DOI:
10.1039/C7RA01778B
(Paper)
RSC Adv., 2017,
7, 19288-19295
Non-swellable self-healing polymer with long-term stability under seawater†
Received
13th February 2017
, Accepted 22nd March 2017
First published on 31st March 2017
Abstract
Polymers are widely used in marine environments due to their unique thermal/mechanical properties. Imparting self-healing ability to those polymers would be a beneficial strategy for improved durability; e.g. extending the life time and reducing the maintenance cost. However, most of the existing self-healing polymers suffer from swelling-induced mechanical instability and loss of self-healing ability because of a severe water uptake in fully submerged conditions. Thus, a judicious design principle to prevent the swelling-induced deterioration of the self-healing polymers is greatly required for the long-term use in water-related applications. In this study, we report a polymer that is non-swellable (<2 wt% of swelling) and highly self-healable (91% based on toughness) under seawater. Dynamic crosslinking of catechol-functionalized polymers with p-phenyldiboronic acid (PDBA) through non-ionic boronate ester bonds is the key to realizing these two properties simultaneously. Atomic force microscopy and transmission electron microscopy reveal that the ionically crosslinked catechol polymers contain hydrophilic aggregates of metal (Ca2+)–catechol complexes, referred to as ion-clusters. In contrast, we find the non-ionic boronate ester crosslinkers are not aggregated but finely dispersed in hydrophobic catechol polymer matrix, allowing materials to self-heal with high water stability. Even in seawater, the PDBA-containing catechol polymer retained its mechanical properties for at least 1 month. The polymer described here could provide a valuable strategy for further development of polymeric materials used in marine industries.
Introduction
Polymers have been widely used in marine environments (e.g. the fishing industry, the shipping industry, offshore facilities, etc.) due to their lightweight nature, good processability, and unique thermal/mechanical properties.1 To date, research on long-term stability,2 antifouling,3 and biodegradation4 abilities for polymeric materials used in seawater applications has been conducted to meet the increasing demand for materials with high durability and advanced functionality.
Imparting a self-healing ability to polymers, which causes them to be able to repair damage without human intervention, would be a beneficial strategy for extending the lifetime and reducing the maintenance cost for polymers used in marine environments. Over the past few decades, the ability to self-heal in dry conditions has been extensively studied for polymers with reversible bonds.5–10 Recently, self-healing in wet conditions was also achieved in a few reports in which the water triggered dynamics of a hydrogen bond interaction,11 a boronic ester bond12 and metal–ligand co-ordinations13,14 were used to facilitate the repair. Ahn and co-workers proved that the hydrogen bonding interaction can be used in self-healable bulk polymers under wet conditions.11 By deprotection of silyl-protecting groups in acidic condition, the newly generated hydrogen bonds between catechol groups led cracked surfaces to heal. Cash and co-workers12 reported that a boronic ester bond was used as a dynamic crosslinker within hydrophobic network polymers, and successful self-healing was achieved with water treatment at the fractured surfaces.
Despite these advances, accomplishing self-healing in fully submerged environments without any external stimuli is still challenging because water-assisted healing is usually accompanied by swelling. In the case of hydrophilic polymers, swelling with water enhances the polymer chain mobility, leading to a higher probability of reformation of dynamic bonds and to successful self-healing at cracked interfaces. However, this swelling also induces severe structural deformation15 and mechanical property changes,16 causing these materials to fail during underwater use. Recently, our group13 and Xia and co-workers14 investigated catechol polymers in the bulk state and revealed relatively high self-healing efficiencies triggered by seawater. Enhanced chain mobility by water absorption and the dynamics of the catechol–metal (i.e., Ca2+, Mg2+, and Fe3+) complexes of these polymers led to successful healing efficiency, suggesting that catechol-containing polymers can be used as tough, self-healing materials in wet conditions. Nevertheless, severe water uptake occurred when these materials were immersed in seawater (∼20 wt%),13,14 indicating that a significant challenge remains for their use under the sea.
Although hydrophobic polymers maintain their intrinsic properties in water without swelling, the formation of a water boundary layer at cracked surfaces drastically weakens the van der Waals interactions, which results in interruption of the reformation of the dynamic bonds.17 Therefore, a judicious polymer design with an appropriate hydrophobicity along with water-triggered dynamic bonds is crucial for obtaining underwater self-healing elastomers with high stability.
To overcome this challenge, we herein develop a highly self-healable tough polymer which can be used under seawater. For achieving such properties, the effect of the electrical nature of dynamic bonds on the water stability of self-healing elastomers is considered. The catechol-based polymer was synthesized as a hydrophobic matrix, and two different crosslinkers were chosen; one is a metalloid (boron) and the other is a metal (calcium). The elastomer networked with the p-phenyldiboronic acid (PDBA) shows non-swellable (<2 wt% water absorption) and self-healable (91% efficiency) properties under seawater with stable mechanical properties. Such healable properties in fully submerged conditions have never been achieved before. Contrary, the one networked with the Ca2+ suffered from the swelling-induced mechanical deterioration. We relate these phenomena to the mesoscopic structure differences revealed by atomic force microscopy (AFM) and transmission electron microscopy (TEM).
Experimental section
Reagents and materials
As solvents, we used methanol, dichloromethane, chloroform, acetone, hexane, ethyl acetate, N,N-dimethylformamide (DMF, dehydrated), pyridine (dehydrated), ethanol (dehydrated), and tetrahydrofuran (THF, dehydrated, stabilizer-free), all of which were obtained from Wako Pure Chemical Industries, Ltd. Acryloyl chloride, triethylamine (TEA), butyl acrylate, dimethyl 2,2′-azobis(2-methylpropionate), p-phenyldiboronic acid and calcium chloride were also obtained from Wako Pure Chemical Industries, Ltd. Acrylic acid was purchased from Sigma-Aldrich. Dopamine hydrochloride was purchased from Tokyo Chemical Industry Co., Ltd. Butyl acrylate was purified by passing it through basic alumina before use. The artificial seawater of which contents are listed in Table S1† was used for self-healing, swelling and long-term stability tests.
Characterization
1H NMR spectra were recorded on a JNM-AL 400 (JEOL RESONANCE Inc.) in deuterated DMSO at 20 °C. Solid-state 11B NMR spectra were recorded on an ECA-500 (JEOL RESONANCE Inc.) with a 4 mm zirconia rotor. The magic angle spinning speed was 16 kHz. The saturated H3BO3 aqueous solution (−19.49 ppm) served as an external chemical shift standard. Gel permeation chromatography (GPC) data were recorded on a HLC-8220 GPC (TOSOH Co.) using 10 mM LiCl in DMF as the eluent. A calibration curve was prepared by using polystyrene standards. Attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectra were obtained with a Thermo Scientific Nicolet iS10 FTIR spectrometer equipped with a Thermo Scientific Smart iTR with a diamond crystal. Differential scanning calorimetry (DSC) data were measured using a SEIKO DSC6220N at a heating rate of 20 °C min−1 under nitrogen (flow rate = 5 mL min−1). The data were recorded from the second heating process (−50 °C to 150 °C) after a single scan (−50 °C to 80 °C). Tensile tests were conducted by SHIMADZU AGS-X 100 N and the dimensions of the dumbbell-shaped sample used are shown in Fig. S5.† The strain rate of 30 mm min−1 was adopted under ambient condition (25 °C, 60% of relative humidity). Each measurement was repeated at least three times. Water contact angle measurements were operated by DMs-401 (Kyowa Interface Science Co., Ltd.). A droplet of deionized water (1 μL) was used for each measurement with high-resolution video. The initial contact angle was measured at 100 ms after water dropping. Data are mean of more than 3 measurements. AFM measurements were operated by a Nanocute (SII Nano Technology, Inc.) with a self-sensitive micro-cantilever PRC-DF40P, using the dynamic force mode. TEM images were obtained with a JEM-2010 TEM (JEOL, Ltd.), operating at 200 kV. Self-healing image and the surface roughness of films were observed using a laser scanning microscopy (VK-X210, Keyence).
Preparation of catechol containing polymers
The procedure was slightly modified from our previous report.13 To a nitrogen-purged 200 mL Schlenk flask, 7.46 g of dopamine acrylamide (DA, 36 mmol), 410 mg of dimethyl 2,2′-azobis(2-methylpropionate) (1.8 mmol), 46.7 mL of n-butyl acrylate (BA, 324 mmol), and 150 mL of anhydrous ethanol was added. The solution was degassed via three freeze–pump–thaw cycles and backfilled with nitrogen. Then the solution was placed in an oil bath at 75 °C and stirred for 24 h. The solution was quenched by liquid nitrogen and reduced in volume to ∼50 mL by rotary evaporation under reduced pressure. THF (30 mL) was then added, and the resulting solution was precipitated by pouring into 0 °C hexane. The precipitate was collected and dried under vacuum to yield a yellowish liquid-like product (42.5 g, yield 87%). 1H NMR (400 MHz, DMSO-d6, δ): 2.17–0.88 (93H, aliphatic region), 2.47 (2H, CONH–CH2–CH2), 3.12 (2H, CONH–CH2–CH2), 3.97 (18H, COO–CH2–CH2–CH2–CH3), 6.62–6.40 (3H, Ar), 7.86 (1H, NH), 8.71 (2H, ArOH). GPC (10 mM LiCl in DMF as eluent, polystyrene standards): Mn = 79![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Mw/Mn = 1.9.
000, Mw/Mn = 1.9.
Preparation of network polymers
In a typical film preparation of P-PDBA 100% and P-Ca2+ 100%, 1.8 g of P(DA-co-BA) (containing 1.33 mmol catechol) was dissolved in 60 mL of chloroform. Then 3 mL of a mixture of cross-linker in methanol (PDBA 110 mg or CaCl2 74 mg) and 0.185 mL of TEA (1.33 mmol) was added. The solution was stirred for 24 h, poured into a Teflon dish, and allowed to evaporate for 24 h at room temperature. The sample was then put in a vacuum oven at 50 °C for another 24 h to remove the remaining solvent. A film with an average thickness of 0.3 mm was prepared by thermal annealing (60 °C, 30 min) with compression between two sheets of Teflon (12.5 MPa).
Sample preparation for AFM
Glass slides (18 × 18 mm) were first washed with deionized (DI) water under sonication and then immersed in Piranha solution (3/1 v/v mixture of 96% sulfuric acid and 30% H2O2) for 1 h to remove organic impurities. (CAUTION: Piranha solution can react violently with organic compounds, and should be handled with extreme care.) The glass slides were rinsed with DI water, sonicated in DI water, and then dried in air. The casting solution for bulk film is diluted 3-fold with CHCl3 and spin-coated onto the washed glass slide at 2000 rpm for 20 s and then dried in air for 2 h, in vacuum chamber at room temperature for 2 h and annealed at 60 °C for 30 min. The dried sample was kept inside the desiccator until AFM observation.
Sample preparation for TEM
The solution for bulk film is diluted 60-fold with CHCl3 and drop-casted onto the carbon coated copper grid. After drying out the solvent under ambient air for 24 h, sample was annealed at 60 °C for 30 min. The dried sample was kept inside the vacuum chamber until TEM observation.
Self-healing tests
Dumbbell-shaped samples were cut into two separate sections, and exposed surfaces were activated with a drop of seawater then rejoined gently under air. Reattached samples were fully immersed in seawater. After the desired period of time (1–3 days), samples were taken out from seawater and dried under vacuum for 12 h and then in a desiccator for another 12 h to remove any remaining water. Before tensile tests, samples were placed in ambient air (25 °C, 60% of relative humidity) for at least 30 min then subjected to tensile tests.
Swelling tests
Square-shaped samples (10 × 10 mm) were cut from films with a pair of scissors. The samples were immersed in artificial seawater or deionized water for 14 days. Before weighing, the samples were carefully wiped with absorbent paper. Swelling ratios were calculated according to the equation: 100 × (Wmeasured − Winitial)/Winitial, where Wmeasured is the measured weight and Winitial is the initial weight.
Results and discussion
Polymer design
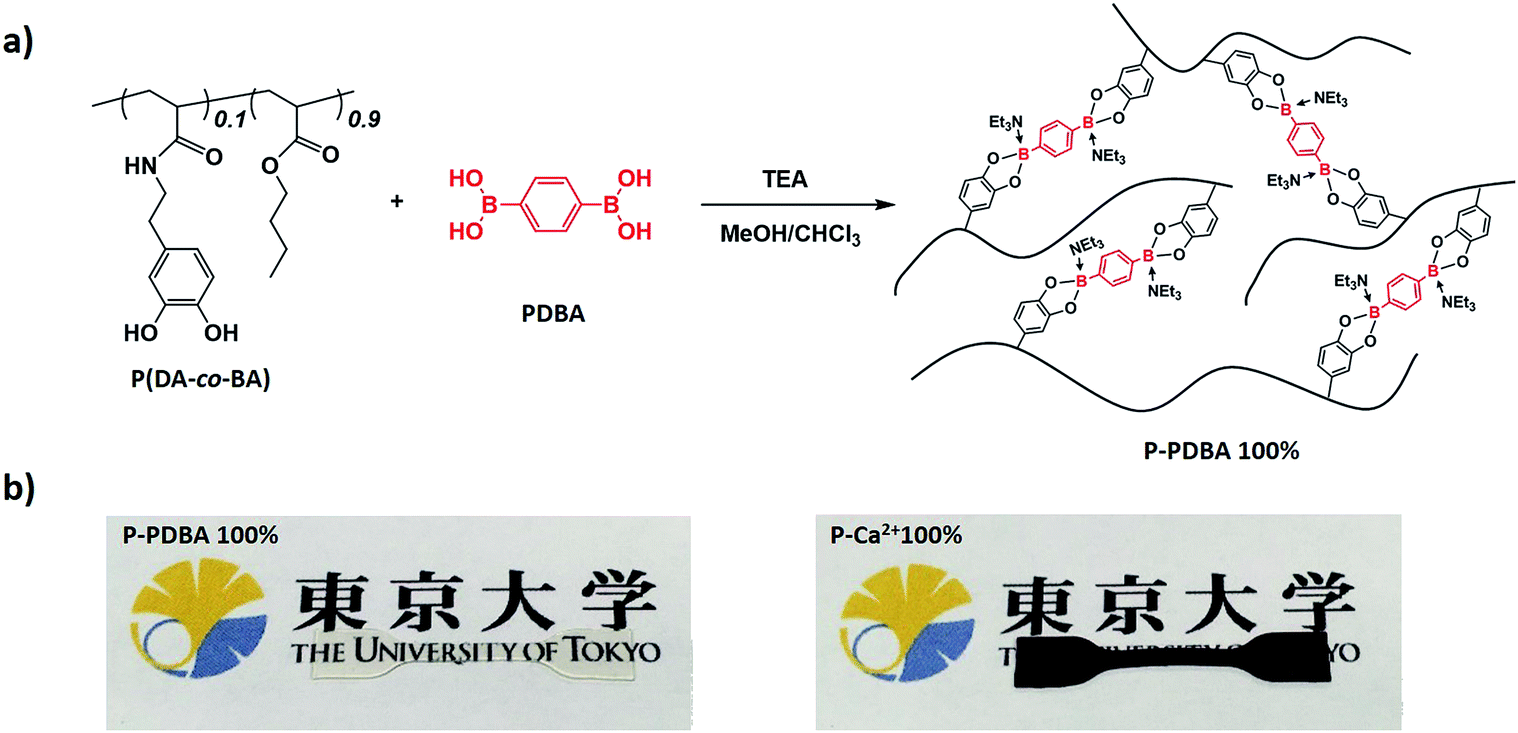
The catechol containing polymer was synthesized as a hydrophobic polymer chain, because catechol is an adaptable ligand which can chelate with various metals18–21 and metalloids.22 For this purpose, poly(dopamine acrylamide-co-n-butyl acrylate) [P(DA-co-BA)] was prepared by a conventional free radical copolymerization of n-butyl acrylate (BA) and dopamine acrylamide (DA) (Scheme S1†). 1H NMR revealed that the comonomer composition of 9:1 (BA:DA) in the resulting product, closely corresponded to the feed ratio of the monomers (Fig. S1†). For the preparation of the crosslinked polymer, the P(DA-co-BA) (1.8 g, containing 1.33 mmol catechol) in chloroform (60 mL) was mixed with a PDBA (110 mg, 0.67 mmol) in methanol (3 mL). Triethylamine (TEA, 1.33 mmol, 2 equiv. of PDBA) was also added to this solution for the effective formation of non-ionic boronate ester bonds.23 This sample is abbreviated P-PDBA 100% based on a bidentate coordination type between catechol moieties in the polymer chain and PDBA. Compositions containing 80 and 60% feed ratios of PDBA were also prepared. A polymer film was then prepared by solution casting on a Teflon dish followed by thermal annealing with compression to yield cross-linked bulk films with high transparency (Fig. 1a). A P(DA-co-BA) film crosslinked with Ca2+ was also prepared by adding CaCl2 in place of PDBA, as a control which forms typical ionic metal–ligand interactions in the polymer matrix (abbreviated P-Ca2+ 100%). Unlike the transparent P-PDBA 100%, the P-Ca2+ 100% film was opaque and dark brown in color (Fig. 1b). The Tg values of all the network polymers were below −10 °C, as determined by differential scanning calorimetry (DSC) measurements (Table 1).
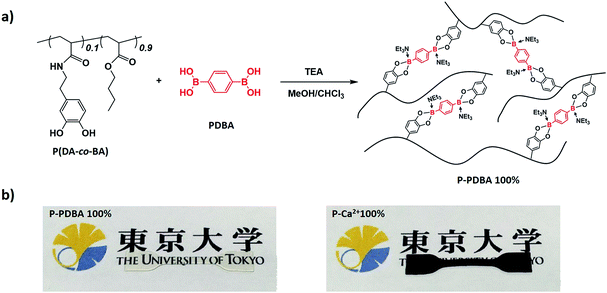 |
| | Fig. 1 (a) Synthesis of a catechol-containing polymer crosslinked with PDBA. (b) P-PDBA 100% has high transparency (left), whereas P-Ca2+ 100% shows opaque and dark brown in color (right). | |
Table 1 Summary of mechanical and thermal properties of polymers
| Sample |
Young's modulusa [MPa] |
Break strength [MPa] |
Strain at break [mm mm−1] |
Toughnessb [MJ m−3] |
Tg [°C] |
| Calculated from the initial slope of stress strain curves. Integration of the area under the stress–strain curves. 100% indicates 100% coordination feed ratio. Unable to measure because it is too soft. |
| P-PDBAc 100% |
2.3 ± 0.3 |
2.8 ± 0.2 |
5.1 ± 0.4 |
10.6 ± 1.4 |
−11.8 |
| P-PDBA 80% |
1.3 ± 0.2 |
2.2 ± 0.2 |
11.1 ± 1.5 |
10.0 ± 0.4 |
−13.5 |
| P-PDBA 60% |
0.2 ± 0.03 |
1.2 ± 0.04 |
13.9 ± 3.7 |
9.2 ± 1.7 |
−17.3 |
| P-Ca2+ 100% |
0.6 ± 0.01 |
2.3 ± 0.1 |
4.5 ± 0.5 |
5.4 ± 0.8 |
−14.1 |
| P(DA-co-BA) |
—d |
—d |
—d |
—d |
−19.3 |
Network characterization
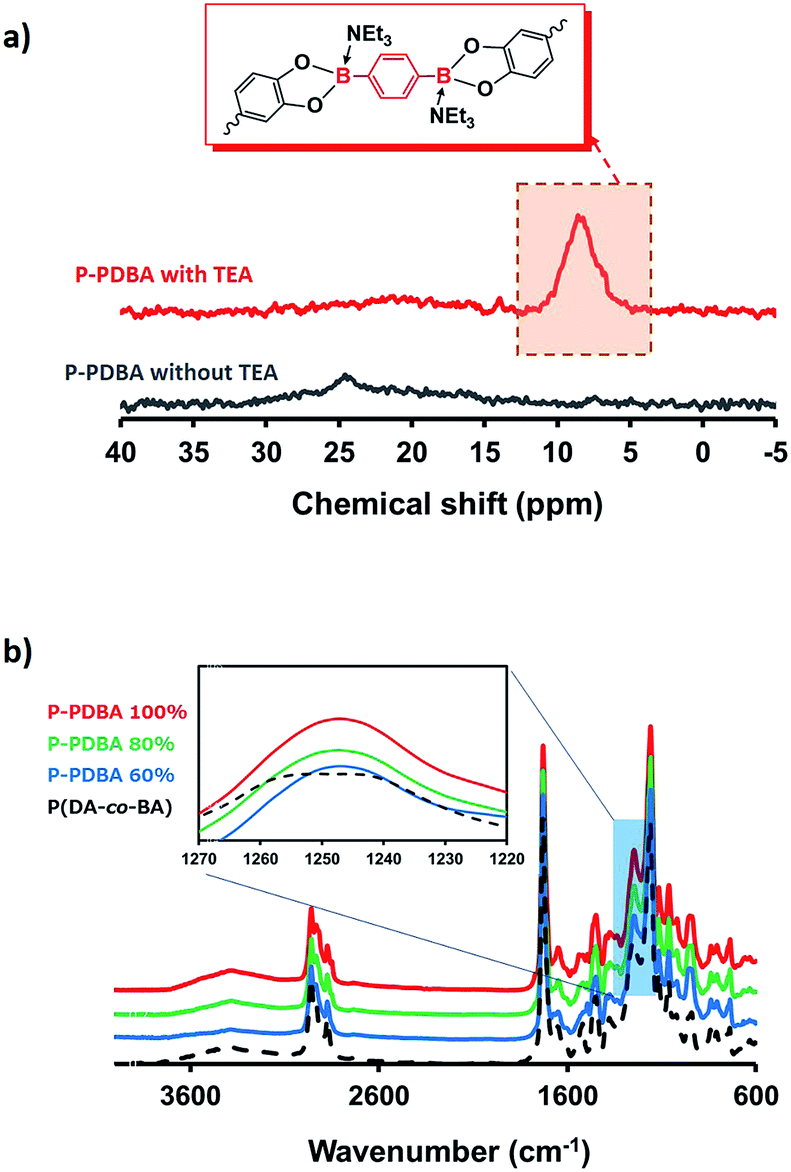
Characterization using solid-state NMR. Formation of the boronate ester bonds in P-PDBA 100% was confirmed by solid-state single-pulse 11B NMR spectroscopy (Fig. 2a). A sharp signal was detected at 8.6 ppm, which supported the formation of the tetrahedral sp3 boron center by B–N dative bond.24–26 When a mixture of P(DA-co-BA) and PDBA was prepared without TEA, a robust film was not formed (Fig. S2†), and only a broad signal at 24 ppm was observed in the 11B NMR spectrum (Fig. 2a). These results indicate that most of the PDBA was only reacted in the presence of TEA to contribute to the crosslinking.
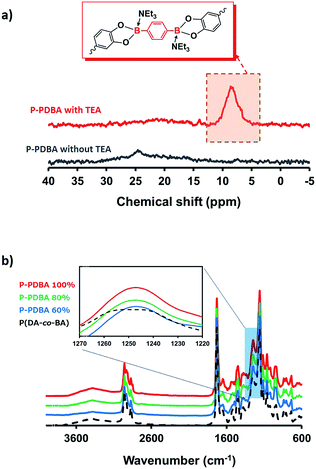 |
| | Fig. 2 (a) Solid-state 11B NMR of P-PDBA 100% with and without TEA. (b) ATR-FTIR spectra of P-PDBA polymers containing different amounts of the cross-linker (PDBA). | |
Characterization using attenuated total reflectance-Fourier transform-infrared spectrometry (ATR-FTIR). ATR-FTIR spectroscopy of polymers with varying feed ratios of PDBA (60, 80 and 100%) also proved coordination between the catechol and boronic acid moieties. The ATR-FTIR spectra in Fig. 2b shows that two δ(C–OH) bands at 1243 and 1254 cm−1 resulting from the phenolic groups of catechol in P(DA-co-BA) were merged into a single peak at 1248 cm−1 in the P-PDBA samples as a result of esterification between the catechol and boronic acid groups.27
Mechanical properties of P-PDBA polymers
The mechanical properties of the P-PDBA samples were determined by tensile tests and dynamic mechanical studies. Table 1 shows the tensile test results of the P-PDBA polymers. As the amount of added PDBA increased, higher Young's modulus and break strength values were obtained. Among the P-PDBAs, P-PDBA 100% displayed the toughest mechanical properties (10.6 MJ m−3), while P-PDBA 80% and 60% still exhibited fairly high toughness (10.0 and 9.2 MJ m−3, respectively). In Fig. S3,† the heights of the loss tangent peaks, which relate to the relaxation of the polymer chain, were compared. By increasing the feed ratio of PDBA, the height of the peak decreased, suggesting that relaxation of the polymer chain was restricted to a larger degree by higher crosslinking densities.
Self-healing properties of P-PDBA polymers
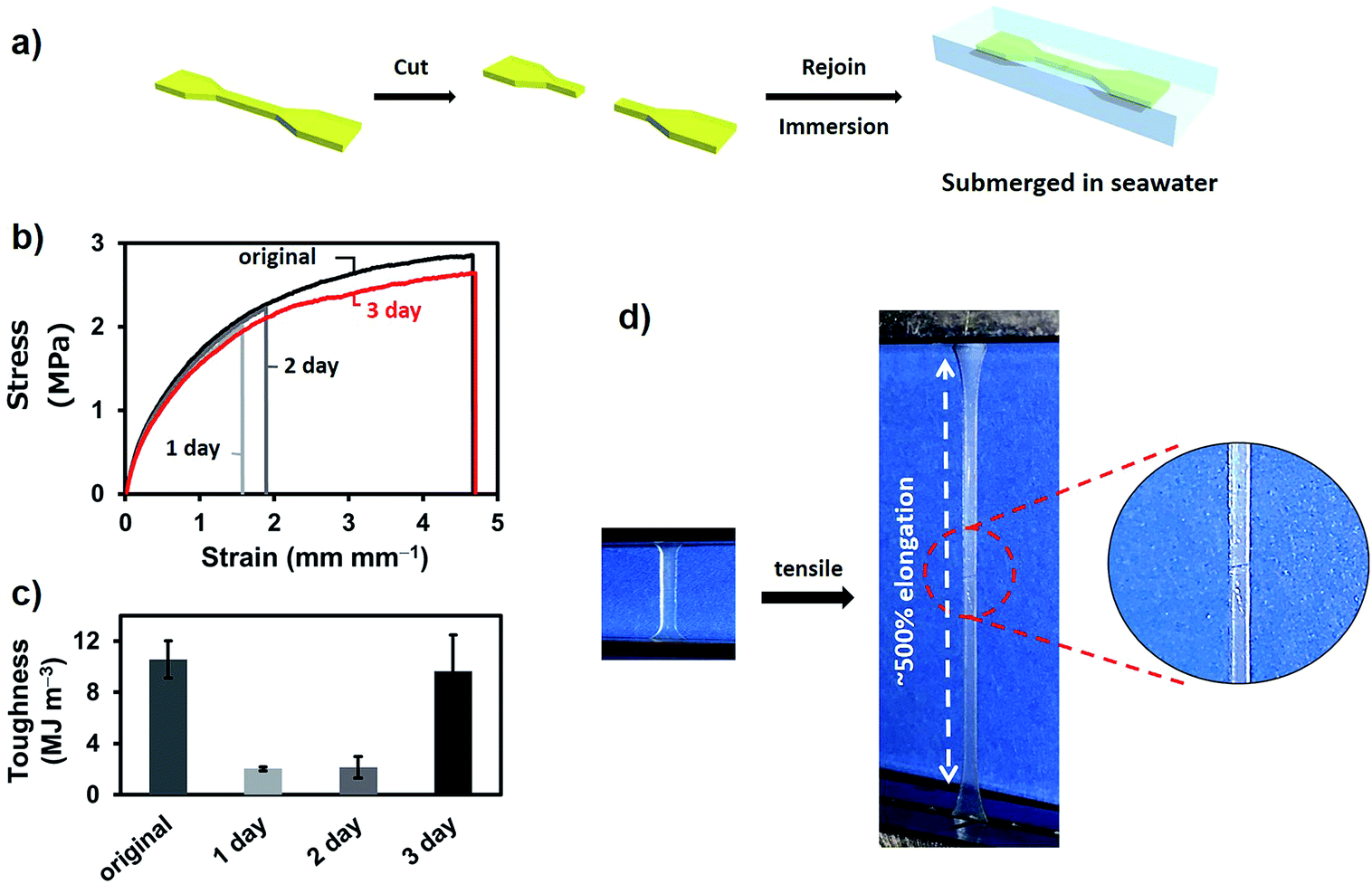
The self-healing test in seawater was performed with P-PDBA 100%. Because this polymer possessed the highest strength and the highest Young's modulus measured in this study, P-PDBA 100% was considered to be the most challenging when it came to displaying self-healing properties. Dumbbell-shaped films were cut completely into two pieces, and the exposed surfaces were wetted with seawater before gently rejoining the two pieces. The rejoined films were then fully immersed in seawater for 1–3 days (Fig. 3a). After drying, tensile tests of the films were conducted. The strain–stress curve of the P-PDBA 100% sample that had healed for 3 days showed almost quantitative recovery of the mechanical properties as compared to the original uncut sample (Fig. 3b). The efficiency of healing, which was quantified as the percent recovery of toughness, was 91% (Fig. 3c). As a control test excluding the water effect in self-healing, some cut P-PDBA 100% samples were rejoined without exposure to seawater and kept in a desiccator (<20% relative humidity) for 3 days. As shown in Table S2,† the healing efficiency for these samples was less than 2%.
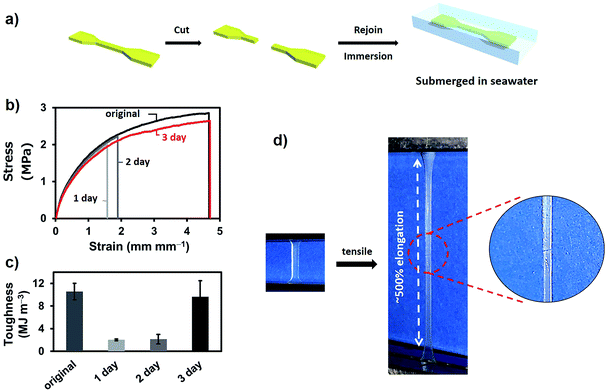 |
| | Fig. 3 (a) The self-healing test protocol. (b) Tensile stress–strain curves of original and healed polymers (1–3 days). (c) Toughness of the original and healed (for 1–3 days) samples. (d) Photographs of the tensile test of P-PDBA 100% after 3 days of healing in seawater. | |
Water stability studies of network polymers
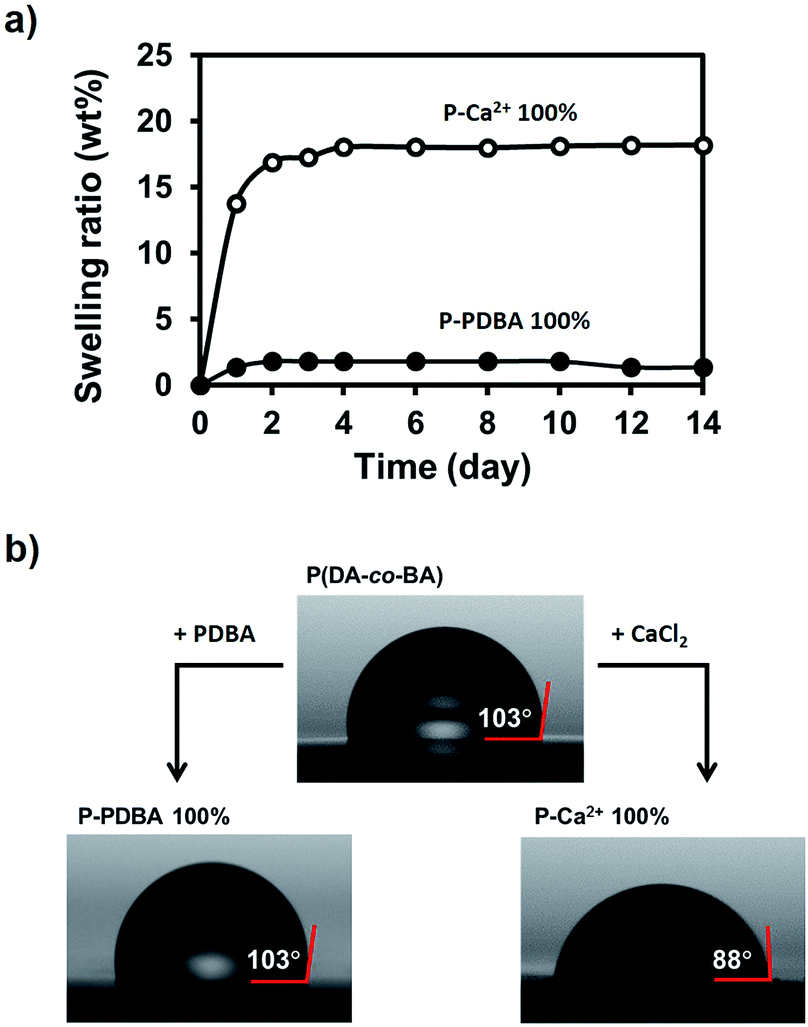
Swelling tests under seawater. The water stability of P-PDBA 100% and P-Ca2+ 100% in seawater was also examined through the estimation of the swelling ratio and water contact angle. For swelling tests, P-PDBA 100% and P-Ca2+ 100% were completely immersed into seawater, and the weight change was measured regularly until no further change in weight was observed. As shown in Fig. 4a, the P-PDBA 100% film reached an equilibrium after 2 days and was swollen at most up to 2 wt%, whereas P-Ca2+ 100% was swollen up to ∼20 wt%.
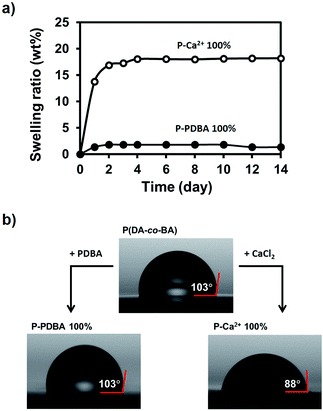 |
| | Fig. 4 (a) The swelling ratio of P-PDBA 100% and P-Ca2+ 100% in seawater over a 2 week period. (b) Water contact angles of P(DA-co-BA), P-PDBA 100%, and P-Ca2+ 100%. | |
Water contact angle tests. To measure the water contact angle, a droplet of deionized water (1 μL) was dropped onto the surfaces of the films, and the contact angles were measured (Fig. 4b). The contact angle of P-PDBA 100% was 103°, revealing the hydrophobicity of its surface. In comparison, the contact angles of P-Ca2+ 100% and non-crosslinked P(DA-co-BA) were 88° and 103°, respectively. Hence, the hydrophobicity of P-PDBA 100% was no different from P(DA-co-BA), while P-Ca2+ 100% was found to be less hydrophobic. The swelling tests and water contact angle measurements indicate that P-PDBA 100% possesses water stability with remarkably lower water absorption and higher hydrophobicity than P-Ca2+ 100%, even though they are made from the same polymer; thus the high water stability of P-PDBA 100% cannot be fully explained by the hydrophobicity of the polymer chain.
Morphological characterizations of network polymers
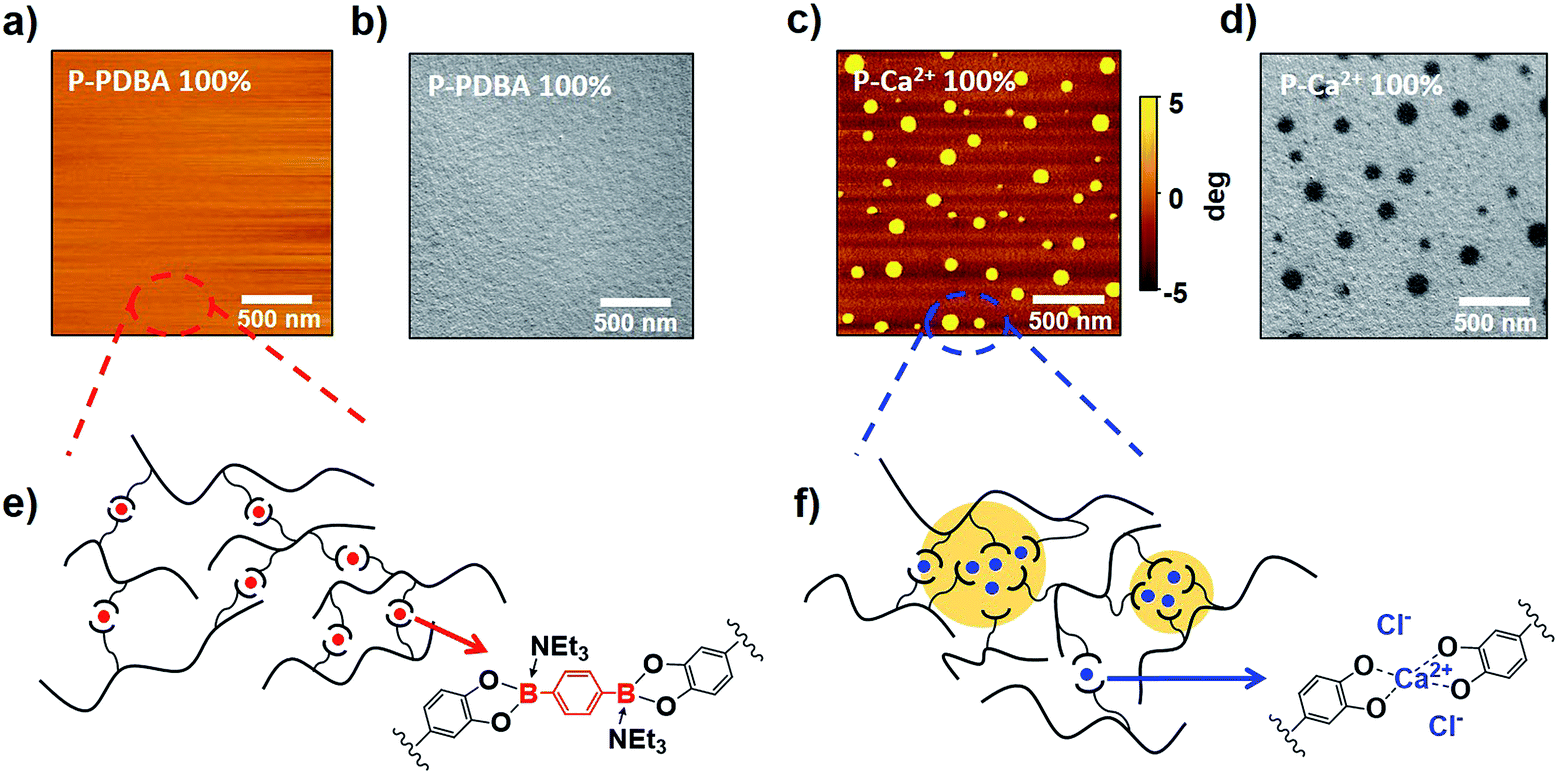
To study the origin of the superior water stability of P-PDBA 100% and the different water-resistivity of the two similar polymers, their mesoscopic morphologies were visualized by atomic-force microscopy (AFM, Fig. 5a and c) and transmission electron microscopy (TEM, Fig. 5b and d). Interestingly, the TEM and AFM images clearly showed a microphase-separated morphology in the P-Ca2+ 100% thin films. As granular domains (<100 nm) appeared darker in the TEM images (Fig. 5d), these domains had higher electron density, presumably due to the concentrated Ca2+ aggregates in the polymer matrix. In contrast, a notable phase-separation was not observed in the images of P-PDBA 100%. These results indicate that the PDBA crosslinkers were finely dispersed in the polymer matrix as crosslinks, and the hydrophobicity of the polymer backbone itself could be well-maintained in the P-PDBA 100% structure. On the contrary, the electrostatic interactions between ions (i.e. catechol–Ca2+ complexes and Cl−) in P-Ca2+ 100% could generate metal-concentrated domains (ion clusters) where the water uptake likely takes place due to the high dielectric constants, even though the polymer chain possessed hydrophobic properties (Fig. 5f).28,29 In the case of P-PDBA 100%, covalently cross-linked PDBAs form sterically hindered tetrahedral boronate ester moieties with fully occupied sp3 orbitals; thus, no strong interactions could be formed between crosslink points in the solid state. Therefore, the PDBA crosslinkers were finely dispersed into the local hydrophobic environment, leading to highly suppressed water absorption (Fig. 5e).
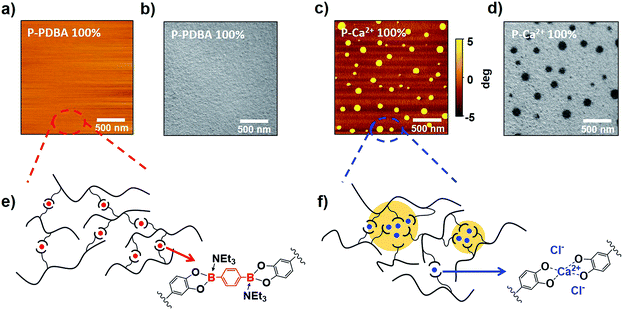 |
| | Fig. 5 (a) AFM phase images of P-PDBA 100%. (b) TEM images of P-PDBA 100%. (c) AFM phase images of P-Ca2+ 100%. (d) TEM images of P-Ca2+ 100%. (e) Covalently crosslinked P-PDBA 100% with finely dispersed crosslinkers. (f) Ionically crosslinked P-Ca2+ 100% with aggregation of crosslinkers (ion clusters). | |
Stability of mechanical properties under seawater
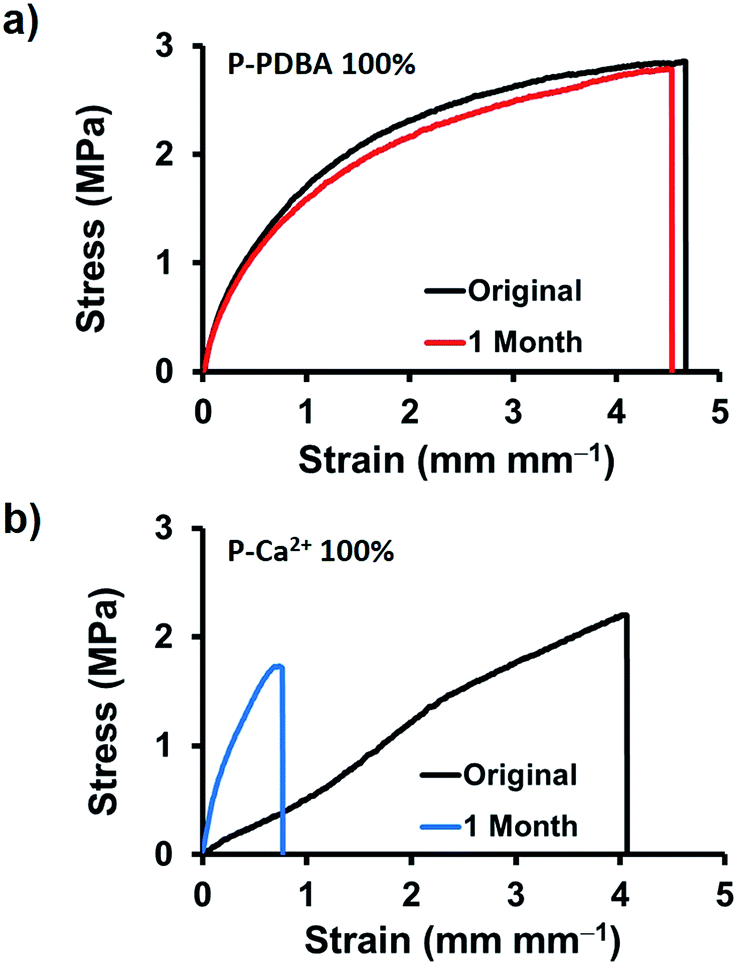
The stability of the mechanical properties of network polymers under seawater was examined. First, dumbbell-shaped films were stored in seawater for 3 days then tensile tests were conducted without drying process. As shown in Table S4,† even though Young's modulus and break strength were decreased when compared with their original properties, the strain at break of P-PDBA 100% was not changed with relatively high toughness (∼2 MJ m−3). However, when the submerged samples were dried before tensile tests, only P-PDBA 100% showed nearly recovered mechanical properties. For further checking the under seawater stability of network polymers, 1 month storage tests were also carried out with drying process. Remarkably, as shown in Fig. 6a, the mechanical properties were completely unchanged (Young's modulus, break strength, and strain at break; see Table S5†), and its toughness was fully maintained (∼100% of original sample). In contrast, the P-Ca2+ 100% showed deteriorated mechanical properties (increased Young's modulus but significantly decreased strain at break) in the storage under seawater (Fig. 6b). These results indicate that the network changes of P-PDBA 100% (e.g. degradation or oxidation of polymer matrix) are highly suppressed under seawater conditions and even though mechanical properties can be affected by the hydrolysis of boron–catechol crosslinks through marginal water absorption (∼2 wt%), they can be fully recovered with dry process.
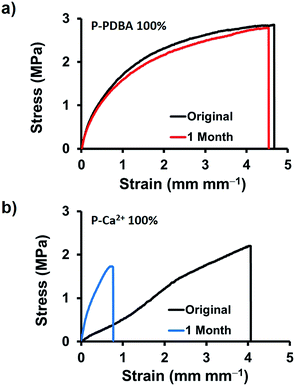 |
| | Fig. 6 Long-term stability under seawater. Tensile stress–strain curves of (a) P-PDBA 100%, and (b) P-Ca2+ films incubated in seawater for 1 month. | |
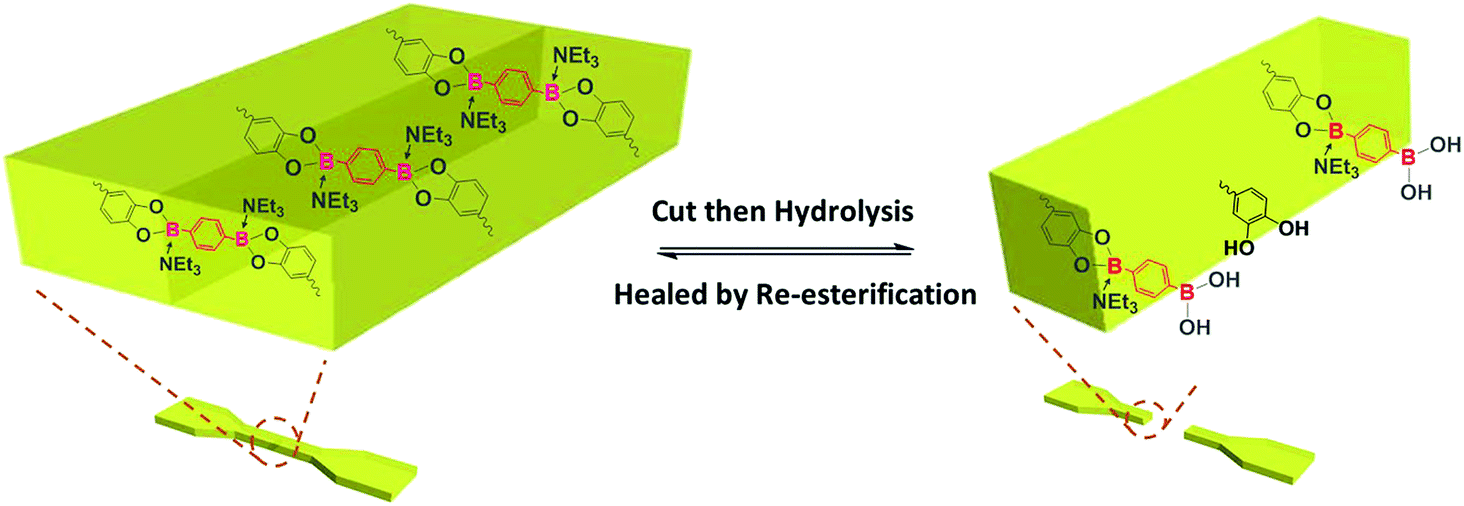
Self-healing mechanism without water absorption
Taken together, the shape and mechanical properties of P-PDBA 100% were well-maintained in seawater because of the suppressed water absorption. The surface of P-PDBA 100%, however, slightly swells with marginal water absorption, which is the key factor for its successful self-healing. The mean surface roughness (Ra) of P-PDBA 100% film was increased after incubation in seawater for three days (Fig. S4†). A possible mechanism for healing behavior is illustrated in Fig. 7. Absorbed water enhances the dynamics of esterification between the catechol and boronic acid moieties and shifts the equilibrium of the reactions slightly toward hydrolysis, resulting in the exposure of free catechol and boronic acid groups on the surface. By attaching the cut surfaces, the functional groups are allowed to form the boronate ester bonds again by esterification. This healing mechanism only works at the outermost surfaces of the crack, where the polymer is water-swollen.
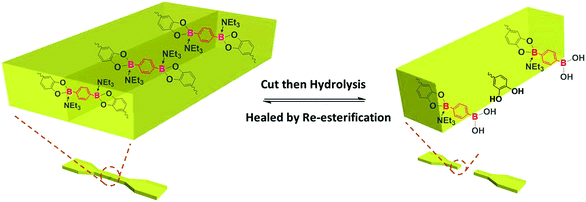 |
| | Fig. 7 Illustration of the water-triggered self-healing process. | |
Conclusions
In summary, we designed and synthesized tough, self-healing, and non-swellable materials from a catechol-based polymer crosslinked with PDBA. P-PDBA 100% showed good stability and remarkable self-healing ability under seawater. Moreover, by comparing with a metal–catechol based polymer (P-Ca2+ 100%), it was proven that the boron-containing catechol polymer has superior self-healing abilities, stability, and transparency under seawater. Water stability and morphology studies revealed that the water affinity of the catechol-based polymers is affected not only by the hydrophobicity of polymer chain, but also by their mesoscopic morphologies which can be controlled by judiciously selecting crosslinkers. We envisage that the design concept of the water-assisted self-healable P-PDBA 100% with high water stability provides a valuable starting point for further development of self-healing polymeric materials used in diverse wet conditions.
Acknowledgements
This work was financially supported by JSPS KAKENHI Grant No. 15K17440.
Notes and references
- M. Doble, R. Venkatesan and N. V. R. Kumar, Polymers in a Marine Environment, Smithers, 2014 Search PubMed.
- P. Le Gac, P. Davies and D. Choqueuse, Oil Gas Sci. Technol., 2015, 70, 279–289 CrossRef.
- A. Akthakul, R. F. Salinaro and A. M. Mayes, Macromolecules, 2004, 37, 7663–7668 CrossRef CAS.
- R. T. Martin, L. P. Camargo and S. A. Miller, Green Chem., 2014, 16, 1768–1773 RSC.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467–472 CrossRef CAS PubMed.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS PubMed.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218–3226 Search PubMed.
- V. K. Thakur and M. R. Kessler, Polymer, 2015, 69, 369–383 CrossRef CAS.
- B. K. Ahn, D. W. Lee, J. N. Israelachvili and J. H. Waite, Nat. Mater., 2014, 13, 867–872 CrossRef CAS PubMed.
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Macromolecules, 2015, 48, 2098–2106 CrossRef CAS.
- J. Li, H. Ejima and N. Yoshie, ACS Appl. Mater. Interfaces, 2016, 8, 19047–19053 CAS.
- N. N. Xia, X. M. Xiong, J. Wang, M. Z. Rong and M. Q. Zhang, Chem. Sci., 2016, 7, 2736–2742 RSC.
- H. Kamata, K. Kushiro, M. Takai, U. Chung and T. Sakai, Angew. Chem., Int. Ed., 2016, 55, 9282–9286 CrossRef CAS PubMed.
- K.-L. G. Ho, A. L. Pometto and P. N. Hinz, J. Environ. Polym. Degrad., 1999, 7, 83–92 CrossRef CAS.
- J. H. Waite, Int. J. Adhes. Adhes., 1987, 7, 9–14 CrossRef CAS.
- H. Shao and R. J. Stewart, Adv. Mater., 2010, 22, 729–733 CrossRef CAS PubMed.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- M. Krogsgaard, M. A. Behrens, J. S. Pedersen and H. Birkedal, Biomacromolecules, 2013, 14, 297–301 CrossRef CAS PubMed.
- M. J. Harrington, A. Masic, N. Holten-Andersen, J. H. Waite and P. Fratzl, Science, 2010, 328, 216–220 CrossRef CAS PubMed.
- L. He, D. E. Fullenkamp, J. G. Rivera and P. B. Messersmith, Chem. Commun., 2011, 47, 7497–7499 RSC.
- R. Nishiyabu, Y. Kubo, T. D. James and J. S. Fossey, Chem. Commun., 2011, 47, 1124–1150 RSC.
- S. L. Diemer, M. Kristensen, B. Rasmussen, S. R. Beeren and M. Pittelkow, Int. J. Mol. Sci., 2015, 16, 21858–21872 CrossRef CAS PubMed.
- R. p. Singhal, B. Ramamurhy, N. Govindraj and Y. Sarwar, J. Chromatogr. A, 1991, 543, 17–38 CrossRef CAS.
- N. Luisier, R. Scopelliti and K. Severin, Soft Matter, 2016, 12, 588–593 RSC.
- I. A. Janković, Z. V. Šaponjić, E. S. Džunuzović and J. M. Nedeljković, Nanoscale Res. Lett., 2009, 5, 81–88 CrossRef PubMed.
- A. Eisenberg and M. Navratil, Macromolecules, 1974, 7, 90–94 CrossRef CAS.
- S. R. Lowry and K. A. Mauritz, J. Am. Chem. Soc., 1980, 102, 4665–4667 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic scheme of polymer (Scheme S1), 1H NMR spectra (Fig. S1), photographs of casted films with/without TEA (Fig. S2), DMA results (Fig. S3), laser scanning microscopy images (Fig. S4), the dimensions of the dumbbell shaped sample (Fig. S5), the composition of the artificial seawater (Table S1), self-healing results (Table S2), the results of swelling ratio (Table S3) and long-term stability results (Table S4). See DOI: 10.1039/c7ra01778b |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a