
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetic metal–organic frameworks as scaffolds for spatial co-location and positional assembly of multi-enzyme systems enabling enhanced cascade biocatalysis
Sijia Chena,
Liyin Wena,
Frantisek Svecb,
Tianwei Tana and
Yongqin Lv *a
*a
aBeijing Key Laboratory of Bioprocess, College of Life Science and Technology, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: lvyq@mail.buct.edu.cn
bBeijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, China
First published on 13th April 2017
Abstract
Magnetic multi-enzyme nanosystems have been prepared via co-precipitation of enzymes and metal–organic framework HKUST-1 precursors in the presence of magnetic Fe3O4 nanoparticles. The spatial co-localization of two enzymes was achieved using a layer-by-layer positional assembly strategy. Glucose oxidase (GOx) and horseradish peroxidase (HRP) were used as the model enzymes for cascade biocatalysis. By controlling the spatial positions of enzymes, three bienzyme nanosystems GOx@HRP@HKUST-1@Fe3O4, GOx–HRP@HKUST-1@Fe3O4 and HRP@GOx@HKUST-1@Fe3O4 were prepared in which GOx and HRP containing layers were in close proximity, either encapsulated in the HKUST-1 inner layer, or immobilized on the HKUST-1 outer shell, or randomly distributed in the two MOF layers. Their properties were characterized by transmission electron microscopy, energy-dispersive X-ray spectroscopy, Fourier transform infrared spectroscopy, X-ray diffraction, thermal gravimetric analysis, and zeta potential measurements. The highest activity was observed at pH = 6 and a temperature of 20 °C. Thanks to the favorable positioning of enzymes, the GOx@HRP@HKUST-1@Fe3O4 nanosystem revealed superior kinetics with a Michaelis constant Km = 0.8 mmol L−1 and the maximum reaction rate Vmax = 11.3 μmol L−1 min−1. The enzyme–HKUST-1 conjugates exhibited remarkably high operational stability compared to the free enzymes. This work provides a facile and versatile approach to spatially organized multienzyme systems with well-defined nanostructures and greatly enhanced the overall biocatalytic efficiency.
Introduction
To achieve cellular transformations and signal transduction pathways, living systems utilize a series of synergistic protein catalysts that simultaneously process cascade reactions using the precise localization and spatial ordering of biocatalytic sites.1,2 Using the strategy of substrate channelling along the specific pathways, the reaction intermediate produced by one enzyme migrates without any isolation, directly to the active site of the downstream enzyme where it acts as a substrate.3Enlightened by the natural cellular metabolism, a series of multienzyme conjugates were developed, and significant enhancements in the cascade reaction kinetics and efficiency both in vivo and in vitro have been reported.4–21 Despite the massive interests, the assembly of “in vivo” multienzyme architecture and its full understanding are always difficult due to the extreme complexity of the intracellular environment.6–8 A fascinating alternative to the complex in vivo architecture is to biomimetically construct an immobilized multienzyme system that would enable efficient cascade biotransformations “in vitro”. Several attractive materials and successful strategies have been proposed leading to the design of an arsenal of multienzyme systems for mimicking the natural metabolic pathways in vitro. Examples are the preparations of hydrogels,15,22 hourglass shaped nanochannel reactor,23 mesoporous silica nanoparticles,18,24 formation of inorganic nanocrystal–protein complexes25 and self-assembled crystals,26 microbeads,27,28 DNA nanostructures,4,5,11,29–31 polymersomes,14,32 nanoparticles,33 nanofibers,13,16,34 graphene,9,35 and metal–organic frameworks (MOFs).17,36,37
In contrast to other scaffolds, MOFs, which are formed by the self-assembly of metal ions and organic linkers, feature ultrahigh surface area and porosity, uniform pores with tunable sizes, surfaces with variable chemistries, and structural diversity.38 The large surface area and uniform pore sizes of MOFs enhance capacity for enzyme immobilization and facilitate mass transport. Furthermore, MOFs can exhibit strong hydrophobic interactions with immobilized enzymes that avert their leakage during operation.39 Most importantly, MOFs enable the retention of enzyme activity upon harsh conditions that normally cause loss of the catalytic activity.40
Our previous study focused on protein encapsulation mechanism in MOFs using molecular dynamic simulations also revealed that MOFs can function as a new category of host materials and molecular chaperones that support the protein folding and prevent their deactivations.41 However, restricted by the microporous regime (<2 nm) of most reported MOFs, the direct encapsulation of enzymes in MOFs remains a significant challenge since enzymes have typically at least one dimension in a range of 3–5 nm. This drawback can be circumvented using a “one-pot” co-precipitation approach in which enzymes are mixed with MOF precursors. The MOF scaffold is then formed in an aqueous buffer at room temperature.17,36,42–44 Particularly, Wu et al. used successfully this strategy to the preparation of a bienzyme nanosystem containing glucose oxidase (GOx) and horseradish peroxidase (HRP) encapsulated in zeolitic imidazolate framework ZIF-8.17 At the same time, Hou et al. reported a mimic GOx@mZIF-8 multienzyme system in which magnetic MOF acted as a mimetic peroxidase.36
Although the use of multienzyme conjugates is well demonstrated, many challenges remain in the development of new general approaches enabling the accurate control of positioning and orientation of enzymes, creation of spatially organized multienzyme systems with well-defined nanostructures, as well as in the investigation of the comprehensive kinetics.3,29,45 Several successful attempts have been proposed to attain these goals. For example, Vriezema et al. designed polymersome nanoreactors to selectively encapsulate three enzymes, Candida antarctica lipase B (CALB), GOx, and HRP.32 Garcia adopted a layer-by-layer assembly strategy using biotin–avidin interactions to achieve the desired spatial co-localization of GOx and HRP on magnetic Fe3O4 nanoparticles.33 Liu's group constructed the same bienzyme system based on the formation of the Cu2+–protein nanocrystal complexes.25 Polyelectrolyte doped hollow fibers were prepared by Zhang's group to accomplish the accurate control of enzyme positions in two multienzyme systems.13,34
Here we report the construction of magnetic metal–organic framework HKUST-1 as scaffold for spatial co-localization and positional assembly of two model enzymes, GOx and HRP. As opposed to previously reported work17 in which the multienzymes were randomly distributed in ZIF-8, in our work, GOx and HRP were spatially co-localized in the HKUST-1 MOF structure via a layer-by-layer positional assembly strategy, with GOx on the outer shell and HRP within the pores of the MOF crystal and vice versa. The use of magnetic nanoparticles enables easy separation of biocatalyst from products using a magnet thus facilitating reusability of the catalyst. We systematically investigated the effects of MOF design, optimized the reaction conditions for each bienzyme nanosystem, and studied effects of spatial co-localization of enzymes.
Experimental
Materials and reagents
Ferric chloride hexahydrate, 1,3,5-benzenetricarboxylic acid (H3BTC), potassium sodium tartrate tetrahydrate, 3,5-dinitrosalicylic acid, phenol, o-phenylenediamine (OPD), 2,3-diaminophenazine (DAP), and D-(+)-glucose were all purchased from J&K Scientific Ltd. (Beijing, China). Sodium citrate, ethylene glycol, sodium acetate anhydrous, and sodium hydroxide were obtained from Sinopharm Chemical Reagent Co., Ltd (Beijing, China). Horseradish peroxidase (lyophilized, activity ∼ 179.2 U mg−1) and glucose oxidase (Aspergillus niger, activity ∼ 174.9 U mg−1) were purchased from Sigma-Aldrich (St. Louis, MO, USA).Instrumentation
The transmission electron microscopy (TEM) images of magnetic nanoparticles were obtained using a HITACHI-H800 transmission electron microscope (Hitachi, Ltd., Japan) with an electron acceleration energy of 200 kV. Scanning electron microscopy (SEM) images of magnetic nanoparticles were examined using a JEOL JSM-6700F field emission scanning electron microscope (SEM) (Hitachi High-Technologies, Tokyo, Japan) operated at 5 kV. X-ray powder diffraction (XRD) measurements were performed on a D/max-UltimaIII (Rigaku Corporation, Japan). Fourier transform infrared spectroscopy (FTIR) was carried out using a Nicolet 6700 (Thermo Fisher Scientific, USA). The zeta potentials were determined using a Zetaplus instrument (BI-90Plus Brookhaven Instruments Corp). The thermal gravimetric analysis was carried out using a Thermogravimetric Analyzer (TGA) (Perkin Elmer, Pyris Diamond S(II), USA) at a heating rate of 10 °C min−1 in temperatures range 25–800 °C under nitrogen atmosphere (nitrogen flow rate 20 mL min−1). A ThermoFisher Ultimate 3000 LC system was used for the reversed-phase chromatographic determination of protein concentrations using a C18 stainless steel column (25 cm × 4.6 mm i.d.), and the eluted compounds were detected at 280 nm.Preparation of HKUST-1@Fe3O4 nanoparticles
The preparation of citric acid-coated Fe3O4 nanoparticles was carried out according to previous work.46,47 To obtain HKUST-1@Fe3O4 nanoparticles, 50 mg citric acid-coated Fe3O4 nanoparticles were well dispersed in 1 mL deionized water followed by the addition of 1 mL 10 mmol L−1 copper(II) acetate. After mechanical stirring at 1200 rpm and 25 °C for 1 h, the Cu2+@Fe3O4 product was thoroughly washed with water and separated using a magnet. The Cu2+@Fe3O4 nanoparticles were then re-dispersed in 1 mL water followed by the addition of 1 mL 10 mmol L−1 1,3,5-benzenetricarboxylic acid (H3BTC). The mixture was mechanically stirred at 1200 rpm and 25 °C for 1 h to obtain the H3BTC@Cu2+@Fe3O4 nanoparticles. After washing with methanol and water, the H3BTC@Cu2+@Fe3O4 nanoparticles were re-dispersed in 1 mL water and mixed with 10 mg copper(II) acetate and 21 mg H3BTC. The reaction was carried out at 25 °C for 8 h by mechanical stirring at 1200 rpm to generate the first HKUST-1 layer. The second layer of HKUST-1 was formed using the same synthetic approach. The final product was washed thoroughly with water and separated using a magnet.Design of GOx/HRP bienzyme nanosystems
For the preparation of GOx@HRP@HKUST-1@Fe3O4 nanosystem in which GOx was immobilized on the outer HKUST-1 shell and HRP was encapsulated in the inner HKUST-1 layer, the H3BTC@Cu2+@Fe3O4 nanoparticles were first mixed with 1 mL aqueous solution containing 10 mg copper(II) acetate, 21 mg H3BTC, and 3 mg HRP, and then reacted under mechanical stirring at 1200 rpm and 25 °C for 8 h to produce the first HRP inner layer. After flushing with water, the nanoparticles were mixed with 1 mL aqueous solution consisting of 10 mg copper(II) acetate, 21 mg H3BTC, and a specific amount of GOx, and reacted for another 8 h to form the GOx outer layer. The final product was washed thoroughly with water and separated using a magnet.For HRP@GOx@HKUST-1@Fe3O4 nanosystem in which HRP was immobilized at the outer HKUST-1 shell and GOx encapsulated in the inner HKUST-1 layer, the H3BTC@Cu2+@Fe3O4 nanoparticles were first mixed with 1 mL aqueous solution containing 10 mg copper(II) acetate, 21 mg H3BTC, a specific amount of GOx, and then reacted under mechanical stirring at 1200 rpm and 25 °C for 8 h to produce the first GOx inner layer. After flushing with water, the nanoparticles were mixed with an aqueous solution comprising 10 mg copper(II) acetate, 21 mg H3BTC, and 3 mg HRP, and reacted for 8 h to generate the HRP outer layer. The final product was washed thoroughly with water and separated using a magnet.
With GOx–HRP@HKUST-1@Fe3O4 nanosystem in which the two enzymes were randomly distributed within the two HKUST-1 layers, the H3BTC@Cu2+@Fe3O4 nanoparticles were first mixed with an aqueous solution containing 10 mg copper(II) acetate, 21 mg H3BTC, 1.5 mg HRP, and a certain amount of GOx, and then reacted under mechanical stirring at 1200 rpm and 25 °C for 8 h to produce the first GOx–HRP layer. The second GOx–HRP layer was prepared using the same synthetic approach. The final product was washed thoroughly with water and separated using a magnet.
The GOx@HKUST-1@Fe3O4 nanosystem was prepared by mixing 10 mg copper(II) acetate, 21 mg H3BTC, and 3 mg GOx with H3BTC@Cu2+@Fe3O4 nanoparticles, and then reacting under mechanical stirring at 1200 rpm and 25 °C for 8 h. The final product was washed thoroughly with water and separated using a magnet.
The HRP@HKUST-1@Fe3O4 nanosystem was prepared by mixing 10 mg copper(II) acetate, 21 mg H3BTC, and 3 mg HRP with H3BTC@Cu2+@Fe3O4 nanoparticles, and then reacting under mechanical stirring at 1200 rpm and 25 °C for 8 h. The final product was washed thoroughly with water and separated using a magnet.
All the experiments were performed in triplicates.
Calculation of enzyme loading
The enzyme loading (Q) of different bienzyme nanosystems was calculated according to eqn (1):
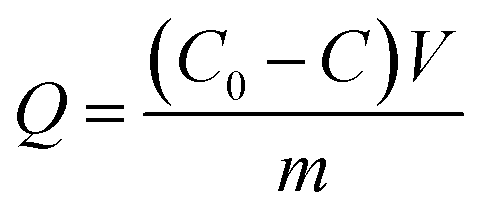 | (1) |
Determination of residual glucose concentrations using DNS reagent
The DNS reagent was first prepared by mixing 0.65 g 3,5-dinitrosalicylic acid, 2.1 g sodium hydroxide, and 0.5 g phenol in 100 mL hot aqueous solution containing 1.29 mol L−1 potassium sodium tartrate. The mixture was then allowed to cool in an ice bath. For the determination of residual glucose concentrations, 1 mL reaction solution was mixed with 1 mL DNS reagent, and kept in a boiling water bath for 15 min. The glucose derivative was determined using Thermo Scientific Microplate Reader (Thermo Labsystems Multiskan Spectrum) with detection at 540 nm.Cascade kinetics study
The cascade kinetics studies for the bienzyme nanosystems were performed using glucose and o-phenylenediamine (OPD) as substrates. The oxidation of glucose by GOx/O2 produces gluconic acid and H2O2 intermediate. The HRP then catalyzes the oxidation reaction of OPD with H2O2 to 2,3-diaminophenazine (DAP), which can be recorded by spectrophotometer at 418 nm. The 1 mL glucose solution with concentrations ranging from 0.56 mM to 111.1 mM was added to the bienzyme systems containing 101 mM OPD in phosphate buffer (pH = 7). The reaction mixture was then incubated at 25 °C while protecting from light for a defined reaction time. The assay solution was then analyzed immediately by UV/Vis spectrophotometry at 418 nm to determine the concentration of produced DAP.Enzyme activity
Enzyme activity studies were carried out following Zore et al. work.9 The activity unit was defined as the amount of enzymes required to convert 1 μmol glucose to gluconic acid in 1 min at pH 6 and 20 °C.13 All experiments were performed in triplicates.Results and discussion
Preparation and characterization of magnetic metal–organic framework
We first prepared magnetic metal–organic framework HKUST-1. This MOF was selected since it features good solvent resistance and stability.46,47 HKUST-1 is prepared under mild conditions that are well suited for encapsulation of enzymes. The citric acid-coated magnetic Fe3O4 nanoparticles were prepared using a modified solvothermal reaction including reduction of Fe(III) ions with trisodium citrate in the presence of ethylene glycol. The magnetic HKUST-1 was then prepared using the “one-pot” co-precipitation approach in which Cu(II) ions coordinated with 1,3,5-benzenetricarboxylic acid in the presence of the magnetic Fe3O4 nanoparticles.The Fe3O4 and HKUST-1@Fe3O4 were imaged in transmission electron microscopy and scanning electron microscopy. Fig. 1a shows the spherical Fe3O4 magnetic nanoparticles that had an average diameter of 216 nm. The co-precipitation approach successfully generated a HKUST-1 MOF shell with an average thickness of 19 nm on the surface of Fe3O4 nanoparticles (Fig. 1b and c). The HKUST-1@Fe3O4 nanocomposites were well dispersed. Comparing Fig. 1b and c revealed that repeating the co-precipitation step for another 8 h led to the formation of a much thicker MOF layer with an average thickness increased to 40 nm. The SEM images in Fig. 2a and b also clearly demonstrate the successful creation of MOF nanocrystal aggregates and significantly increased particle size of the HKUST-1@Fe3O4 nanocomposites.
 | ||
| Fig. 1 TEM images of citric acid-coated Fe3O4 magnetic nanoparticles (a), HKUST-1@Fe3O4 (single layer of HKUST-1) (b), HKUST-1@Fe3O4 (two layers of HKUST-1) (c), GOx@HRP@HKUST-1@Fe3O4 (d), GOx–HRP@HKUST-1@Fe3O4 (e), and HRP@GOx@HKUST-1@Fe3O4 (f). | ||
 | ||
| Fig. 2 SEM images of citric acid-coated Fe3O4 magnetic nanoparticles (a), HKUST-1@Fe3O4 (two layers of HKUST-1) (b), GOx@HRP@HKUST-1@Fe3O4 (c), GOx–HRP@HKUST-1@Fe3O4 (d), and HRP@GOx@HKUST-1@Fe3O4 (e). | ||
X-ray diffraction (XRD) patterns illustrated in Fig. 3a include all diffraction peaks typical of magnetite (JCPDS card no. 19-0629). After the formation of HKUST-1 layers, new peaks were observed which corresponded to the formation of HKUST-1 crystals. The FTIR spectra of Fe3O4 nanoparticles and their modified counterparts shown in Fig. 3b exhibit characteristic peak centered at 1630 cm−1 (black line) assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of citric acid groups at Fe3O4.47 The peak centered at 582 cm−1 corresponds to Fe–O stretching.36 A large broad band at 3400–3500 cm−1 is attributed to the O–H bond stretching. The formation of HKUST-1 layer shifted the peak of CO stretching vibrations to 1710 cm−1. The prominent peaks centered at 1570 cm−1 and 1623 cm−1 are characteristic of the aromatic rings of HKUST-1. The peaks in the range of 1100–1600 cm−1 are ascribed to the vibrational and bending modes of H–O–H, the vibrational peaks of C–H, and stretching vibration peak of C–O, respectively.48,49 Owing to the deprotonation of citric acid layers, the citric acid-coated magnetic Fe3O4 nanoparticles exhibited a negative zeta potential of −22.7 mV at pH 7 (Fig. 3c). While the zeta potential of the original HKUST-1 crystals was −12.7 mV, it increased to a negative value of −17.5 mV after the formation of HKUST-1@Fe3O4 nanocomposite.
O stretching vibrations of citric acid groups at Fe3O4.47 The peak centered at 582 cm−1 corresponds to Fe–O stretching.36 A large broad band at 3400–3500 cm−1 is attributed to the O–H bond stretching. The formation of HKUST-1 layer shifted the peak of CO stretching vibrations to 1710 cm−1. The prominent peaks centered at 1570 cm−1 and 1623 cm−1 are characteristic of the aromatic rings of HKUST-1. The peaks in the range of 1100–1600 cm−1 are ascribed to the vibrational and bending modes of H–O–H, the vibrational peaks of C–H, and stretching vibration peak of C–O, respectively.48,49 Owing to the deprotonation of citric acid layers, the citric acid-coated magnetic Fe3O4 nanoparticles exhibited a negative zeta potential of −22.7 mV at pH 7 (Fig. 3c). While the zeta potential of the original HKUST-1 crystals was −12.7 mV, it increased to a negative value of −17.5 mV after the formation of HKUST-1@Fe3O4 nanocomposite.
 | ||
| Fig. 3 X-ray diffraction patterns (a), FTIR spectra (b), zeta potentials (c), and thermal gravimetric analysis (d) of citric acid-coated Fe3O4 magnetic nanoparticles (1), HKUST-1@Fe3O4 (two layers of HKUST-1) (2), GOx@HRP@HKUST-1@Fe3O4 (3), GOx–HRP@HKUST-1@Fe3O4 (4), and HRP@GOx@HKUST-1@Fe3O4 (5). | ||
The TGA plots presented in Fig. 3d indicate that the magnetic Fe3O4 nanoparticles lost 16.5% of their weight at 800 °C, which corresponds to the removal of water and citric acid functionalities. TGA of HKUST-1@Fe3O4 demonstrates multistage decompositions. The first stage characterized by a weight loss of 5.5% at 152 °C is due to the removal of residual coordinated solvent or adsorbed molecules within the HKUST-1 framework. The second weight loss amounting 29.6% observed between 152 and 500 °C occurs due to the decomposition of HKUST-1. The third stage observed between 500 and 800 °C includes the removal of carbon and represents a weight loss of 13%. The residual 48.9% of weight is copper and iron. Energy-dispersive X-ray spectroscopy data shown in Table 1 revealed the presence of 23.55 at% C, 50.63 at% O, and 25.82 at% Fe in citric acid-coated Fe3O4 nanoparticles. The incorporation of HKUST-1 layers changed the elemental compositions to 48.35 at% C, 40.63 at% O, 10.16 at% Fe, and 0.86 at% Cu. Additionally, due to the porous structures of MOF, the HKUST-1@Fe3O4 nanocomposites exhibited a surface area of 327.6 m2 g−1, an average pore size of 0.67 nm, and a pore volume of 0.24 cm3 g−1, all calculated using BET equation.
| Nanoparticles | C (at%) | O (at%) | N (at%) | Fe (at%) | Cu (at%) | S (at%) |
|---|---|---|---|---|---|---|
| Fe3O4 | 23.55 | 50.63 | — | 25.82 | — | — |
| HKUST-1@Fe3O4 | 48.35 | 40.63 | — | 10.16 | 0.86 | — |
| GOx@HRP@HKUST-1@Fe3O4 | 48.58 | 35.18 | 7.11 | 8.35 | 0.51 | 0.27 |
| GOx–HRP@HKUST-1@Fe3O4 | 46.55 | 38.33 | 6.73 | 7.56 | 0.54 | 0.29 |
| HRP@GOx@HKUST-1@Fe3O4 | 47.46 | 37.22 | 6.36 | 7.93 | 0.69 | 0.34 |
These results confirmed the successful formation of HKUST-1@Fe3O4 core–shell structures.
Spatial co-localization and positional assembly of GOx/HRP bienzymes using magnetic HKUST-1
The bienzyme systems were designed to include glucose oxidase and horseradish peroxidase as model enzymes thanks to their robustness and capability to maintain their activity during the immobilization process.3 As we demonstrated earlier, the use of multienzyme conjugates for multistage biotransformations is favorable and accelerates the reaction kinetics since a reaction intermediate produced from one enzyme can be directly utilized by the second enzyme without any long distance diffusion or penetration.50 However, the cascade reaction kinetics and catalytic efficiency rely on the design of the spatially co-localized multienzyme systems. In this respect, we prepared three different enzymatic nanosystems using spatially co-localization of HRP and GOx at different sites of HKUST-1 via a layer-by-layer self-assembly approach. The synthetic routes are illustrated in Scheme 1. The three bienzyme nanosystems GOx@HRP@HKUST-1@Fe3O4, HRP@GOx@HKUST-1@Fe3O4, and GOx–HRP@HKUST-1@Fe3O4 vary through GOx and HRP location. The enzymes were either encapsulated in the HKUST-1 inner layer, or immobilized at the HKUST-1 outer shell, or randomly distributed within the two MOF layers. The TEM and SEM images of the three bienzyme conjugates containing GOx and HRP at different locations shown in Fig. 1d–f and 2c–e confirm that there is no difference in morphologies. MOF shells with an average thickness of 46 nm were formed via the aggregation of HKUST-1 nanocrystals with encapsulated enzymes. Fig. 3a shows that the crystal structure and crystallinity did not exhibit any remarkable differences that could be observed between immobilized enzyme nanosystems and generic Fe3O4 nanoparticles. The FTIR spectra of three bienzyme nanosystems also correspond well with the HKUST-1@Fe3O4 nanocomposites (Fig. 3b). The successful immobilization of enzymes was further confirmed by changes in zeta potential. Although GOx (pI ∼ 4.2) is negatively charged and HRP (pI ∼ 9.0) bears a net positive charge at pH 7.0,16,51 the three bienzyme nanosystems revealed similar zeta potentials of −11.1, −12.4, and −12.4 mV, respectively (Fig. 3c). The thermal gravimetric analysis plots shown in Fig. 3d are also similar for all three bienzyme nanosystems. The enzyme decomposition occurred between 152 and 500 °C in which weight losses of 35.8%, 37.6%, and 38.5% were observed for GOx@HRP@HKUST-1@Fe3O4, GOx–HRP@HKUST-1@Fe3O4, and HRP@GOx@HKUST-1@Fe3O4, respectively. Furthermore, EDS measurements shown in Table 1 revealed the appearance of sulfur after encapsulation of enzymes in MOF layers. | ||
| Scheme 1 Schematic illustrations of the three different strategies for the positional assembly and spatial co-localization of GOx and HRP, and their cascade biocatalysis reaction. | ||
Enzyme loading and specific activities are summarized in Table 2. For each HKUST-1 layer, the enzyme loading was at the same order of magnitude ranging from 50.9 to 58.9 mg enzyme per gram Fe3O4 nanoparticles, which was consistent with the TGA results. This is equal to 85% to 98% enzyme that were immobilized to the bioconjugates taking into consideration that 3 mg of each free enzyme were added to the system. However, the bienzyme nanosystems with diverse locations of enzymes exhibited significant differences in activity. The GOx@HRP@HKUST-1@Fe3O4 nanosystem featured the highest activity, which was 79 U mg−1 for GOx. For each multi-enzyme assembly system, the yield is higher than 89%.
| Positionally assembled strategies | Enzyme | Enzyme loading capacity (mg per g Fe3O4) | Specific activity (U mg−1) |
|---|---|---|---|
| GOx@HRP@HKUST-1@Fe3O4 | GOx | 58.2 | 79 |
| HRP | 58.9 | ||
| GOx–HRP@HKUST-1@Fe3O4 | GOx | 56.2 | 61 |
| HRP | 57.4 | ||
| HRP@GOx@HKUST-1@Fe3O4 | HRP | 55.2 | 69 |
| GOx | 50.9 |
Cascade enzymatic catalysis
The cascade reaction catalyzed by GOx and HRP includes two steps: first, GOx catalyzes the oxidation of glucose to gluconic acid and hydrogen peroxide by using molecular oxygen as an electron acceptor. Then, HRP converts H2O2 intermediate to water and oxygen. The continuous decomposition of hydrogen peroxide is favorable since it protects GOx from deactivation and also accelerates the oxidation reaction.We first tested the cascade biocatalysis performances with GOx–HRP@HKUST-1@Fe3O4 bienzyme nanosystem by comparing the glucose conversion at different pH (4–7) and temperature (15–50 °C) ranges. As illustrated in Fig. 4a and b, the highest consumption of glucose was observed at pH 6 and the optimum temperature was found to be 20 °C. This result is consistent with previous reports.52–54
 | ||
| Fig. 4 Effects of different reaction conditions including temperature (a), pH (b), and reaction time (c) on the consumption of glucose substrate. The reaction was carried out at pH 7 for 3 h in (a), at a temperature of 20 °C for 3 h in (b), and at pH 6 and a temperature of 20 °C for (c). Glucose concentration 1 mg mL−1. | ||
Another factor affecting the cascade reaction is the reaction time. The three bienzyme nanosystems were used to catalyze reaction of 2 mL glucose solution with a concentration of 5.55 mmol L−1. Fig. 4c shows that the GOx@HRP@HKUST-1@Fe3O4 nanosystem enabled an almost 100% conversion of glucose after 15 h. In contrast, 36 and 48 h were needed to achieve the complete conversion of glucose to gluconic acid reaction conditions of pH 6 and a temperature of 20 °C. The reaction rate was assayed by calculating the consumption of using the GOx–HRP@HKUST-1@Fe3O4 and HRP@GOx@HKUST-1@Fe3O4 nanosystems, respectively. We assume that this remarkable difference in reaction rate can be attributed to the difference in spatial co-localization of enzymes.
We also studied the effects of GOx/HRP ratio on enzymatic activity. As expected, the bienzyme nanosystem with a GOx/HRP ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 produced the highest activity 102 U mg−1 (Table 3).
:4 produced the highest activity 102 U mg−1 (Table 3).
| Bienzyme nanosystems | Activity (U mg−1) |
|---|---|
| GOx/HRP (1:1) |
77 |
| GOx/HRP (1:2) |
87 |
| GOx/HRP (1:3) |
87 |
| GOx/HRP (1:4) |
102 |
| GOx/HRP (1:5) |
86 |
Kinetics study
To further elucidate the effect of spatial co-localization of both enzymes on the biocatalysis kinetics, the enzymatic reactions were carried out under the optimized glucose substrate within a concentration range of 0.56–111.1 mmol L−1 at the initial stage of reaction up to consumption of 5% of glucose substrate. The kinetics curves were drawn by plotting enzymatic reaction rate versus substrate concentration. The Michaelis constant Km and maximum reaction rate Vmax were calculated using glucose and OPD as substrates. Fig. 5 shows that the reaction rate increased with the increase in glucose concentration. At a certain glucose concentration, the bienzyme system became saturated with glucose and reached the maximum reaction rate Vmax. The Michaelis constant Km is defined as the substrate concentration at which the enzyme reaction rate is half Vmax. A lower Km value corresponds to a higher affinity between the substrate and enzyme, while a higher Vmax value reflects a faster reaction rate, and vice versa. As shown in Table 4, the Km for all three bienzyme nanosystems was similar with a value of 0.8, 0.9, and 0.7 mmol L−1, respectively. However, the HRP@GOx@HKUST-1@Fe3O4 nanosystem with GOx inside and HRP outside exhibited the lowest Vmax value of 10.2 μmol L−1 min−1, whereas the GOx@HRP@HKUST-1@Fe3O4 nanosystem with HRP inside and GOx outside featured the highest Vmax value of 11.3 μmol L−1 min−1. This result was consistent with the data obtained from optimization of reaction time in which the GOx@HRP@HKUST-1@Fe3O4 nanosystem required the shortest time of 15 h to achieve the complete conversion of glucose to gluconic acid, complete conversion of glucose to gluconic acid. In contrast, the HRP@GOx@HKUST-1@Fe3O4 nanosystem needed 48 h to reach the same conversion. | ||
| Fig. 5 Kinetic plots depicting the oxidation of glucose catalyzed using our bienzyme nanosystems GOx@HRP@HKUST-1@Fe3O4 (a), GOx–HRP@HKUST-1@Fe3O4 (b), HRP@GOx@HKUST-1@Fe3O4 (c), individually immobilized enzymes (d), and free enzymes (e). Conditions: pH 6, reaction temperature 20 °C. | ||
| Km (mM) | Vmax (μM min−1) | |
|---|---|---|
| GOx@HRP@HKUST-1@Fe3O4 | 0.8 | 11.3 |
| GOx–HRP@HKUST-1@Fe3O4 | 0.9 | 10.5 |
| HRP@GOx@HKUST-1@Fe3O4 | 0.7 | 10.2 |
| GOx@HKUST-1@Fe3O4 + HRP@HKUST-1@Fe3O4 | 2.4 | 8.4 |
| Free enzymes | 2.3 | 7.2 |
The kinetic parameters listed in Table 4 clearly demonstrate that the reaction rates of the bienzyme nanosystems were closely related to the location of the two enzymes. Scheme 1 illustrates the mechanism of the cascade reaction catalyzed by the three bienzyme nanosystems studied. With regard to the GOx@HRP@HKUST-1@Fe3O4 complex in which GOx is localized at the outer shell and HRP in the inner layer of the complex, glucose and oxygen in the solution first access GOx, and are converted to gluconic acid and H2O2. The H2O2 intermediate then migrates directly to the HRP enzyme and oxidizes OPD to DAP. This route accelerates the cascade biocatalysis reaction rate.
In contrast, in the case of HRP@GOx@HKUST-1@Fe3O4 nanosystem, both glucose and oxygen substrates must first diffuse through the HRP outer layer to approach the GOx inner layer where the first reaction occurs. As a result, the reaction rate is reduced and Vmax is much lower. For the GOx–HRP@HKUST-1@Fe3O4 nanosystem in which GOx and HRP were randomly distributed in both MOF layers, the reaction rate laid between those observed for each system with separated enzyme layers. It is worth of noting that the regular pore structures of MOF are favorable to facilitate substrate channeling which enhances cascade biocatalytic performance.
For comparison, we also prepared MOF layers including only a single immobilized enzyme, i.e. GOx@HKUST-1@Fe3O4 and HRP@HKUST-1@Fe3O4. The cascade reaction was carried out by mixing immobilized GOx with immobilized HRP in glucose solutions at different concentrations. The kinetic curve is presented in Fig. 5d, which as expected reveals a dramatically higher Km value of 2.4 mmol L−1 and a lower Vmax value of 8.4 μmol L−1 min−1 (Table 4). Meanwhile, enzymatic cascade catalysis of glucose and OPD using free GOx and HRP exhibits a much lower Vmax value of 7.2 μmol L−1 min−1 as shown in Fig. 5e. This result is consistent with our conclusion that the channeling of the substrate in bienzyme nanosystem enables higher rate of the cascade reaction compared to the situation in which the enzymes are randomly distributed or separately immobilized.
Operational stability and reusability
The operational stability of the immobilized enzymes is another important parameter that affects the real-life applications. Here, we estimated the operational stability via testing the residual activities of immobilized enzymes in comparison with free enzymes after incubation in phosphate buffer under denaturing conditions including pH (2, 6, and 9) and temperature (40, 65, and 95 °C) for 24 h. The initial and residual activities were calculated using glucose as substrate at 20 °C and pH 6. We discovered that the HKUST-1 immobilized bienzyme system exhibited exceptional stability under harsh conditions, whereas activity of free enzymes rapidly deteriorated. Fig. 6a confirms that free enzymes lost at least 80% of their initial activities after incubation in phosphate buffer at pH 2 or 9. In contrast, the enzyme–HKUST-1 conjugates retained more than 70% of their initial activities after treatment under the same conditions. For example, 93% and 92% of residual activity was retained in HRP@GOx@HKUST-1@Fe3O4 nanosystem at pH 2 and 9, respectively. At a higher temperature of 65 °C, free enzymes lost 52% of their initial activities as opposed to only 10% loss for HRP@GOx@HKUST-1@Fe3O4 (Fig. 6b). At a high temperature of 95 °C, free enzymes lost their activity completely while the HRP@GOx@HKUST-1@Fe3O4 nanosystem retained 21% residual activity. The superiority in the high chemical and thermal stability of immobilized enzymes can be ascribed to the protecting effects of HKUST-1. Under harsh conditions that typically result in enzyme denaturation and deactivation, MOFs that act as solid scaffolds for enzyme immobilization are favorable since they help to preserve protein folding thus maintaining the activity. This finding is in agreement with previously reported studies.40,41 Comparing the GOx@HRP@HKUST-1@Fe3O4 and HRP@GOx@HKUST-1@Fe3O4 bioconjugate nanosystems, the latter exhibited better thermal and chemical tolerance. This result can be explained by the fact that the rate controlling reaction is oxidation of glucose catalyzed by GOx and the HKUST-1 outer layer protects better the inner layer containing GOx.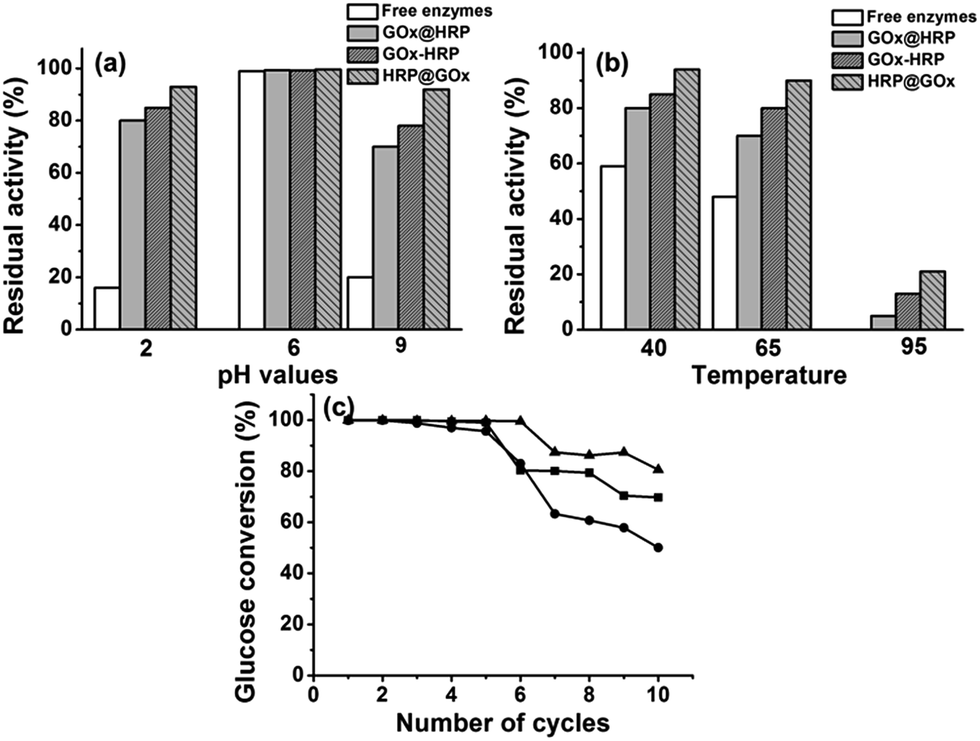 | ||
| Fig. 6 Operational stabilities of free enzymes in comparison to three bienzyme nanosystems at different pH values (2, 6, and 9) (a) and different temperatures (40, 65, and 95 °C) (b), and reusability of three bienzyme nanosystems with respect to the number of cycles the conjugates were used for (c). | ||
The utilization of magnetic Fe3O4 nanoparticles facilitates the reuse of immobilized enzymes. In this respect, we also determined the reusability of our bienzyme nanosystems by repeatedly catalyzing 2 mL 0.5 mg mL−1 glucose substrate solution under the same reaction conditions of 20 °C, pH 6, and a reaction time of 15 h. As illustrated in Fig. 6c, the HRP@GOx@HKUST-1@Fe3O4 nanosystem again had the best operational stability with a glucose conversion of 80.6% even after repeated use for ten cycles. This result further proves our previous conclusion that the positioning of GOx in the inner HKUST-1 layer is more beneficial to retain its activity. Meanwhile, all three bienzyme nanosystems kept up to 96% of their initial activities after storing at 4 °C for more than three weeks.
Conclusions
We demonstrated the preparation of three recyclable multi-enzyme nanosystems using magnetic metal–organic framework HKUST-1 as scaffold. These nanosystems contained GOx and HRP with different spatial co-localizations that clearly influenced the cascade reaction kinetics and operational stability of immobilized enzymes. We discovered that the bienzyme nanosystem with GOx on the outer MOF layer and HRP in the inner HKUST-1 layer enabled a faster reaction rate and enhanced the cascade biocatalysis, whereas, the opposite positioning approach is advantageous to improve the thermal and chemical resistance of enzymes. This suggests an interesting strategy that enables precise positioning of multi-enzymes and possible control of active site orientation. Our results confirm that immobilization of enzymes using MOFs is a promising approach that can lead to design of the artificial biomimetic reactors simulating processes in living cells and to achieve extracellular biosynthesis.Acknowledgements
The authors gratefully acknowledge the financial support from National Natural Science Foundation of China (21576017, 21436002, and 21606016), 973 programs (2014CB745100), 863 programs (2015BAD15B07), the Higher Education and High-Quality and World-Class Universities (PY201613), and the Public Hatching Platform for Recruited Talents of Beijing University of Chemical Technology.Notes and references
- G. G. Hammes and C. W. Wu, Science, 1971, 172, 1205 CAS.
- A. S. Shaw and E. L. Filbert, Nat. Rev. Immunol., 2009, 9, 47 CrossRef CAS PubMed.
- J. L. Lin, L. Palomec and I. Wheeldon, ACS Catal., 2014, 4, 505 CrossRef CAS.
- J. Fu, M. Liu, Y. Liu, N. W. Woodbury and H. Yan, J. Am. Chem. Soc., 2012, 134, 5516 CrossRef CAS PubMed.
- O. I. Wilner, Y. Weizmann, R. Gill, O. Lioubashevski, R. Freeman and I. Willner, Nat. Nanotechnol., 2009, 4, 249 CrossRef CAS PubMed.
- J. E. Dueber, G. C. Wu, G. R. Malmirchegini, T. S. Moon, C. J. Petzold, A. V. Ullal, K. L. J. Prather and J. D. Keasling, Nat. Biotechnol., 2009, 27, 753 CrossRef CAS PubMed.
- C. J. Delebecque, A. B. Lindner, P. A. Silver and F. A. Aldaye, Science, 2011, 333, 470 CrossRef CAS PubMed.
- H. Cheng, L. Zhang, J. He, W. Guo, Z. Zhou, X. Zhang, S. Nie and H. Wei, Anal. Chem., 2016, 88, 5489 CrossRef CAS PubMed.
- O. V. Zore, A. Pattammattel, S. Gnanaguru, C. V. Kumar and R. M. Kasi, ACS Catal., 2015, 5, 4979 CrossRef CAS.
- R. Qu, L. Shen, A. Qu, R. Wang, Y. An and L. Shi, ACS Appl. Mater. Interfaces, 2015, 7, 16694 CAS.
- V. Linko, M. Eerikainen and M. A. Kostiainen, Chem. Commun., 2015, 51, 5351 RSC.
- L. Lin, J. Yan and J. Li, Anal. Chem., 2014, 86, 10546 CrossRef CAS PubMed.
- X. Ji, Z. Su, P. Wang, G. Ma and S. Zhang, ACS Catal., 2014, 4, 4548 CrossRef CAS.
- D. Grafe, J. Gaitzsch, D. Appelhans and B. Voit, Nanoscale, 2014, 6, 10752 RSC.
- X. Wang, Z. Li, J. Shi, H. Wu, Z. Jiang, W. Zhang, X. Song and Q. Ai, ACS Catal., 2014, 4, 962 CrossRef CAS.
- H. Liang, S. Jiang, Q. Yuan, G. Li, F. Wang, Z. Zhang and J. Liu, Nanoscale, 2016, 8, 6071 RSC.
- X. Wu, J. Ge, C. Yang, M. Hou and Z. Liu, Chem. Commun., 2015, 51, 13408 RSC.
- H. Gustafsson, A. Kuchler, K. Holmberg and P. Walde, J. Mater. Chem. B, 2015, 3, 6174 RSC.
- L. Zhou, W. Tang, Y. Jiang, L. Ma, Y. He and J. Gao, RSC Adv., 2016, 6, 90061 RSC.
- G. Begum, W. B. Goodwin, B. M. deGlee, K. H. Sandhage and N. Kroger, J. Mater. Chem. B, 2015, 3, 5232 RSC.
- W. Siti, H.-P. M. de Hoog, O. Fischer, W. Y. Shan, N. Tomczak, M. Nallani and B. Liedberg, J. Mater. Chem. B, 2014, 2, 2733 RSC.
- C. A. Liao, Q. Wu, Q. C. Wei and Q. G. Wang, Chem.–Eur. J., 2015, 21, 12620 CrossRef CAS PubMed.
- M. Onda, Y. Lvov, K. Ariga and T. Kunitake, Biotechnol. Bioeng., 1996, 51, 163 CrossRef PubMed.
- B. El-Zahab, H. Jia and P. Wang, Biotechnol. Bioeng., 2004, 87, 178 CrossRef CAS PubMed.
- Z. Li, Y. Zhang, Y. Su, P. Ouyang, J. Ge and Z. Liu, Chem. Commun., 2014, 50, 12465 RSC.
- V. Liljeström, J. Mikkilä and M. A. Kostiainen, Nat. Commun., 2014, 5, 4445 Search PubMed.
- J. Rocha-Martin, S. Velasco-Lozano, J. M. Guisan and F. Lopez-Gallego, Green Chem., 2014, 16, 303 RSC.
- E. T. Hwang, B. K. Seo, M. B. Gu and A. P. Zeng, Catal. Sci. Technol., 2016, 6, 7267 CAS.
- J. Fu, M. Liu, Y. Liu and H. Yan, Acc. Chem. Res., 2012, 45, 1215 CrossRef CAS PubMed.
- Y. Hu, F. Wang, C.-H. Lu, J. Girsh, E. Golub and I. Willner, Chem.–Eur. J., 2014, 20, 16203 CrossRef CAS PubMed.
- E. A. Jeffrey, D. Brodin and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4564 CrossRef PubMed.
- D. M. Vriezema, P. M. L. Garcia, N. Sancho Oltra, N. S. Hatzakis, S. M. Kuiper, R. J. M. Nolte, A. E. Rowan and J. C. M. van Hest, Angew. Chem., Int. Ed., 2007, 119, 7522 CrossRef.
- J. Garcia, Y. Zhang, H. Taylor, O. Cespedes, M. E. Webb and D. Zhou, Nanoscale, 2011, 3, 3721 RSC.
- X. Ji, Z. Su, P. Wang, G. Ma and S. Zhang, ACS Nano, 2015, 9, 4600 CrossRef CAS PubMed.
- A. Alshammari, M. G. Posner, A. Upadhyay, F. Marken, S. Bagby and A. Ilie, ACS Appl. Mater. Interfaces, 2016, 8, 21077 CAS.
- C. Hou, Y. Wang, Q. Ding, L. Jiang, M. Li, W. Zhu, D. Pan, H. Zhu and M. Liu, Nanoscale, 2015, 7, 18770 RSC.
- X. Wu, M. Hou and J. Ge, Catal. Sci. Technol., 2015, 5, 5077 CAS.
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213 RSC.
- Y. Chen, S. Han, X. Li, Z. Zhang and S. Ma, Inorg. Chem., 2014, 53, 10006 CrossRef CAS PubMed.
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 7240 CrossRef CAS PubMed.
- H. Zhang, Y. Lv, T. Tan and D. van der Spoel, J. Phys. Chem. B, 2016, 120, 477 CrossRef CAS PubMed.
- F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761 CrossRef CAS PubMed.
- X. Wu, C. Yang, J. Ge and Z. Liu, Nanoscale, 2015, 7, 18883 RSC.
- F. K. Shieh, S. C. Wang, C. I. Yen, C. C. Wu, S. Dutta, L. Y. Chou, J. V. Morabito, P. Hu, M. H. Hsu, K. C. W. Wu and C. K. Tsung, J. Am. Chem. Soc., 2015, 137, 4276 CrossRef CAS PubMed.
- S. Schoffelen and J. C. M. van Hest, Soft Matter, 2012, 8, 1736 RSC.
- J. Liu, Z. Sun, Y. Deng, Y. Zou, C. Li, X. Guo, L. Xiong, Y. Gao, F. Li and D. Zhao, Angew. Chem., Int. Ed., 2009, 121, 5989 CrossRef.
- Y. Cao, L. Wen, F. Svec, T. Tan and Y. Lv, Chem. Eng. J., 2016, 286, 272 CrossRef CAS.
- F. N. Azad, M. Ghaedi, K. Dashtian, S. Hajati and V. Pezeshkpour, Ultrason. Sonochem., 2016, 31, 383 CrossRef CAS PubMed.
- S. Mosleh, M. R. Rahimi, M. Ghaedi, K. Dashtian and S. Hajati, RSC Adv., 2016, 6, 17204 RSC.
- C. Garcia-Galan, Á. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885 CrossRef CAS.
- J. H. Pazur and K. Kleppe, Biochemistry, 1964, 3, 578 CrossRef CAS PubMed.
- C. M. Wong, K. H. Wong and X. D. Chen, Appl. Microbiol. Biotechnol., 2008, 78, 927 CrossRef CAS PubMed.
- J. L. Munoz-Munoz, F. Garcia-Molina, R. Varon, J. N. Rodriguez-Lopez, F. Garcia-Canovas and J. Tudela, Biosci., Biotechnol., Biochem., 2007, 71, 390 CrossRef CAS PubMed.
- D. Lan, B. Li and Z. Zhang, Biosens. Bioelectron., 2008, 24, 934 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |