DOI:
10.1039/C7RA04196A
(Paper)
RSC Adv., 2017,
7, 28788-28796
Facile synthesis of low band-gap DPP–EDOT containing small molecules for solar cell applications†
Received
13th April 2017
, Accepted 18th May 2017
First published on 31st May 2017
Abstract
Four donor–donor–acceptor–donor–donor (D1–D2–A–D2–D1) type small molecules (SM1, SM2, SM3 and SM4), in which diketopyrrolopyrrole (DPP) was used as an acceptor (DPP) core and 3,4-ethylenedioxythiophene (EDOT), triphenylamine (TPA) or alkyl thiophene (AT) acted as a donor, were synthesized (by direct alkylation) for photovoltaic applications. The benzene flanked DPPs (SM1 and SM2) show more limited absorption and a lower-lying HOMO energy level compared to the thiophene flanked DPPs (SM3 and SM4, respectively), due to benzene being a poor donor compared to a thiophene moiety. SM3 and SM4 have absorptions extending to 750 nm (in CH2Cl2) and can be regarded as low band-gap small molecules. The inverted small molecule solar cell (using PCBM as an acceptor, and ZnO and MoO3 as the electron and hole transporters, respectively) based on SM2 has the highest value of Voc (0.82 V) due to SM2 having the lowest HOMO level. An SM3 based device achieves the highest value of Jsc (5.56 mA cm−2), due to the wide absorption profile and proper LUMO level. The poor photovoltaic performances of SM1 and SM4 (in which D1 is a triphenyl amine group) are due to the serious self-aggregation of the molecules in the SMx/PCBM active films. Two acceptor–donor–acceptor type (A–D–A) molecules (SM5 and SM6) were also synthesized by Stille coupling, in which DPP and EDOT were used as the acceptor and donor, respectively, for the structure–photovoltaic performance study. The SM5 and SM6 films, having on-set absorption of up to 1000 nm, can also be regarded as low band-gap small molecules. SM5 and SM6 are structural isomers, however the inverted cell based on SM6 has a much higher PCE than that based on SM5, due to SM6 having better solubility and a lower HOMO energy level. This study provides a guideline for the rational design and preparation of conjugated small molecules for organic solar cells.
1. Introduction
The solution processed organic solar cell is considered to be a viable route for realizing low-cost renewable energy.1,2 Organic small molecules for solution processed bulk heterojunction (BHJ) solar cells (SMSCs) have attracted significant attention due to their well-defined molecular structures, easy purification, small batch to batch variations in their properties, and intrinsic monodispersity.3–5 A large number of papers have been published in the past several years regarding the synthesis, device engineering, and device physics of solution-processed SMSCs, and the devices based on small molecules have not achieved PCEs of higher than 10%.6 Further progress relies on molecular design and advanced device processing techniques. Diketopyrrolopyrrole (DPP), an industrial pigment with strong π–π interaction, electron withdrawing characteristics and an efficient charge transport property, has emerged recently as one of the most widely used organic building blocks to construct molecules for organic field effect transistors (OFETs)7–10 and organic solar cells (OPVs).11–19 The relatively low lying HOMO levels make the DPP-containing molecules especially promising candidates for applications in organic solar cells to achieve high values of Voc.20–23
Synthesizing new molecules with high yield and low waste for specific applications is a major challenge for the synthetic chemist. Recently, a new synthetic approach, namely the direct arylation (DCA) of C–H bonds, was reported for preparing DPP derivatives.24–28 The direct C–H arylation reaction has advantages over the traditional cross coupling reaction in terms of avoiding the use of organometallic precursor reagents, higher atom economy, fewer synthetic steps, higher yields and better compatibility with the functional groups. As a result, DCA has the advantages of a faster reaction rate, less expensive process and less waste production, hence making a substantial contribution to the development of sustainable chemistry.29–35 On the other hand, 3,4-ethylenedioxythiophene (EDOT) shows a strong electron donating ability. It can provide a noncovalent intermolecular interaction with the adjacent aryl units, thus inducing the self-rigidification of the conjugated system.36–45 Therefore it was used widely as an electron donating moiety to combine with the DPP acceptor to construct push–pull or donor acceptor (D–A) p-type organic semiconductors.46–49
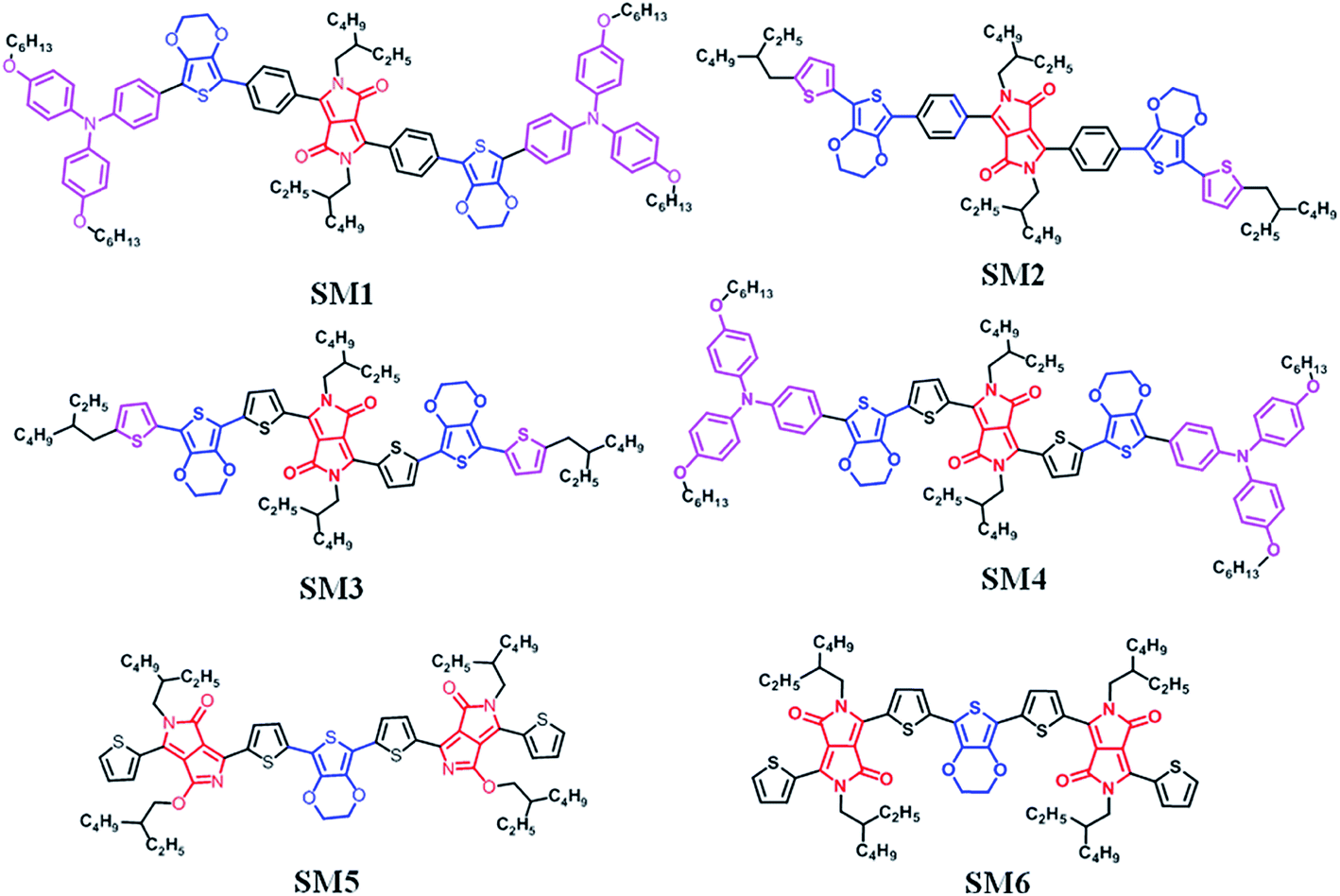
In the present work, we report the synthesis and physical properties of six new DPP containing small molecules, SM1–6, which were originally designed to be used in SMSCs. SM1–4, with D1–D2–A–D2–D1 molecular architectures, were synthesized by direct C–H arylation, in which triphenylamine–EDOT or 2-ethylhexylthiophene–EDOT was used as a donating unit. Benzene flanked DPP (DPP[Ph]2) or thiophene flanked DPP (DPP[Th]2) was used as an acceptor. SM5 and SM6 (which are isomers) have an A–D–A type molecular architecture and were prepared by the Stille coupling reaction, in which EDOT was used as a donor and DPP was used as an acceptor. The chemical structures of the six molecules are shown in Fig. 1. The high photovoltaic small p-type molecule generally has a planarized molecular architecture, which was formed by large fused-ring moieties or intermolecular interactions. In this paper we used EDOT (a pseudo two dimension conjugated system50) as a donor unit to combine with the DDP acceptor and construct a planar small p-type molecule for SMSCs. The structure–physicochemical properties–photovoltaic performance relationship of these new compounds can provide valuable guidelines for designing new organic electronic materials. Moreover, we also demonstrated here that the green direct C–H arylation reaction can be used to prepare organic compounds containing both EDOT and DPP units.
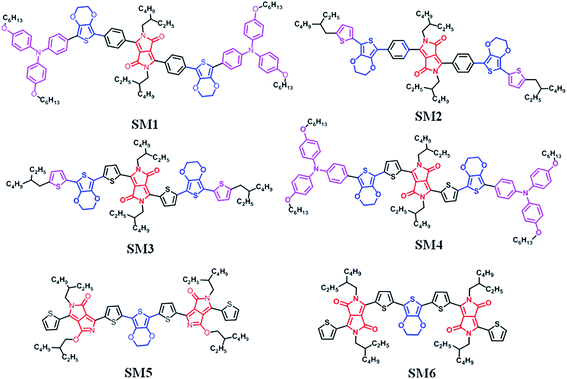 |
| | Fig. 1 Chemical structure of the DPP-based molecules SM1–6. | |
2. Experimental section
2.1 Materials and physicochemical studies
All chemicals, unless otherwise specified, were purchased from commercial resources and used as received. The 1H NMR and 13C NMR spectra were recorded with a Bruker 300 MHz NMR spectrometer using CDCl3 as a solvent. The FAB-MS spectra were obtained using JMS-700 HR-MS. The UV/Vis absorption spectra were recorded using a Cary 300 Bio spectrometer. HPLC grade CH2Cl2 was purchased from ACROS Co. and distilled from CaH2 under an argon (Ar) atmosphere for electrochemical study. Tetra-n-butylammonium hexafluorophosphate (TBAPF6) was recrystallized from ethanol twice and then stored in a desiccator before use. The glassy carbon electrode was polished with 0.05 mm alumina on Buehler felt pads and ultrasonicated for 2 min to remove the residue contamination. A platinum wire was used as the auxiliary electrode and the reference electrode was a home-made Ag/AgCl (in KCl (sat’d)) electrode. The supporting electrolyte was 0.1 M tetrabutylhexafluorophosphate (TBAPF6) CH2Cl2 solution. The square-wave voltammograms (frequency: 25 Hz; step potential: 0.0051 V; amplitude: 0.01995 V) were recorded using a potentiostat/galvanostat (PGSTAT 30, Autolab, Eco-Chemie, the Netherlands) and ferrocene was used as a calibration standard (0.54 V versus Ag/AgCl reference electrode).
2.2 Synthesis of SM1–SM6
SM1–4 were synthesized by the newly developed direct C–H arylation method. SM5 and SM6 were prepared with the well-known Stille coupling reaction. The detailed synthetic procedures and structure analyses of the molecules are collected in the ESI.†
2.3 Device fabrication and photovoltaic properties characterization
Inverted BHJ SMSCs with ITO/ZnO/SMx:PCBM/MoO3/Ag architectures were fabricated in this study. The ITO-coated glass substrates were cleaned by ultrasonicating in deionized water, followed by acetone and isopropanol for 10 min each, and then dried under a stream of dry nitrogen. The cleaned ITO substrates were treated with UV–ozone for 10 min. The ZnO film was prepared from its precursor solution of zinc acetate dihydrate [Zn(CH3COO)2·2H2O] in 2-ethoxyethanol and monoethanolamine (MEA) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in volume) using the spin-coating method in air. After spin-coating the SMx:PCBM active layer in a glove box, a thin layer of molybdenum oxide was deposited on top of the active layer with a high vacuum thermal evaporator at an evaporation rate of 0.1 Å s−1. Ultimately, the 100 nm silver film was deposited on top of the molybdenum oxide layer as an electrode. The processes (composition and fabrication condition screening) for the device fabrication and photovoltaic property characterization are very similar to what we reported previously, unless specified.51
:1 in volume) using the spin-coating method in air. After spin-coating the SMx:PCBM active layer in a glove box, a thin layer of molybdenum oxide was deposited on top of the active layer with a high vacuum thermal evaporator at an evaporation rate of 0.1 Å s−1. Ultimately, the 100 nm silver film was deposited on top of the molybdenum oxide layer as an electrode. The processes (composition and fabrication condition screening) for the device fabrication and photovoltaic property characterization are very similar to what we reported previously, unless specified.51
3. Results and discussion
3.1 Synthesis
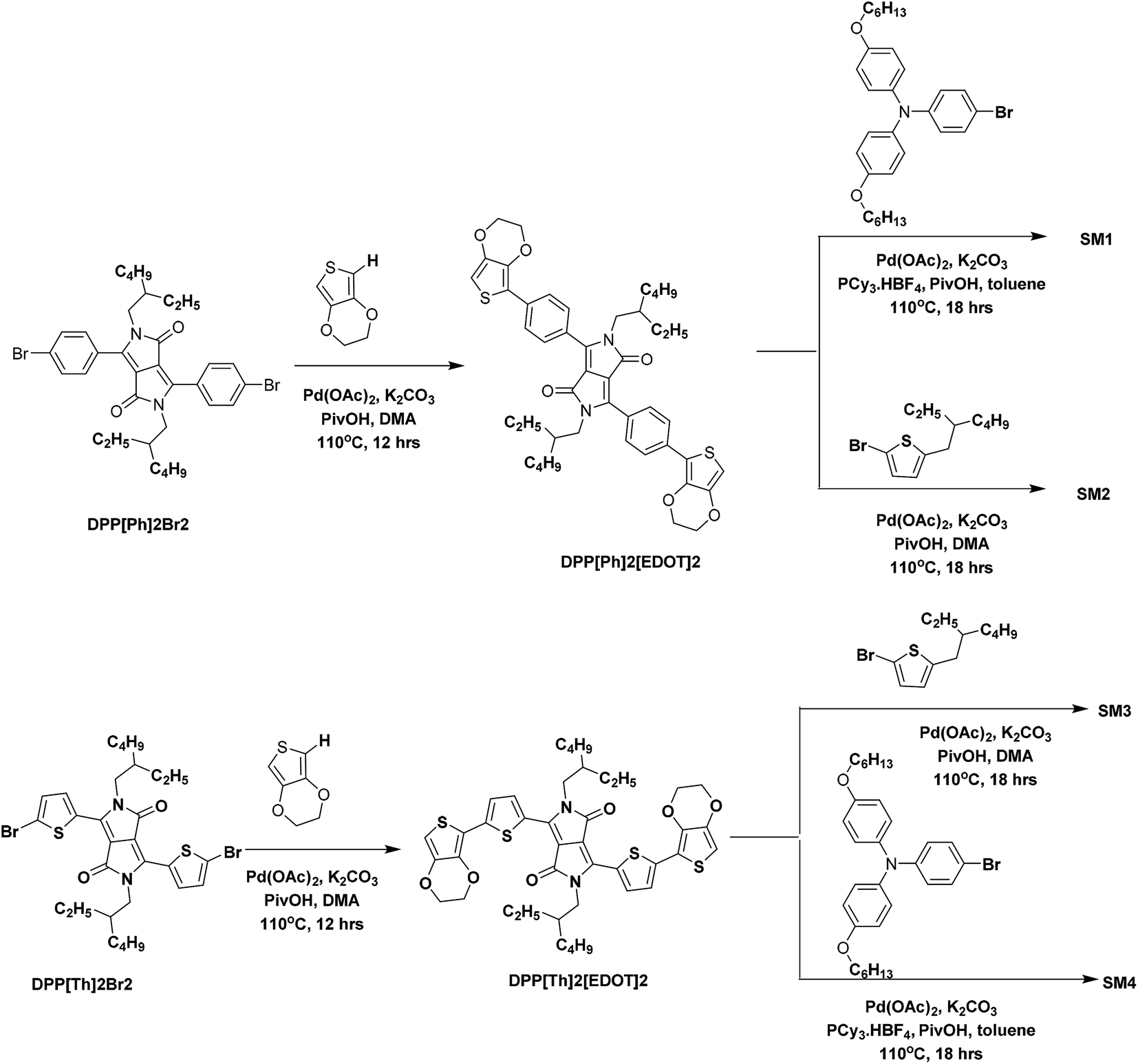
The bromides of phenyl flanked DPP (DPP[Ph]2Br2), thienyl flanked DPP (DPP[Th]2Br2), and thienyl flanked DPPiso (DPPiso[Th]2Br) were synthesized and purified following the reported procedures52–55 with slight modifications. The procedures for preparing SM1–4 are outlined in Scheme 1. The direct C–H arylation reaction was performed using Pd(OAc)2 as a catalyst and K2CO3 as a base at a temperature of 110 °C. DPP[Ph2][EDOT2] and DPP[Th2][EDOT2] were synthesized by the C–H arylation of DPP[Ph]2Br2 and DPP[Th]2Br2 in the presence of EDOT to get yields of 38% and 44%, respectively. The molar ratio of DPP[Ph]2Br2/EDOT was controlled to be 1:2 to avoid the formation of a polymer. A similar catalyst system, combining Pd(OAc)2 and PCy3·HBF4, was used to proceed the reaction between RO–TPA–Br and the DPP derivatives to obtain SM1 and SM4 with yields of 46% and 41%, respectively. The alkyl thiophene end capped SM2 and SM3 were prepared by brominating alkylthiophene with the corresponding DPP unit in dimethyl amine (DMA). The yields of SM2 and SM3 are 66% and 62%, respectively.
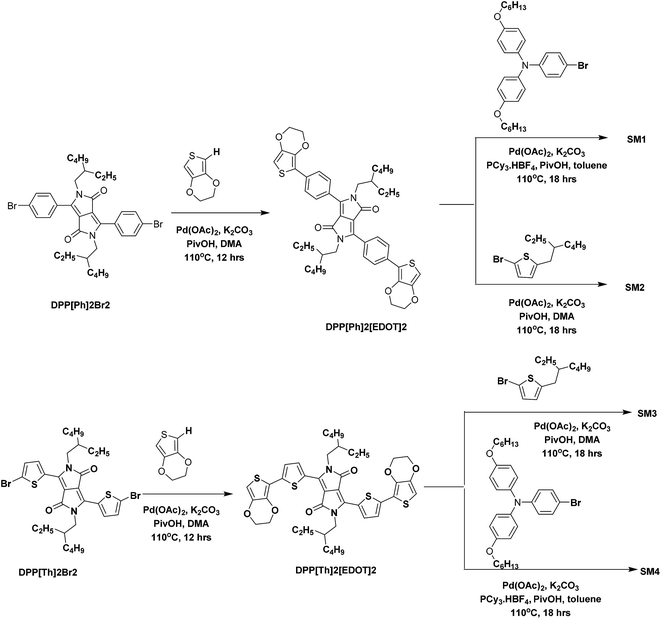 |
| | Scheme 1 Synthesis of SM1–SM4 via the direct C–H arylation reaction. | |
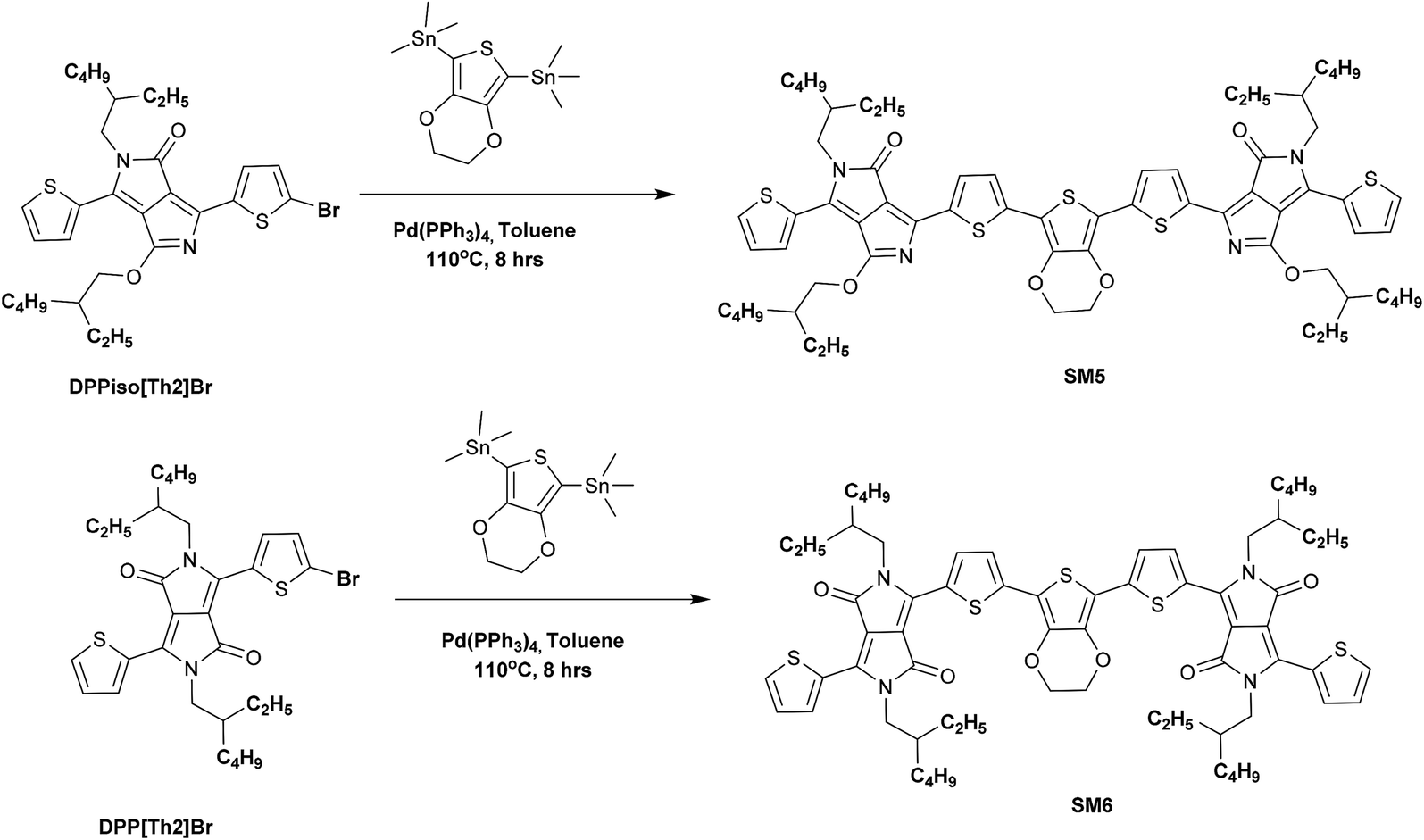
SM5 and SM6 were synthesized by Stille coupling using Pd(PPh3)4 as a catalyst and toluene as a solvent and using the processes displayed in Scheme 2. The Stille coupling between DPPiso[Th]2Br and stannated EDOT afforded SM5 with a yield of 83%. The same method was used for reacting DPP[Th]2Br with stannated EDOT to obtain SM6 (yield: 78%). The SM2, SM3 and SM6 molecules process good solubility in common organic solvents but the other three molecules only have fair solubility, including chloroform, toluene, tetrahydrofuran, o-dichlorobenzene and dichloromethane, which is attributed to the long branched alkyl chains on the DPP unit.
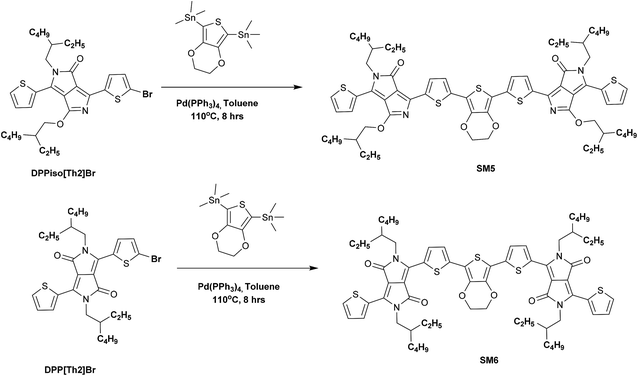 |
| | Scheme 2 Synthesis of SM5 and SM6 via the Stille coupling reaction. | |
3.2 Optical properties
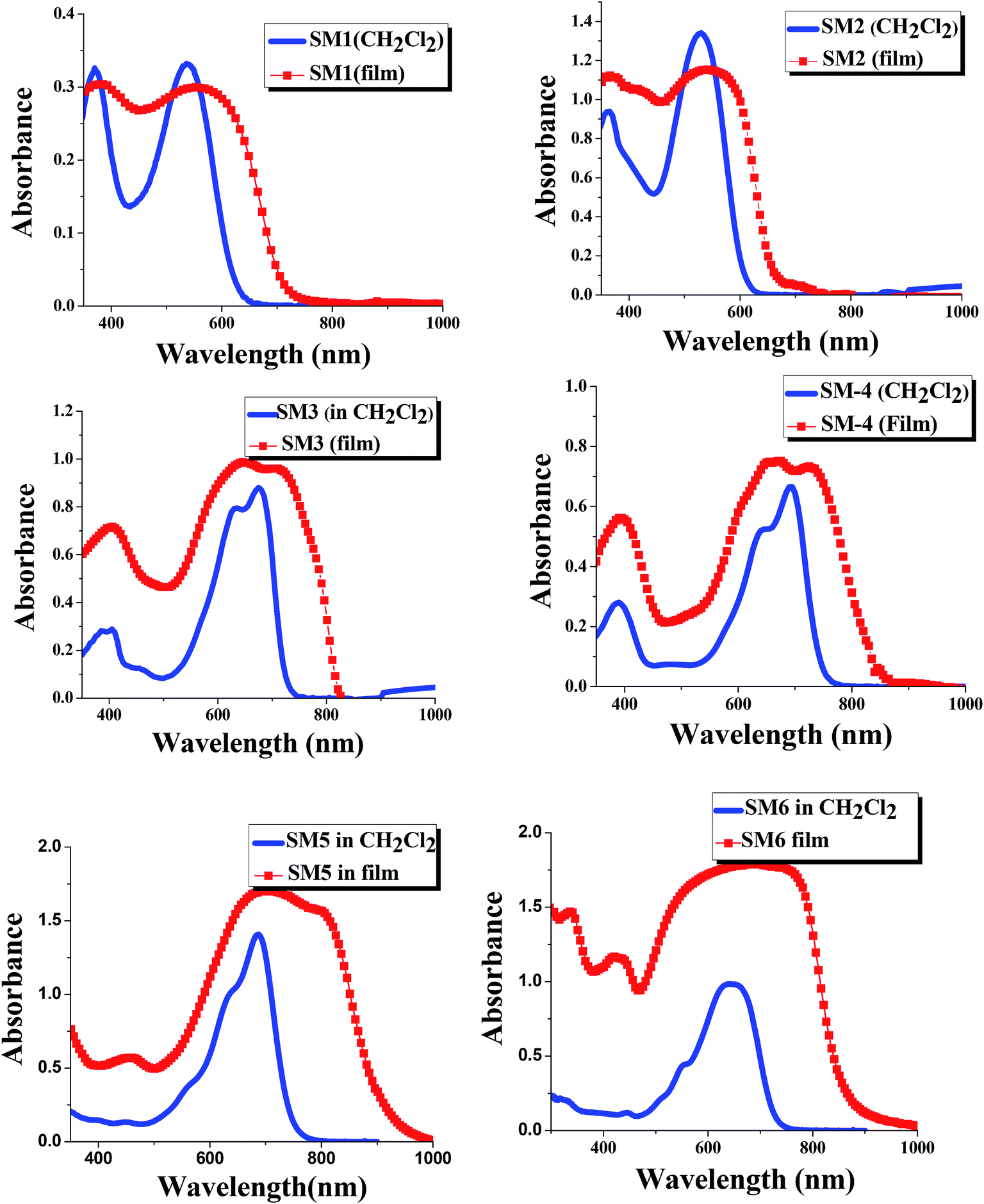
The UV-Vis absorption spectra of SM1–6 in dichloromethane and as a film state are displayed in Fig. 2. The optical band gaps calculated from the onset wavelength of the absorption spectra (in CH2Cl2) are listed in Table 1. The absorption peak at 350 to 500 nm could be attributed to the π–π* electronic transitions of the conjugated backbone and the broad distinct peak in the wavelength range of 500 to 800 nm arose from the intramolecular charge transfer transitions (ICT) between the donor and acceptor moieties in the molecules. DPP flanked by benzene units shows a high molar absorption coefficient with absorption maxima at 541 nm and 529 nm for SM1 and SM2, respectively. TPA end capped SM1 shows a broader absorption than alkyl thiophene end capped SM2, which is because the former contains two bulk triphenyl amine units. SM3 and SM4 exhibit maximum absorptions at longer wavelengths (600–750 nm) compared to SM1 and SM2, due to the fact that in general11–19 diketopyrrolopyrrole (DPP) flanked with a thiophene unit has better conjugation (it forms a more planar conformation) than that flanked with benzene. A red shifted absorption was observed for triphenylamine end capped SM4 (λmax = 692 nm) compared to alkyl thiophene end capped SM3 (λmax = 676 nm), due to triphenylamine being a stronger donor than alkyl thiophene.
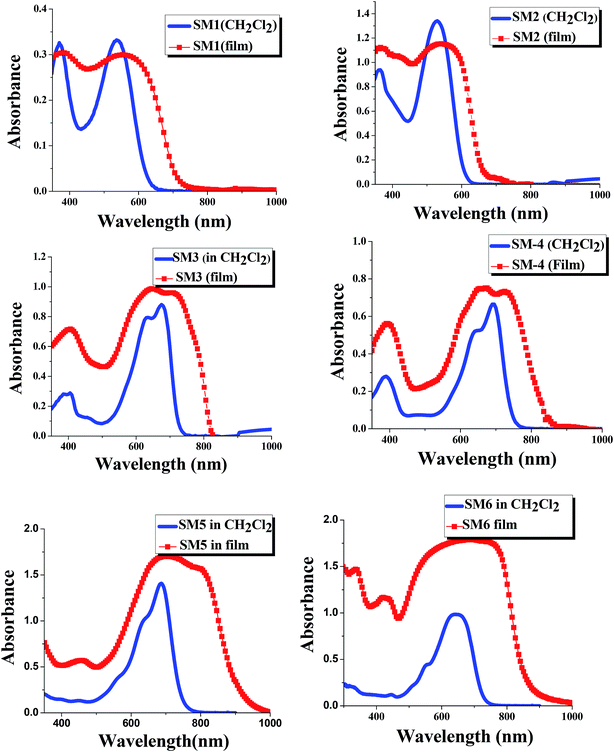 |
| | Fig. 2 UV-Vis absorption spectra of SM1–6 in dichloromethane and in the film state (the concentration of all molecules is 6 × 10−5 M). | |
Table 1 Optical and electrochemical properties of SM1–SM6 molecules
| Small molecules |
UV-vis absorption |
1st E1/2a (V vs. Ag/AgCl) |
HOMOb (eV) |
λonset (nm) |
E0–0c (eV) |
LUMOd (eV) |
| λmax in CH2Cl2 (nm) |
λmax in film (nm) |
| FeCp2+/0 = +0.50 V (vs. Ag/AgCl) in TBAPF6/CH2Cl2. The HOMO energy level was calculated from the half wave potential of oxidation according to the empirical equation EHOMO = −(E1/2 + 4.5) V by assuming the energy level of ferrocene to be −4.5 eV below the vacuum level (E1/2 (V) vs. NHE). The energy gap (E0–0) was calculated from the onset wavelength of the UV-Vis spectrum according to the equation Egap = 1240/λonset. LUMO = HOMO + E0–0. |
| SM1 |
369, 541 |
377, 559 |
0.59 |
−5.29 |
681 |
1.82 |
−3.47 |
| SM2 |
361, 529 |
362, 554 |
0.86 |
−5.56 |
641 |
1.93 |
−3.63 |
| SM3 |
403, 676 |
405, 651 |
0.51 |
−5.21 |
757 |
1.63 |
−3.58 |
| SM4 |
406, 692 |
392, 667 |
0.37 |
−5.07 |
771 |
1.57 |
−3.5 |
| SM5 |
685 |
709 |
0.64 |
−5.32 |
795 |
1.55 |
−3.77 |
| SM6 |
644 |
689 |
0.80 |
−5.49 |
785 |
1.60 |
−3.89 |
SM5 and SM6 (with an A–D–A architecture) are isomers with the alkyl chains connected to different atoms (O or N) in the DPP core. SM5 with one alkyl chain connected to O of the DPP core shows an absorption maximum at 685 nm, which is red shifted compared to that (644 nm) of the other isomer, SM6, which has both alkyl chains connected to two N in DPP. The results indicate that the substituents on the diketopyrrolopyrrole backbone also affect the optical properties of the DPP containing molecules. Surprisingly, when one of the alkyl chains is attached to O of the ketone in DPP, the conjugation length of the whole molecule increases. It seems that the ketone group on DPP will disrupt the conjugation of the backbone.
The SM1–SM6 films on quartz substrates were prepared by the drop casting method and their normalized absorption spectra are also displayed in Fig. 2. A strong red shift is observed in the absorption of the film samples compared to the corresponding solution spectra for all molecules. In addition, the absorption peaks of the films are also broader than those of the solutions, providing evidence that the DPP containing molecules have strong π–π stacking in the solid state. Moreover, the absorption profiles of the SM5 and SM6 films extend to close to 1000 nm, therefore they can be regarded as low band-gap small molecules. As expected, phenyl flanked DPP (SM1 and SM2) shows larger Eg values (1.82 eV and 1.93 eV, respectively) than those of thienyl flanked DPP SM3 and SM4 (1.63 eV and 1.57 eV, respectively).56 SM5 and SM6 have smaller Eg values (1.57 eV and 1.55 eV, respectively, close to other A–D–A type DPP containing molecules reported before56) than SM1–4, due to these molecules containing two DPP units.
3.3 Electrochemical properties
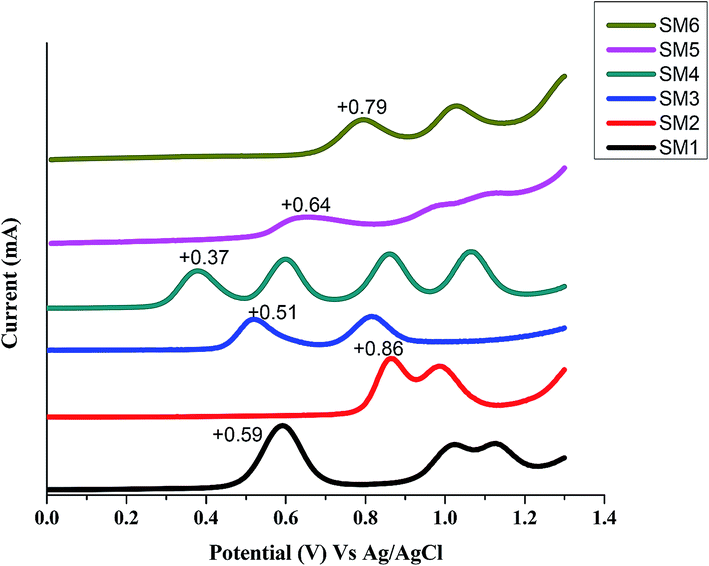
The square-wave voltammograms of SMx molecules measured in TBAPF6/CH2Cl2 electrolyte solution are displayed in Fig. 3. SM4 containing the strongly electron donating triphenylamine (TPA) and easily oxidized thiophene conjugated connector has the lowest first oxidation potential of +0.39 V. The first oxidation peaks for SM1 (containing electron donating triphenylamine) and SM3 (thiophene connector) are +0.59 V and +0.51 V, respectively, which are lower than those of the other 3 molecules. Interestingly, all SM molecules have more than 1 oxidation peak at a potential between +0.37–1.2 V (vs. Ag/AgCl): SM4 even has four oxidation peaks at this potential range. Therefore besides the oxidation of the π electron on the molecular backbone, oxidation also occurs on the DPP and triphenylamine units. SM5 and SM6 are isomers, SM5 has three oxidation peaks but SM6 has only two peaks. The results suggest that on the DPP unit, when the alkyl chains connect to the O atoms of the ketone groups, the non-bonded electrons on the N of the pyrrole ring will also be oxidized.
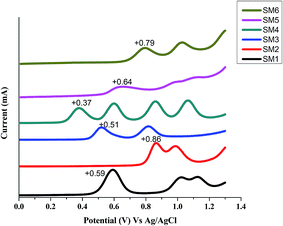 |
| | Fig. 3 Square wave voltammograms of SM1–SM6 in CH2Cl2 (electrolyte: 0.1 M TBAPF6 in CH2Cl2; working electrode: platinum. Scan rate: 0.1 V s−1). | |
The energies of the highest-occupied molecular orbital (HOMO), calculated from the first oxidation potential of the molecules, are illustrated in Fig. 4. The LUMO (lowest-unoccupied molecular orbital) levels were calculated from the HOMO + Eg (Eg is the on-set energy of the UV/Vis absorption spectrum (in CH2Cl2)) and both the HOMO and LUMO are also summarized in Fig. 4 and Table 1. The benzene flanked DPP molecules (SM1 and SM2) have HOMO levels at −5.29 eV and −5.56 eV, respectively, whereas the HOMO energy levels of thiophene flanked DPP (SM3 and SM4) are −5.21 eV and 5.07 eV, respectively. Comparing the HOMO levels of the SM1/SM2 and SM4/SM3 pairs, it exhibits obviously that the molecules containing TPA as an ending group have higher HOMO levels, consistent with TPA being a stronger donor (than alkyl thiophene) in leveling the HOMO energy of the molecule. The HOMO energy levels for A–D–A type molecules (SM5 and SM6) are −5.32 eV and −5.49 eV, respectively, due to both compounds containing two acceptors. Furthermore, the alkyl chain substituted on the ketone group of DPP will reduce the band gap energy as revealed by the absorption spectra (see Fig. 2) but it also raises both the HOMO and LUMO energy levels, which may not be a good property since the value of Voc of the resulting OPV cell will decrease. Overall the LUMO energy levels of all molecules (SM1–6) are higher than the LUMO energy levels of PC61BM, which was used generally as an acceptor in organic photovoltaics, indicating that the photo-generated excitons can efficiently dissociate at the p-type small molecules/PCBM acceptor interface and then transport the electron to the cathode.57,58 For applications in organic photovoltaics (OPVs), the HOMO level of p-type molecules is also critical, the molecule with lower HOMO may result in lowering the value of Jsc but increasing the value of Voc of the corresponding cell. It is a dilemma for designing p-type molecules for OPV applications. However, if the HOMO level is lower than that of the hole transporting material, (such as MoO3 in this study), the hole cannot move to the anode through the hole transporting layer. The data illustrated in Fig. 4 reveal that only SM4 has the HOMO level higher than the HOMO of the hole transporting materials. In other words, the dissociated hole cannot transport to the anode efficiently for the cell based on SM4.
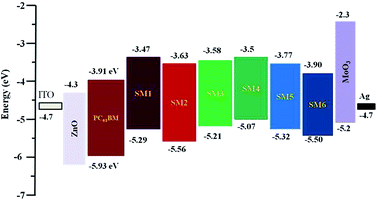 |
| | Fig. 4 Frontier orbital energy levels of SM1–6, PC61BM as well as the electron and hole transporting materials used in this study. | |
3.4 Density functional theory (DFT) calculations
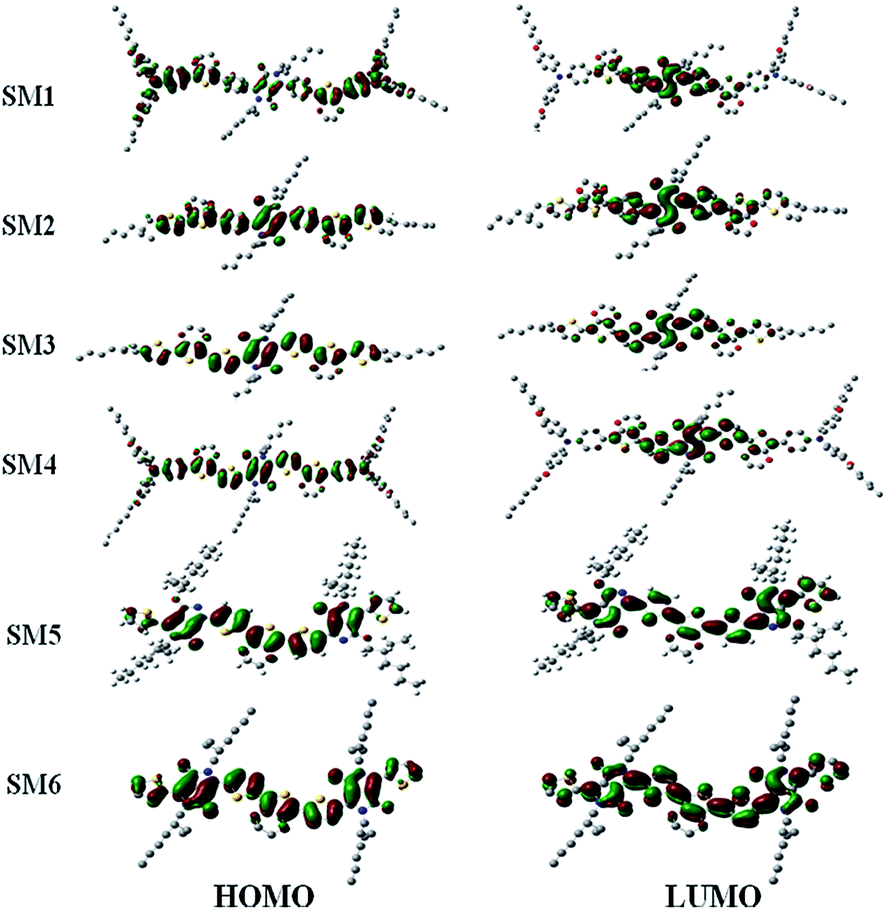
To assess the optimized molecular geometry and frontier orbital (HOMO and LUMO) distribution of these SMx molecules, density functional theory (DFT) calculations were carried out using Gaussian09 at the B3LYP/6-31G* basic set, and the results are displayed in Fig. 5. The electron density of the HOMO distributes all over the backbone of the molecule (which includes triphenylamine, EDOT and thiophene), whereas the LUMO is contributed to mainly from the electron withdrawing DPP units. A DPP moiety with strong π–π stacking and planar structure would synergistically reinforce the intermolecular interaction to enhance the transport properties. The HOMO and LUMO distributions also conclude that an effective intermolecular charge transfer occurs in the SMx films, which is consistent with the results observed in the absorption spectra in CH2Cl2 solution and film state (Fig. 2). A similar frontier orbital distribution was observed in other DPP containing molecules that were reported in the literature.12,59–62 The frontier orbital distributions for these six molecules are very similar. Therefore, the photovoltaic performance of these small molecules will not be attributed to the distribution of their frontier orbitals even SM1–4 have D1–D2–A–D2–D1 structures but the structures of SM5 and SM6 are A–D–A type.
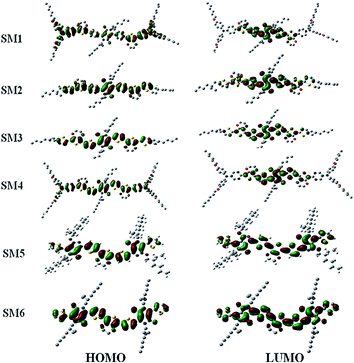 |
| | Fig. 5 Frontier orbital distribution of the SM1–6 molecules. | |
3.5 Photovoltaic performance
The inverted solar cells based on these SMx small molecule donors (as well as the P3HT reference) were fabricated with a device architecture of ITO/ZnO/SMx:PC61BM/MoO3/Al, and the current density–voltage (J–V) curves of the best performance devices for each molecule are displayed in Fig. S1, (ESI†). The corresponding photovoltaic data of the optimized cells are summarized in Table 2. All cells show rather poor power conversion efficiency (PCE) even after optimizing the compositions (the donor/acceptor ratios, solvents and concentrations) and processes (such as spin coating rate, adding additive, solvent and thermal annealing). SM1 and SM4, having the triphenyl amine group on the edge, exhibit extremely low values of Jsc of 0.2–0.3 mA cm−2. That is why the high efficiency small molecules recently reviewed by Nguyen6 et al. contain no triphenyl amine groups. This is probably due to their molecule planarity, therefore a serious aggregation occurs at the active layer as reported in other TPA–DPP containing molecules.20,59,63 We found that the SM1 (or SM4)/PCBM active film is rather rough as observed by the naked eye. The poor film morphology as well as high HOMO could be the reasons for the low Jsc. As expected, SM2 and SM6 with low HOMO levels (compatible to MoO3, see Fig. 4) have better photovoltaic performances and SM2 having the lowest HOMO resulted in the highest value of Voc of the cell. Nevertheless, the values of Jsc and FF of SM2 and SM6 based cells are much lower than those based on P3HT, probably due to the LUMO of these two molecules not being high enough for efficient exciton dissociation.12,61,64 The inverted cell based on SM3 achieves the highest value of Jsc but the value of Voc is lower than that for those based on SM2 and SM6, due to SM3 having broad absorption (see Fig. 2) and the highest LUMO, but the HOMO is also very high. SM6 although has very broad absorption but its LUMO level is not higher enough (only 0.02 eV higher than the n-type PCBM), therefore the Jsc of the corresponding cell is not as high as that for the cell based on SM3. SM6 and SM5 are isomers however the cell based on SM6 has much higher PCE than that based on SM5. SM5 although has proper frontier orbitals energy level (compatible with the electron and hole transporting layers) and broad absorption profile but exhibits very poor photovoltaic performance. It is because of its low solubility (in chlorobenzene or dichlorobenzene), therefore the SM5/PCBM active film is very rough, resulting in low PCE. Overall the highest PCE of the inverted cells based on these SMx molecules is only about 1%.
Table 2 Device characteristics of the solar cells based on SMx moleculesa
| Donor (D) |
SMx/PC61BM ratio (w/w) |
Annealing temperature (°C) |
Jsc (mA cm−2) |
Voc (V) |
FF (%) |
PCE (%) |
| Solvent: chlorobenzene; concentration of SMx: 2 wt%; spin rate: 500–1000 rpm, 60 s; additive (DIO) is not good for this donor system. |
| SM1 |
1:1 |
As-spun |
0.21 |
0.73 |
29.24 |
0.04 |
| SM2 |
1:3 |
As-spun |
2.83 |
0.78 |
29.68 |
0.65 |
| 80 |
3.13 |
0.73 |
33.28 |
0.76 |
| 100 |
2.88 |
0.82 |
29.72 |
0.70 |
| 120 |
3.02 |
0.81 |
29.97 |
0.73 |
| SM3 |
3:1 |
As-spun |
2.3 |
0.49 |
37.46 |
0.42 |
| 80 |
2.57 |
0.54 |
38.28 |
0.53 |
| 100 |
2.87 |
0.53 |
35.64 |
0.54 |
| 120 |
5.56 |
0.53 |
34.35 |
1.01 |
| SM4 |
1:1 |
As-spun |
0.32 |
0.44 |
27.81 |
0.04 |
| SM5 |
1:1 |
140 |
1.04 |
0.66 |
26.27 |
0.18 |
| SM6 |
1:1 |
As-spun |
2.72 |
0.54 |
43.5 |
0.67 |
| 140 |
3.78 |
0.63 |
43.75 |
1.04 |
| P3HT |
1:1 |
140 |
7.83 |
0.62 |
69.92 |
3.39 |
4. Conclusion
In conclusion, we have synthesized four D1–D2–A–D2–D1 type DPP containing small molecules, SM1–4, using an effective direct C–H activation reaction, which has the advantage of easy purification and few synthetic steps. DPP-containing SM5 and SM6, which have an A–D–A structure, were synthesized by the Stille coupling reaction. All molecules were designed for application in bulk heterojunction small molecule solar cells (SMSCs). The physicochemical properties of these small molecule donors revealed that a change of the terminal group or DPP core impacts their solubility, and their optical and electrochemical properties. All molecules showed broad absorption and (besides SM4) the HOMO and LUMO energy levels were suitable to be used as p-type molecules for PCBM based organic photovoltaic devices. However, due to the relatively low lying LUMO and serious self-aggregation, the highest PCE of the best performance molecules was only ca. 1%. Nevertheless, the results clearly demonstrate that the DPP unit represents an interesting building block for the design of push–pull molecules with various optical and electronic properties.
References
- L. Dou, J. You, Z. Hong, Z. Xu, G. Li, R. A. Street and Y. Yang, Adv. Mater., 2013, 25, 6642–6671 CrossRef CAS PubMed.
- R. F. Service, Science, 2011, 332, 293 CrossRef CAS PubMed.
- J. Roncali, Acc. Chem. Res., 2009, 42, 1719–1730 CrossRef CAS PubMed.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245–4272 RSC.
- Y. Chen, X. Wan and G. Long, Acc. Chem. Res., 2013, 46, 2645–2655 CrossRef CAS PubMed.
- S. D. Collins, N. A. Ran, M. C. Heiber and T.-Q. Nguyen, Adv. Energy Mater., 2017, 7, 1602242–1602287 CrossRef.
- Y. Qiao, Y. Guo, C. Yu, F. Zhang, W. Xu, Y. Liu and D. Zhu, J. Am. Chem. Soc., 2012, 134, 4084–4087 CrossRef CAS PubMed.
- H. Zhong, J. Smith, S. Rossbauer, A. J. P. White, T. D. Anthopoulos and M. Heeney, Adv. Mater., 2012, 24, 3205–3211 CrossRef CAS PubMed.
- A. K. Palai, J. Lee, S. Das, J. Lee, H. Cho, S. U. Park and S. A. Pyo, Org. Electron., 2012, 13, 2553–2560 CrossRef CAS.
- S. L. Suraru, U. Zschieschang, H. Klauk and F. Wurthner, Chem. Commun., 2011, 47, 1767–1769 RSC.
- B. Walker, A. B. Tamayo, X. D. Dang, P. Zalar, J. H. Seo, A. Garcia, M. Tantiwiwat and T. Q. Nguyen, Adv. Funct. Mater., 2009, 19, 3063–3069 CrossRef CAS.
- A. B. Tamayo, B. Walker and T. Q. Nguyen, J. Phys. Chem. C, 2008, 112, 11545–11551 CAS.
- S. Loser, C. J. Bruns, H. Miyauchi, R. P. Ortiz, A. Facchetti, S. I. Stupp and T. J. Marks, J. Am. Chem. Soc., 2011, 133, 8142–8145 CrossRef CAS PubMed.
- J. Huang, C. Zhan, X. Zhang, Y. Zhao, Z. Lu, H. Jia, B. Jiang, J. Ye, S. Zhang, A. Tang, Y. Liu, Q. Pei and J. Yao, ACS Appl. Mater. Interfaces, 2013, 5, 2033–2039 CAS.
- M. Chen, W. Fu, M. Shi, X. Hu, J. Pan, J. Ling, H. Li and H. Z. Chen, J. Mater. Chem. A, 2013, 1, 105–111 CAS.
- J. Liu, B. Walker, A. Tamayo, Y. Zhang and T. Q. Nguyen, Adv. Funct. Mater., 2013, 23, 47–56 CrossRef CAS.
- B. Walker, J. Liu, C. Kim, G. C. Welch, J. K. Park, J. Lin, P. Zalar, C. M. Proctor, J. H. Seo, G. C. Bazan and T. Q. Nguyen, Energy Environ. Sci., 2013, 6, 952–962 CAS.
- Y. Lin, P. Cheng, Y. Li and X. A. Zhan, Chem. Commun., 2012, 48, 4773–4775 RSC.
- J. Liu, Y. Sun, P. Moonsin, M. Kuik, C. M. Proctor, J. Lin, B. B. Hsu, V. Promarak, A. J. Heeger and T. Q. Nguyen, Adv. Mater., 2013, 25, 5898–5903 CrossRef CAS PubMed.
- O. P. Lee, A. T. Yiu, P. M. Beaujuge, C. H. Woo, T. W. Holcombe, J. E. Millstone, J. D. Douglas, M. S. Chen and J. M. Frechet, Adv. Mater., 2011, 23, 5359–5363 CrossRef CAS PubMed.
- J. Huang, H. Jia, L. Li, Z. Lu, W. Zhang, W. He, B. Jiang, A. Tang, Z. Tan, C. Zhan, Y. Li and J. Yao, Phys. Chem. Chem. Phys., 2012, 14, 14238–14242 RSC.
- E. Ripaud, D. Demeter, T. Rousseau, E. Boucard-Cétol, M. Allain, R. Po, P. Leriche and J. Roncali, Dyes Pigm., 2012, 95, 126–133 CrossRef CAS.
- L. Biniek, B. C. Schroeder, C. B. Nielsen and I. J. McCulloch, Mater. Chem., 2012, 22, 14803–14813 RSC.
- S. Y. Liu, M. Shi, J. Huang, Z. Jin, X. Hu, J. Pan, H. Li, A. K. Y. Jen and H. Z. Chen, J. Mater. Chem. A, 2013, 1, 2795–2805 CAS.
- J. Zhang, D. Y. Kang, S. Barlow and S. R. Marder, J. Mater. Chem., 2012, 22, 21392 RSC.
- Q. Guo, J. Dong, D. Wan, D. Wu and J. You, Macromol. Rapid Commun., 2013, 34, 522–527 CrossRef CAS PubMed.
- S.-Y. Liu, W.-Q. Liu, J.-Q. Xu, C.-C. Fan, W.-F. Fu, J. Ling, J.-Y. Wu, M.-M. Shi, A. K. Y. Jen and H.-Z. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 6765–6775 CAS.
- S. Y. Liu, W. F. Fu, J. Q. Xu, C. C. Fan, H. Jiang, M. Shi, H. Y. Li, J. W. Chen, Y. Cao and H. Z. Chen, Nanotechnology, 2014, 25, 014006 CrossRef PubMed.
- A. Facchetti, L. Vaccaro and A. Marrocchi, Angew. Chem., Int. Ed., 2012, 51, 3520–3523 CrossRef CAS PubMed.
- L. G. Mercier and M. Leclerc, Acc. Chem. Res., 2013, 46, 1597–1605 CrossRef CAS PubMed.
- Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 3308–3311 CrossRef CAS PubMed.
- M. Wakioka, Y. Nakamura, Y. Hihara, F. Ozawa and S. Sakaki, Organometallics, 2013, 32, 4423–4430 CrossRef CAS.
- D. J. Schipper and K. Fagnou, Chem. Mater., 2011, 23, 1594–1600 CrossRef CAS.
- D. J. Burke and D. J. Lipomi, Energy Environ. Sci., 2013, 6, 2053–2066 CAS.
- S. Y. Liu, H. Y. Li, M. M. Shi, H. Jiang, X. L. Hu, W. Q. Li, L. Fu and H. Z. Chen, Macromolecules, 2012, 45, 9004–9009 CrossRef CAS.
- S. M. Tuladhar, M. Azzouzi, F. Delval, J. Yao, A. A. Y. Guilbert, T. Kirchartz, N. F. Montcada, R. Dominguez, F. Langa, E. Palomares and J. Nelson, ACS Energy Lett., 2016, 1, 302–308 CrossRef CAS.
- J. Roncali, P. Blanchard and P. Frère, J.Mater.
Chem., 2005, 15, 1589–1610 RSC.
- D. Demeter, T. Rousseau and J. Roncali, RSC Adv., 2013, 3, 704–707 RSC.
- N. F. Montcada, R. Domínguez, B. Pelado, P. de la Cruz, E. Palomares and F. Lang, J. Mater. Chem. A, 2015, 3, 11340–11348 CAS.
- C. Wang, C. J. Mueller, E. Gann, A. C. Y. Liu, M. Thelakkat and C. R. McNeill, J. Mater. Chem. A, 2016, 4, 3477–3486 CAS.
- K. Zhang, B. Tieke, J. C. Forgie and P. J. Skabara, Macromol. Rapid Commun., 2009, 30, 1834–1840 CrossRef CAS PubMed.
- Y. Zhu, I. Heim and B. Tieke, Macromol. Chem. Phys., 2006, 207, 2206–2214 CrossRef CAS.
- I. Perepichka, E. Levillain and J. Roncali, J. Mater. Chem., 2004, 14, 1679–1681 RSC.
- M. Turbiez, P. Frure, M. Allain, C. Videlot, J. Ackermann and J. Roncali, Chem.–Eur. J., 2005, 11, 3742–3752 CrossRef CAS PubMed.
- C. J. Mueller, C. R. Singh and M. Thelakkat, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 639–648 CrossRef CAS.
- F. Grenier, B. Réda Aïch, Y.-Y. Lai, M. Guérette, A. B. Holmes, Y. Tao, W. W. H. Wong and M. Leclerc, Chem. Mater., 2015, 27, 2137–2143 CrossRef CAS.
- M. Shaker, J.-H. Lee, C. K. Trinh, W. Kim, K. Lee and J.-S. Lee, RSC Adv., 2015, 5, 66005–66012 RSC.
- M. Shaker, C. K. Trinh, W. Kim, H. Kim, K. Lee and J.-S. Lee, New J. Chem., 2015, 39, 4957–4964 RSC.
- C. Y. Liu, H. Zhao and H. Yu, Org. Lett., 2011, 13, 4068–4071 CrossRef CAS PubMed.
- C.-Y. Chen, S.-J. Wu, J.-Y. Li, C.-G. Wu, J.-G. Chen and K.-C. Ho, Adv. Mater., 2007, 19, 3888–3891 CrossRef CAS.
- Y.-Q. Wu, H.-C. Chen, Y.-S. Yang, S. H. Chang and C.-G. Wu, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1822–1833 CrossRef CAS.
- W. Zeng, Y. Cao, Y. Bai, Y. Wang, Y. Shi and M. Zhang, Chem. Mater., 2010, 22, 1915–1925 CrossRef CAS.
- F. Zhang, K. Jiang, J. Huang, C. Yu, S. Li and S. M. Chen, J. Mater. Chem. A, 2013, 1, 4858–4863 CAS.
- G. L. Zhang, H. Bala, Y. M. Cheng, D. Shi, X. J. Lv and Q. J. Yu, Chem. Commun., 2009, 45, 2198–2200 RSC.
- B. Zhao, K. Sun, F. Xue and J. Ouyang, Org. Electron., 2012, 13, 2516–2524 CrossRef CAS.
- J. W. Lee, Y. S. Choi and W. H. Jo, Org. Electron., 2012, 13, 3060–3066 CrossRef CAS.
- Y. He and Y. F. Li, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC.
- C. J. Brabec, C. Winder, N. S. Sariciftic, J. C. Hummelen, A. Dhanabalan, P. A. van Hal and R. A. J. Janssen, Adv. Funct. Mater., 2002, 12, 709–712 CrossRef CAS.
- C. Ji, L. Yin, L. Wang, T. Jia, S. Meng, Y. Sun and Y. Li, J. Mater. Chem. C, 2014, 2, 4019–4026 RSC.
- B. Walker, X. Han, C. Kim, A. Sellinger and T.-Q. Nguyen, ACS Appl. Mater. Interfaces, 2012, 4, 244–250 CAS.
- B. Sun, W. Hong, Z. Yan, H. Aziz and Y. Li, Adv. Mater., 2014, 26, 2636–2642 CrossRef CAS PubMed.
- C. Li, Y. Chen, Y. Zhao, H. Wang, W. Zhang, Y. Li, X. Yang, C. Ma, L. Chen, X. Zhua and Y. Tu, Nanoscale, 2013, 5, 9536–9540 RSC.
- J. Chen, M. Xiao, F. Meng, L. Duan, H. Tan, Y. Wang, Y. Liu, R. Yang and W. Zhu, Synth. Met., 2015, 199, 400–407 CrossRef CAS.
- J. H. Seo, Synth. Met., 2012, 162, 748–752 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04196a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*