
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organoboron copolymers containing thienothiophene and selenophenothiophene analogues: optical, electrochemical and fluoride sensing properties†
Gulsen Turkoglua,
M. Emin Cinar ab and
Turan Ozturk*ac
ab and
Turan Ozturk*ac
aDepartment of Chemistry, Istanbul Technical University, Maslak, Istanbul 34469, Turkey. E-mail: ozturktur@itu.edu.tr
bDepartment Chemie-Biologie, OC1, Universität Siegen, Adolf-Reichwein-Str., 57068 Siegen, Germany
cChemistry Group Laboratories, TUBITAK UME, PO Box 54, 41470, Gebze-Kocaeli, Turkey
First published on 27th April 2017
Abstract
Conjugated donor–acceptor (D–A) copolymers possessing alternating fused bicyclic aromatic rings thieno[3,2-b]thiophene, selenopheno[3,2-b]thiophene, thieno[2,3-b]thiophene and selenopheno[2,3-b]thiophene as donors, thiophene as a π-conjugated bridge and mesitylboron as an acceptor were synthesized and characterized by spectroscopic methods. Large Stokes shifts of 96–166 nm were recorded and the solution quantum yields of the polymers were in the range of 5–18%. Their ionization potentials and electron affinities were investigated by cyclic voltammetry. Optical band gaps varied between 2.26 and 2.78 eV. (TD)-DFT studies were performed to unveil their electronic structures, Kohn–Sham orbitals and electronic transitions. Absorption and emission measurements revealed that these polymers have high sensitivity to fluoride anions, among which the polymer possessing cross-conjugated thienothiophene units had the best sensing properties supported by orbital composition analysis with Mulliken partition. Thus, copolymers P1–P4 could be used as colorimetric and fluorescent sensors for small fluoride anions.
Introduction
Like thiophenes, selenophenes are categorized in the class of chalcogenophenes.1 Fused selenophenes have been the focus of scientists owing to their promising use in optoelectronic materials.2 In spite of the extensive laboratory work devoted to the synthesis and investigation of organic thin film transistors (OTFTs), to the best of our knowledge, the properties of oligomeric fused selenium heterocycles and fused selenophene containing polymers have not been thoroughly explored, possibly due to lack of synthetic methodologies and their low solubilities.3 Replacement of sulphur with selenium enhances the optoelectronic properties, such as an increase of conductivity as observed with tetraselenafulvalene (TSF) with respect to tetrathiafulvalene (TTF) derivatives at room temperature.4 Moreover, polyselenophenes have higher conductivity and mobility which might be based on better p-orbital overlap in Se compounds.1,5 Furthermore, polyselenophenes have smaller band gaps according to both computational6 and experimental studies.7 In addition, organic materials having “Se” atom in their structures are supposed to have higher frontier molecular orbitals (FMO), i.e. lower oxidation and reduction potentials, and easier polarizability. Existence of intermolecular Se⋯Se interactions in their structures leads to plausible inter-chain charge transfers,8 accepted as gorgeous properties for optoelectronics.It is well known that fused thiophene containing polymers, as an active layer, provide high charge mobilities in field effect transistors (FET).9 Besides, fused rings afford a high degree of planarity and rigidity resulting in higher crystallinity and extended pi overlap. Additionally, they do not only hinder unfavourable chain folding but also diminish reorganization energy of the molecules, and make feasible intermolecular charge hopping, resulting in the increase of the mobility.
Heteroaromatic compounds with sulphur have been the one of the best candidates as electron-donating scaffolds for low band gap polymers. Their Se analogues afford high charge mobilities arisen from strong intermolecular interactions through Se atoms.10 Exchange of sulphur with selenium, known as a less electronegative and more polarizable atom with respect to sulphur, affects the electronic and optical properties.11
The low band gap polymers can be successfully achieved through D–π–A approach, which requires copolymerization of electron-rich donor and strong acceptor monomers.12 LUMO of the obtained polymers is predominantly located on the electron accepting unit, whereas HOMO is mainly placed on the donor material. In addition to the fused heteroaromatic systems with sulphur and/or selenium atoms, conjugated organic molecules and polymers containing three-coordinate boron have also drawn considerable attention owing to their convincing optoelectronic and sensor properties.13 As boron is an electron deficient atom, it has a strong electron acceptor character due to its empty p orbital. Its use in polymers having conjugated electron donor units has generated a new type of donor–acceptor conjugated polymeric systems for material science.14 The bulky mesityl group attached to the boron protects it from any nucleophilic attacks. Thus, boron containing compounds with vacant pπ orbital can also be used as Lewis acids to coordinate Lewis bases, such as fluoride ions (F−), leading to a remarkable colour and/or luminescence changes, which have been extensively exploited in chemical sensors.15
Herein, we report the synthesis, optical and electrochemical properties of four novels D–π–A copolymers P1–P4, possessing acceptor mesityl boron units and donor fused conjugated and cross conjugated selenophenothiophene heteroaromatic linkers. A detailed structure–property correlation suggests insights into the influence of linkers on the electronic structures and photophysical properties of organoboron polymers. Finally, all the copolymers have been successfully applied to detect fluoride anion (F−) in order to probe their sensing properties.
Results and discussion
Synthesis of monomers and polymers
The syntheses of the polymers P1–P4 are shown in Scheme 1. Dibrominated monomers 1–4 were synthesized according to the established methods.3b,16–18 The precursors for the polymerizations, 2,5-di(thiophen-2-yl)selenopheno[3,2-b]thiophene (5), 2,5-di(thiophen-2-yl)selenopheno[2,3-b]thiophene (6), 2,5-di(thiophen-2-yl)thieno[3,2-b]thiophene (7) and 2,5-di(thiophen-2-yl)thieno[2,3-b]thiophene (8) were synthesized via a Suzuki coupling in moderate to good yields (40–72%). Syntheses of the polymers were performed through the reactions of the monomers 5–8 with mesityldimethoxyborane (Scheme 1). Lithiation of the monomers with n-BuLi at −78 °C was followed by addition of mestiyldimethoxyborane in THF at room temperature providing the polymers P1–P4. They were purified by precipitation from n-hexane leading to reddish-orange (P1), yellow (P2), reddish-brown (P3) and orange (P4) powdery solids in the yields of 39–50%. The polymers were found to be stable to air and moisture that no special precaution was taken during the purification processes. The number-average molecular weights (Mn) of P1–P4 were determined using light scattering technique and found to be 97.9 kDa, 11.2 kDa, 40.5 kDa, and 40.7 kDa, respectively. As thermal gravimetric analysis (TGA) provides valuable information on thermal stability of materials, TGA analyses of the polymers P1–P4 were conducted, which indicated the relative thermal stability of P1 and P3 with respect to P2 and P4 (Fig. S7†). While main thermal decomposition steps of P1 and P3 was observed around 178 and 170 °C, respectively, decompositions of P2 and P4 were recorded to be at lower temperatures (78 and 154 °C, respectively). Lower molecular weight could be a reason for the lower decomposition temperature of the polymer P2. | ||
| Scheme 1 Preparation of the polymers P1–P4. | ||
Photophysical properties
The UV-Vis absorption spectra of the polymers in THF and spin-coated thin-films were recorded (Fig. 1 and S8 in ESI†) and therefrom obtained corresponding data are summarized in Table 1. The lowest energy absorptions were assigned to be localized π–π* transitions with a strong intramolecular charge transfer (ICT) to boron acceptor. UV-Vis absorption maxima of both P1 and P3 polymers were significantly red-shifted implying that they had effective conjugation lengths with respect to P2 and P4. While the maxima were recorded for P1 and P3 to be 406 and 400 nm, respectively, those were 362 nm for P2 and 348 nm for P4, which have cross-conjugated selenopheno[2,3-b]thiophene and thieno[2,3-b]-thiophene, respectively (Fig. 1). This large hypsochromic shifts of P2 (44 nm) and P4 (52 nm) are consistent with a diminished effective conjugation length compared to P1 and P3. Furthermore, the comparatively low absorptions of P2 and P4 could be connected to the poor electronic couplings of cross-conjugated donors and mesitylboron acceptor in the polymer chain. In addition, the absorption maxima obtained from thin-films of P1–P4 were about 6 to 12 nm red-shifted compared to solution state, supporting well organized intermolecular packings and sulfur–sulfur interactions in solid states in solid states.19 Optical HOMO–LUMO gaps of the polymers were determined from the onsets of UV-Vis absorption spectra in solution state to be 2.26 eV for P1 as the smallest one, and 2.60, 2.52 and 2.78 eV for P2–P4, respectively. The smallest band gap of P1 mainly emerges from the strong D–A interaction between the electron rich selenopheno[3,2-b]thiophene, more efficiently increasing the HOMO level than the corresponding conjugated sulphur containing thieno[3,2-b]thiophene, and the electron poor mesitylboron units, diminishing the LUMO level and prompting strong ICT.13d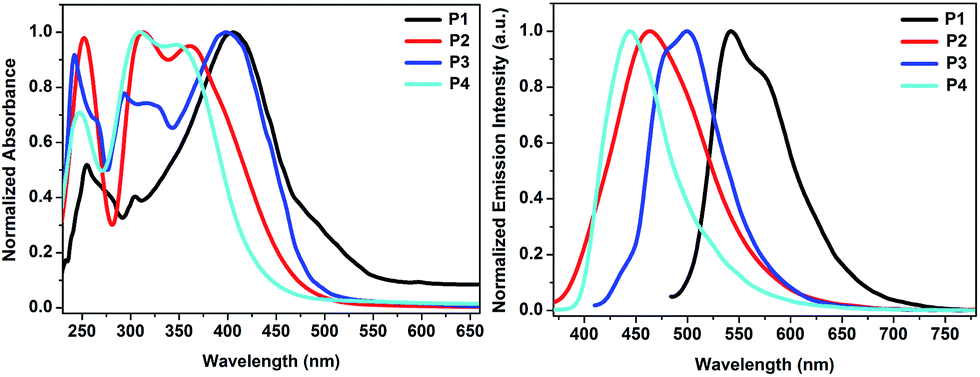 | ||
| Fig. 1 UV-Vis (left) and emission (right) spectra of P1–P4 in THF. | ||
| λabsmax a [nm] | λemmax b [nm] | λabsmax c [nm] | λemmax c [nm] | ΦF [%] | Sg [nm] | Eoptg [eV] | ECVox h [V] | ECVred h [V] | HOMOi [eV] | LUMOi [eV] | ECVg [eV] | HOMOj [eV] | EDFTg k [eV] | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Only longest wavelength absorption in THF.b Emission in THF.c Absorption and emission maxima of the polymer films spin coated on glass.d Using rodhamine 6G as a standard in ethanol.e Anthracene as a standard in cyclohexane.f Coumarin1 as a standard in ethanol.g Stokes shift.h All data reported vs. Fc/Fc+. For polymers P1–P4 recorded on thin films at scan rates of 100 mV s−1 with CH3CN/TBAPF6 (0.1 M) as supporting electrolyte; Eox and Ered were estimated from the oxidation and reduction peaks.i HOMO = −Eoxonset + 4.40 (eV), LUMO = −Eredonset + 4.40 (eV).j At (PCM:THF)B3LYP/6-31G(d) level.k From (CPCM:THF)-CAM-TD-B3LYP/6-311++G(d,p)//B3LYP/6-31G(d) level computations. | ||||||||||||||
| P1 | 406 | 572(s) | 412 | 653(s) | 10d | 166 | 2.26 | 1.28 | −1.59 | −5.22 | −3.45 | 1.77 | −5.13 | 2.66 |
| 542 | 547 | |||||||||||||
| P2 | 362 | 464 | 437(s) | 504 | 5e | 102 | 2.60 | 1.62 | −1.79 | −5.35 | −3.28 | 2.09 | −5.30 | 2.97 |
| 370 | 430 | |||||||||||||
| P3 | 400 | 480(s) | 410 | 511 | 18f | 100 | 2.52 | 1.17 1.89 | −1.36 | −5.30 | −3.44 | 1.86 | −5.15 | 2.70 |
| 500 | 345 | |||||||||||||
| P4 | 348 | 444 | 360 | 502 | 12f | 96 | 2.78 | 1.32 | −1.41 | −5.37 | −3.26 | 2.11 | −5.33 | 3.00 |
| 476 | ||||||||||||||
The fluorescence spectra of the solutions, having the same concentration used for the UV-Vis measurements and thin films, were recorded (Fig. 1, S8,† and Table 1). While a strong yellow-green emission at 542 nm with a shoulder peak at around 572 nm was recorded for P1, polymer P3 emitted a green colour with λem maximum of 500 nm, which is about 42 nm blue shifted compared to that of P1 pointing out the more effective conjugation and charge transfer in P1. Presence of cross-conjugated selenopheno[2,3-b]thiophene and thieno[2,3-b]thiophene in the main backbones of P2 and P4 caused a significant blue-shift with respect to P1 and P3. While P2 exhibited a deep-blue emission at 464 nm, P4 gave a blue-violet emission at around 444 nm. Thus, the photophysical properties of these emitters are strongly affected primarily by the π-conjugation length and then with the presence of “Se” atom in the ring. Absorption spectra of P2 and P4 had two maxima above 300 nm. While low energy absorption bands arose from π–π* transitions, high energy absorption bands were trigged by charge transfer to mesitylboron unit. The fluorescence quantum yields (ΦF) of both copolymers were measured in THF and found to be 0.18 for P3 and 0.12 for P4, whereas the other polymers P1 and P2 exhibited slightly lower quantum yields of 0.10 and 0.05, respectively, due to higher rate of nonradiative decays emerging from high degree of stabilization of charge transfer state, by means of that making vicinity of this state to ground charge transfer states. Moreover, P1 and P3 had the same longest absorption wavelengths, the emission of P1 was found to be much longer than P3, which had the largest Stokes shift of 166 nm. While the polymers P1 and P2, having selenophenothiophene units, exhibited the Stokes shift of 166 and 102 nm, respectively, those of the other polymers P3 and P4, possessing thienothiophene units, had the Stokes shits of 100 and 96 nm, respectively. These results clearly indicated that cross-conjugated ring of selenophenothiophene, i.e. selenopheno[3,2-b]thiophene, is more effective in generating red shift in fluorescence, which is obviously due to a better contribution of big selenium atom to the available proper conjugation compare with sulphur atom.
The solid state fluorescence of P1–P4 showed an intense green fluorescence with the maxima of 547, 504, 511, and 502 nm, respectively (Fig. S8,† Table 1). Such a clear red-shift from 5 to 66 nm in comparison to the solution state could be attributed to boron acceptor and rigid fused aromatic donor core units having better interactions in solid state.
Electrochemical properties
The redox properties of all the polymers were determined by cyclic voltammetry on thin films. The CV diagrams of the polymers P1–P4 are depicted in Fig. 2 and electrochemical results are listed in Table 1. While the oxidation processes were irreversible for P1, P2 and P4, two quasi-reversible characteristics (Eox = +1.17 and +1.89 V, vs. Fc/Fc+) were recorded for P3, the first oxidation process of which was found to occur at the lowest potential among the four polymers. Slightly lower oxidation potential of P1 (Eox = +1.28 V), having a cross-conjugated selenophenothiophene unit, than P2 (+1.62 V) indicated a low laying HOMO of P2 compared to P1. The same trend was observed with P3 and P4. The oxidation of P4 (+1.32 V), possessing a cross-conjugated thienothiophene, was found to be more difficult than P3. The reason for the difference could be explained not only with the presence of the different donor units in the polymer back-bones but also with the structures of the fused rings. | ||
| Fig. 2 Cyclic voltammograms of the polymers P1–P4 on glassy carbon disk electrode in acetonitrile, 0.1 M Bu4NPF6, 100 mV s−1 scan rate, vs. Fc/Fc+. | ||
All the polymers displayed irreversible reduction peaks in the CV diagrams, which is possibly due to the known relaxation effects generating hysteresis phenomena (Fig. 2). The reduction potentials of P1 and P2 were recorded to be −1.59 and −1.79 V, whereas those of P3 and P4 were measured as −1.36 and −1.41 V, respectively, indicating a more difficult reduction in the case of P1 and P2 possessing electron rich and more polarizable Se atoms with respect to P3 and P4 having more electronegative and less polarizable S atoms. In addition, compared to P1 and P2, the boron-copolymers P3 and P4 showed more positive shifts in their first reduction potentials, supporting improved electron-accepting ability.
From the electrochemical data, the HOMO and LUMO energy levels and there from calculated energy gap of P1–P4 are listed in Table 1. The HOMOs of P1–P4 were found to be in the range of −5.22 to −5.37 eV, while LUMO energy levels were obtained at −3.45 to −3.26 eV. The HOMO energy levels of P1 and P2 are lower than the corresponding S containing cross-conjugated P3 and P4 pointing out a better stability of P2 and P4 against oxidative doping. The calculated electrochemical HOMO–LUMO gaps of selenophenothiophene derivatives (1.77 eV for P1 and 2.09 eV for P2) are considerably smaller than those of corresponding thienothiophene analogues (1.86 eV for P3 and 2.11 eV for P4).
Fluoride sensing properties
Presence of electron-deficient boron centres could be a good advantage for anion bindings such as fluorides and cyanides.15c,15e,15g,20 Triarylboranes have been widely studied as chemo-sensors for fluoride anions with high selectivity and sensitivity.21 Complexation of boron with anions typically leads to a distinct change in the photophysical properties, and polymeric species are particularly interesting because of their potential for reciprocal interactions with different binding sites.15b,15g,22 Thus, tetrabutylammonium fluoride (TBAF) as a fluoride source was gradually added to a THF solution of each polymer and the response of P1–P4 to fluoride anions was investigated by UV-Vis absorption and fluorescence spectroscopy. Titration of organoborane polymers P1–P4 with fluoride ion displayed an interesting binding process of fluoride ion to boron atom in the absorption spectra. Upon addition of less than 1.0 equiv. of TBAF to the polymer P1, while the absorption at 406 nm started to decrease, new bands gradually appeared in the high-energy region at 267, 322 and 354 nm, intensities of which enhanced upon continuing the addition of TBAF (Fig. 3a). An isosbestic point at 373 nm was observed. On the other hand, when more than 1.0 equiv. of TBAF was added, intensities of the new absorption bands started to decrease and the absorption band at 406 nm, which already decreased upon addition of TBAF, almost disappeared along with the disappearance of the isosbestic point. This process was reflected in a colour change of the solution from yellow to complete colourless, which allowed a naked-eye detection of fluoride anion. Decrease of the band at 406 nm and appearance of the new bands at 267, 322 and 354 nm may indicate the interaction of boron atoms with fluoride ions and interruption of electron flow to boron, which causes shortening of the conjugation and lowering of the absorption at 406 nm. Further addition of TBAF, i.e. above 1.0 equiv., may result in the quenching of the polymer and decrease of absorptions even at 267, 322 and 354 nm.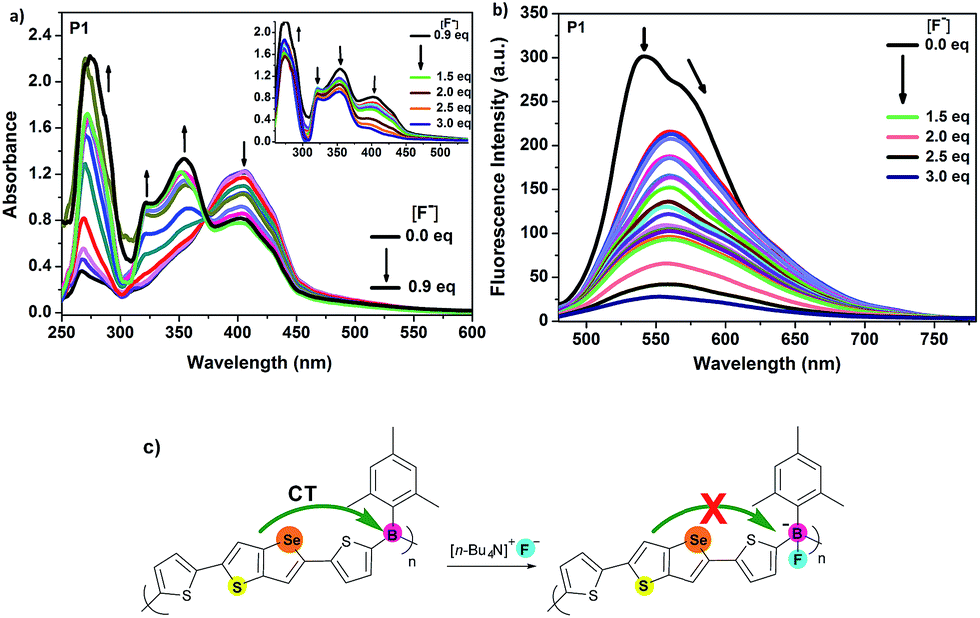 | ||
| Fig. 3 (a) Absorption spectra and (b) emission spectra (λexc = 470 nm) of P1 (1 × 10−4 M based on boron sites) in THF in the presence of various amounts of TBAF. (c) Pictorial view of interruption of charge transfer (CT) by fluoride binding. | ||
Fig. 3b shows the change in fluorescence spectrum of P1 upon addition of various concentrations of TBAF. After the addition of first 0.05 equiv. of TBAF, a sharp decrease of emission intensity at 542 nm with a shoulder at 572 nm was observed with an appearance of a new band with lower intensity at 560 nm. When reached the addition of 3.0 equiv. of TBAF, almost a complete quenching took place. Thus, P1 works as a “turn-off” sensor for fluoride ion. This result is attributable to the apparent amplified quenching effect, which has also been described for other organoborane polymers,15b,22a–f involving the efficient energy transfer to lower energy charge transfer states which are generated by binding of fluoride anion to boron.
Next, fluoride sensing properties of P3 were examined (Fig. S9a†). Similarly, upon addition of TBAF, the characteristic intense charge-transfer absorption band at 400 nm steadily decreased, while the absorption maximum at 242, 329 and 293 nm appeared and further addition up to 0.7 equiv. made them intense bands. An isosbestic point at 350 nm was obtained at this stage. Continuing the addition from 0.7 to 2.4 equiv. caused all the absorption bands gradually decrease, except the band at 242 nm, which slightly increased, and a new weak band at 370 nm appeared. Colour of the solution turned to colourless from yellow. Furthermore, the emission intensity of P3 was also quenched in this process (Fig. S9b†).
The stepwise addition of TBAF resulted in a steady decrease of the broad emission band between 375–600 nm, which was almost completely quenched with 3.0 equiv. of TBAF; hence it is also a “turn-off” sensor for fluoride ion. Quenching of fluorescence intensity was observed to be more effective with the polymer P3 (78%) compare with the polymer P1 (69%) at the addition of the same amount of TBAF (1.5 equiv.). Absorption and emission changes of the polymers (P1 and P3) clearly indicated the disruption of the π-conjugation through the boron empty orbital by the formation of the corresponding fluoroborate in the polymer chain.
Considering the specific interactions between F− and the boron centres of P1 and P3, the response of P2 and P4 to F− was also investigated through absorption and fluorescence spectra (Fig. S10† and 4). With the initial addition of fluoride ions up to 0.6 equiv. the main absorption peaks of P2 at 252, 312 and 364 nm increased dramatically, while the absorption peaks at 312 and 364 nm were slightly blue shifted by 26 and 12 nm, respectively. When the addition of the ion was continued from 0.6 to 2.4 equiv., all the bands were gradually decreased as a result of coordination of the fluoride ion to the empty p orbital of the boron atom. A similar tendency was observed with the polymer P4 in absorption spectra upon addition of fluoride anion (Fig. 4a). Moreover, in both P2 and P4, a colour change of the solution from yellow to complete colourless was observed. In the fluorescence spectra, although initially P2 exhibited a deep blue ICT emission at 464 nm, intensity of the emission was steadily decreased resulting in 60% of fluorescence quenching efficiency with the increase of fluoride ion concentration and a violet-blue emission peak at 431 nm emerged (Fig. S10†). This indicated the donor–acceptor interaction between cross conjugated selenophenothiophene and boron has stopped. In contrast to the behaviour of P2, the emission of P4 at 444 nm was almost completely quenched and a red-shifted band at 480 nm, having weak intensity at 475 nm was developed with an addition of small amount of fluoride ion (0.05 equiv.) (Fig. 4b). It was disappeared with further addition of TBAF. That is, addition of only 0.05 equiv. of TBAF led to about 82% fluorescence quenching of P4 and the emission intensity was almost completely quenched ca. 94% with 1.5 equiv. of TBAF. Stern–Volmer plots shown in Fig. S11† for both P1 and P2 are nearly identical, indicating that both molecules have a similar affinity toward fluoride ion in fluorescence quenching mechanism. On the other hand, the polymers P3 and P4 had higher sensitivities toward fluoride ions compare with P1 and P2. Thus, polarizability difference of Se and S atoms and their orientation in the ring could be affecting the sensing power of the whole material. Comparison of the both systems, i.e. “Se” (P1, P2) and “S” (P3, P4), indicated that as Se is a better electron donor compare with S atom, it provides more electrons to the empty orbital of boron making the material less sensitive toward fluoride ions. Thus, the systems containing Se atoms are less sensitive accepting fluoride ions. Concerning the best sensitivity of P4, it has the least electron donation among the four polymers due to both having “S” atom instead of “Se” and its cross-conjugated structure.
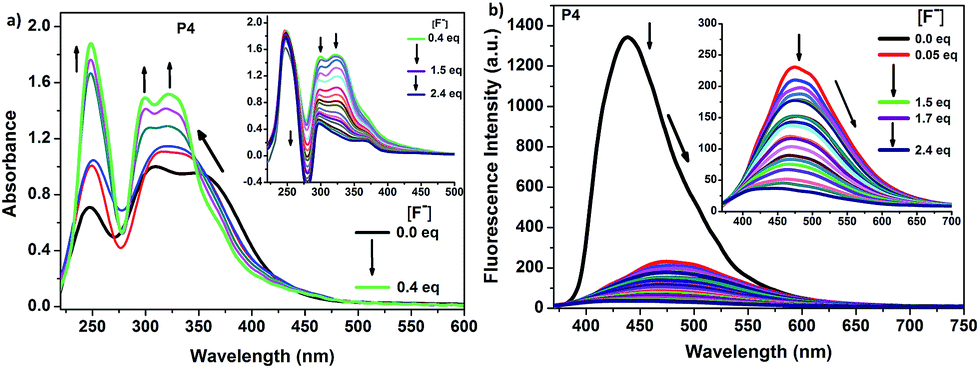 | ||
| Fig. 4 (a) Absorption spectra and (b) emission spectra (λexc = 350 nm) of P4 (1 × 10−5 M based on boron sites) in THF in the presence of various amounts of TBAF. | ||
Computation
The properties of the polymers were investigated further by performing density functional theory (DFT) calculations on monomers (M) and segments of polymers (S), possessing mesitylboron acceptor between thienothiophene (TT) and selenophenothiophene (SeT) donor units (Fig. 5). Geometry optimizations of their S0 and S1 states were realized without any symmetry constraints by means of Gaussian 09 package program.23 B3LYP24 method with 6-31G(d) basis set was chosen because of its good performance in the estimation of structures and properties of optoelectronic compounds.25 Solvent effect (THF) was included by using polarizable continuum model (PCM).26 Analysis of the harmonic vibrational frequencies using analytical second derivatives was performed to confirm the minima. Orbital composition analysis with Mulliken partition was carried out using Multiwfn program (a multifunctional wavefunction analyser).27 TD-DFT calculations with solvent effect (CPCM)28 were conducted on polymer segments at coulomb-attenuating density functional theory (CAM-TD-B3LYP)29 level with 6-311++G(d,p) basis set to obtain the lowest singlet–singlet vertical excitations owing to the good performance of CAM-B3LYP in the calculations of excitation energies.30 Absorption bands were collected for singlets with N states of 50. While visualization of MOs with an isosurface value of 0.04 au was accomplished with GaussView 5.0, absorption and emission spectra were produced with GaussSum 3.0.31 | ||
| Fig. 5 FMOs (isosurface value = 0.04 au) of monomers M1–M4 and segments S1–S4 at (PCM:THF)B3LYP/6-31G(d) level. | ||
Computations performed on the monomers depicted that TT and SeT units attached to the mesityl boron are almost planar to the boron plane with a dihedral angle of less than 10°, whereas the mesityl group attached to boron atom stayed vertical with respect to TT and SeT groups (>60°) (Fig. 5). The localized HOMOs are observed on TT and SeT groups of all the molecules, spreading marginally over the thiophene substituents. On the other hand, the LUMOs of the molecules are entirely located on boron atoms with slight contributions of TT and SeT units. Therefore, TT and SeT play as electron rich groups and mesityl boron as an electron poor unit. In the case of monomers M2 and M4 with cross-conjugated systems, LUMO orbitals are spread over only half of the TT and SeT units. MOs undoubtedly demonstrate an ICT between donor and acceptor units. The presence of nodes on electron poor boron atoms in HOMOs results in the charge density separation and consequently hampers the delocalization of HOMOs over all the whole system witnessed with the constructed HOMO surfaces for S1–S4.
The predicted HOMO energy levels of −5.13, −5.30, −5.15, and −5.33 for S1–S4 are matching well with those of P1–P4 (Table 1), respectively, emphasizing the increase of HOMO levels with incorporation of Se atoms in P1 and P3 compared to P2 and P4. This phenomenon was reflected to HOMO–LUMO gaps. That is, Egap values of 2.66, 2.97, 2.70, and 3.00 eV were slightly overestimated at TD-TDFT level of theory (Table 1), but the trend well aligns with those obtained from the onset of UV-Vis absorption spectra, demonstrating the smaller band gaps for S1 and S3 with Se than their corresponding S2 and S4 having S atoms.
Orbital composition analysis provided the highest LUMO orbital composition of 22.0% for boron atom of S4 and the lowest one for boron atom of S1 with 17.0% orbital composition supporting the experimentally obtained fluoride quenching efficiencies. The larger the LUMO orbital composition on boron atom, the higher the sensitivity of boron atom toward Lewis base.
The estimated UV-Vis absorption spectra of S1–S4 and vertical excitation energies are provided in Fig. S13† and Table 2, respectively. The computed spectral properties are matching well with the experimentally observed values. The λmax values of compounds arise from the HOMO to LUMO transitions pointing out the fact that the low energy transitions emerge from HOMO → LUMO. Emission spectra of the polymer segments were predicted by performing the calculations on the optimized excited state geometries with solvent effect (CPCM:THF). The obtained emission spectra are illustrated in Fig. S14.† In all segments (S1–S4), the emission predominantly originates from LUMO → HOMO transition, well in alignment with the experimental outcome (Table S1†). Fluoride attachment on the electron deficient boron atom (S1-F–S4-F) switches the planar geometry of boron to tetrahedral structure. While the HOMOs and LUMOs were observed on the whole molecule, excluding mesityl boron units in S1-F and S3-F, HOMOs were speaded over the system up to cross-conjugated rings and LUMOs were localized on one of the terminal thiophene separated by cross-conjugated systems in S2-F and S4-F (Fig. S12†). This results in the interruption of charge transfer. As a consequent, it gives rise to a blue shift in the predicted absorption spectra by 51–70 nm, which is matching well with the results recorded in the fluoride sensing investigations (Table 2).
| States | λabs [nm] | E [eV] | Fa | Major contributionb [%] | Exp. [nm] | |
|---|---|---|---|---|---|---|
| a Oscillator strength.b H: HOMO, L: LUMO. | ||||||
| S1 | S1 | 466 | 2.66 | 2.311 | H → L (77%) | 470 |
| S2 | 408 | 3.04 | 0.628 | H−1 → L (55%), H → L+1 (36%), H−1 → L+1 (30%) | 406 | |
| S3 | 336 | 3.68 | 0.199 | H → L+2 (37%) | 303 | |
| S24 | 244 | 5.08 | 0.124 | H−10 → L (14%), H−10 → L+1 (13%), H−10 → L+2 (12%) | 256 | |
| S2 | S1 | 418 | 2.97 | 1.734 | H → L (76%) | — |
| S2 | 355 | 3.49 | 0.362 | H → L+1 (16%), H−1 → L (55%) | 362 | |
| S5 | 322 | 3.85 | 0.447 | H → L+3 (23%), H−1 → L+3 (24%) | 312 | |
| S16 | 263 | 4.71 | 0.148 | H−11 → L (32%), H−3 → L+3 (10%) | 252 | |
| S3 | S1 | 459 | 2.70 | 2.343 | H → L (77%) | 400 |
| S2 | 400 | 3.10 | 0.582 | H−1 → L (56%), H → L+1 (34%) | 320 | |
| S3 | 331 | 3.75 | 0.276 | H−1 → L+1 (32%), H → L+2 (38%) | 292 | |
| S15 | 252 | 4.92 | 0.084 | H−1 → L+1 (13%), H → L+2 (19%) | 243 | |
| S4 | S1 | 413 | 3.00 | 1.755 | H → L (76%) | — |
| S2 | 351 | 3.54 | 0.438 | H → L+1 (21%), H−1 → L (54%) | 348 | |
| S4 | 318 | 3.90 | 0.586 | H → L+2 (30%), H−1 → L+3 (29%) | 308 | |
| S12 | 268 | 4.63 | 0.308 | H−3 → L+3 (19%), H−2 → L+2 (22%), | 248 | |
| S21 | 234 | 5.29 | 0.127 | H−9 → L+2 (26%), H−8 → L+3 (37%) H−11 → L (20%) | — | |
| S1-F | S1 | 415 | 2.99 | 2.144 | H → L (49%) | — |
| S2 | 400 | 3.09 | 0.663 | H → L+1 (45%), H−1 → L (44%) | 354 | |
| S5 | 291 | 4.26 | 0.101 | H−6 → L (62%), H−6 → L+1 (25%) | 322 | |
| S6 | 290 | 4.27 | 0.094 | H−5 → L+1 (23%), H−7 → L+1 (19%) | 267 | |
| S2-F | S1 | 350 | 3.53 | 1.048 | H → L+2 (38%) | 364 |
| S2 | 343 | 3.61 | 0.152 | H → L (27%), H−1 → L (30%) | — | |
| S3 | 337 | 3.68 | 1.097 | H → L+3 (18%), H−1 → L+3 (17%), H → L+1 (12%) | — | |
| S4 | 331 | 3.74 | 0.714 | H−1 → L+2 (18%), H → L+1 (15%), H → L+3 (14%) | — | |
| S5 | 294 | 4.21 | 0.188 | H−3 → L (51%), H−4 → L (22%) | 312 | |
| S6 | 292 | 4.25 | 0.174 | H−2 → L+1 (30%), H−4 → L+1 (28%) | 252 | |
| S3-F | S1 | 406 | 3.06 | 2.238 | H → L (49%) | — |
| S2 | 392 | 3.16 | 0.640 | H → L+1 (46%), H−1 → L (46%) | 329 | |
| S7 | 277 | 4.48 | 0.083 | H−6 → L+1 (34%), H−6 → L (29%) | 293 | |
| S8 | 276 | 4.49 | 0.101 | H−7 → L (38%), H−7 → L+1 (26%) | 243 | |
| S4-F | S1 | 343 | 3.62 | 1.231 | H → L+2 (50%) | 321 |
| S3 | 330 | 3.76 | 0.460 | H−1 → L (28%), H → L (20%), H → L+1 (14%) | 300 | |
| S4 | 329 | 3.77 | 0.973 | H → L+1 (20%), H−1 → L+1 (12%), H−1 → L (11%) | — | |
| S5 | 284 | 4.36 | 0.196 | H−3 → L+1 (27%), H−4 → L+1 (25%) | 248 | |
Conclusions
Synthesis of new four D–π–A polymers was realized, which possess fused bicyclic aromatic rings thieno[3,2-b]thiophene, selenopheno[3,2-b]thiophene, thieno[2,3-b]thiophene and selenopheno[2,3-b]thiophene as donors and mesitylboron as an acceptor connected through a thiophene π-spacer. Their properties were investigated optically, electrochemically and as fluoride ion sensors, comparing their structures and interactions of S and Se atoms with boron. As selenium atoms improve the conjugation and facilitate charge-transfer processes due to their more polarizable and less electronegative properties, polymer P1, having better optical properties compared to P3, exhibited a broader absorption band with a larger red shift emission band.Moreover, P1 exhibited the longest emission wavelength, and thus the largest Stokes shift among the four organoboron-based polymers. Quantum efficiencies of the polymers were obtained to be in the range of 5–18%. DFT studies shed light on their electronic structures and frontier molecular orbitals, which supported the experimental outcome. CAM-TD-B3LYP calculations provided vertical excitations from HOMO to LUMO with energies varying from 2.66 to 3.00 eV. Fluoride sensing properties of these polymers P1–P4 were investigated through absorption and emission measurements, owing to the high Lewis acidity of the boron centres to coordinate fluoride anions. The enhancement of strong sensitivity with P4 was detected in comparison to the other polymers P1–P3, which was interpreted to be due to the presence of less polarizable sulfur atom compare with selenium and its cross-conjugated structure. Upon addition of TBAF as a fluoride ion source, P1–P4 exhibited a “turn off” sensor properties, observed by a colour change of the solution from yellow to completely colourless. Thus, the present polymers can be used as fluorescent or colorimetric fluoride anion sensors. On the basis of the optical and electrochemical studies, these polymers can be promising candidates for the large area device fabrication as functional materials.
Experimental
All reagents were used as received from commercial sources without further purification. Tetrahydrofuran (THF) and diethyl ether (Et2O) were dried over sodium in the presence of benzophenone. All chemical reactions were performed under N2 atmosphere. 2,5-Dibromoselenopheno[3,2-b]thiophene (1),3b,16,32,33 2,5-dibromoselenopheno[2,3-b]thiophene (2),17,18,32 2,5-dibromothieno[3,2-b]thiophene (3),16,34 2,5-dibromothieno[2,3-b]thiophene (4),16,34b 4,4,5,5-tetramethyl-2-(thiophen-2-yl)-[1,3,2]dioxaborolane35 and mesityldimethoxyborane36 were synthesized according to published procedures. 1H and 13C NMR spectra were recorded on a Varian model NMR (500 MHz). Proton and carbon chemical shifts are reported in ppm downfield from tetramethylsilane (TMS). UV-Vis measurements were studied on HITACHI U-0080D. Mass spectra were recorded on Bruker MICROTOFQ and Thermo LCQ-Deca ion trap mass instruments. CH-Instruments Model 400A was used as a potentiostat for the CV studies. Cyclic voltammetry was performed on a glassy carbon disk electrode coated with a polymer thin film was used as the working electrode, a platinum wire as the counter electrode and an Ag wire as the reference electrode. Tetrabutylammonium hexafluorophosphate (Bu4NPF6) (0.1 M) in acetonitrile, saturated with nitrogen, was used as the supporting electrolyte. A ferrocene internal standard was used to calibrate the results. The molar mass and molar mass distribution of the polymers were determined using gel permeation chromatography, equipped with Perkin-Elmer 200 GPC high pressure pump, injector, THF columns (guard column + Styragel HR 2 + Styragel HR 3 + Styragel HR 4E + Styragel HR 5E), Wyatt Optilab differential refractive index detector (654 nm) and Dawn Heleos multi angle light-scattering detector. The mobile phase was THF (flow rate: 0.7 mL min−1). Measurements were conducted at 25 °C. Polymer concentrations were in the range of 0.5–2.0 mg mL−1 and all the samples were filtered through 0.2 μm filter prior to use. Thermal gravimetric analyses (TGA) of the polymers were performed by EXSTAR-SII TG/DTA7300 thermal gravimetry-differential thermal analysis (TG-DTA) under dynamic argon atmosphere. Temperature calibration of the instrument was conducted according to indium melting point and enthalpy.2,5-Di(thiophen-2-yl)selenopheno[3,2-b]thiophene (5)
In a Schlenk tube, a mixture of 2-(thiophen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (687 mg, 3.27 mmol), 2,5-dibromoselenopheno[3,2-b]thiophene (1) (451 mg, 1.30 mmol) and 2 M aqueous K2CO3 (2 mL) in a stirred THF (30 mL) was flushed with N2 for 15 min. Then, to this mixture was added Pd(PPh3)2Cl2 (69.0 mg, 5% mmol) as catalyst and it was refluxed with vigorous stirring for 60 h. The excess solvent was distilled and the compound was extracted with dichloromethane (DCM) (3 × 20 mL). The organic phase was washed with water, dried over anhydrous MgSO4 and solvent was evaporated. The residue was purified by column chromatography using n-hexane/DCM (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, Rf = 0.78) to obtain an orange solid in 55% (253 mg) yield. M.p. 220–221 °C; 1H NMR (500 MHz, CDCl3): δ 7.45 (s, 1H), 7.33 (s, 1H), 7.24 (d, J = 5.5 Hz, 2H), 7.21 (d, J = 3.5 Hz, 1H), 7.15 (d, J = 3.5 Hz, 1H), 7.04–7.01 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 141.9, 139.7, 139.5, 138.5, 137.6, 137.4, 127.9, 126.8, 124.9, 124.7, 124.3, 123.8, 118.9, 117.9. MS (m/z): [M + 2]+ calcd for C14H8S3Se, 351.89; found, 353.10.
:1, Rf = 0.78) to obtain an orange solid in 55% (253 mg) yield. M.p. 220–221 °C; 1H NMR (500 MHz, CDCl3): δ 7.45 (s, 1H), 7.33 (s, 1H), 7.24 (d, J = 5.5 Hz, 2H), 7.21 (d, J = 3.5 Hz, 1H), 7.15 (d, J = 3.5 Hz, 1H), 7.04–7.01 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 141.9, 139.7, 139.5, 138.5, 137.6, 137.4, 127.9, 126.8, 124.9, 124.7, 124.3, 123.8, 118.9, 117.9. MS (m/z): [M + 2]+ calcd for C14H8S3Se, 351.89; found, 353.10.
Following the same procedure of 5, compounds 6–8 were prepared.
2,5-Di(thiophen-2-yl)selenopheno[2,3-b]thiophene (6)
Compound 2 (736 mg, 2.10 mmol) was reacted with 2-(thiophen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (942 mg 4.48 mmol) in the presence of 2 M aqueous K2CO3 (10.7 mL) and PdCl2(PPh3)2 (212 mg, 5% mol) in THF (35 mL). The residue was purified by column chromatography using n-hexane/DCM (5:1, Rf = 0.70) affording a pale yellow solid in 48% (357 mg) yield. M.p. 169–170 °C; 1H NMR (500 MHz, CDCl3): δ 7.40 (dd, J = 5.0 and 1.0 Hz, 1H), 7.38 (dd, J = 3.8 and 2.5 Hz, 1H), 7.32 (s, 1H), 7.27 (d, J = 1.0 Hz, 1H), 7.26 (d, J = 1.5 Hz, 1H), 7.21 (dd, J = 3.8 and 1.2 Hz, 1H), 7.10 (dd, J = 5.2 and 3.8 Hz, 1H), 7.04 (dd, J = 5.2 and 3.8 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 148.8, 140.9, 136.8, 136.3, 135.7, 130.6, 127.8, 127.4, 127.2, 126.7, 125.0, 124.3, 118.5, 101.8. MS (m/z): [M + 1]+ calcd for C14H8S3Se, 351.89; found, 352.00.
2,5-Di(thiophen-2-yl)thieno[3,2-b]thiophene (7)37
Compound 3 (585 mg 1.90 mmol) was treated with 2-(thiophen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (866 mg, 4.12 mmol) in the presence of 2 M aqueous K2CO3 (10 mL) and PdCl2(PPh3)2 (140 mg, 5% mol) in THF (40 mL). The residue was purified by column chromatography using n-hexane/DCM (5:1, Rf = 0.75) furnishing a yellow solid in 72% (433 mg) yield. M.p. 255–256 °C; 1H NMR (500 MHz, CDCl3): δ 7.46 (dd, J = 3.5 and 1.2 Hz, 2H), 7.39 (dd, J = 5.2 and 1.2 Hz, 2H), 7.28 (s, 2H), 7.11 (dd, J = 5.2 and 3.5 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 147.3, 140.7, 137.5, 135.5, 133.6, 128.0, 126.6, 125.2, 124.4, 116.2.
2,5-Di(thiophen-2-yl)thieno[2,3-b]thiophene (8)
Compound 4 (414 mg 1.40 mmol) was reacted with 2-(thiophen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (650 mg, 3.10 mmol) in the presence of 2 M aqueous K2CO3 (7 mL) and PdCl2(PPh3)2 (100 mg, 5% mol) in THF (40 mL) following the above procedure. The residue was purified by column chromatography using n-hexane/DCM (5:1, Rf = 0.68) providing a yellowish orange solid in 40% (166 mg) yield. M.p. 195–196 °C; 1H NMR (500 MHz, CDCl3): δ 7.27 (s, 2H), 7.25 (dd, J = 5.2 and 1.0 Hz, 2H), 7.21 (dd, J = 3.5 and 1.0 Hz, 2H), 7.04 (dd, J = 5.2 and 3.5 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 146.9, 139.8, 137.3, 134.9, 127.8, 124.6, 124.3, 123.7, 116.3.
General method for the syntheses of the polymers P1–P4
To a solution of 2,5-di(thiophen-2-yl)selenopheno[3,2-b]thiophene (5) (272 mg, 770 μmol) in 60 mL of dry THF was added 1.06 mL n-BuLi (1.6 M, 1.70 mmol) dropwise at −78 °C under nitrogen atmosphere. After 1 h, the temperature was warmed up to room temperature. Stirring was then continued for another 1 h. The mixture was then cooled down to −78 °C and MesB(OMe)2 (168 μL, 900 μmol) was added and the solution was allowed to warm up to room temperature and the mixture was stirred for 3 days. After removal of the solvent under reduced pressure, the crude product was dissolved in a minimum amount of THF and precipitated in n-hexane. The precipitate was filtered, washed repeatedly with n-hexane and dried under vacuum to afford P1 as a reddish-orange solid in yield of 50% (198 mg). Mn = 97.9 kDa, Mw = 110.0 kDa, Mw/Mn (Đ) = 1.12.Synthesis of P2
Polymerization of compound 6 (257 mg, 730 μmol), with MesB(OMe)2 (125 μL, 800 μmol) in the presence of n-BuLi (960 μL, 1.6 M, 1.50 mmol) in 40 mL of dry THF gave P2 as a yellow solid in 40% yield (150 mg). Mn = 11.2 kDa, Mw = 28.7 kDa, Mw/Mn (Đ) = 2.55.Synthesis of P3
Polymerization of compound 7 (273 mg 900 μmol) with MesB(OMe)2 (147 μL, 1.00 mmol) in the presence of n-BuLi (1.17 mL, 1.6 M, 1.88 mmol) in 40 mL of dry THF furnished P3 as a reddish-brown solid in yield of 39% (165 mg). Mn = 40.5 kDa, Mw = 55.6 kDa, Mw/Mn (Đ) = 1.37.Synthesis of P4
Polymerization of compound 8 (240 mg, 800 μmol) with MesB(OMe)2 (185 μL, 900 μmol) in the presence of n-BuLi (1.00 mL, 1.6 M, 1.60 mmol), in 40 mL of dry THF afforded P4 as an orange solid in 44% yield (160 mg). Mn = 40.7 kDa, Mw = 62.7 kDa, Mw/Mn (Đ) = 1.54.Acknowledgements
G. T. and M. E. C. thank the Scientific and Technological Research Council of Turkey (TUBITAK) for Postdoctoral Research Fellowships BIDEB 2218 and 2216 programs, respectively. We thank Prof. M. S. Eroglu for Light Scattering measurements. We are indebted to National Center for High Performance Computing (UYBHM) (tvddvb) and the High-Performance-Computing (HPC) Linux Cluster HorUS of University of Siegen for the computer time provided. Unsped Global Logistic is gratefully acknowledged for financial support.References
- (a) Y. M. Kim, E. Lim, I. N. Kang, B. J. Jung, J. Lee, B. W. Koo, L. M. Do and H. K. Shim, Macromolecules, 2006, 39, 4081–4085 CrossRef CAS; (b) J. Hollinger, D. Gao and D. S. Seferos, Isr. J. Chem., 2014, 54, 440–453 CrossRef CAS; (c) A. Patra, M. Bendikov and S. Chand, Acc. Chem. Res., 2014, 47, 1465–1474 CrossRef CAS PubMed.
- (a) T. Yamamoto and K. Takimiya, J. Am. Chem. Soc., 2007, 129, 2224–2225 CrossRef CAS PubMed; (b) H. Ebata, E. Miyazaki, T. Yamamoto and K. Takimiya, Org. Lett., 2007, 9, 4499–4502 CrossRef CAS PubMed.
- (a) A. M. Ballantyne, L. Chen, J. Nelson, D. D. C. Bradley, Y. Astuti, A. Maurano, C. G. Shuttle, J. R. Durrant, M. Heeney, W. Duffy and I. McCulloch, Adv. Mater., 2007, 19, 4544–4547 CrossRef CAS; (b) S. P. Mishra, A. E. Javier, R. Zhang, J. Liu, J. A. Belot, I. Osaka and R. D. McCullough, J. Mater. Chem., 2011, 21, 1551–1561 RSC; (c) K. Takimiya, Y. Kunugi, Y. Konda, H. Ebata, Y. Toyoshima and T. Otsubo, J. Am. Chem. Soc., 2006, 128, 3044–3050 CrossRef CAS PubMed; (d) K. Takimiya, Y. Konda, H. Ebata, N. Niihara and T. Otsubo, J. Org. Chem., 2005, 70, 10569–10571 CrossRef CAS PubMed; (e) Y. A. Getmanenko, P. Tongwa, T. V. Timofeeva and S. R. Marder, Org. Lett., 2010, 12, 2136–2139 CrossRef CAS PubMed.
- (a) M. R. Bryce, J. Mater. Chem., 1995, 5, 1481–1496 RSC; (b) E. Ertas, İ. Demirtas and T. Ozturk, Beilstein J. Org. Chem., 2015, 11, 403–415 CrossRef CAS PubMed.
- (a) Y. Kunugi, K. Takimiya, Y. Toyoshima, K. Yamashita, Y. Aso and T. Otsubo, J. Mater. Chem., 2004, 14, 1367–1369 RSC; (b) D. J. Crouch, P. J. Skabara, J. E. Lohr, J. J. W. McDouall, M. Heeney, I. McCulloch, D. Sparrowe, M. Shkunov, S. J. Coles, P. N. Horton and M. B. Hursthouse, Chem. Mater., 2005, 17, 6567–6568 CrossRef CAS.
- (a) S. S. Zade and M. Bendikov, Org. Lett., 2006, 8, 5243–5246 CrossRef CAS PubMed; (b) U. Salzner, J. B. Lagowski, P. G. Pickup and R. A. Poirier, Synth. Met., 1998, 96, 177–189 CrossRef CAS.
- H. Pang, P. J. Skabara, S. Gordeyev and J. J. W. McDouall, Chem. Mater., 2007, 19, 301–307 CrossRef CAS.
- A. Patra, Y. H. Wijsboom, S. S. Zade, M. Li, Y. Sheynin, G. Leitus and M. Bendikov, J. Am. Chem. Soc., 2008, 130, 6734–6736 CrossRef CAS PubMed.
- (a) M. E. Cinar and T. Ozturk, in Thiophenes, ed. J. A. Joule, Springer-Verlag, Berlin Heidelberg, 2014, pp. 161–202 Search PubMed; (b) M. E. Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036–3140 CrossRef CAS PubMed; (c) S. T. Cankaya, A. Capan, M. E. Cinar, E. Akin, S. Topal and T. Ozturk, J. Sulfur Chem., 2013, 34, 638–645 CrossRef CAS; (d) A. Buyruk, M. E. Cinar, M. S. Eroglu and T. Ozturk, ChemistrySelect, 2016, 1, 3028–3032 CrossRef CAS.
- (a) W.-H. Lee, S. K. Son, K. Kim, S. K. Lee, W. S. Shin, S.-J. Moon and I.-N. Kang, Macromolecules, 2012, 45, 1303–1312 CrossRef CAS; (b) M. Heeney, W. Zhang, D. J. Crouch, M. L. Chabinyc, S. Gordeyev, R. Hamilton, S. J. Higgins, I. McCulloch, P. J. Skabara, D. Sparrowe and S. Tierney, Chem. Commun., 2007, 47, 5061–5063 RSC.
- (a) D. S. Chung, H. Kong, W. M. Yun, H. Cha, H.-K. Shim, Y.-H. Kim and C. E. Park, Org. Electron., 2010, 11, 899–904 CrossRef CAS; (b) S. Das and S. S. Zade, Chem. Commun., 2010, 46, 1168–1170 RSC; (c) S. Haid, A. Mishra, C. Uhrich, M. Pfeiffer and P. Bauerle, Chem. Mater., 2011, 23, 4435–4444 CrossRef CAS; (d) S. Das, P. B. Pati and S. S. Zade, Macromolecules, 2012, 45, 5410–5417 CrossRef CAS.
- (a) Y. J. Cheng, S. H. Yang and C. S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed; (b) G. Turkoglu, M. E. Cinar, A. Buyruk, E. Tekin, S. P. Mucur, K. Kaya and T. Ozturk, J. Mater. Chem. C, 2016, 4, 6045–6053 RSC; (c) E. B. Sevinis, C. Sahin, M. E. Cinar, M. S. Eroglu and T. Ozturk, Polym. Eng. Sci., 2016, 56, 1390–1398 CrossRef CAS; (d) E. Sezer, I. Osken, M. E. Cinar, O. Demirel, B. Ustamehmetoğlu and T. Ozturk, Electrochim. Acta, 2016, 222, 1592–1603 CrossRef CAS.
- (a) F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed; (b) P. Chen and F. Jäkle, J. Am. Chem. Soc., 2011, 133, 20142–20145 CrossRef CAS PubMed; (c) A. M. Priegert, B. W. Rawe, S. C. Serin and D. P. Gates, Chem. Soc. Rev., 2016, 45, 922–953 RSC; (d) Y. Ren and F. Jäkle, Dalton Trans., 2016, 45, 13996–14007 RSC.
- (a) N. Matsumi, K. Naka and Y. Chujo, J. Am. Chem. Soc., 1998, 120, 10776–10777 CrossRef CAS; (b) J. F. Mike and J. L. Lutkenhaus, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 468–480 CrossRef CAS; (c) J. Winsberg, T. Hagemann, S. Muench, C. Friebe, B. Häupler, T. Janoschka, S. Morgenstern, M. D. Hager and U. S. Schubert, Chem. Mater., 2016, 28, 3401–3405 CrossRef CAS; (d) X. Yin, F. Guo, R. A. Lalancette and F. Jäkle, Macromolecules, 2016, 49, 537–546 CrossRef CAS.
- (a) G. Zhou, M. Baumgarten and K. Mullen, J. Am. Chem. Soc., 2008, 130, 12477–12484 CrossRef CAS PubMed; (b) H. Li and F. Jäkle, Macromol. Rapid Commun., 2010, 31, 915–920 CrossRef CAS PubMed; (c) Z. M. Hudson and S. Wang, Acc. Chem. Res., 2009, 42, 1584–1596 CrossRef CAS PubMed; (d) S. Yamaguchi, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2001, 123, 11372–11375 CrossRef CAS PubMed; (e) C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbaï, Chem. Rev., 2010, 110, 3958–3984 CrossRef CAS PubMed; (f) H. Zhao, L. A. Leamer and F. P. Gabbaï, Dalton Trans., 2013, 42, 8164–8178 RSC; (g) F. Cheng, E. M. Bonder and F. Jäkle, J. Am. Chem. Soc., 2013, 135, 17286–17289 CrossRef CAS PubMed; (h) S.-B. Zhao, P. Wucher, Z. M. Hudson, T. M. McCormik, X.-Y. Liu, S. Wang, X.-D. Feng and Z.-H. Lu, Organometallics, 2008, 27, 6446–6456 CrossRef CAS; (i) S. K. Mellerup, Y.-L. Rao, H. Amarne and S. Wang, Org. Lett., 2016, 18, 4436–4439 CrossRef CAS PubMed; (j) Y. Hu, Z. Zhao, X. Bai, X. Yuan, X. Zhang and T. Masuda, RSC Adv., 2014, 4, 55179–55186 RSC; (k) H. Li and F. Jäkle, Angew. Chem., Int. Ed., 2009, 48, 2313–2316 CrossRef CAS PubMed.
- M. Heeney, C. Bailey, K. Genevicius, M. Giles, M. Shkunov, D. Sparrowe, S. Tierney, W. Zhang and I. McCulloch, Proc. SPIE, 2005, 5940, 1–9 CrossRef.
- S. Yasuike, J. Kurita and T. Tsuchiya, Heterocycles, 1997, 45, 1891–1894 CrossRef CAS.
- V. P. Litvinov, Y. L. Gol'dfarb and I. P. Konyaeva, Izv. Akad. Nauk SSSR, Ser. Khim., 1980, 2, 372–377 Search PubMed.
- (a) H. Kong, Y. K. Jung, N. S. Cho, I.-N. Kang, J.-H. Park, S. Cho and H.-K. Shim, Chem. Mater., 2009, 21, 2650–2660 CrossRef CAS; (b) M. D. Curtis, J. Cao and J. W. Kampf, J. Am. Chem. Soc., 2004, 126, 4318–4328 CrossRef CAS PubMed; (c) E. G. Kim, V. Coropceanu, N. E. Gruhn, R. S. Sanchez-Carrera, R. Snoeberger, A. J. Matzger and J. L. Bredas, J. Am. Chem. Soc., 2007, 129, 13072–13081 CrossRef CAS PubMed.
- (a) S. Yamaguchi, S. Akiyama and K. Tamao, J. Organomet. Chem., 2002, 652, 3–9 CrossRef CAS; (b) P. Chen and F. Jäkle, J. Am. Chem. Soc., 2011, 133, 20142–20145 CrossRef CAS PubMed; (c) P. Chen, X. Yin, N. Baser-Kirazli and F. Jäkle, Angew. Chem., Int. Ed., 2015, 54, 10768–10772 CrossRef CAS PubMed.
- (a) T. W. Hudnall, C.-W. Chiu and F. P. Gabbaï, Acc. Chem. Res., 2009, 42, 388–397 CrossRef CAS PubMed; (b) T. Agou, J. Kobayashi and T. Kawashima, Chem.–Eur. J., 2007, 13, 8051–8060 CrossRef CAS PubMed; (c) S. Miyasaka, J. Kobayashi and T. Kawashima, Tetrahedron Lett., 2009, 50, 3467–3469 CrossRef CAS; (d) Z. Zhang, R. M. Edkins, J. Nitsch, K. Fucke, A. Eichhorn, A. Steffen, Y. Wang and T. B. Marder, Chem.–Eur. J., 2015, 21, 177–190 CrossRef CAS PubMed; (e) D. R. Levine, M. A. Siegler and J. D. Tovar, J. Am. Chem. Soc., 2014, 136, 7132–7139 CrossRef CAS PubMed; (f) H. Shia, J. Yanga, X. Donga, L. Fanga, C. Donga and M. M. F. Choib, Synth. Met., 2013, 179, 42–48 CrossRef; (g) Z.-Q. Liu, M. Shi, F.-Y. Li, Q. Fang, Z.-H. Chen, T. Yi and C.-H. Huang, Org. Lett., 2005, 7, 5481–5484 CrossRef CAS PubMed.
- (a) M. Miyata and Y. Chujo, Polym. J., 2002, 34, 967–969 CrossRef CAS; (b) A. Sundararaman, M. Victor, R. Varughese and F. Jäkle, J. Am. Chem. Soc., 2005, 127, 13748–13749 CrossRef CAS PubMed; (c) V. D. B. Bonifacio, J. Morgado and U. Scherf, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2878–2883 CrossRef CAS; (d) W. Liu, M. Pink and D. Lee, J. Am. Chem. Soc., 2009, 131, 8703–8707 CrossRef CAS PubMed; (e) K. Parab, K. Venkatasubbaiah and F. Jäkle, J. Am. Chem. Soc., 2006, 128, 12879–12885 CrossRef CAS PubMed; (f) K. Parab and F. Jäkle, Macromolecules, 2009, 42, 4002–4007 CrossRef CAS; (g) F. Guo, X. Yin, F. Pammer, F. Cheng, D. Fernandez, R. A. Lalancette and F. Jäkle, Macromolecules, 2014, 47, 7831–7841 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (c) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- E. Ravindran, S. J. Ananthakrishnan, E. Varathan, V. Subramanian and N. Somanathan, J. Mater. Chem. C, 2015, 3, 4359–4371 RSC and references 65–68 therein.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- (a) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS; (b) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- I. Borges, E. Uhl, L. Modesto-Costa, A. J. A. Aquino and H. Lischka, J. Phys. Chem. C, 2016, 120, 21818–21826 CAS.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- (a) A. F. Holleman, E. Wiberg and N. Wiberg, Lehrbuch der Anorganischen Chemie, Walter de Gruyter & Co., Berlin, 2007 Search PubMed; (b) H. Pang, P. J. Skabara, S. Grodeyev, J. J. W. McDouall, S. J. Coles and M. B. Hursthouse, Chem. Mater., 2007, 19, 301–307 CrossRef CAS.
- (a) M. Heeney, W. Zhang, S. Tierney and I. McCulloch, PCT Int. Appl., WO 2008 077465 A2 20080703, 2008; (b) S. P. Mishra, A. E. Javier, R. Zhang, J. Liu, J. A. Belot, I. Osaka and R. D. McCullough, J. Mater. Chem., 2011, 21, 1551–1561 RSC.
- (a) G. H. V. Bertrand, V. K. Michaelis, T.-C. Ong, R. G. Griffin and M. Dinca, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4923–4928 CrossRef CAS PubMed; (b) M. Heeney, W. Zhang, S. Tierney and I. McCulloch, PCT Int. Appl., WO 2005121150A1 20051222, 2005; (c) X. Zhang, K. Markus and J. M. Adam, Macromolecules, 2004, 37, 6306–6315 CrossRef CAS; (d) M. O. Ahmed, W. Pisula and S. G. Mhaisalkar, Molecules, 2012, 17, 12163–12171 CrossRef CAS PubMed.
- C. Lo and C. Hsu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3355–3365 CrossRef CAS.
- (a) J. B. Heilmann, M. Scheibitz, Y. Qin, A. Sundararaman, F. Jäkle, T. Kretz, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2006, 45, 920–925 CrossRef CAS PubMed; (b) M. Scheibitz, H. Li, J. Schnorr, A. S. Perucha, M. Bolte, H.-W. Lerner, F. Jäkle and M. Wagner, J. Am. Chem. Soc., 2009, 131, 16319–16329 CrossRef CAS PubMed.
- X. Zhang and A. J. Matzger, J. Org. Chem., 2003, 68, 9813–9815 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01793f |
| This journal is © The Royal Society of Chemistry 2017 |