reen-Yen, Q. Keduo, S. L. Morris-Natschke and L. Kuo-Hsiung, Nat. Prod. Rep., 2009, 26, 1321–1344 RSC.
- H. Sheng and H. Sun, Nat. Prod. Rep., 2011, 42, 543–593 RSC.
- D. P. Gossan, A. A. Magid, P. A. Yao-Kouassi, J. Josse, S. C. Gangloff, H. Morjani and L. Voutquenne-Nazabadioko, Fitoterapia, 2016, 110, 89–95 CrossRef CAS PubMed.
- M. G. Ponnapalli, S. Sukki, S. C. V. A. R. Annam, M. Ankireddy, H. Tirunagari, V. R. Tuniki and V. V. P. Bobbili, ChemInform, 2015, 46, 1570–1574 CrossRef.
- R. Azerad, Fitoterapia, 2016, 114, 168–187 CrossRef CAS PubMed.
- X. Wen, H. Sun, J. Liu, K. Cheng, P. Zhang, L. Zhang, J. Hao, L. Zhang, P. Ni and S. E. Zographos, J. Med. Chem., 2008, 51, 3540–3554 CrossRef CAS PubMed.
- F. Yu, Q. Wang, Z. Zhang, Y. Peng, Y. Qiu, Y. Shi, Y. Zheng, S. Xiao, H. Wang and X. Huang, J. Med. Chem., 2013, 56, 4300–4319 CrossRef CAS PubMed.
- P. Y. Gao, RSC Adv., 2015, 6, 2431–2435 RSC.
- E. M. F. Khusnutdinova, N. I. Medvedeva, D. V. Kazakov, O. S. Kukovinets, A. N. Lobov, K. Y. Suponitsky and O. B. Kazakova, Tetrahedron Lett., 2015, 57, 148–151 CrossRef.
- B. A. Dar, A. M. Lone, W. A. Shah and M. A. Qurishi, Eur. J. Med. Chem., 2016, 111, 26–32 CrossRef CAS PubMed.
- L. Fu and G. W. Gribble, Org. Lett., 2013, 15, 1622–1625 CrossRef CAS PubMed.
- T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC.
- O. V. Nesterova, M. N. Kopylovich and D. S. Nesterov, RSC Adv., 2016, 6, 93756–93767 RSC.
- G. Y. Ruan, Z. H. Qi, Y. Zhang, W. Liu and Y. Wang, RSC Adv., 2016, 6, 35855–35858 RSC.
- A. T. Nelson, A. M. Camelio, K. R. Claussen, J. Cho, L. Tremmel, J. Digiovanni and D. Siegel, Bioorg. Med. Chem. Lett., 2015, 25, 4342 CrossRef CAS PubMed.
- L. Fu and G. W. Gribble, Org. Lett., 2013, 15, 1622 CrossRef CAS PubMed.
- M. Schrewe, M. K. Julsing, B. Bühler and A. Schmid, Chem. Soc. Rev., 2013, 42, 6346–6377 RSC.
- H. L. T. Syed Adnan Ali Shah, S. Sultan, M. A. B. M. Faridz, M. A. B. M. Shah, S. Nurfazilah and M. Hussain, Int. J. Mol. Sci., 2014, 15, 12027–12060 CrossRef PubMed.
- D. Zhou, N. Li, Y. Zhang, C. Yan, K. Jiao, Y. Sun, H. Ni, B. Lin and Y. Hou, RSC Adv., 2016, 6, 97302–97312 RSC.
- F. Xu, K. Liao, Y. Liu, Z. Zhang, D. Guo, Z. Su and B. Liu, Fitoterapia, 2016, 109, 201–205 CrossRef CAS PubMed.
- J. L. Tian, Y. Chen, Y. X. Wang, X. Huang, X. Sun, K. C. Liu and S. J. Song, RSC Adv., 2016, 6, 112712–112720 RSC.
- L.-W. Qian, J. Zhang, J.-H. Liu and B.-Y. Yu, Tetrahedron Lett., 2009, 50, 2193–2195 CrossRef CAS.
- Y. Y. Zhu, L. W. Qian, J. Zhang, J. H. Liu and B. Y. Yu, Tetrahedron, 2011, 67, 4206–4211 CrossRef CAS.
- F. F. Guo, X. Feng, Z. Y. Chu, D. P. Li, L. Zhang and Z. S. Zhang, J. Asian Nat. Prod. Res., 2013, 15, 15–21 CrossRef CAS PubMed.
- M. J. Acerson, B. S. Bingham, C. A. Allred and M. B. Andrus, Tetrahedron Lett., 2015, 56, 3277–3280 CrossRef CAS.
- C. Hui and J. Xu, Tetrahedron Lett., 2016, 57, 2692–2696 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01811h |
| This journal is © The Royal Society of Chemistry 2017 |