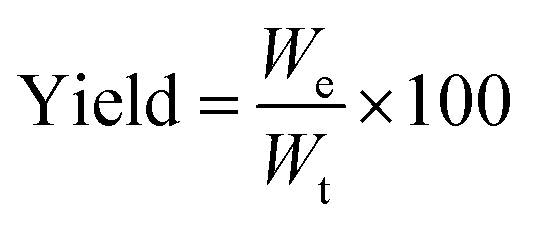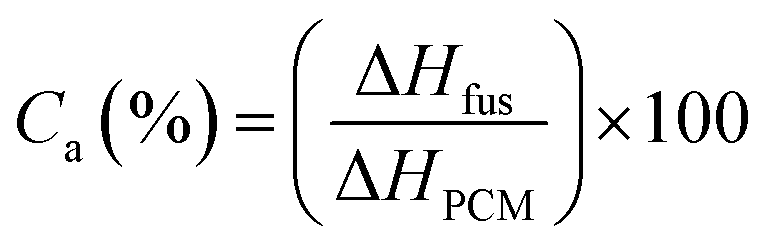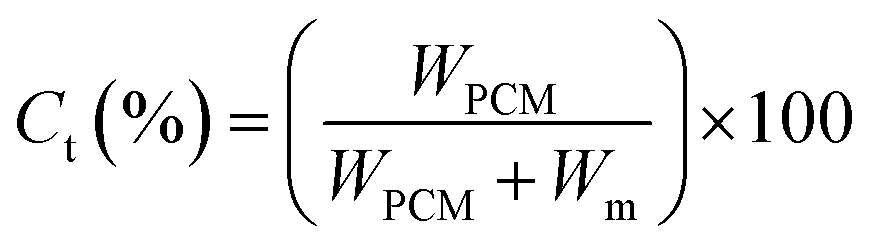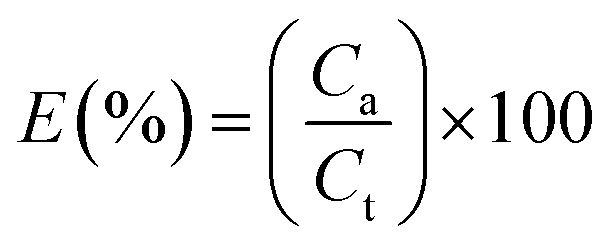DOI:
10.1039/C7RA01805C
(Paper)
RSC Adv., 2017,
7, 21196-21204
Preparation of polyurea microcapsules containing phase change materials in a rotating packed bed†
Received
13th February 2017
, Accepted 3rd April 2017
First published on 13th April 2017
Abstract
A series of polyurea (PUA) hollow microcapsules (HMCs) and microcapsules containing phase change materials (microPCMs) were synthesized with a rotating packed bed (RPB) via interfacial polymerization between isophorone diisocyanate (IPDI) and diethylenetriamine (DETA). Phase change materials (PCMs) were proved to be successfully encapsulated by a PUA shell with FT-IR spectroscopy. The morphologies and microstructures of the microcapsules were observed by optical microscopy and SEM, and the results showed that the morphologies of the microcapsules were all spherical geometries, and their surfaces were smooth and compact. The effect of the mass ratio of the core-to-monomer (C/M), emulsifier dosage and rotational speed (N) on the latent heat of the PUA microcapsules was investigated with DSC. Compared to a conventional stirring tank reactor, the RPB shows much better performance for encapsulating PCM via interfacial polymerization. The suggested C/M ratio, emulsifier dosage and N in our research conditions were 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 0.5 wt% and 1200 rpm, respectively.
:1, 0.5 wt% and 1200 rpm, respectively.
Introduction
In the last few decades, phase change materials (PCMs) have attracted great attention due to their high energy storage density, narrow operating temperature1,2 and near isothermal heat storage.3 PCMs can absorb energy from the environment in a heating process as phase change takes place, and release the latent energy to the environment in a reverse cooling process.4 Meanwhile they also present some inherent drawbacks in practical applications, such as volume expansion, liquid migration and supercooling.5 In order to overcome the shortcomings of PCMs, the preparation of microencapsulated PCMs (microPCMs) has been widely studied.6 Microencapsulation is a technique by which liquid droplets, solid particles or gas bubbles can be coated with a continuous film of synthetic or natural polymers.7 Among various methods of microencapsulation, interfacial polymerization is an important technique owing to its relatively rapid reaction speed, mild reaction course and low penetrability.8 Many predecessors have worked on microencapsulation of phase change materials by interfacial polymerization, and a lot of output has been published.4,8–13 In these literatures, material recipes and technological parameters were all carefully investigated, and their effects on the structure and properties of microPCMs were also thoroughly reported. But in these researches, the preparation time of microPCMs was found to be relatively long, usually several hours, even if high speed homogenizer was used.13 This is because that the generation of shells mainly depends on the migration speed of monomers. But the micro-mixing action of conventional stirring tank reactors is not effective enough for the diffusion of hydrophilic monomer through the interface. Thus, to shorten the preparation time of microPCMs and avoid using high power apparatus, a kind of high-efficiency technique which can intensify monomers' diffusion during interfacial polymerization process is very attractive.
Rotating packed bed (RPB) is a reactor creating higher gravity which is orders of magnitude larger than that on the earth through the action of centrifugal force, as so called “Higee”.14–16 It's a novel and high-efficiency reactor utilized to intensify the mass transfer. When liquid reactant goes through the rotating packing in the RPB, the fluid will be split into fine droplets, threads and thin films, and result in significant intensification of micro-mixing and mass transfer.17–19 The volumetric mass transfer coefficient of fluid in a PRB is 1 to 3 orders of magnitude higher than that in a conventional stirring tank reactor.20,21 Compared to conventional reactors, the RPB also has the advantages of smaller equipment size, thereby benefiting a reduction of capital and operating costs.15,22 Based on these advantages, the RPB has been applied in process intensification in many aspects such as adsorption,23–28 distillation,29–31 synthesis of nanoparticles,32,33 nanofibers,34,35 butyl rubber36,37 and degradation of organic pollutants.38–40 Therefore, RPB would be suitable for the preparation of microPCMs through interfacial polymerization.
As far as we know, there were barely no researches on preparing microcapsules in the RPB. In this research, PUA was selected as a capsule shell material because the reaction between –NCO and –NH2 occurs quickly at room temperature. Isophorone diisocyanate (IPDI) was chosen as the isocyanate monomers as its relatively low hydrolyzing reactivity in comparison of aromatic isocyanates. A series of PUA hollow microcapsules and microPCMs were successfully prepared in the RPB. Effects of mass ratio of core-to-monomer (C/M), emulsifier dosage and rotational speed (N) on the properties of PUA microcapsules were discussed in detail. The structure and properties of the microcapsules were investigated by FT-IR, DSC, optical microscope and SEM.
Materials and methods
Chemicals
Isophorone diisocyanate (IPDI, purity: 99.0 wt%), diethylenetriamine (DETA, purity: 99.0 wt%) and polyoxyethylene(10) nonyl phenyl ether (OP-10, purity: 99.0 wt%) were analytical pure and purchased from Aladdin Industrial Corporation, Shanghai, China. Cyclohexane (purity: 99.5 wt%) was analytical pure and purchased from Kelong Chemical Reagent Factory, Sichuan, China. Industrial grade paraffin was used as PCM and purchased from Shanghai Tianlan New Material Technology Co., Ltd, Shanghai, China.
RPB equipment
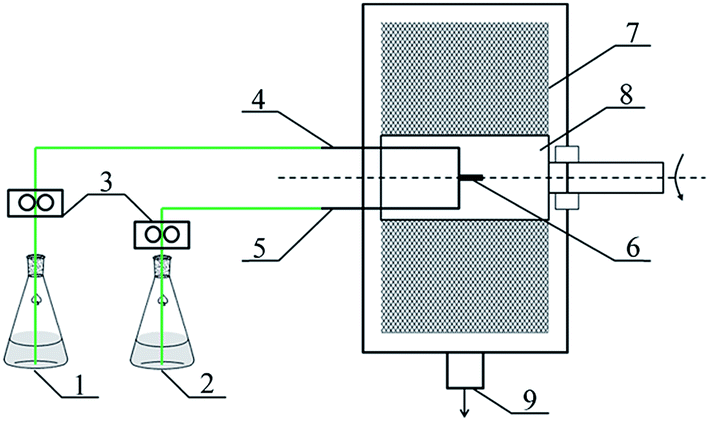
The RPB used in this research was homemade by Research Center of Ministry of Education for High Gravity Engineering and Technology, Beijing University of Chemical Technology, and its structural parameters were listed in Table 1. The schematic diagram of RPB is illustrated in Fig. 1.
Table 1 Structural parameters of RPB
| Subjects |
Parameters |
| Mean inner diameter of the casing |
75 mm |
| Axial length of the casing |
60 mm |
| Inner diameter of the rotor |
40 mm |
| Outer diameter of the rotor |
100 mm |
| Packing material |
Stainless steel wire mesh |
| Mesh size of the packing material |
50–70 mesh |
| Voidage |
0.96 |
| Surface area per unit volume of the dry packing |
500 m2 m−3 |
| Packing radial thickness |
30 mm |
| Diameter of the wire |
0.22 mm |
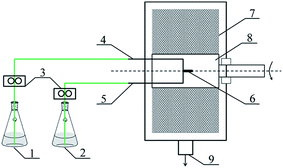 |
| | Fig. 1 Schematic diagram of PUA microcapsules synthesis equipment (1) reagent A, (2) reagent B, (3) metering pump, (4) reagent A inlet, (5) reagent B inlet, (6) liquid distributor, (7) packing, (8) rotator, (9) products outlet. | |
General procedures for preparation of microcapsules by RPB
1.92 g IPDI and different dosages of PCM were dissolved in cyclohexane and diluted with cyclohexane to 25 ml (marked as solution S1). The mass of S1 was measured. Then, different dosages of OP-10 (based on the mass of S1, similar hereinafter) was dissolved in deionized water and diluted with water to 50 ml (marked as solution S2). The IPDI-PCM O/W emulsion (reagent A) was obtained by emulsifying S1 and S2 in a 250 ml conical flask with a magnetic stirrer at 2500 rpm for 5 min. 0.9 g DETA was dissolved in deionized water and diluted with water to 25 ml (reagent B). The mole ratio between DETA and IPDI was fixed as 1.01 in all experiments. Reagent A and B were fed into the RPB (see Fig. 1) at a flow rate of 15 ml min−1 and 5 ml min−1, respectively. The reaction was conducted at a certain rotational speed (N), and the products were simultaneously collected from outlet. The whole process was completed in 6 minutes. The formed microcapsules were filtered and washed in hot water (60 °C) to remove the unreacted monomers and core materials, and then were freeze-dried for 12 h.
The hollow microcapsule (labelled as 1200r-HMC) was prepared as the general procedures described above. The only exception is that PCM was not involved in the synthetic recipe. OP-10 dosage was 0.5 wt%. The rotational speed (N) was 1200 rpm, which means the actual centrifugal acceleration (a) was 1183.2 m s−2 (calculated by formula S1 and S2, see ESI†).
Preparation of microPCMs at different N. The preparation process was conducted as the general procedures above. The dosage of PCM was 2.82 g in order that the mass ratio of C/M (C means PCM, and M means the sum of IPDI and DETA) was 1:1. OP-10 dosage was 0.5 wt%. The RPB was running at different N (600 rpm, 900 rpm, 1200 rpm, 1500 rpm and 1800 rpm). The microcapsules obtained were labelled as 600r-microPCM, 900r-microPCM, 1200r-microPCM, 1500r-microPCM and 1800r-microPCM, respectively.
Preparation of microPCMs with different mass ratio of C/M. The preparation process was conducted as the general procedures above. The mass ratio of C/M was 1:1, 2:1, 3:1 and 4:1, respectively. OP-10 dosage was 2.0 wt%. The RPB was running at 900 rpm. The microcapsules obtained were labelled as C/M-1-microPCM, C/M-2-microPCM, C/M-3-microPCM and C/M-4-microPCM, in accordance with C/M ratio changing from 1:1 to 4:1.
Preparation of microPCMs with different emulsifier dosage. The preparation process was conducted as the general procedures above. The mass ratio of C/M was 1:1. The dosage of OP-10 was 0.5 wt%, 1.0 wt%, 2.0 wt%, 3.0 wt% and 5.0 wt%, respectively. The RPB was running at 900 rpm. The microcapsules obtained were labelled as OP-0.5-microPCM, OP-1-microPCM, OP-2-microPCM, OP-3-microPCM and OP-5-microPCM, respectively.
Preparation of control group. Reagent A and B were prepared the same as described in the preparation of microPCMs at different N. Then reagent A (IPDI-PCM O/W emulsion) was transferred to a 500 ml beaker, and reagent B was directly added into the O/W emulsion in a time span of 10 s and stirred by a mechanical stirrer (IKA EUROSTAR 20 high speed digital, Germany) with a propeller blade at 1200 rpm for 1 h. The resultant microPCM (labelled as control-microPCM) was filtered, washed and freeze-dried as the general procedures above.
Characterization
FT-IR spectroscopy analysis. FT-IR analyses of microPCMs were performed in a FT-IR spectroscopy (IS10, Thermo Fisher Scientific, USA). PCM and PUA microcapsules (1200r-HMC, 1200r-microPCM) were ground and mixed with KBr to make pellets, and then all samples were scanned from 4000 to 400 cm−1.
Morphology observation. The IPDI-PCM O/W emulsion (cyclohexane: 20 ml, deionized water: 40 ml, IPDI: 1.55 g, PCM: 2.27 g, OP-10: 0.2 g, preparation procedure as same as described before) and microcapsules (before washing and drying) were directly dripped on a glass slide and then observed by an optical microscope (CX41, Olympus, Japan). The morphology of microcapsules in a powder state was examined at 15 kV after coating with platinum using a scanning electron microscope (SU3500, HITACHI, Japan). The thickness of shells can be achieved from the SEM images of cross-section of microcapsules.
Size and size distribution analysis. The mean diameter and size distribution of microPCMs were estimated with Nano measurer 1.2 (measured from more than 100 microcapsules on optical microscope images).
Overall yield. The dry weight of microPCMs obtained was used to assess the overall yield of our experiments. The yield of microPCMs was calculated by formula (1).| |
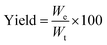 | (1) |
where We and Wt represent experimental dry weight and theoretical dry weight of microcapsules, respectively. Therein, Wt = WIPDI + WDETA + WPCM + WOP-10.
Actual core content. The actual core content (Ca) of microPCMs is defined as Lu11 reported.| |
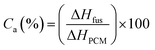 | (2) |
where, ΔHfus (J g−1) and ΔHPCM (J g−1) represent the fusing enthalpy of PUA microPCMs and pure PCM respectively. All ΔHf were determined by a DSC (200PC, NETZSCH Instruments, Germany) with a sealed aluminum pan under nitrogen purge. The heating progress was as follows: weighed samples were heated at a rate of 10 °C min−1 from 0 °C to 60 °C firstly to eliminate thermal history, and then cooled to 0 °C at a rate of 5 °C min−1 for the crystallization of PCM, followed by temperature-programming from 0 °C to 60 °C with a rate of 5 °C min−1 to detect ΔH.
Theoretical core content. The theoretical core content (Ct) is defined as Lu11 reported.| |
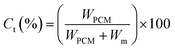 | (3) |
where, WPCM and Wm (Wm = WIPDI + WDETA, 2.82 g) represent, the weight of PCM and the total weight of monomers, respectively.
Encapsulation efficiency. The encapsulation efficiency (E) is defined as Lu11 reported.| |
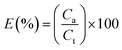 | (4) |
Results and discussion
Mechanism of shell formation
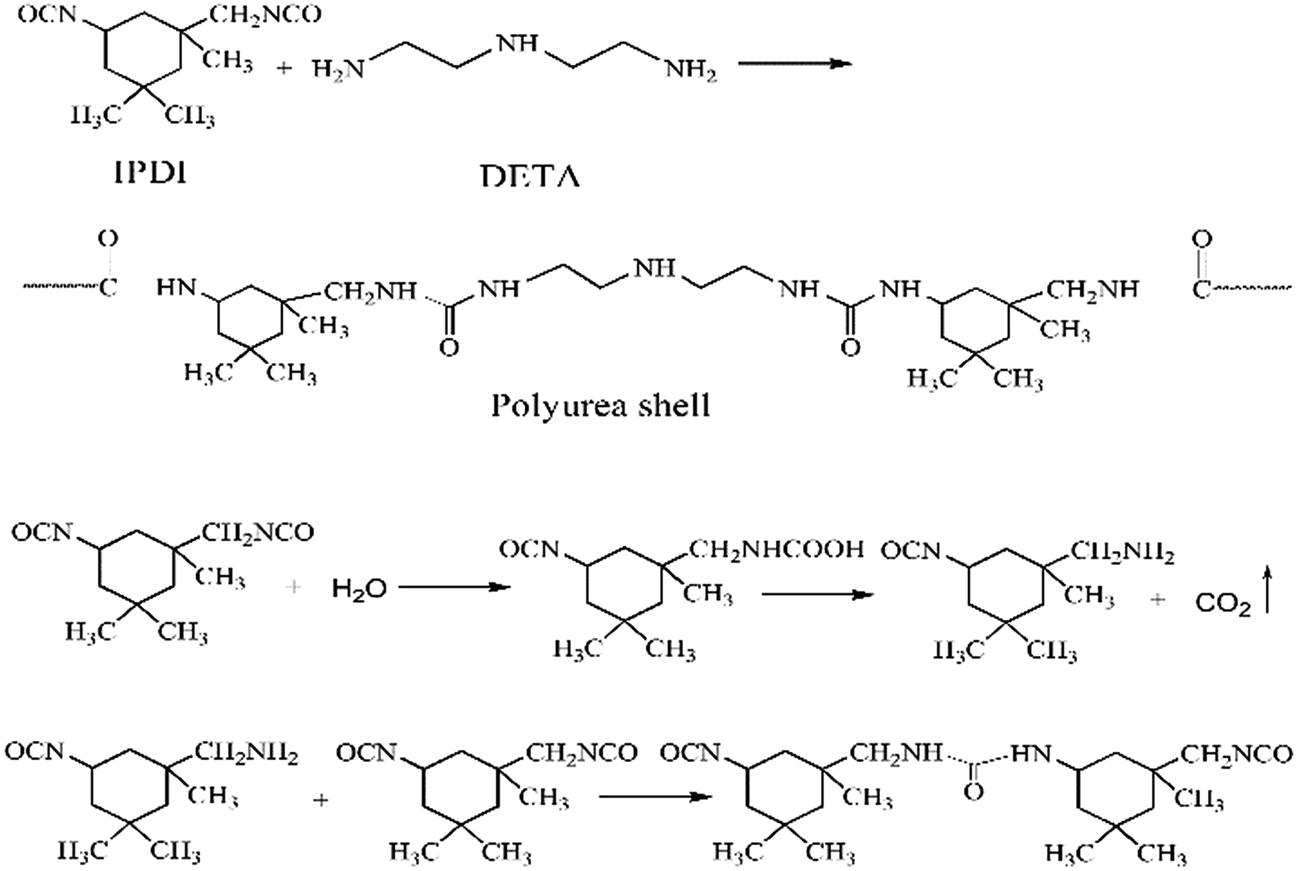
The PUA shell is formed by the reaction between –NCO groups of IPDI and –NH2 groups of DETA at the O/W interface.9 Meanwhile, the –NCO groups of IPDI may be hydrolyzed to –NH2 groups that also reacts with IPDI to form the PUA shell. The reaction scheme is shown in Fig. 2. In the running RPB, the reaction solutions cross through the wire mesh packing from the inner surface to the external surface under the action of the strong centrifugal force, which lead to full micro-mixing of monomers and faster interfacial polymerization. Therefore, the application of RPB in preparation of microcapsule through interfacial polymerization may largely affect the characters of microcapsule obtained.
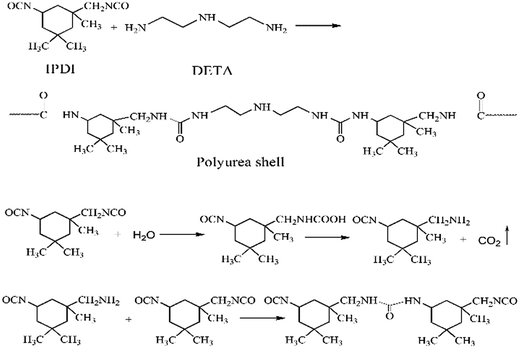 |
| | Fig. 2 The reaction scheme of polyurea shell formation. | |
Yield of microPCMs
The experimental and theoretical dry weights of the control-microPCM and microPCMs prepared by RPB at different N are shown in Table 2. The yields of microPCMs using the N of higher than 900 rpm are higher than that of control-microPCM (73.87%). However, when N is 1800 rpm, the yield of microPCM is obviously lower than that of control. This is because too strong centrifugal force generated under high rotational speed is not beneficial for shell formation, and thus leads to the breakage of microcapsules.
Table 2 The yields of control-microPCM and microPCMs prepared by RPB
| Sample numbers |
We (g) |
Wt (g) |
Yield (%) |
| Control-microPCM |
4.24 ± 0.08 |
5.74 |
73.86 ± 1.39 |
| 600r-microPCM |
3.97 ± 0.02 |
5.74 |
69.16 ± 0.69 |
| 900r-microPCM |
4.33 ± 0.03 |
5.74 |
75.44 ± 0.52 |
| 1200r-microPCM |
4.81 ± 0.06 |
5.74 |
83.74 ± 1.13 |
| 1500r-microPCM |
4.60 ± 0.04 |
5.74 |
80.14 ± 0.70 |
| 1800r-microPCM |
3.56 ± 0.06 |
5.74 |
62.02 ± 1.05 |
FT-IR spectra
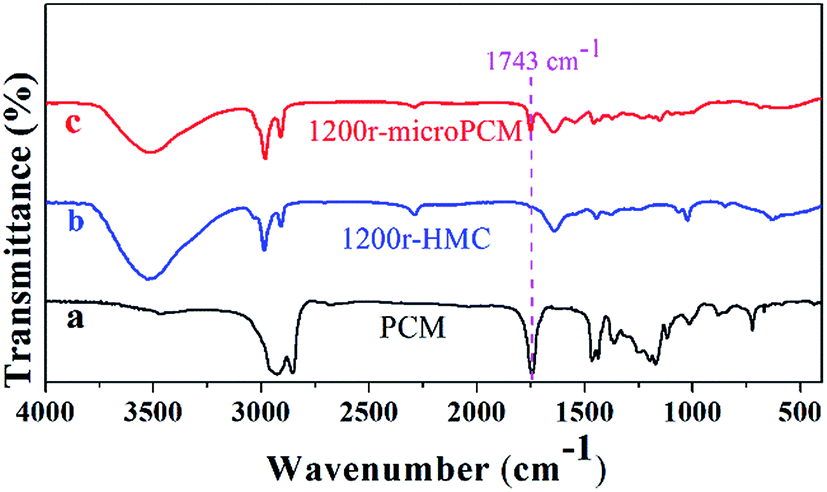
The FT-IR spectra of PCM (line a), HMC (line b) and microPCMs (line c) are illustrated in Fig. 3. It can be seen in line a, two strong absorption bands at 2924 and 2854 cm−1 are associated with aliphatic C–H stretching vibrations of methyl and methylene groups, respectively. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration is found around 1743 cm−1 which belongs to the ester component of PCM. The peaks at 1466 and 1362 cm−1 are assigned to the asymmetric and symmetric vibrations of the methyl groups. The in-plane rocking vibrations of methylene groups are observed at 722 cm−1. In line b, the spectrum of 1200r-HMC shows strong absorption bands at 3446 cm−1 and 1646 cm−1 corresponding to hydrogen-bonded N–H and CO stretching vibrations, respectively. These results confirm the formation of urea linkage. The weak peak at 2270 cm−1 is attributed to very few unreacted –NCO groups, which indicates the reaction between –NCO and –NH2 has been almost completed. The peaks at 2927 and 2852 cm−1 belong to the asymmetric and symmetric C–H stretching vibrations. These characteristic peaks prove the formation of PUA shell. Comparing line a and line b, the peak at 1743 cm−1 is regarded as the characteristic peak for PCM. In line c, the formation of urea linkage in 1200r-microPCM is also confirmed by strong absorption bands at 3446 cm−1 and 1646 cm−1 corresponding to hydrogen-bonded N–H and CO stretching vibrations. The asymmetric and symmetric C–H stretching vibrations are also found at 2927 and 2852 cm−1. In comparison with line b, the characteristic peak at 1743 cm−1 proves that PCM has been successfully encapsulated in PUA shell via interfacial polymerization proceeded in RPB.
O stretching vibration is found around 1743 cm−1 which belongs to the ester component of PCM. The peaks at 1466 and 1362 cm−1 are assigned to the asymmetric and symmetric vibrations of the methyl groups. The in-plane rocking vibrations of methylene groups are observed at 722 cm−1. In line b, the spectrum of 1200r-HMC shows strong absorption bands at 3446 cm−1 and 1646 cm−1 corresponding to hydrogen-bonded N–H and CO stretching vibrations, respectively. These results confirm the formation of urea linkage. The weak peak at 2270 cm−1 is attributed to very few unreacted –NCO groups, which indicates the reaction between –NCO and –NH2 has been almost completed. The peaks at 2927 and 2852 cm−1 belong to the asymmetric and symmetric C–H stretching vibrations. These characteristic peaks prove the formation of PUA shell. Comparing line a and line b, the peak at 1743 cm−1 is regarded as the characteristic peak for PCM. In line c, the formation of urea linkage in 1200r-microPCM is also confirmed by strong absorption bands at 3446 cm−1 and 1646 cm−1 corresponding to hydrogen-bonded N–H and CO stretching vibrations. The asymmetric and symmetric C–H stretching vibrations are also found at 2927 and 2852 cm−1. In comparison with line b, the characteristic peak at 1743 cm−1 proves that PCM has been successfully encapsulated in PUA shell via interfacial polymerization proceeded in RPB.
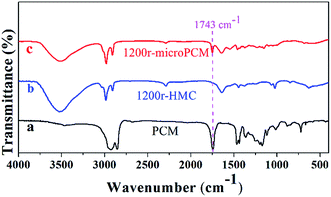 |
| | Fig. 3 The FT-IR spectra of PCM, HMC (1200r-HMC) and microPCM (1200r-microPCM). | |
Morphology
At first, the effect of RPB on IPDI-PCM O/W emulsion was observed. Fig. 4 is the optical microphotographs of IPDI-PCM O/W emulsion after treating by RPB with different N. The control group is the IPDI-PCM O/W emulsion without disposing with RPB. Compared with the control group, the particle size of emulsion after treating with RPB is smaller and the size distribution is more uniform. When N is relatively low, the particle size of emulsion decreases quickly with increasing N. When N is higher than 1200 rpm, the particle size does not change obviously. It seems that the liquids experience a re-dispersing effect after transferring into RPB. Chen36 applied a coalescence-redispersion model to describe the exchange of reactants among droplets on every cage of RPB, considering that mixing was realized by collision, coalescence, and redispersion of two droplets.41 In the coalescence-redispersion model, all droplets have the same size and the coalescence of any two droplets is followed instantaneously by re-dispersing into two identical droplets,36 through which, the intensified mass transfer and high micro-mixing efficiency in RPB should be beneficial to monomers' diffusion during interfacial polymerization. The results obtained in this research are consistent with this model, and imply that the interfacial polymerization for preparation of microcapsule might be enhanced by RPB.
 |
| | Fig. 4 Optical microphotographs of IPDI-PCM O/W emulsion treated by RPB under different N (×100). | |
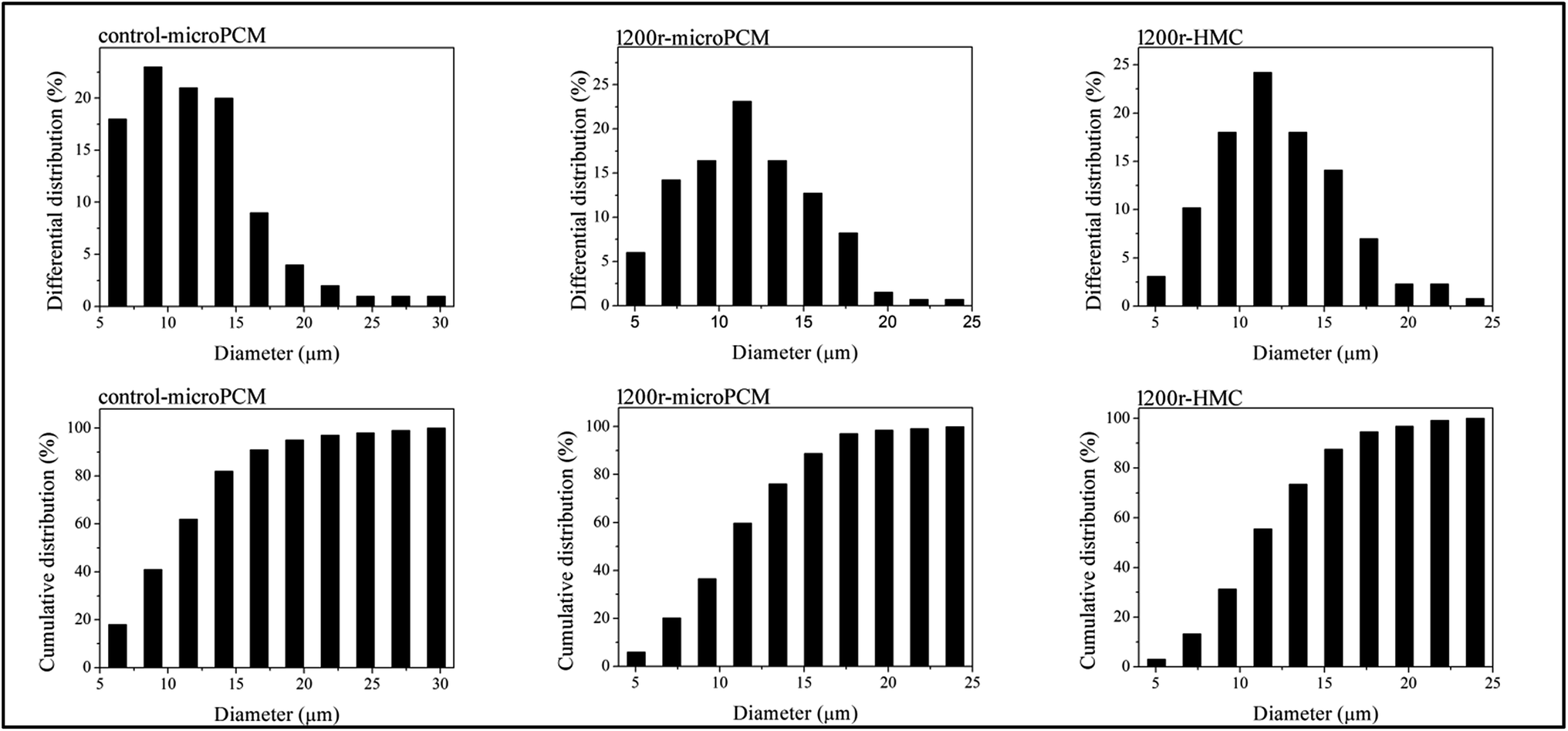
Optical microphotographs of control-microPCM, 1200r-HMC and 1200r-microPCM are shown in Fig. 5. All the three groups of microcapsules show clear spherical morphology, while a few of control-microPCMs and 1200r-microPCMs show a subsidence phenomenon. The mean diameters of control-microPCM, 1200r-HMC and 1200r-microPCM measured from optical microphotographs are about 11.88 μm, 12.31 μm and 11.83 μm, respectively. Interestingly, the mean diameter of 1200r-HMC is a little larger than that of 1200r-microPCM. This may be explained on the fact that cyclohexane droplets may coalesce faster during encapsulation process due to their very low viscosity.11 Fig. 6 shows the differential distributions and cumulative distributions of prepared microcapsules. In comparison to control-microPCM, the size distributions of both 1200r-HMC and 1200r-microPCM are much narrower. From the cumulative distribution analysis, it is found that more than 94.6% 1200r-HMC and 97.0% 1200r-microPCM are smaller than 17.65 μm. Normally, the particle size and size distribution of microcapsule is depending on the emulsifying process, such as stirring speed, emulsifier dosage and emulsifying time. The difference of size distribution between control-microPCM and microcapsules fabricated in RPB should be explained by the fact that the liquids reagents experience a re-dispersing effect as described above when running in RPB.
 |
| | Fig. 5 Optical microphotographs of PUA microcapsules (×200). | |
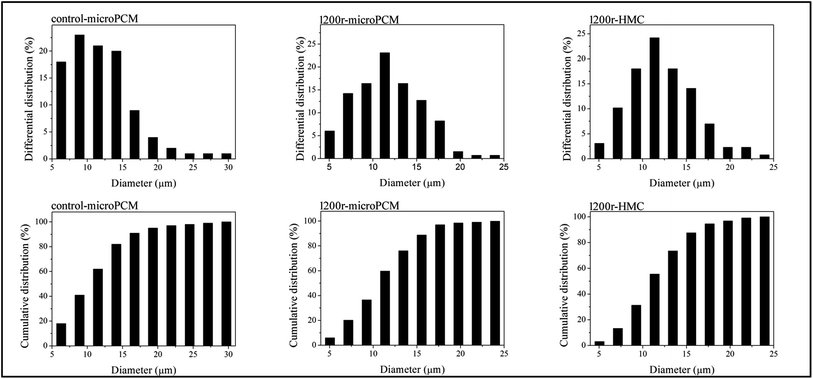 |
| | Fig. 6 Size distributions of synthesized microcapsules (contro-microPCM, 1200r-microPCM and 1200r-HMC). | |
Fig. 7 shows the SEM photographs of control-microPCM, 1200r-HMC and 1200r-microPCM in the powder state. In Fig. 7A and B, the rough surfaces of control-microPCM are observed, and some polymer filled in the interspace between microcapsules can be observed as well, which is possibly due to the incomplete encapsulation of the core material or the shell material can not cover the cores absolutely.9 It can be clearly seen that both 1200r-HMC (Fig. 7D and E) and 1200r-microPCM (Fig. 7G and H) are spheres with a smooth and compact surface. Furthermore, as shown in Fig. 7C, F and I, the internal surface of shell was smooth. And the thickness of prepared microcapsules can also be estimated from the cross-section of Fig. 7C, F and I. Compared with 1200r-HMC and 1200r-microPCM, whose shell thicknesses are 100 nm and 90 nm, respectively, the control-microPCM shows a thinner shell (70 nm). This difference may be caused by that the micro-mixing efficiency of RPB is higher up to orders of magnitudes in comparison of conventional stirring tank reactors. In RPB, the O/W interface regenerates quickly and DETA diffuses fast into oil phase, leading to high polymerization rate, which directly influences the properties of obtained PUA microcapsules. In conventional reactors, relatively low micro-mixing efficiency is not sufficient for the polycondensation reaction between IPDI and DETA, and thus results in defective PUA shell.
 |
| | Fig. 7 SEM of PUA microcapsules (1200r-HMC: (A–C); 1200r-microPCM: (D–F); control-microPCM: (G–I)). | |
Thermal storage performance of microPCMs
The morphology, chemical structure and surface characteristic of microcapsule shell decide the functional performance of microcapsules. Some researchers have reported the effects of technological parameters on the shell in conventional stirring tank reactors. For example, Zhan5 used three kinds of surfactants to prepare PUA microcapsules applied in textile industry and they pointed out that emulsifier content showed great influence on thermal properties and morphologies of microcapsules. Siddhan10 studied the effect of C/M ratio and PCM-to-cyclohexane ratio on core content and encapsulation efficiency of PUA microPCMs. In our research, PUA microPCMs were fabricated by using RPB with different C/M, emulsifier dosage and N, and their latent heats were detected with DSC. The latent heat of phase change transition and fusing temperature of the PCM used in this paper were 217.7 J g−1 and 32.6 °C (DSC curve of pure PCM is shown in Fig. S1†).
Effect of C/M on the thermal storage performance. Table 3 shows the phase change behaviours of PUA microPCMs with different C/M ratio (1:1 to 4:1). All ΔHfus of samples can be detected with DSC, which means PUA microPCMs have been successfully prepared with RPB. When C/M increases from 1:1 to 4:1, ΔHfus of PUA microPCMs increases from 92.52 J g−1 to 138.60 J g−1. Based on formula (3), the Ct of PUA microPCMs rises from 50.0% to 80.0% with increasing C/M, and calculated Ca based on formula (2) also increases. While, E calculated from formula (4) shows a decreasing trend. This indicates that when the core content is high enough, the polymer shell can not encapsulate the core material perfectly anymore, resulting in a little decrease of E. A higher C/M ratio often leads to form improperly walls around capsules and decreases the shell thickness.10 As a result, the encapsulated PCM strength deteriorates and the chances of PCM spillage increases.42 The fusing temperature increases with increasing C/M ratio from 1:1 to 3:1. This probably owes to a decreased ratio of surface to bulk paraffin inside the microcapsule, as the amount of paraffin encapsulated is increased.9 Meanwhile, Tfus of all samples are lower than that of pure paraffin. This is because there are no strong interactions between the paraffin molecules and the PUA shell, which leads to a depression of the phase change temperatures of the paraffin in the microPCM.43,44 Furthermore, the effect of the spatial confinement on the thermal characteristics of PCM may also lead to the difference in temperature between encapsulated and bulk paraffin.45 Ca and E are two of most important parameters to evaluate thermal storage performance of microPCMs. In order to gain higher Ca and E simultaneously, the most suitable C/M is 3:1 for preparing PUA microPCMs in RPB.
Table 3 Thermal properties of microPCMs prepared with different mass ratio of C/M
| Sample number |
Tfus (°C) |
ΔHfus (J g−1) |
Ct (%) |
Ca (%) |
E (%) |
| C/M-1-microPCM |
27.4 |
92.52 |
50.0 |
42.50 ± 0.50 |
85.0 ± 0.92 |
| C/M-2-microPCM |
29.7 |
122.90 |
66.7 |
56.45 ± 0.87 |
84.63 ± 1.30 |
| C/M-3-microPCM |
32.3 |
137.80 |
75.0 |
63.30 ± 0.37 |
83.90 ± 0.49 |
| C/M-4-microPCM |
32.2 |
138.60 |
80.0 |
63.67 ± 0.14 |
79.60 ± 0.17 |
Effect of emulsifier dosage on the thermal storage performance. Table 4 shows the thermal properties of microPCMs fabricated with different emulsifier dosage in RPB. With the emulsifier amount increasing from 0.5 wt% to 5.0 wt% (based on the mass of S1), ΔHfus of PUA microPCMs decreases from 96.18 J g−1 to 53.28 J g−1. According to formula (2), their Ca are 44.18%, 43.35%, 42.50%, 28.03% and 24.47%, respectively. As Ct is 50.0% (C/M = 1:1), correlated E of 88.36%, 86.70%, 85.0%, 52.40% and 49.0% are presented. It is obvious that both Ca and E of microPCMs are decreased when more emulsifier is added. Interestingly, when the dosage of OP-10 is 0.5 wt%, the resultant Ca and E display the maximum, 44.18% and 88.36%, respectively. The droplet size of emulsion decreases with the increase of emulsifier concentration46 and the diameter of microcapsules usually depends on the particle size of initial emulsion. The small particle size of the microcapsules only supplies small space, which inhibits the motion of PCM chains to some extent, resulting in a significant decline of the crystallinity of PCM.12 Meanwhile, emulsifier is the impurity for PCM and will affect the perfection of the crystallization course.47 Furthermore, the addition of emulsifier can reduce weight percentage of PCM, and the emulsifier contributes nothing to the enthalpy, which leads to the decline of enthalpy. Therefore, lower latent heat of fusion for microPCMs is presented with increasing emulsifier dosage. Compared to the enthalpy, the phase change temperatures are affected by the amount of emulsifier in a more complicated way. As Table 5 shows, the fusing temperature firstly decreases and then increases when emulsifier dosage increases from 0.5 wt% to 5.0 wt%. For one thing, as discussed above, the decline of crystallinity and perfection of crystallization may lead to a decrease of phase change temperatures. On the other hand, more amount of emulsifier contributes to more compact shell structure with fewer defects, such as pores and leaks on shells, which bring out higher phase change temperatures.4 The comprehensive effect results in this kind of shift in our research.
Table 4 Thermal properties of microPCMs prepared with different dosage of emulsifier
| Sample number |
Tfus (°C) |
ΔHfus (J g−1) |
Ct (%) |
Ca (%) |
E (%) |
| OP-0.5-microPCM |
29.9 |
96.18 |
50.0 |
44.18 ± 0.69 |
88.36 ± 1.38 |
| OP-1-microPCM |
29.0 |
94.38 |
50.0 |
43.35 ± 0.28 |
86.70 ± 0.55 |
| OP-2-microPCM |
27.4 |
92.52 |
50.0 |
42.50 ± 0.33 |
85.0 ± 0.66 |
| OP-3-microPCM |
28.0 |
61.02 |
50.0 |
28.03 ± 1.81 |
52.40 ± 3.61 |
| OP-5-microPCM |
29.3 |
53.28 |
50.0 |
24.47 ± 0.65 |
49.0 ± 1.30 |
Table 5 Thermal properties of control-microPCM and microPCMs prepared in RPB at different N
| Sample number |
Tfus (°C) |
ΔHfus (J g−1) |
Ct (%) |
Ca (%) |
E (%) |
| Control-microPCM |
29.4 |
84.50 |
50.0 |
38.80 ± 0.92 |
77.60 ± 1.84 |
| 600 rpm-microPCM |
31.0 |
93.74 |
50.0 |
43.06 ± 0.48 |
86.12 ± 0.97 |
| 900 rpm-microPCM |
29.9 |
96.18 |
50.0 |
44.18 ± 0.69 |
88.36 ± 1.38 |
| 1200 rpm-microPCM |
31.7 |
105.59 |
50.0 |
48.5 ± 0.20 |
97.0 ± 0.41 |
| 1500 rpm-microPCM |
30.9 |
100.88 |
50.0 |
46.34 ± 1.38 |
92.68 ± 2.76 |
| 1800 rpm-microPCM |
30.1 |
84.76 |
50.0 |
38.93 ± 1.01 |
77.86 ± 2.02 |
Effect of N in RPB on the thermal storage performance. Hereinbefore, the most rational C/M is 3:1. However, a further increase of N in RPB will increase the shear force, which may cause collapsing of microcapsules with the C/M of 3:1. So the C/M of 1:1 was employed when we investigated the effect of N on the property of latent heat of microPCMs. The DSC curves of control-microPCM and microPCMs fabricated in RPB at different N were determined, and the results are shown in Table 5. ΔHfus of resultant microPCMs firstly increases and then decreases with the increasing N. Tfus of microPCMs are lower than that of pure PCM. The highest ΔHfus of 105.59 J g−1 is obtained when N is 1200 rpm. According to formula (2), the calculated Ca of microPCMs at 600 rpm, 900 rpm, 1200 rpm, 1500 rpm and 1800 rpm are 43.06%, 44.18%, 48.50%, 46.34% and 38.93%, respectively. As N is ranging from 600–1800 rpm, resultant E based on formula (4) are 86.12%, 88.36%, 97.0%, 92.68% and 77.86%, respectively. Compared to control-microPCM, whose Ca and E are 38.80% and 77.60%, it's easy to find that all of microPCMs synthesized in RPB show better thermal storage performance. The reasons may be explained as follows. In comparison of conventional stirring tank reactor, micro-mixing efficiency of RPB is obviously higher, leading to better encapsulation performance. With an increase of N, more vigorous impingement between liquid and the packing is achieved, and the increase in collision probability among the liquid elements results in better mixing effects.36 Therefore, Ca and E of prepared microPCMs increase obviously when N increases in a relatively low range. But overly rapid rotating speed may lead to collapse of initial polymer shell and thus result in imperfectness of microPCMs. Meanwhile, according to Shi,17 the mean resident time of liquid droplets in RPB would decrease with increasing N, which may cause incomplete reaction between monomers at the interface. Therefore, as N is higher than 1200 rpm, Ca and E of microPCMs show a reducing trend. Herein, the chosen optimum N is 1200 rpm for this work.RPB is a novel and high-efficiency reactor used to intensify the micro-mixing and mass transfer through creating higher gravity which is orders of magnitude larger than that on the earth. This new technology was firstly applied to microencapsulation of phase change materials via interfacial polymerization. In comparison to conventional methods using high speed homogenizer for preparing microcapsules, RPB shows significant advantages, such as much shorter preparation time, lower reaction temperature, higher core content and encapsulation efficiency (see ESI, Table S1†). As a result, RPB shows great potential in microencapsulation area.
Conclusions
PUA hollow microcapsules and microPCMs can be successfully synthesized in RPB via interfacial polymerization between IPDI and DETA, through which PUA can effectively cover the PCM. Technological parameters i.e. C/M ratio, emulsifier dosage and N show significant effects on the thermal storage performance of PUA microPCMs. Compared to the control-microPCM synthesized in a conventional stirring tank reactor, microPCMs prepared with RPB under proper conditions show much better morphologies and higher Ca and E. Meanwhile, the reaction time can be minimized obviously. Hence, RPB shows great potential on scaling up of microPCMs and applying in other interfacial polymerization.
Acknowledgements
This project is financially supported by South Wisdom Valley Innovative Research Team Program and Key Technology R&D Program of Sichuan Province (2015GZ0064). We sincerely thank Research Center of the Ministry of Education for High Gravity Engineering and Technology (Beijing University of Chemical Technology, Beijing, China) for assistance on design and manufacture of rotating packed bed used in this research.
References
- A. M. Khudhair and M. M. Farid, Energy Convers. Manage., 2004, 45, 263–275 CrossRef CAS.
- M. M. Farid, A. M. Khudhair, S. A. K. Razack and S. Al-Hallaj, Energy Convers. Manage., 2004, 45, 1597–1615 CrossRef CAS.
- R. K. Sharma, P. Ganesan, V. V. Tyagi, H. S. C. Metselaar and S. C. Sandaran, Energy Convers. Manage., 2015, 95, 193–228 CrossRef CAS.
- J. F. Su, L. X. Wang and L. Ren, Colloids Surf., A, 2007, 299, 268–275 CrossRef CAS.
- S. P. Zhan, S. H. Chen, L. Chen and W. M. Hou, Powder Technol., 2016, 292, 217–222 CrossRef CAS.
- E. Fallahi, M. Barmar and M. H. Kish, Iran. Polym. J., 2010, 19, 277–286 CAS.
- P. Ni, M. Zhang and N. Yan, J. Membr. Sci., 1995, 103, 51–55 CrossRef CAS.
- J. S. Cho, A. Kwon and C. G. Cho, Colloid Polym. Sci., 2002, 280, 260–266 CAS.
- J. F. Su, L. X. Wang, L. Ren, Z. Huang and X. W. Meng, J. Appl. Polym. Sci., 2006, 102, 4996–5006 CrossRef CAS.
- P. Siddhan, M. Jassal and A. K. Agrawal, J. Appl. Polym. Sci., 2007, 106, 786–792 CrossRef CAS.
- S. Lu, J. Xing, Z. Zhang and G. Jia, J. Appl. Polym. Sci., 2011, 121, 3377–3383 CrossRef CAS.
- H. Zhang and X. Wang, Sol. Energy Mater. Sol. Cells, 2009, 93, 1366–1376 CrossRef CAS.
- F. Sala, G. Bedek, E. Devaux, D. Dupont and L. Gengembre, J. Membr. Sci., 2011, 370, 23–33 CrossRef.
- C. Ramshaw, Heat Recovery Syst. CHP, 1993, 13, 493–513 CrossRef CAS.
- H. Zhao, L. Shao and J. F. Chen, Chem. Eng. J., 2010, 156, 588–593 CrossRef CAS.
- Y. S. Chen, Ind. Eng. Chem. Res., 2011, 50, 1778–1785 CrossRef CAS.
- X. Shi, Y. Xiang, L. X. Wen and J. F. Chen, Chem. Eng. J., 2013, 228, 1040–1049 CrossRef CAS.
- H. J. Yang, G. W. Chu, J. W. Zhang, A. Zhigang Shen and J. F. Chen, Ind. Eng. Chem. Res., 2005, 44, 7730–7737 CrossRef CAS.
- K. Yang, G. Chu, H. Zou, B. Sun, L. Shao and J. F. Chen, Chem. Eng. J., 2011, 168, 1377–1382 CrossRef CAS.
- Z. Qian, L. Xu, H. Cao and K. Guo, Ind. Eng. Chem. Res., 2009, 48, 9261–9267 CrossRef CAS.
- X. L. Ding, X. Y. Hu, Y. G. Ding, Y. X. Wu and D. H. Li, Chem. Eng. Commun., 2000, 178, 249–256 CrossRef.
- W. Wang, H. K. Zou, G. W. Chu, Z. Weng and J. F. Chen, Chem. Eng. J., 2014, 240, 503–508 CrossRef CAS.
- C. C. Lin and H. S. Liu, Ind. Eng. Chem. Res., 1999, 16, 161–167 Search PubMed.
- C. C. Lin, R. O. C. Yu, S. Chen and H. S. Liu, J. Chin. Inst. Chem. Eng., 2004, 35, 531–538 CAS.
- C. F. Chang and S. C. Lee, Water Res., 2012, 46, 2869 CrossRef CAS PubMed.
- Y. S. Chen, Y. C. Hsu, C. C. Lin, C. Y. Tai and H. S. Liu, Environ. Sci. Technol., 2008, 42, 2631–2636 CrossRef CAS PubMed.
- L. L. Zhang, J. X. Wang, Q. Sun, X. F. Zeng and J. F. Chen, Chem. Eng. J., 2012, 181–182, 624–629 CrossRef CAS.
- K. Guo, J. Wen, Y. Zhao, Y. Wang, Z. Zhang, Z. Li and Z. Qian, Environ. Sci. Technol., 2014, 48, 6844–6849 CrossRef CAS PubMed.
- K. Trevour and R. F. James, Ind. Eng. Chem. Res., 1996, 35, 4646–4655 CrossRef.
- C. C. Lin, J. Chem. Eng. Jpn., 2002, 35, 1298–1304 CrossRef CAS.
- G.-W. Chu, X. Gao, Y. Luo, H.-K. Zou, L. Shao and J.-F. Chen, Sep. Purif. Technol., 2013, 102, 62–66 CrossRef CAS.
- J. F. Chen, J. Y. Zhang, Z. G. Shen, J. Zhong and J. Yun, Ind. Eng. Chem. Res., 2006, 45, 8723–8727 CrossRef CAS.
- X. Han, Z. Liang, W. Wang, J. Chen, C. Xue and H. Zhao, Ceram. Int., 2015, 41, 3568–3573 CrossRef CAS.
- B. C. Guo, Y. B. Zhao, W. Wu, H. Meng, H. K. Zou, J. F. Chen and G. W. Chu, Chem. Eng. Process., 2013, 70, 1–8 CrossRef CAS.
- Y. B. Zhao, M. Arowo, W. Wu, H. K. Zou, J. F. Chen and G. W. Chu, J. Ind. Eng. Chem., 2015, 25, 280–287 CrossRef CAS.
- J. F. Chen, H. Gao, H. K. Zou, G. W. Chu, L. Zhang, L. Shao, Y. Xiang and Y. X. Wu, AIChE J., 2010, 56, 1053–1062 CrossRef CAS.
- W. Wang, H. Zou, G. Chu, Y. Xiang, H. Peng and J. Chen, Chin. J. Chem. Eng., 2014, 22, 398–404 CrossRef CAS.
- C. C. Chang, C. Y. Chiu, C. Y. Chang, C. F. Chang, Y. H. Chen, D. R. Ji, J. Y. Tseng and Y. H. Yu, J. Hazard. Mater., 2009, 168, 649–655 CrossRef CAS PubMed.
- C. C. Chang, C. Y. Chiu, C. Y. Chang, C. F. Chang, Y. H. Chen, D. R. Ji, Y. H. Yu and P. C. Chiang, J. Hazard. Mater., 2009, 161, 287–293 CrossRef CAS PubMed.
- Z. Zeng, H. Zou, X. Li, B. Sun, J. Chen and L. Shao, Chem. Eng. Process., 2012, 60, 1–8 CrossRef CAS.
- R. L. Curl, AIChE J., 1963, 9, 175–181 CrossRef CAS.
- P. B. Salunkhe and P. S. Shembekar, Renewable Sustainable Energy Rev., 2012, 16, 5603–5616 CrossRef CAS.
- G. Fang, Z. Chen and H. Li, Chem. Eng. J., 2010, 163, 154–159 CrossRef CAS.
- A. Sarı and A. Karaipekli, Sol. Energy Mater. Sol. Cells, 2009, 93, 571–576 CrossRef.
- F. D. C. Paula and D. G. Shchukin, Chem.–Eur. J., 2015, 21, 11174 CrossRef PubMed.
- F. Gaudin and N. Sintes-Zydowicz, Colloids Surf., A, 2008, 331, 133–142 CrossRef CAS.
- J. Yong, E. Ding and G. Li, Polymer, 2002, 43, 117–122 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01805c |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a and
Bi Shi
a and
Bi Shi